

Abstract
This multicenter, open‐label phase 1/2 study evaluated single‐agent carfilzomib in 50 heavily pretreated Japanese patients with relapsed/refractory multiple myeloma (median of five prior treatments). In phase 1, patients were dosed at three levels: 15, 20, or 20/27 mg/m2. Maximum tolerated dosage was not reached at the tolerability evaluation. Patients in phase 2 were treated with 20/27 mg/m2 carfilzomib. Median duration of exposure to carfilzomib in the 20/27 mg/m2 group at this final analysis was 4.7 months (range: 0.3‐39.4). Overall response rate in the 20/27 mg/m2 group, primary endpoint of the study, was 22.5% (n = 9) (95% confidence interval, 12.3‐37.5) with 2.5% (n = 1) stringent complete response. Median progression‐free survival and overall survival in the 20/27 mg/m2 group were 5.1 months (95% CI, 2.8‐13.6) and 22.9 months (95% CI, 14.1‐not estimable), respectively. Frequently occurring grade ≥3 adverse events in the 20/27 mg/m2 group included lymphopenia (72.5%), neutropenia (40.0%), and leukopenia (32.5%). Giving long‐term carfilzomib monotherapy led to long‐term overall survival for heavily pretreated multiple myeloma patients with a favorable safety profile. Carfilzomib monotherapy can be a good option for heavily pretreated multiple myeloma patients.
Keywords: carfilzomib, clinical trial, hematopoiesis, Japanese, multiple myeloma
Abbreviations
- AE
adverse events
- CBR
clinical benefit rate
- CI
confidence interval
- CR
complete response
- DLT
dose‐limiting toxicities
- DOR
duration of response
- G‐CSF
granulocyte colony‐stimulating factor
- MM
multiple myeloma
- MTD
maximum tolerated dose
- ORR
overall response rate
- OS
overall survival
- PFS
progression‐free survival
- PK
pharmacokinetics
- PN
peripheral neuropathy
- PR
partial response
- RRMM
relapsed/refractory MM
- sCR
stringent complete response
- VGPR
very good partial response
1. INTRODUCTION
Multiple myeloma is a disease characterized by the proliferation of differentiated plasma cells leading to hematopoietic tumors. The incidence of MM in the USA is 6.6 cases per 100 000 people, with a lifetime risk of developing MM of 0.8%.1 In Japan, the incidence is slightly lower, at 5.5 per 100 000, but this has increased dramatically since 1975 when the incidence per 100 000 people was 0.9.2 Several combination regimens including melphalan, prednisolone, and bortezomib base combination, lenalidomide base combination, and intensive chemotherapy with hematopoietic stem cell transplantation have been evaluated as first‐line treatments, and the outcome for MM patients has improved.3 Although these treatment options have improved OS markedly in MM patients,3 MM usually relapses and remains incurable in most cases.
Carfilzomib is a second‐generation epoxyketone protease inhibitor that irreversibly inhibits the chymotrypsin‐like activity of the proteasome.4, 5 In a phase 2 study (PX‐171‐003‐A1) of patients with RRMM, carfilzomib monotherapy of 20‐27 mg/m2 showed an ORR of 23.7%, a PFS of 3.7 months, and an OS of 15.6 months.6 Notably, carfilzomib expressed efficacy for bortezomib‐resistant patients and was not associated with PN. Consequently, the United States Food and Drug Administration approved carfilzomib monotherapy through the accelerated approval process based on this phase 2 study. Phase 3 studies such as ASPIRE7 (evaluating carfilzomib, lenalidomide, and dexamethasone) and ENDEAVOR8 (evaluating carfilzomib and dexamethasone) showed clinically meaningful efficacy and safety profiles for the treatment of RRMM. Based on these phase 3 results, carfilzomib was approved worldwide for the treatment of RRMM. Moreover, recent long‐term analyses of the ASPIRE9 and ENDEAVOR10 studies expressed significant and clinically meaningful OS improvement versus standard therapies. To our knowledge, carfilzomib is the first and only MM treatment that extends OS in the relapsed setting over the current standard of care. Since 2010, carfilzomib has been studied in Japan as monotherapy, as two‐drug combination with dexamethasone, and as three‐drug combination with lenalidomide and dexamethasone, and these studies yielded promising results for Japanese RRMM patients.11, 12, 13 In an interim analysis of this study,13 we reported the short follow‐up period data of single‐agent carfilzomib in Japanese patients with RRMM. Here, we present the final results of the study with long‐term follow‐up data.
2. MATERIALS AND METHODS
2.1. Ethics
The study was conducted in accordance with the principles of the Declaration of Helsinki, Article 14 and Article 80‐2 of the Pharmaceutical Affairs Law, and Good Clinical Practice. The study protocol was approved by the institutional review board of each institution. All patients provided written, informed consent to participate.
2.2. Overall study design
The study design has been previously reported in detail.13 Briefly, this was a 15‐center, open‐label phase 1/2 study (ONO‐7057‐01) of single‐agent carfilzomib in Japanese patients with RRMM. In phase 1, safety, tolerability, efficacy, PK, and pharmacodynamics were examined for carfilzomib given i.v. at doses of 15, 20, and 20/27 mg/m2. The PK data have been previously reported.13 During phase 2, the efficacy and safety of carfilzomib monotherapy were evaluated at the recommended dosage determined in phase 1. The primary endpoint of this study was ORR.
2.3. Study patients
Full inclusion and exclusion criteria have previously been published.13 Patients were inpatients aged ≥20 years with symptomatic RRMM and a life expectancy of ≥3 months. Key exclusion criteria were Waldenstrom's macroglobulinemia or IgM myeloma, POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes), plasma cell leukemia, and chemotherapy within 21 days prior to first dose of the study drug. Concomitant use of the following agents was prohibited: anticancer agents with demonstrated efficacy against MM, irradiation of bone marrow, and other investigational products throughout the study; and hematopoietic growth factor, bisphosphonates, and denosumab during cycle 1 in phase 1.
2.4. Treatments
In the phase 1 dose escalation part, carfilzomib was given i.v. over 10 minutes at dosages of 15, 20, and 20/27 mg/m2/d on days 1, 2, 8, 9, 15, and 16 of each 28‐day cycle until the withdrawal of consent, disease progression, or the occurrence of unacceptable toxicity. For the 20/27 mg/m2 dosage, 20 mg/m2 was given on days 1 and 2 of cycle 1 and escalated to 27 mg/m2 on day 8 of cycle 1 and thereafter. Oral or i.v. dexamethasone (4 mg) and oral or i.v. hydration were given to prevent infusion reactions. Antibacterial and/or antiviral drugs were given as necessary. DLT were defined as specific types of AE occurring within 28 days after the start of administration in phase 1 and for which a drug relationship could not be ruled out. Non‐hematologic AE included grade ≥2 neuropathy with pain; grade ≥3 non‐hematological toxicity (excluding nausea; vomiting; diarrhea; hyperphosphatemia associated with new bone formation; tumor lysis syndrome; and transient increase in lactic acid dehydrogenase, electrolyte imbalance, hyperuricemia, and renal impairment secondary to tumor lysis syndrome); and grade ≥3 nausea, vomiting, or diarrhea that could not be controlled by giving adequate and appropriate antiemetics or antidiarrheal drugs. The hematological AE predefined as DLT (including AE pertaining to hematocyte abnormalities and changes such as leukopenia and decreased lymphocyte count) included grade 4 neutropenia (neutrophil count <500/mm3) persisting for ≥8 days that occurred in the absence of supportive therapy with G‐CSF product (G‐CSF products were prohibited during DLT evaluation), febrile neutropenia (neutrophil count <1000/mm3 and fever >38.3°C), and grade 4 thrombocytopenia requiring platelet transfusion or accompanied by bleeding.
When three patients enrolled in the same cohort completed evaluation for the 28‐day period (1 cycle) and no study drug‐related DLT occurred, the next dose‐level cohort was initiated. If a study drug‐related DLT occurred in one of three patients, three more patients were to be enrolled in the same cohort. If a study drug‐related DLT occurred in two or more of the three patients (or three or more of the six patients after patients have been added), enrollment in the same cohort was suspended, and the dose was regarded as the maximum dose. If the dose in cohort 2 or subsequent cohorts was the maximum dose, the dose from the previous cohort was defined as the MTD. The MTD was used in phase 2. In cases where the dose could be increased to cohort 3 and the number of patients with DLT in cohort 3 was ≤2 of six patients, the dose for cohort 3 was planned to be the dose for phase 2. The final decision on the dose for phase 2 was made through deliberation by the sponsor and coordinating investigator. The opinion of the Efficacy and Safety Monitoring Committee was sought as necessary.
2.5. Efficacy assessment
Efficacy was analyzed in the full analysis set, comprising patients who received at least one dose of the study drug and underwent at least one efficacy or pharmacodynamics evaluation after receiving the study drug. The primary endpoint of the study was the ORR (defined as sCR + CR + VGPR + PR). Response was evaluated based on central laboratory data according to the International Myeloma Working Group uniform response criteria.14 The minimal response was evaluated using the European Group for Blood and Marrow Transplantation criteria.15 Other endpoints were PFS, OS, and DOR. Subgroup analysis of PFS was carried out to identify factors that influence efficacy.
2.6. Safety assessment
Safety was evaluated in the safety analysis set, comprising all patients who had received at least one dose of the study drug. AE were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0.
2.7. Statistical analyses
Statistical analysis was carried out by Ono Pharmaceutical Co., Ltd. The ORR, primary endpoint for phase 2, was calculated for evaluable patients whose best responses were classified as sCR, CR, VGPR, or PR and the corresponding two‐sided 95% CI was calculated using the Wilson method. Median OS and PFS were calculated using the Kaplan‐Meier method. No significance level was specified, and no statistical tests were carried out for safety and efficacy measures. Data were analyzed using SAS version 9.3 or later (SAS Institute Inc.).
3. RESULTS
3.1. Baseline demographic and clinical characteristics
In the present study, the patients’ last visit was on May 12, 2017, and the median follow‐up period of this final analysis was 21.7 months. The data cutoff date of the interim analysis was June 11, 2014, and the median follow‐up period was 6.3 months. The baseline demographic and clinical characteristics were previously reported.13 Briefly, 50 RRMM patients who had received a median of five (range: 3‐10) previous treatments were enrolled in this study between August 2011 and January 2014. Fifteen (30%) patients had high‐risk cytogenetics, that is, were positive for del(17p) in ≥20% of screened plasma cells, t(4;14), t(14;16), or hypodiploidy.
3.2. Dose escalation
Description of the dose escalation was previously reported.13 Seventeen patients were enrolled in phase 1. Although one out of six patients in the 20 mg/m2 cohort experienced a DLT (thrombotic microangiopathy, cardiomyopathy, hepatic disorder, and sensorimotor disorder), no further DLT were observed in the four patients in the 15 mg/m2 cohort or the seven patients in the 20/27 mg/m2 cohort. Thus, 20/27 mg/m2 was chosen as the dosage for phase 2.
3.3. Carfilzomib exposure in the 20/27 mg/m2 group
Forty of the 50 patients enrolled in this study (seven patients enrolled for phase 1 who received the 20/27 mg/m2 dosage and 33 additional patients enrolled for phase 2) received a carfilzomib dosage of 20/27 mg/m2. Median number of treatment cycles was 6.0 (range: 1‐38), and median duration of exposure was 4.7 months (range: 0.3‐39.4). The longest treatment duration was >3 years.
3.4. Overall response rate and clinical benefit rate
The efficacy analysis set comprised all 50 patients enrolled in this study. ORR in the 20/27 mg/m2 group was 22.5% (9/40) (95% CI, 12.3‐37.5) (Table 1). One patient (2.5%) achieved sCR and two patients (5.0%) achieved VGPR. CBR was 32.5% (n = 13) (95% CI, 20.1‐48.0).
Table 1.
Treatment response to carfilzomib monotherapy in Japanese patients with relapsed or refractory multiple myeloma
| Carfilzomib | ||||||||
|---|---|---|---|---|---|---|---|---|
| 15 mg/m2 (n = 4) | 20 mg/m2 (n = 6) | 20/27 mg/m2 (n = 40) | Total (N = 50) | |||||
| No. of patients (%) | 95% CI | No. of patients (%) | 95% CI | No. of patients (%) | 95% CI | No. of patients (%) | 95% CI | |
| ORR (sCR + CR + VGPR + PR) | 1 (25.0) | 4.6, 69.9 | 0 (0.0) | 0.0, 39.0 | 9 (22.5) | 12.3, 37.5 | 10 (20.0) | 11.2, 33.0 |
| sCR | 0 | 0.0, 49.0 | 0 | 0.0, 39.0 | 1 (2.5) | 0.4, 12.9 | 1 (2.0) | 0.4, 10.5 |
| CR | 0 | 0.0, 49.0 | 0 | 0.0, 39.0 | 0 | 0.0, 8.8 | 0 | 0.0, 7.1 |
| VGPR | 0 | 0.0, 49.0 | 0 | 0.0, 39.0 | 2 (5.0) | 1.4, 16.5 | 2 (4.0) | 1.1, 13.5 |
| PR | 1 (25.0) | 4.6, 69.9 | 0 | 0.0, 39.0 | 6 (15.0) | 7.1, 29.1 | 7 (14.0) | 7.0, 26.2 |
| SD | 1 (25.0) | 4.6, 69.9 | 3 (50.0) | 18.8, 81.2 | 18 (45.0) | 30.7, 60.2 | 22 (44.0) | 31.2, 57.7 |
| PD | 0 | 0.0, 49.0 | 1 (16.7) | 3.0, 56.4 | 9 (22.5) | 12.3, 37.5 | 10 (20.0) | 11.2, 33.0 |
| NE | 2 (50.0) | 15.0, 85.0 | 2 (33.3) | 9.7, 70.0 | 4 (10.0) | 4.0, 23.1 | 8 (16.0) | 8.3, 28.5 |
| CBR (sCR + CR + VGPR + PR + MR) | 1 (25.0) | 4.6, 69.9 | 2 (33.3) | 9.7, 70.0 | 13 (32.5) | 20.1, 48.0 | 16 (32.0) | 20.8, 45.8 |
CBR, clinical benefit rate; CI, confidence interval; CR, complete response; MR, minimal response; NE, not evaluable; ORR, overall response rate; PD, progressive disease; PR, partial response; sCR, stringent complete response; SD, stable disease; VGPR, very good partial response.
3.5. Progression‐free survival, overall survival, and duration of response
Median PFS and the OS in the 20/27 mg/m2 group were 5.1 months (95% CI, 2.8‐13.6) and 22.9 months (95% CI, 14.1‐not estimable), respectively, at the final analysis. Kaplan‐Meier estimates of PFS are shown in Figure 1 and OS in Figure 2. Median DOR was 16.3 months (95% CI, 2.3‐not estimable). Subgroup analysis of PFS in the 20/27 mg/m2 group is shown in Table 2.
Figure 1.
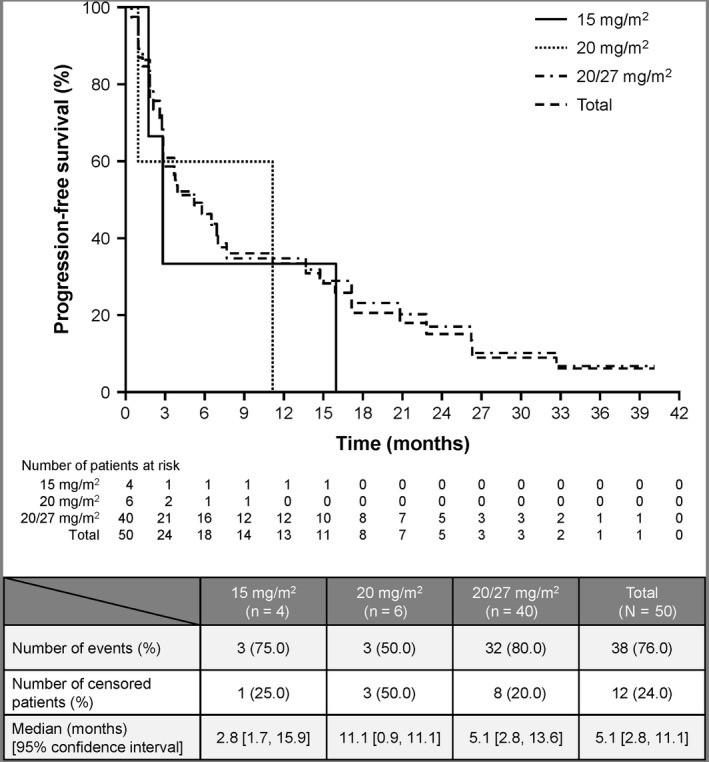
Progression‐free survival of patients treated with carfilzomib monotherapy
Figure 2.
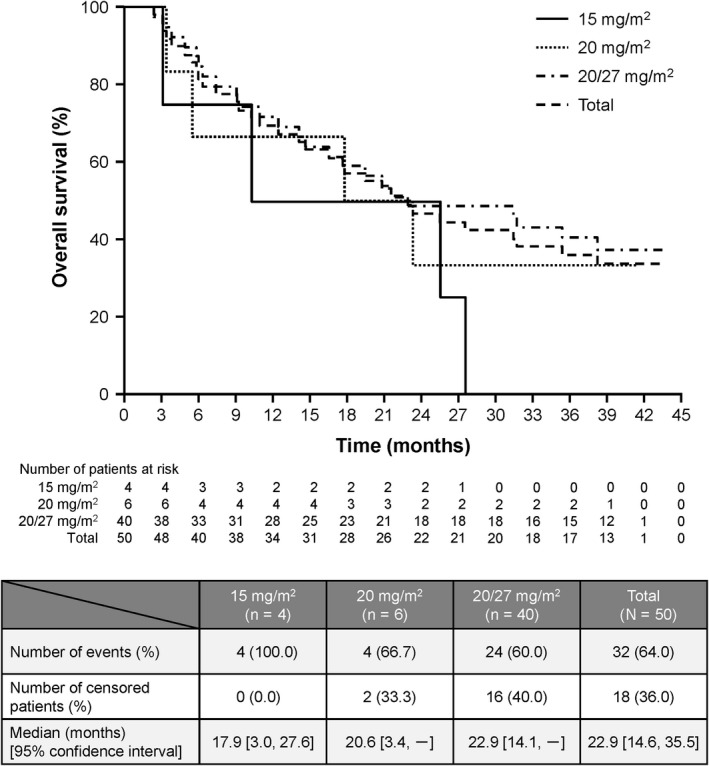
Overall survival of patients treated with carfilzomib monotherapy
Table 2.
Subgroup analysis of progression‐free survival in the 20/27 mg/m2 group
| Carfilzomib, 20/27 mg/m2 | |||
|---|---|---|---|
| Characteristic | Category | Event/total (%) | Median [95% CI] (mo) |
| Overall | 32/40 (80.0) | 5.1 [2.8, 13.6] | |
| Sex | Male | 15/18 (83.3) | 7.0 [0.9, 17.1] |
| Female | 17/22 (77.3) | 3.8 [2.8, 7.7] | |
| Age (y) 1 | <65 | 10/12 (83.3) | 6.9 [0.9, 17.1] |
| ≥65 | 22/28 (78.6) | 5.1 [2.6, 14.7] | |
| Age (y) 2 | <75 | 27/34 (79.4) | 5.1 [2.8, 13.6] |
| ≥75 | 5/6 (83.3) | 6.5 [1.3, 20.8] | |
| ECOG performance status | 0 | 16/23 (69.6) | 5.8 [2.1, 22.8] |
| 1 | 16/17 (94.1) | 4.5 [2.6, 13.6] | |
| Stage (Durie‐Salmon) | I | 7/7 (100.0) | 5.1 [0.9, 26.3] |
| II | 9/10 (90.0) | 2.8 [0.5, 22.8] | |
| III | 14/18 (77.8) | 3.7 [2.1, 13.6] | |
| Stage (ISS) | 1 | 10/12 (83.3) | 3.9 [1.8, 22.8] |
| 2 | 10/14 (71.4) | 5.1 [2.1, 13.6] | |
| 3 | 7/9 (77.8) | 6.9 [0.9, 26.2] | |
| Stage (R‐ISS) | 1 | 2/3 (66.7) | 7.0 [0.9, 22.8] |
| 2 | 10/23 (43.5) | 3.9 [2.8, 13.6] | |
| 3 | 0/0 | Not evaluable | |
| No. of prior treatments | <5 | 14/16 (87.5) | 2.9 [1.3, 7.0] |
| ≥5 | 18/24 (75.0) | 6.9 [2.8, 22.8] | |
| No. of prior bortezomib treatments | 1 | 15/20 (75.0) | 3.8 [2.6, 7.7] |
| ≥2 | 17/20 (85.0) | 5.8 [1.8, 20.8] | |
| Bortezomib at most recent treatment | Yes | 13/13 (100.0) | 3.9 [1.8, 7.7] |
| No | 19/27 (70.4) | 6.9 [2.8, 17.1] | |
| Prior lenalidomide treatment | Yes | 26/33 (78.8) | 5.8 [2.8, 14.7] |
| No | 6/7 (85.7) | 2.9 [1.3, 20.8] | |
| Prior thalidomide treatment | Yes | 19/23 (82.6) | 3.9 [2.1, 13.6] |
| No | 13/17 (76.5) | 6.5 [1.8, 17.1] | |
| Prior lenalidomide and thalidomide treatments | Yes | 13/16 (81.3) | 5.8 [1.8, 17.1] |
| No | 19/24 (79.2) | 5.1 [2.6, 14.7] | |
| Prior stem cell transplantation | Yes | 16/17 (94.1) | 3.4 [1.8, 13.6] |
| No | 16/23 (69.6) | 5.8 [2.8, 17.1] | |
| High‐risk cytogeneticsa | Yes | 8/11 (72.7) | 2.9 [0.5, 17.1] |
| No | 21/26 (80.8) | 5.1 [2.1, 17.1] | |
CI, confidence interval; ECOG, European Cooperative Oncology Group; ISS, International Staging System; R‐ISS, revised ISS stage.
High‐risk cytogenetics were defined as positive for del(17p) in ≥20% of screened plasma cells, t(4;14), t(14;16), or hypodiploidy.
3.6. Safety
Fifty patients who received at least one dose of carfilzomib were included in the safety analysis set. All 50 enrolled patients experienced at least one AE, and 90.0% had at least one AE of grade ≥3. Of the 40 patients who received 20/27 mg/m2, 38 (95.0%) experienced at least one AE of grade ≥3. AE of any grade occurring in ≥30% of patients in the 20/27 mg/m2 group were lymphopenia (85.0%), thrombocytopenia (75.0%), neutropenia (57.5%), leukopenia (55.0%), increased serum creatinine (45.0%), decreased hemoglobin (45.0%), hypophosphatemia (40.0%), increased serum lactate dehydrogenase (32.5%), increased white blood cell count (32.5%), pyrexia (30.0%), nasopharyngitis (30.0%), and anemia (30.0%). PN‐related events occurred in nine (18.0%) patients, of whom eight were in the 20/27 mg/m2 cohort. Additionally, weight gain was observed in three of 40 patients (7.5%), edema was observed in one of 40 patients (2.5%), and peripheral edema was observed in two of 40 patients (5.0%) in the carfilzomib 20/27 mg/m2 group. Peripheral edema also occurred in one (25.0%) patient in the 15 mg group. No grade ≥3 weight gain, edema, or peripheral edema occurred.
Adverse events of any grade and grade ≥3 occurring in at least 20% of patients are shown in Table 3. There were no deaths resulting from AE within 30 days of the last dose.
Table 3.
Adverse events of any grade and grade ≥3 adverse events with an incidence of ≥20%
| Carfilzomib | ||||||||
|---|---|---|---|---|---|---|---|---|
| 15 mg/m2 | 20 mg/m2 | 20/27 mg/m2 | Total | |||||
| No. of patients | 4 | 6 | 40 | 50 | ||||
| All grades | Grade ≥3 | All grades | Grade ≥3 | All grades | Grade ≥3 | All grades | Grade ≥3 | |
| Hematological | ||||||||
| Lymphopeniaa | 3 (75.0) | 1 (25.0) | 6 (100.0) | 5 (83.3) | 34 (85.0) | 29 (72.5) | 43 (86.0) | 35 (70.0) |
| Thrombocytopenia | 0 | 0 | 4 (66.7) | 1 (16.7) | 30 (75.0) | 12 (30.0) | 34 (68.0) | 13 (26.0) |
| Neutropenia | 3 (75.0) | 1 (25.0) | 2 (33.3) | 2 (33.3) | 23 (57.5) | 16 (40.0) | 28 (56.0) | 19 (38.0) |
| Leukopenia | 2 (50.0) | 0 | 2 (33.3) | 1 (16.7) | 22 (55.0) | 13 (32.5) | 26 (52.0) | 14 (28.0) |
| Decreased hemoglobin | 1 (25.0) | 0 | 2 (33.3) | 1 (16.7) | 18 (45.0) | 11 (27.5) | 21 (42.0) | 12 (24.0) |
| Increased white blood cell count | 1 (25.0) | 0 | 3 (50.0) | 0 | 13 (32.5) | 0 | 17 (34.0) | 0 |
| Anemia | 0 | 0 | 0 | 0 | 12 (30.0) | 6 (15.0) | 12 (24.0) | 6 (12.0) |
| Increased neutrophil count | 0 | 0 | 2 (33.3) | 0 | 9 (22.5) | 0 | 11 (22.0) | 0 |
| Non‐hematological | ||||||||
| Increased serum creatinine | 2 (50.0) | 0 | 2 (33.3) | 1 (16.7) | 18 (45.0) | 2 (5.0) | 22 (44.0) | 3 (6.0) |
| Increased serum lactate dehydrogenase | 0 | 0 | 4 (66.7) | 1 (16.7) | 13 (32.5) | 0 | 17 (34.0) | 1 (2.0) |
| Hypophosphatemia | 0 | 0 | 1 (16.7) | 0 | 16 (40.0) | 3 (7.5) | 17 (34.0) | 3 (6.0) |
| Hyperglycemia | 2 (50.0) | 0 | 4 (66.7) | 2 (33.3) | 10 (25.0) | 1 (2.5) | 16 (32.0) | 3 (6.0) |
| Increased aspartate aminotransferase | 0 | 0 | 4 (66.7) | 2 (33.3) | 10 (25.0) | 3 (7.5) | 14 (28.0) | 5 (10.0) |
| Hypertensionb | 2 (50.0) | 0 | 2 (33.3) | 0 | 10 (25.0) | 5 (12.5) | 14 (28.0) | 5 (10.0) |
| Pyrexia | 0 | 0 | 2 (33.3) | 0 | 12 (30.0) | 2 (5.0) | 14 (28.0) | 2 (4.0) |
| Increased alanine aminotransferase | 0 | 0 | 3 (50.0) | 1 (16.7) | 10 (25.0) | 2 (5.0) | 13 (26.0) | 3 (6.0) |
| Nasopharyngitis | 0 | 0 | 1 (16.7) | 0 | 12 (30.0) | 0 | 13 (26.0) | 0 |
| Increased blood urea | 0 | 0 | 3 (50.0) | 0 | 9 (22.5) | 0 | 12 (24.0) | 0 |
| Malaise | 2 (50.0) | 0 | 2 (33.3) | 0 | 8 (20.0) | 0 | 12 (24.0) | 0 |
| Peripheral neuropathyc | 0 | 0 | 1 (16.7) | 0 | 8 (20.0) | 0 | 9 (18.0) | 0 |
Data are n (%).
Lymphopenia includes lymphopenia and decreased lymphocyte counts.
Hypertension includes hypertension and increased blood pressure.
Peripheral neuropathy includes peripheral neuropathy, peripheral sensory neuropathy, and trigeminal nerve disorder.
Adverse events resulting in treatment withdrawal occurred in seven (17.5%) patients in the 20/27 mg/m2 group. The events were (all occurring in one patient [2.5%] each) as follows: infection, ejection fraction decreased, hypercalcemia, muscular weakness, plasma cell myeloma, spinal cord compression, and hypertension. AE resulting in dose reduction or treatment interruption occurred in 25 (62.5%) patients. The most common events were neutropenia (17.5%), nasopharyngitis (12.5%), pyrexia (10.0%), upper respiratory tract inflammation (7.5%), increased aspartate aminotransferase (7.5%), increased serum creatinine (5.0%), increased alanine aminotransferase (5.0%), anemia (5.0%), influenza (5.0%), upper respiratory tract infection (5.0%), and hypophosphatemia (5.0%).
4. DISCUSSION
Results of the present study show the long‐term benefit of carfilzomib 20/27 mg/m2 monotherapy in patients with RRMM. ORR (the primary endpoint for phase 2) in the carfilzomib 20/27 mg/m2 group was 22.5% (95% CI, 12.3‐37.5) at the final analysis. This ORR was the same result as in the interim analysis of this study.13 One patient (2.5%) achieved sCR, four (10.0%) achieved a minimal response. The CBR increased from 27.5% at the interim analysis to 32.5% after long‐term exposure to carfilzomib 20/27 mg/m2. The median PFS with carfilzomib 20/27 mg/m2 was 5.1 months (95% CI, 2.8‐13.6), and the median OS was 22.9 months (95% CI, 14.1‐not estimable). According to the subgroup analysis of PFS, prior bortezomib exposure seemed to influence the PFS results. This trend was similar to that reported in the PX‐171‐003‐A1 study.6 Although carfilzomib is an active drug for patients who are refractory to previous bortezomib, the exposure to bortezomib may slightly impact the response.
In 13 patients whose most recent treatment included bortezomib, the median PFS was 3.9 months. A favorable efficacy profile of carfilzomib monotherapy was observed.
Importantly, no new safety concerns were raised during the long‐term follow up compared with the interim analysis.13 None of the grade ≥3 AE increased by 5% or more compared with those reported in the interim analysis. Although PN‐related events occurred in eight patients (20%) in the 20/27 mg/m2 group in the final analysis, no patients developed grade 3 or higher PN. Hypertension or increased blood pressure occurred in 14 patients. In most of the cases, blood pressure normalized after discontinuation of carfilzomib treatment and most events were managed with/without pharmacotherapy.
Carfilzomib monotherapy has previously been evaluated in MM patients in a phase 2 study (PX‐171‐003‐A1)6 and in a randomized phase 3 study (FOCUS).16 In the PX‐171‐003‐A1 study,6 ORR was 23.7%, median PFS was 3.7 months, and median OS was 15.6 months. In the FOCUS study, ORR was 19.1%, median PFS was 3.7 months, and median OS was 10.2 months. The current efficacy results are consistent with these previous study data. Median duration of treatment was 3.0 (range, 0.03‐16.9) months in the PX‐171‐003‐A1 study6 and 3.8 (range, 0.07‐31.9) months in the FOCUS study.16 In the present study, treatment with carfilzomib was more prolonged (4.7 months [range, 0.3‐39.4]) than in these previous studies.6, 16 We presume that the investigators’ adequate management of the emerging AE during carfilzomib treatment contributed to the long‐term administration of carfilzomib, which, in turn, led to good efficacy. Regarding carfilzomib safety, the incidences of most of the AEs were similar to those reported in previous studies; however, the incidences of laboratory abnormalities, such as abnormal blood cell count and increased serum creatinine, seemed higher than in previous studies.6, 16 We have noticed that in Japanese studies, laboratory abnormalities tend to be reported more frequently than in studies conducted elsewhere; we presume this may be because Japanese physicians tend to report laboratory abnormalities very carefully. In particular, on the day after giving carfilzomib, creatinine levels tended to transiently increase, but then returned to baseline levels by the time of the next dose. Therefore, during carfilzomib monotherapy (without dexamethasone), administration of the drug may be related to an acute and transient increase in creatinine. Moreover, acute kidney injury was reported as the most common adverse event leading to carfilzomib discontinuation (1.7%) when carfilzomib 20/27 mg/m2 was given with dexamethasone in the phase 3 A.R.R.O.W. study.17
Additionally, in the present study, weight gain, edema, and peripheral edema were observed in 7.5%, 2.5%, and 5% of patients, respectively, in the carfilzomib 20/27 mg/m2 group. Peripheral edema also occurred in 25.0% of patients in the 15 mg group. No grade ≥3 weight gain, edema, or peripheral edema occurred. As with the increase in creatinine, we consider that weight gain and edema were mainly attributable to acute dose effects.
A previous phase 3 study of pomalidomide and low‐dose dexamethasone included a similar population (median of five prior treatments) to the current study. In that study, ORR was 31%, median PFS was 4.0 months, and median OS was 12.7 months,18 which are similar to the results of the present study.
Although the results of the present study need to be interpreted in light of the relatively small number of patients, the inclusion of only Japanese patients, and the lack of a comparator group, this study confirmed that long‐term (>3 years) monotherapy with carfilzomib (20/27 mg/m2) is feasible in Japanese patients with heavily pretreated RRMM. Thus, we consider that carfilzomib monotherapy could be an effective maintenance therapy for RRMM, even for patients who have previously received intensive therapy with the triplet regimen (carfilzomib 20/27 mg/m2, lenalidomide, and dexamethasone)9 or the high‐dose regimen (carfilzomib 20/56 mg/m2 and dexamethasone).10 Further studies are required to address the above limitations.
DISCLOSURE
Dr Iida has received honoraria and research funds from Ono Pharmaceutical, Janssen Pharmaceutical, Celgene, Bristol‐Myers Squibb, Takeda Pharmaceutical, and Novartis, and research funds from Chugai Pharmaceutical, Kyowa Hakko Kirin, Sanofi, MSD, Daiichi Sankyo, Gilead, and AbbVie. Dr Watanabe has no conflicts of interest. Dr Matsumoto has received honoraria and research funds from Ono Pharmaceutical and honoraria from Celgene, Janssen Pharmaceutical, Takeda Pharmaceutical, and Bristol‐Myers Squibb. Dr Suzuki has received research funds from Ono Pharmaceutical. Dr Sunami has received honoraria and research funds from Ono Pharmaceutical, Bristol‐Myers Squibb, Takeda Pharmaceutical, and Celgene, and research funds from Novartis, GlaxoSmithKline, Janssen Pharmaceutical, AbbVie, Sanofi, MSD, Alexion Pharma, and Daiichi Sankyo. Dr Ishida has received honoraria and research funds from Ono Pharmaceutical, and honoraria from Takeda Pharmaceutical, Celgene, Bristol‐Myers Squibb, Janssen Pharmaceutical, Novartis, Nippon Shinyaku, Chugai Pharmaceutical, and Daiichi Sankyo. Dr Ando has received research funds from Ono Pharmaceutical. Dr Chou has received honoraria and research funds from Ono Pharmaceutical, and honoraria from Takeda Pharmaceutical, Janssen Pharmaceutical, and Bristol‐Myers Squibb. Dr Ozaki has received research funds from Ono Pharmaceutical. Dr Taniwaki has received honoraria and research funds from Bristol‐Myers Squibb, and research funds from Ono Pharmaceutical, Chugai Pharmaceutical with Roche, Celgene, Kyowa Hakko Kirin, Janssen Pharmaceutical, and Astellas Pharma. Dr Uike has received research funds from Ono Pharmaceutical. Dr Shibayama has received honoraria and research funds from Ono Pharmaceutical, and honoraria from Takeda Pharmaceutical, Janssen Pharmaceutical, Celgene, and Novartis. Dr Hatake has received research funds from Ono Pharmaceutical, and honoraria from Eisai, Kyowa Hakko Kirin, Takeda Pharmaceutical, Celgene, Daiichi Sankyo, Meiji Seika, Otsuka, and Towa. Dr Izutsu has received honoraria and research funds from Ono Pharmaceutical, Takeda Pharmaceutical, Gilead, Janssen Pharmaceutical, MSD, Bayer, Daiichi Sankyo, Novartis, Chugai, and Celgene, and honoraria from Kyowa Hakko Kirin, and research funds from Zenyaku Kogyo, Mundipharma, AbbVie, Solasia Pharma, Celtrion, Symbio, Astellas Pharma, Astellas Amgen BioPharma, HUYA Bioscience International, and Sanofi. Dr Ishikawa has received research funds from Ono Pharmaceutical. Mr Shumiya is an employee of Ono Pharmaceutical. Dr Tobinai has received consultant fees from Zenyaku Kogyo, HUYA Bioscience International and Celgene, and honoraria and research funds from Ono Pharmaceutical, Eisai, Takeda Pharmaceutical, Mundipharma, Janssen Pharmaceutical, Kyowa Hakko Kirin, Celgene, and Chugai Pharmaceutical, and honoraria from Zenyaku Kogyo and HUYA Bioscience International, and research funds from GlaxoSmithKline, Servier, and AbbVie. The study was designed under the responsibility of Ono Pharmaceutical in conjunction with the steering committee; the study was funded by Ono Pharmaceutical; carfilzomib was provided by Ono Pharmaceutical; Ono Pharmaceutical collected and analyzed the data and contributed to the interpretation of the study. All authors had full access to all of the data in the study and had final responsibility for the decision to submit for publication.
ACKNOWLEDGMENTS
The authors wish to thank all patients who participated in this study and their families as well as all investigators, physicians, nurses, and clinical research coordinators who helped with this study. The authors are also thankful to Dr Hirokazu Murakami (Gunma University Graduate School of Health Science, Maebashi), who was the medical consultant, as well as Dr Yutaka Ariyoshi (Aichi Cancer Centre Aichi Hospital, Okazaki), Dr Chihiro Shimazaki (Japan Community Healthcare Organization Kyoto Kuramaguchi Medical Center, Kyoto), Dr Masahiro Kizaki (Saitama Medical Center, Saitama Medical University, Kawagoe), Dr Takao Katoh (International University of Health and Welfare, Mita Hospital, Tokyo), Dr Masashi Takahashi (Yujin‐Yamazaki Hospital, Hikone), and Dr Terufumi Kato (Kanagawa Cancer Center, Yokohama) for their strict review of the clinical data as members of the Efficacy and Safety Evaluation Committee. We also acknowledge the statistical support of Naokazu Gion and Toshiaki Ozaki (Ono Pharmaceutical, Osaka) and critical review of the manuscript by Amgen (Thousand Oaks). The authors wish to thank Marion Barnett of Edanz Medical Writing for providing medical writing assistance, which was funded by Ono Pharmaceutical through EMC K.K. in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Iida S, Watanabe T, Matsumoto M, et al. Carfilzomib monotherapy in Japanese patients with relapsed or refractory multiple myeloma: A phase 1/2 study. Cancer Sci. 2019;110:2924‐2932. 10.1111/cas.14139
Clinical trial registry: JapicCTI‐111570
REFERENCES
- 1. Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2014. Bethesda, MD: National Cancer Institute; https://seer.cancer.gov/csr/1975_2014/, based on November 2016 SEER data submission, posted to the SEER web site, April 2017. Accessed February 20, 2019. [Google Scholar]
- 2. Hori M, Matsuda T, Shibata A, et al. Cancer incidence and incidence rates in Japan in 2009: a study of 32 population‐based cancer registries for the Monitoring of Cancer Incidence in Japan (MCIJ) project. Jpn J Clin Oncol. 2015;45:884‐891. [DOI] [PubMed] [Google Scholar]
- 3. Kumar SK, Mikhael JR, Buadi FK, et al. Management of newly diagnosed symptomatic multiple myeloma: updated Mayo Stratification of Myeloma and Risk‐Adapted Therapy (mSMART) consensus guidelines. Mayo Clin Proc. 2009;84:1095‐1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Demo SD, Kirk CJ, Aujay MA, et al. Antitumor activity of PR‐171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007;67:6383‐6391. [DOI] [PubMed] [Google Scholar]
- 5. Parlati F, Lee SJ, Aujay M, et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin‐like activity of the proteasome. Blood. 2009;114:3439‐3447. [DOI] [PubMed] [Google Scholar]
- 6. Siegel DS, Martin T, Wang M, et al. A phase 2 study of single‐agent carfilzomib (PX‐171‐003‐A1) in patients with relapsed and refractory multiple myeloma. Blood. 2012;120:2817‐2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142‐152. [DOI] [PubMed] [Google Scholar]
- 8. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomized, phase 3, open‐label, multicentre study. Lancet Oncol. 2016;17:27‐38. [DOI] [PubMed] [Google Scholar]
- 9. Siegel DS, Dimopoulos MA, Ludwig H, et al. Improvement in overall survival with carfilzomib, lenalidomide, and dexamethasone in patients with relapsed or refractory multiple myeloma. J Clin Oncol. 2018;36:728‐734. [DOI] [PubMed] [Google Scholar]
- 10. Dimopoulos MA, Goldschmidt H, Niesvizky R, et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an open‐label, randomised, phase 3 trial. Lancet Oncol. 2017;18:1327‐1337. [DOI] [PubMed] [Google Scholar]
- 11. Iida S, Tobinai K, Taniwaki M, et al. Phase I dose escalation study of high dose carfilzomib monotherapy for Japanese patients with relapsed or refractory multiple myeloma. Int J Hematol. 2016;104:596‐604. [DOI] [PubMed] [Google Scholar]
- 12. Suzuki K, Ri M, Chou T, et al. Carfilzomib, lenalidomide and dexamethasone in patients with heavily pretreated multiple myeloma: a phase 1 study in Japan. Cancer Sci. 2017;108:461‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Watanabe T, Tobinai K, Matsumoto M, et al. A phase 1/2 study of carfilzomib in Japanese patients with relapsed and/or refractory multiple myeloma. Br J Haematol. 2016;172:745‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Durie BG, Harousseau JL, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20:1467‐1473. [DOI] [PubMed] [Google Scholar]
- 15. Bladé J, Samson D, Reece D, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high‐dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998;102:1115‐1123. [DOI] [PubMed] [Google Scholar]
- 16. Hájek R, Masszi T, Petrucci MT, et al. A randomized phase III study of carfilzomib vs low‐dose corticosteroids with optional cyclophosphamide in relapsed and refractory multiple myeloma (FOCUS). Leukemia. 2017;31:107‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moreau P, Mateos MV, Berenson JR, et al. Once weekly versus twice weekly carfilzomib dosing in patients with relapsed and refractory multiple myeloma (A.R.R.O.W.): interim analysis results of a randomized, phase 3 study. Lancet Oncol. 2018;19:953‐964. [DOI] [PubMed] [Google Scholar]
- 18. San Miguel J, Weisel K, Moreau P, et al. Pomalidomide plus low‐dose dexamethasone versus high‐dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM‐003): a randomised, open‐label, phase 3 trial. Lancet Oncol. 2013;14:1055‐1066. [DOI] [PubMed] [Google Scholar]