

Abstract
Characterization of circulating tumor cells (CTC) is important to prevent death caused by the metastatic spread of cancer cells because CTC are associated with distal metastasis and poor prognosis of breast cancer. We have previously developed suspension cells (SC) using breast cancer cell lines and demonstrated their high metastatic potential. As survival of CTC is highly variable from a few hours to decades, herein we cultured SC for an extended time and named them adapted suspension cells (ASC). Silent mating‐type information regulation 2 homolog 1 (SIRT1) expression increased in ASC, which protected the cells from apoptosis. High SIRT1 expression was responsible for the suppression of nuclear factor kappa B (NF‐κB) activity and downregulation of reactive oxygen species (ROS) in ASC. As the inhibition of NF‐κB and ROS production in SIRT1‐depleted ASC contributed to the development of resistance to apoptotic cell death, maintenance of a low ROS level and NF‐κB activity in ASC is a crucial function of SIRT1. Thus, SIRT1 overexpression may play an important role in growth adaptation of SC because SIRT1 expression is increased in long‐term rather than in short‐term cultures.
Keywords: adapted suspension cell, circulating tumor cell, NF‐κB, reactive oxygen species, SIRT1
Abbreviations
- AD
adherent cell
- ASC
adapted suspension cell
- CSC
cancer stem cell
- CTC
circulating tumor cell
- DCFDA
dichlorofluorescein diacetate
- EMT
epithelial‐mesenchymal transition
- FOXO
Forkhead box O
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- HDAC
histone deacetylase
- miRNA
microRNA
- NAC
N‐acetylcysteine
- NAM
nicotinamide
- NF‐κB
nuclear factor kappa B
- PARP
poly ADP ribose polymerase
- PDTC
pyrrolidine dithiocarbamate
- ROS
reactive oxygen species
- SC
suspension cell
- SIRT
silent mating‐type information regulation 2 homolog 1
- TNF
tumor necrosis factor
1. INTRODUCTION
Metastatic spread of cancer cells to distant specific organs such as bone and lung is the main cause of cancer‐related death.1 Early detection of metastatic cancer is critical to prevent metastasis‐associated death. EMT, an essential event in the spread of cancer cells to other organs, facilitates the penetration of cancer cells into the bloodstream.2 These penetrated cancer cells termed CTC can be detected in the bloodstream of patients.1 Thus, the detailed characterization of CTC is indispensible for the accurate diagnosis, better prognosis, and efficient therapy of metastatic cancers.3 Many studies have been conducted to characterize and efficiently isolate CTC; however, the small number of CTC poses several difficulties. Nonetheless, accumulating evidence has partially revealed the characteristics of CTC. CTC were successfully isolated from patients’ blood samples by positive selection using epithelial phenotype marker and/or by CD133 stem cell marker and cultured ex vivo,4, 5 indicating that CTC potentially include CSC, which can survive for a long time. In addition, CTC lines were established to overcome scarcity, which is a hurdle hampering characterization of CTC.4 These reports support the idea that some CTC can survive for a long time.
In general, suspension cultures leading to spheroid formation are carried out to study features of CSC, and SC cultured for a few weeks were used in this study. However, this short culture period is insufficient for the stabilization of cells. In the present study, we cultured cancer cells in an ultra‐low attachment dish for a long period of more than 6 months to allow stabilization and adaptation in a suspension state. These ASC were compared with adherent parent cancer cells to investigate how cancer cells survive in the bloodstream as CTC. We found that proliferation of SC subjected to more than 150 passages increased as quickly as that of AD and that the expression of silent mating type‐information regulation 2 homolog 1 (SIRT1) increased in ASC. Therefore, we investigated the role of SIRT1 in the promotion of ASC proliferation.
Silent mating‐type information regulation family members are NAD+‐dependent HDAC (class III). Seven SIRT paralogs (SIRT1‐7) have been identified in humans and each SIRT shows different localization, function, and activity.6 SIRT play various roles in cell cycle progression and apoptosis of cancer cells.7 Of these, SIRT1 is significantly overexpressed in various cancers, including breast, prostate, pancreas, and colon cancers,8, 9, 10 and the inhibition of SIRT1 expression results in the arrest of growth and apoptosis of several types of cancer cell.11, 12, 13, 14 Contrary to the protumor function of SIRT1, several studies have shown that SIRT1 may also act as a tumor suppressor and inhibit intestinal tumorigenesis through the suppression of cancer cell growth in a mouse model of colon cancer.15 SIRT1 is involved in the direct DNA damage response, and activation of SIRT1 results in protection against mutant BRCA1‐associated breast cancer.16 SIRT1 suppresses the function of ROS through the stimulation of antioxidants by FOXO pathways.17 Conversely, ROS is also able to suppress SIRT1 expression by oxidative modification of its cysteine residues.17
Nuclear factor kappa B signaling plays an important role in apoptosis, cell proliferation, and tumorigenesis through the regulation of the expression of several genes,18 and is associated with an increase in glycolytic energy flux.19 Phosphorylation of IκB triggers the initiation of NF‐κB signaling and the released p65 subunit moves into the nucleus and becomes acetylated at the Lys310 residue by p300 and p300/CBP‐associated factor, resulting in its complete activation. The acetylated active form of p65 is directly inactivated by deacetylation triggered by SIRT1, resulting in suppression of NF‐κB‐dependent gene expression.19 Aside from the SIRT1‐mediated inhibition of NF‐κB activity, NF‐κB signaling is also able to suppress SIRT1 activity through the expression of miRNAs as well as through an increase in ROS production.20 These antagonistic functions of NF‐κB and SIRT1 play several roles in inflammation and metabolism,19 regulation of autophagy,21 and neuroinflammation and cognitive impairment induced by sepsis.22 Therefore, the outcome of crosstalk between SIRT1, ROS, and NF‐κB should be decoded in the tumor context.
2. MATERIALS AND METHODS
2.1. Antibodies and reagents
The following antibodies and reagents were used: Sirtuin Antibody Sampler Kit #9787T, NF‐κB p65 Antibody Sampler Kit #4767, Death Receptor Antibody Sampler Kit #8356, antibodies against cleaved PARP #5626, cleaved caspase‐3 #9664, cleaved caspase‐8 #8592, cleaved caspase‐9 #9509, Bak #6947, Bax #5023, Bcl‐2 #15071, and Bcl‐X #2764 (Cell Signaling Technology, Danvers, MA, USA); antibodies against HDAC3 #sc‐130319 and β‐actin #sc‐58673 (Santa Cruz Biotechnology, Dallas, TX, USA); nicotinamide (NAM); and ammonium pyrrolidine dithiocarbamate (PDTC) (Sigma‐Aldrich, St Louis, MO, USA).
2.2. Cell culture
MDA‐MB‐468 human breast cancer cells were purchased from the ATCC (Manassas, VA, USA). Cell suspensions were generated according to the previous method23 and cells passaged more than 150 times in ultra‐low attachment plates (Corning, Corning, NY, USA) were named ASC. Both AD and SC were maintained in RPMI‐1640 medium (HyClone, Logan, UT, USA) supplemented with 10% FBS (Equitech‐Bio, Kerrville, TX, USA) at 37°C in a humidified atmosphere of 5% CO2.
2.3. Cell proliferation assay
Adherent cell and SC were seeded in six‐well normal and ultra‐low attachment plates at a density of 5 × 105 cells/well, respectively, and incubated for 72 hours. The cells were stained with 0.3% Trypan blue (Alfa Aesar, Ward Hill, MA, USA), and cell number was counted with a LUNA automated cell counter (Logos Biosystems, Annandale, VA, USA) according to the instructions of the manufacturer. Cell proliferation assays were carried out in triplicate.
2.4. RNA extraction and real‐time quantitative polymerase chain reaction
Total RNA was extracted using the extraction reagent RNAiso Plus (TaKaRa, Shiga, Japan) according to the manufacturer's instructions. Isolated RNA was reverse transcribed into cDNA using M‐MLV Reverse Transcriptase (Thermo Fisher Scientific, Waltham, MA, USA) at 42°C for 1 hour and RT‐qPCR was carried out using Maxima SYBR Green (Thermo Fisher Scientific) with primers optimized for real‐time PCR. A QuantStudio 3 Real‐Time PCR System (Applied Biosystems, Foster City, CA, USA) was used and reaction specificity was confirmed by melting curve analysis. Relative gene expression was analyzed using the comparative Ct method.
Primers used for RT‐PCR amplification were as follows: SIRT1 forward (5′‐TGCTGGCCTAATAGAGTGGCA‐3′) and reverse (5′‐CTCAGCGCCATGGAAAATGT‐3′); SIRT2 forward (5′‐AGAGGCAGAGATGGACTTCCT‐3′) and reverse (5′‐CTCCCACCAAACAGATGACC‐3′); SIRT3 forward (5′‐TGGTATCCCAGACTTCAGAT‐3′) and reverse (5′‐TGTGTGTAGAGCCGCAGAA‐3′); SIRT4 forward (5′‐CAAGTCCGGAGCTTTCAGGT‐3′) and reverse (5′‐TGCAAGGATGATCCCACCAC‐3′); SIRT5 forward (5′‐GTCTAGTGGTGGGCACTTCC‐3′) and reverse (5′‐GGAAGAGTCGTTCCACAGGG‐3′); SIRT6 forward (5′‐GCAGTCTTCCAGTGTGGTGT‐3′) and reverse (5′‐AAGGTGGTGTCGAACTTGGG‐3′); SIRT7 forward (5′‐TGAGTGCTGCCGACCTAA‐3′) and reverse (5′‐TCGCAGTTCTGAGACACC‐3′); and 18s rRNA forward (5′‐AGCTATCAATCTGTCAATCCTGTC‐3′) and reverse (5′‐ CTTAATTGACTCAACACGGGA‐3′).
2.5. Immunoblot assay
Cell lysates were prepared using a lysis buffer (50 mmol/L Tris‐HCl pH 8.0, 150 mmol/L NaCl, 1 mmol/L EDTA, 0.5% NP‐40, a protease inhibitor cocktail tablet). Total cell lysate was mixed with 5 × SDS sample buffer. Proteins were resolved by 10% SDS polyacrylamide gel electrophoresis at 135 mA for 1 hour and the separated protein bands were transferred onto 0.45‐μm nitrocellulose membranes (GE Healthcare, Buckinghamshire, UK) for 2 hours. Membranes were blocked at 25°C in TBST (20 mmol/L Tris‐HCl pH 8.0, 150 mmol/L NaCl, 0.05% Tween 20) with 3% BSA for 20 minutes and subsequently incubated with appropriate primary antibody at 4°C for overnight. The membranes were incubated with HRP‐conjugated secondary antibodies (Assay Designs, Ann Arbor, MI, USA) for 2 hours at room temperature and the proteins were visualized using an ECL substrate (Thermo Fisher Scientific). Detection was carried out using an LAS3000 luminescent image analyzer (Fuji Film, Tokyo, Japan).
2.6. Chromatin immunoprecipitation sequencing
Adherent cells and ASC from MDA‐MB‐468 were fixed with 1% formaldehyde for 10 minutes and treated with 0.125 M glycine to terminate crosslinking. Nuclei were extracted in Farnham lysis buffer (5 mmol/L PIPES pH 8.0, 85 mmol/L KCl, 0.5% NP‐40, and protease inhibitor) and fragmented by sonication. A total of 1 mg fragmented chromatin was used for immunoprecipitation with 4 μg H3K4me3 (Millipore, Temecula, CA, USA), 10 μg H3K27ac, and 10 μg RNA Pol II (Abcam, Burlingame, CA, USA). Libraries for next‐generation sequencing were generated and sequenced using the HiSeq 2500 (Illumina, San Diego, CA, USA).
2.7. Chromatin immunoprecipitation sequencing analysis
FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to check the quality of ChIP‐seq data, and low‐quality reads and leftover adapter sequences were eliminated by Cutadapt ( https://cutadapt.readthedocs.io). To align the sequenced reads to the human reference genome hg19, Bowtie 224 was used. HOMER software25 and Integrative Genomics Viewer (IGV) genome browser (http://software.broadinstitute.org/software/igv/) were used for visualization.
2.8. Plasmid and siRNA transfection
Cells were transfected with 40 ng/mL siRNA using Lipofectamine RNAiMax transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's transfection protocol. To suppress SIRT1 expression, SIRT1‐1 (5′‐ACUUUGCUGUAACCCUGUA‐3′) and SIRT1‐2 (5′‐AGA GUU GCC ACC CAC ACC U‐3′) were used. Cells were transfected with 2 μg/mL plasmid using Lipofectamine 3000 DNA transfection reagent (Invitrogen) according to the manufacturer's transfection protocol.
2.9. Mitochondrial membrane potential analysis
Mitochondrial membrane potential was estimated with JC‐1 staining which detects polarization/depolarization of the mitochondrial membrane. Cells were seeded at a density of 5 × 105 cells/well into six‐well plates and transfected with the indicated siRNAs. JC‐1 staining was carried out 48 hours after transfection using the BD MitoScreen Kit (Becton Dickinson, Franklin Lakes, NJ, USA) according to the manufacturer's instructions. The cells were subsequently examined with a flow cytometer (Becton Dickinson), and the data were analyzed with FlowJo software (LLC, Ashland, OR, USA).
2.10. Intracellular ROS analysis
To measure the intracellular ROS concentration, a DCFDA cellular ROS detection assay kit was used (Abcam) as per the manufacturer's instructions. Briefly, cells were seeded at a density of 2 × 104 cells/well into a black‐sided clear‐bottomed 96‐well plate (Corning). The cells were stained with 25 μmol/L DCFDA for 45 minutes at 37°C for 24 hours after transfection with si‐SIRT and then evaluated using a microplate reader. Data are representative of three independent experiments carried out in triplicate.
2.11. Measurement of reduced GSH/oxidized GSSG ratio
A GSH/GSSG‐Glo Assay kit (Promega, Madison, WI, USA) was used according to the manufacturer's instructions. Briefly, cells were seeded at a density of 1 × 104 cells/well into a white‐sided and clear‐bottomed 96‐well plate (Corning). Cells were treated with or without GSSG reagent, and the luminescence of each well was measured with a microplate reader.
2.12. Luciferase assay
Cells were seeded into six‐well plates and cotransfected with a mixture of 200 ng pGL34xNF‐κB‐luciferase reporter plasmid and 5 ng pRL‐CMV plasmid using Lipofectamine 3000 reagent. Cells were lysed 24 hours after incubation and luciferase activity was determined using the Dual‐Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions.
2.13. Orthotopic mouse model of breast cancer metastasis
MDA‐MB‐468 SC and ASC, which stably expressed firefly luciferase were orthotopically transplanted into mouse mammary fat pad as previously described23 and metastasis was observed with IVIS Lumina XRMS in vivo bioluminescence imaging system (Caliper Life Sciences, Hopkinton, MA, USA). Plans and protocols for animal experiments were approved by the Institutional Animal Care and Use Committee of Sookmyung Women's University, Seoul, Republic of Korea (SMWU‐IACUC‐1608‐018).
2.14. Statistical analysis
A two‐tailed and non‐parametric Mann‐Whitney U test was used to compare the two indicated groups. Maximum level of significance of P < .05 was used for all statistical comparisons. Values of *P < .05, **P < .01, and ***P < .001 indicated significant differences. All data are expressed as mean ± standard deviation (SD). All statistical analyses were carried out using GraphPad Prism version 5.0 for Windows (GraphPad Software).
3. RESULTS
3.1. Inhibition of SIRT1 expression suppresses ASC proliferation
We previously reported that the proliferation of SC is slower than that of AD.23 However, SC subjected to more than 150 passages showed similar proliferation to AD, but higher than that of SC after 40 passages. We named the cells subjected to 150 passages ASC (Figure 1A). As SIRT1 is known to be involved in the regulation of cell proliferation and apoptosis,26 mRNA levels of SIRT family members were examined using qPCR. mRNA expression levels of all SIRT family members increased in ASC, with maximum overexpression reported for SIRT1 mRNA (Figure 1B). Protein levels of all SIRT family members also increased (Figure 1C). However, the protein expression of SIRT1 was not at a maximum. Level of SIRT1 protein was examined to evaluate whether SIRT1 is involved in the increase in the proliferation of ASC in a passage‐dependent way. Maximum overexpression of SIRT1 was observed in the cells subjected to more than 150 passages (Figure 1D). No difference in SIRT1 protein level was observed in more than 150 passaged AD (Figure S1). To examine whether the increase in SIRT1 expression is associated with alteration in promoter region, open promoter markers (H3K4me3 and H3K27ac) were examined; no significant difference was observed (Figure 1E). These results suggest that no changes in the enhancer region are responsible for SIRT1 transcription.
Figure 1.
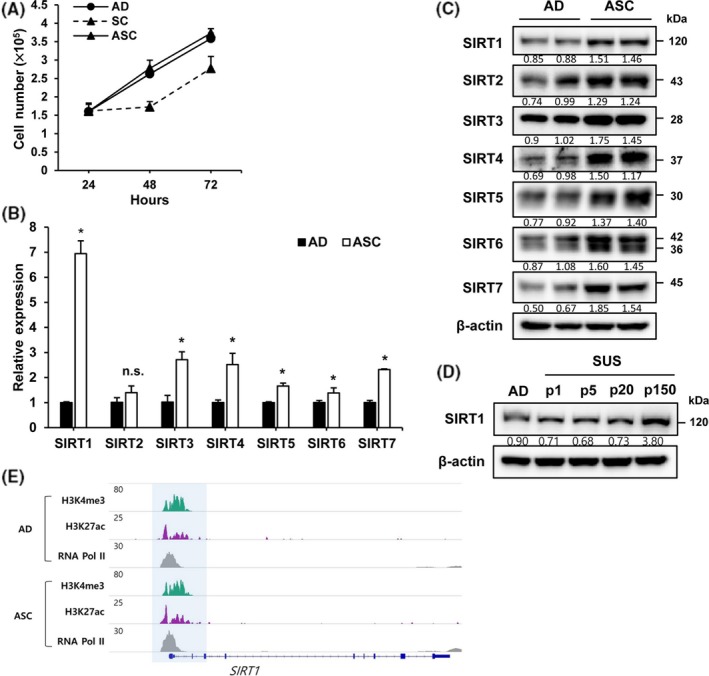
Silent mating‐type information regulation 2 homolog 1 (SIRT1) expression is increased in adapted suspension cells (ASC). A, Numbers of adherent cells (AD), suspension cells (SC) and ASC were measured at the indicted time. B‐C, SIRT levels in AD and ASC were determined by qRT‐PCR and immunoblot assays. Normalization was done using 18S rRNA and β‐actin. D, Level of SIRT1 was analyzed in differently passaged SC by immunoblot assay. E, Genome browser snapshot of H3K4me3, H3K27ac, and RNA Pol II ChIP‐seq at SIRT1 locus. H3K4me3, H3K27ac, and RNA Pol II are co‐occupied at the promoter region of SIRT1 and their enrichment is identical in AD and ASC. *P < .05
We evaluated the effect of SIRT1 inhibition on the proliferation of ASC. Cells were treated with an SIRT inhibitor, NAM, for 72 hours. In comparison with AD, ASC showed a dramatic decrease in proliferation in a dose‐dependent way (Figure 2A). As a remarkable difference in cell number was observed after treatment with 1 mmol/L NAM, we evaluated the time‐dependent effect on cell proliferation using a 1 mmol/L concentration of NAM. A significant decrease in cell number was observed after 48 hours of treatment with NAM (Figure 2B). To confirm that SIRT1 is associated with the increase in the proliferation of ASC, proliferation of cells transfected with siSIRT1 was examined. The number of ASC greatly decreased after 3 days of siSIRT1 treatment, but no effect was observed in siSIRT1‐treated AD and SC (Figure 2C,D). These results indicate that the increase in SIRT1 expression contributes to the enhancement in the proliferation of ASC.
Figure 2.
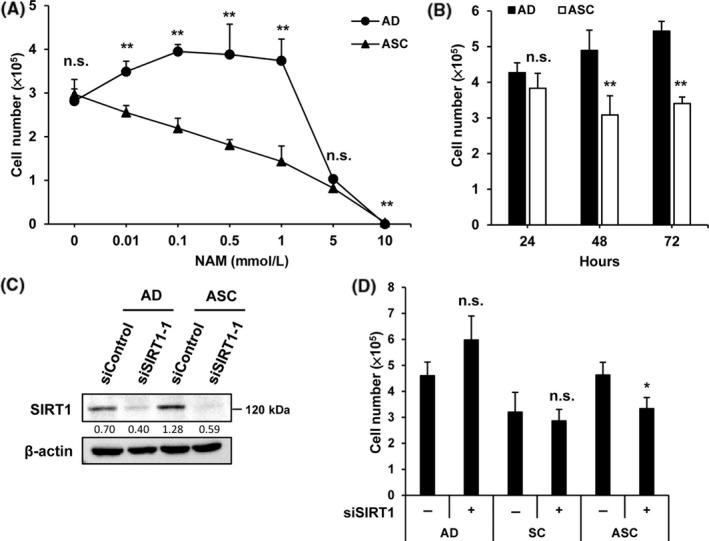
Silent mating‐type information regulation 2 homolog 1 (SIRT1) inhibition suppresses proliferation of adapted suspension cells (ASC). A, Adherent cells (AD) and ASC were plated at a density of 5 × 105 per well onto six‐well plates and cultured for 72 h with the indicated concentrations of nicotinamide (NAM). Number of cells was counted, respectively. B, Number of cells was counted at the indicted time points after treatment with 1 mmol/L NAM. C, Cells transfected with control siRNA or SIRT1 siRNA (40 ng/mL) were cultured for 48 h, and the efficiency of siSIRT was examined by immunoblot assay. D, Number of cells treated with siSIRT1 or siControl was counted 72 h after the treatment. *P < .05, **P < .01; n.s., not significant
3.2. Inhibition of SIRT1 expression results in the induction of apoptotic death in ASC
As the inhibition of SIRT expression induces apoptosis, we examined the effect of SIRT overexpression on ASC. SIRT function was inhibited by the treatment of cells with NAM. Suppression of SIRT function progressively increased the level of cleaved PARP in ASC, whereas AD showed an abrupt increase in PARP cleavage in response to treatment with more than 1 mmol/L concentration of NAM (Figure 3A). SIRT1 expression was depleted by siSIRT1 treatment, and the level of cleaved PARP was examined. Level of cleaved PARP dramatically increased in ASC but not in AD. In addition, levels of cleaved caspases were examined to investigate whether SIRT1 depletion stimulates the intrinsic or the extrinsic apoptosis pathway. Cleavage of caspase‐3 and caspase‐9 dramatically increased, but that of caspase‐8 was mediated by extracellular stimulation and was not observed in ASC (Figure 3B). We further confirmed these observations by evaluating the level of signaling molecules related to the extrinsic apoptosis pathway. No significant difference was observed in both AD and ASC (Figure 3C). These results provide evidence that the increase in SIRT1 expression contributes to the prevention of apoptotic cell death in ASC.
Figure 3.
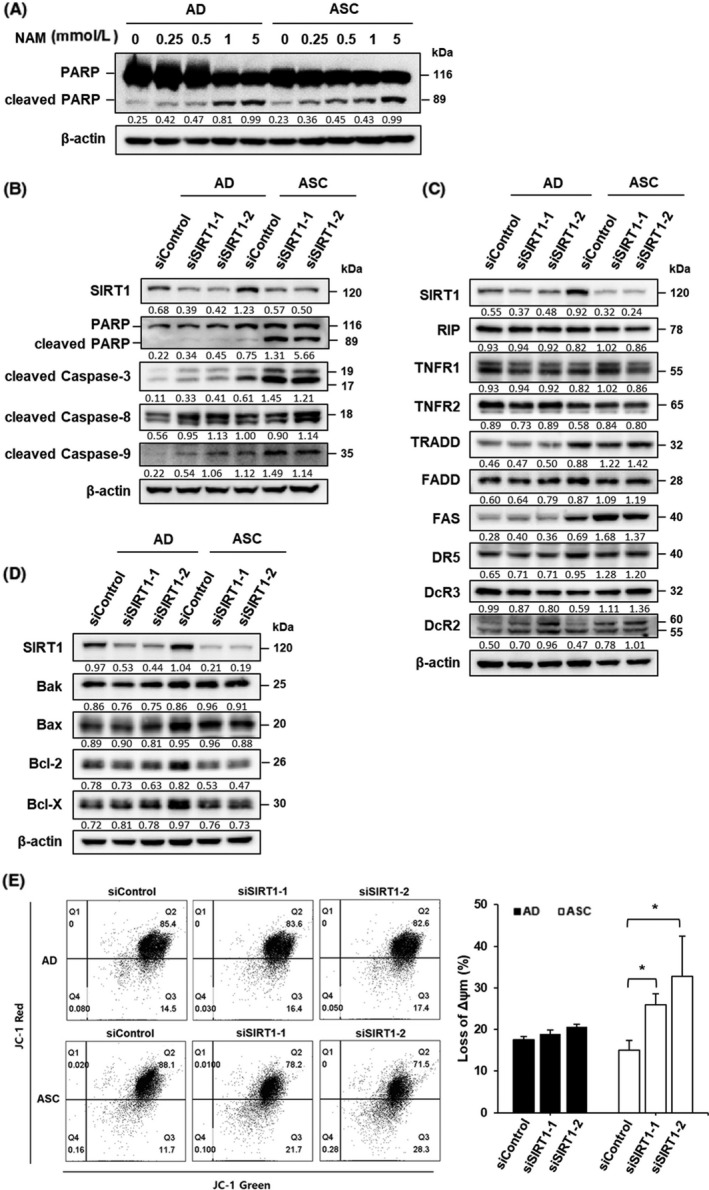
Silent mating‐type information regulation 2 homolog 1 (SIRT1) inhibition induces intrinsic apoptotic signaling in adapted suspension cells (ASC). A, Adherent cells (AD) and ASC were plated at a density of 5 × 105 per well and cultured for 72 h with the indicated concentration of nicotinamide (NAM). Level of cleaved poly ADP ribose polymerase (PARP) as a marker of apoptosis was determined by immunoblot assay. B, Levels of cleaved PARP, caspase‐3, ‐8, and ‐9 were examined in SIRT1‐depleted cells using immunoblot assay. C, Level of death receptor signaling associated protein in SIRT1‐depleted cells was determined by immunoblot assays. D, Level of BCL‐2 family protein in SIRT1‐depleted cells was determined by immunoblot assays. E, Cells transfected with siControl and siSIRT1 were stained with JC‐1 dye and analyzed by flow cytometry. Bar diagram shows percentage of cells showing mitochondrial depolarization. *P < .05
Anti‐apoptotic Bcl‐2 family members prevent apoptosis through inhibition of mitochondrial outer membrane permeabilization, whereas pro‐apoptotic Bcl‐2 family members exert opposite effects. Thus, the levels of these Bcl‐2 family members were measured in both cell types. No significant difference was observed in Bak and Bax pro‐apoptotic protein levels after treatment with siSIRT1. However, the overexpression of anti‐apoptotic proteins such as Bcl‐2 and Bcl‐XL was more prominent in ASC and decreased after the depletion of SIRT expression (Figure 3D), indicating that high expression levels of anti‐apoptotic proteins play a key role in the survival of ASC. We investigated whether SIRT1‐depleted cells show changes in mitochondrial membrane permeability. After SIRT1 siRNA transfection for 48 hours, mitochondrial membrane potential was analyzed with flow cytometry using JC‐1 staining. SIRT1 depletion induced mitochondrial membrane depolarization in ASC as compared with AD (Figure 3E). These data indicate that the increase in the expression of anti‐apoptotic proteins stimulated by SIRT1 could contribute to the prevention of apoptotic death of ASC through the maintenance of mitochondrial membrane depolarization.
3.3. Silent mating‐type information regulation 2 homolog 1 alleviates intracellular ROS levels to prevent apoptotic death of ASC
Reactive oxygen species are the main cause of the decrease in mitochondrial membrane potential, whereas anti‐apoptotic proteins such as Bcl‐2 and Bcl‐X reduce ROS levels.27, 28 Therefore, we thought that SIRT1 depletion may stimulate ROS production. Intracellular ROS levels were measured by DCFDA staining. SIRT1‐depleted ASC showed a significant increase in ROS levels as compared to SIRT1‐depleted AD and SC (Figure 4A). Therefore, ROS is maintained at a low level through an increase in the expression of Bcl‐2 and Bcl‐X in ASC as compared to AD (Figure 3D). As the ratio of GSH and GSSG determines the level of ROS,29 increase in the GSH/GSSG ratio may contribute to the low level of ROS in ASC. ASC showed approximately a two‐fold increase in this ratio as compared with AD, indicating that ROS was maintained at a low level for the survival of ASC by maintenance of a high ratio of GSH/GSSG (Figure 4B). To determine the mechanism underlying the increase in the GSH/GSSG ratio, levels of GSSG‐producing glutathione peroxidase (GPX) and GSH‐producing glutathione reductase (GSR) were quantified by qPCR (Figure 4C). The increase in GSH contributed to a high GSH/GSSG ratio. As SIRT1‐depleted ASC were highly susceptible to proliferation (Figure 2A), we determined whether siSIRT1‐induced ROS expression is responsible for the severe decrease in cell number. siSIRT1‐induced ROS expression was inhibited by treatment with a ROS scavenger, NAC. Cells transfected with siSIRT1 for 24 hours were treated with the indicated concentrations of NAC for an additional 48 hours. Proliferation of SIRT1‐depleted AD decreased after NAC treatment in a dose‐dependent way, whereas the proliferation of SIRT1‐depleted ASC was unaffected after NAC treatment (Figure 4D). Next, the effect of NAC on the siSIRT1‐induced PARP cleavage in both cells was examined. Treatment of ASC with 5 mmol/L NAC for 48 hours after siSIRT1 transfection resulted in a dramatic decrease in the cleavage of PARP (Figure 4E). Taken together, these results indicate that the increase in SIRT1 expression in ASC is responsible for the prevention of apoptotic cell death induced by the excessive production of ROS.
Figure 4.
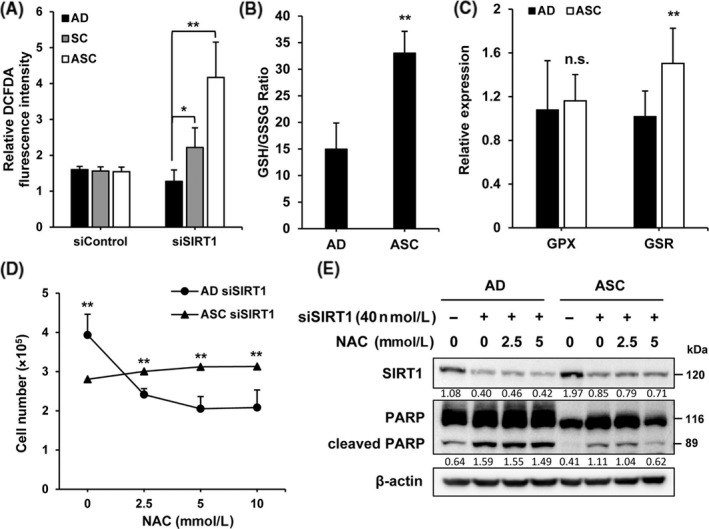
Silent mating‐type information regulation 2 homolog 1 (SIRT1) regulates the level of reactive oxygen species (ROS) in adapted suspension cells (ASC). A, Cells transfected with siControl and siSIRT1 were labeled with dichlorofluorescein diacetate (DCFDA) and analyzed on a fluorescent plate reader. B, Intracellular reduced glutathione (GSH) and oxidized glutathione (GSSG) were measured using a luminescence plate reader in adherent cells (AD) and ASC. Data are given as GSH/GSSG ratio. C, Levels of glutathione‐related enzymes including GPX and GSR in AD and suspension cells (SC) were determined by qRT‐PCR assays. Normalization was carried out using 18S rRNA. D, Cells transfected with siControl and siSIRT1 were treated with N‐acetylcysteine (NAC) at the indicated concentration for 48 h and then the number of cells was counted. E, Cells were transfected with control or SIRT1 siRNA for 24 h and then treated with NAC at the indicated concentrations for 48 h. Level of cleaved poly ADP ribose polymerase (PARP) in SIRT1‐depleted cells was determined by immunoblot assays. *P < .05, **P < .01; n.s., not significant
3.4. Silent mating‐type information regulation 1 overexpression inhibits NF‐κB activity by deacetylation of p65 in ASC
As SIRT1 inhibits the NF‐κB signaling pathway,17 NF‐κB activity was examined in both cell types after transfection with NF‐κB luciferase vector. Luciferase activity of NF‐κB was reduced by approximately three‐fold in ASC compared to SC (Figure 5A). The low NF‐κB activity may be increased by TNF‐α, a representative NF‐κB activator.30 Surprisingly, TNF‐α failed to increase NF‐κB activation in ASC (Figure 5B). Acetylated or phosphorylated NF‐κB p65 level was measured in both cell types to determine the reason underlying the inability of TNF‐α to activate NF‐κB. The acetylated NF‐κB p65 level was markedly decreased in ASC. However, the level of phosphorylated NF‐κB p65 progressively increased according to the degree of adaptation (Figure 5C). As NF‐ κB p65 was deacetylated by HDAC3 and/or SIRT1 at Lys310,31 the HDAC3 level was also examined. No significant changes in the level of HDAC3 were observed in AD and ASC (Figure 5C) as compared to SIRT1‐treated cells (Figure 1D). Thus, we examined whether SIRT1 is responsible for the decrease in NF‐κB activity by reducing the level of acetylated NF‐κB p65 at Lys310. SIRT1 depletion dramatically increased the level of acetylation of NF‐κB p65 at Lys310, but no significant differences were observed in total NF‐κB p65 and phosphorylated NF‐κB p65 (at Ser536) (Figure 5D); thus, SIRT1 overexpression suppresses NF‐κB activity.
Figure 5.
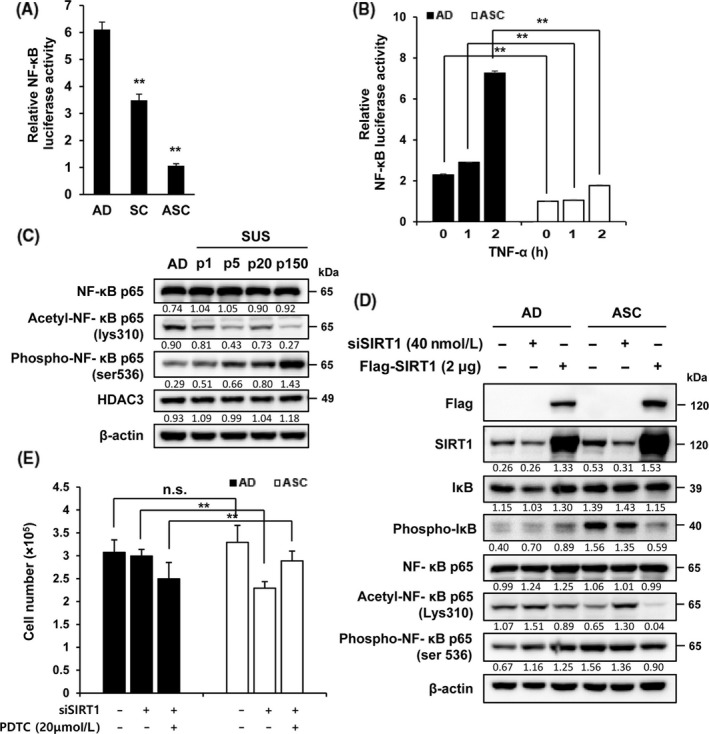
Silent mating‐type information regulation 2 homolog 1 (SIRT1) inhibits the activation of nuclear factor kappa B (NF‐κB) activity in adherent cells (AD) and adapted suspension cells (ASC). A, Cells were transfected with the NF‐κB luciferase plasmid for 24 h and then relative luciferase activity was analyzed. Firefly luciferase was used as a reporter, and Renilla luciferase was used as a control. B, Cells transfected with NF‐κB luciferase plasmid were stimulated with or without tumor necrosis factor (TNF)‐α for 1 or 2 h and then NF‐κB luciferase activity was measured. C, Levels of NF‐κB p65 and HDAC3 were analyzed by immunoblot assay in AD and differently passaged suspension cells (SC). D, Cells were transfected with siSIRT1 or siControl for 48 h and levels of protein associated with NF‐κB signaling were examined using immunoblot assay. E, Cells transfected with siSIRT1 or siControl were treated with or without pyrrolidine dithiocarbamate (PDTC) for 48 h and then the number of cells was counted. **P < .01; n.s., not significant
To determine whether the SIRT1‐mediated inhibition of NF‐κB activity is related to cell proliferation, both SIRT1‐depleted cells were treated with PDTC, an antioxidant and inhibitor of NF‐κB activation. PDTC treatment resulted in a recovery of the loss of ASC number observed in response to SIRT1 depletion (Figure 5E). These results indicate that inhibition of NF‐κB activity and ROS level by SIRT1 overexpression is the underlying mechanism for ASC survival.
4. DISCUSSION
Meng et al32 reported that CTC can survive a decade because they are continuously dying and being replenished by tumor cells shedding from tumor tissues. Most CTC will not be able to survive in anoikis conditions in the turbulence of the circulating bloodstream.33 However, some CTC including CSC can survive. Thus, it is believed that ASC contain CSC. ASC show a high basal level of cleaved caspases and an increase in pro‐apoptotic proteins compared with AD (Figure 3B,D), and their expression was greatly increased in the absence of SIRT1. Hence, SIRT1 overexpression seems to contribute to overcoming the apoptotic death of ASC through an increase in the rate of proliferation, although ASC are more susceptible to apoptotic death than AD.
In an orthotopic xenograft model, we previously showed that the metastatic ability of SC was increased as compared to that of AD.23 In the present study, we determined whether ASC have higher metastatic potential than SC. In the invasion assay, ASC showed a twofold increase in metastatic ability compared to SC (Figure S2A). In an orthotopic xenograft model, ASC injection resulted in the formation of a tumor mass in two of five mice, and the tumors metastasized in both mice, whereas SC injection resulted in the formation of a tumor mass in three of five mice without metastasis (Figure S2B). ASC still maintain high invasive ability, which may be affected by SIRT1 depletion (Figure S2C).
SIRT1 is overexpressed in CSC and plays a critical role in maintaining the characteristics of CSC through sustainable expression of stemness genes such as Oct4 and Nanog.99 We have previously shown that SC contain a CSC‐like population.23 In the present study, we showed that ASC had increased expression of SOX2 and OCT4 as stemness factors (Figure S3). Thus, SIRT1 expression may likely contribute to the survival of ASC as well as to CSC. However, as ZR‐75‐1‐derived ASC, but not T47D‐derived ASC, showed an increase in SIRT1 expression (Figure S4), the role of SIRT1 may be interpreted in the context of tumors. To evaluate whether SIRT1 overexpression affects OS, The Cancer Genome Atlas (TCGA) data of patients with breast cancer were analyzed. OS decreased in patients with breast cancer expressing a high level of SIRT1 to between 50 and 100 months, whereas it abruptly decreased in patients with triple‐negative breast cancer (Figure S5).
SIRT1 overexpression enhances the EMT process and metastasis in osteosarcoma,34 whereas SIRT1 inhibition significantly suppresses the EMT process in esophageal cancer cells.35 As a molecular mechanism, SIRT1 inhibition reduces the level of FOXO3a and matrix MMP2, which degrades extracellular matrices to allow penetration of cancer cells into the basement membrane.36 Therefore, SIRT1 plays a critical role during the EMT process by allowing physical detachment of cancer cells from the primary cancer site. In contrast, SIRT1 plays a positive role in the induction of anoikis resistance after the EMT process in gastric cancer cells.37 In the present study, we showed that SIRT1 inhibition dramatically induced apoptotic death in ASC as compared to AD. Thus, the higher expression of SIRT1 in ASC than in AD may be critical to overcome anoikis and for long‐term survival. Therefore, the increase in SIRT1 expression could be an important event post‐EMT as well as during the EMT process.
Activation of NF‐κB allows cancer cells to evade apoptotic death through the upregulation of anti‐apoptotic proteins such as Bcl‐2, Bcl‐XL, cFLIP, and cIAP2.38, 39 A critical event in NF‐κB activation is acetylation of p65. For suppression of the NF‐κB signaling pathway, SIRT1 deacetylates p65 and inhibits NF‐κB activity. Thus, the antagonistic function of SIRT1 and NF‐κB is very important for balanced regulation of apoptosis. In the present study, we found that NF‐κB activity was downregulated, SIRT1 level increased in ASC, and SIRT1 deficiency caused apoptotic death even after recovery of NF‐κB activity in ASC. Thus, NF‐κB activation is not critical for the survival of ASC. Therefore, it is likely that NF‐κB activation results in death or survival depending on the tumor context. In contrast, ROS interacts with the NF‐κB signaling components and may either activate or inhibit NF‐κB signaling depending on the tumor context.40 As SIRT1 depletion caused an increase in ROS production and apoptotic death regardless of the recovery of NF‐κB activity in ASC, the maintenance of a low level of ROS is important for high SIRT1 expression because it contributes to the increase in the GSH/GSSG ratio. Further studies are warranted to elucidate the importance of suppression of NF‐κB activity as well as to investigate whether NF‐κB suppression is a general event in ASC derived from any breast cancer cell line.
In summary, we report, for the first time, how SIRT1 expression is increased in ASC. SIRT1 expression was higher in ASC and AD, and it protected the cells from apoptotic death. Thus, the increase in SIRT1 expression may play a crucial role in the stable survival of SC by maintaining low levels of ROS because SIRT1 expression was higher after long‐term adaptation. Development of a strategy to suppress SIRT1 activity would be an efficient way to prevent the survival and metastatic spread of CTC in breast cancer cells. In addition, it is important to determine whether this mechanism may be generalized in different types of cancer.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) [NRF‐2016R1A5A1011974].
Park JY, Han S, Ka HI, et al. Silent mating‐type information regulation 2 homolog 1 overexpression is an important strategy for the survival of adapted suspension tumor cells. Cancer Sci. 2019;110:2773–2782. 10.1111/cas.14147
REFERENCES
- 1. Aceto N, Bardia A, Miyamoto DT, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110‐1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Savagner P. The epithelial‐mesenchymal transition (EMT) phenomenon. Ann Oncol 2010;21(Suppl 7):vii89‐vii92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jansson S, Bendahl PO, Larsson AM, Aaltonen KE, Ryden L. Prognostic impact of circulating tumor cell apoptosis and clusters in serial blood samples from patients with metastatic breast cancer in a prospective observational cohort. BMC Cancer. 2016;16:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grillet F, Bayet E, Villeronce O, et al. Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut. 2017;66:1802‐1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Varillas JI, Zhang J, Chen K, et al. Microfluidic isolation of circulating tumor cells and cancer stem‐like cells from patients with pancreatic ductal adenocarcinoma. Theranostics. 2019;9:1417‐1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schemies J, Uciechowska U, Sippl W, Jung M. NAD(+)‐dependent histone deacetylases (sirtuins) as novel therapeutic targets. Med Res Rev. 2010;30:861‐889. [DOI] [PubMed] [Google Scholar]
- 7. Yamamoto H, Schoonjans K, Auwerx J. Sirtuin functions in health and disease. Mol Endocrinol. 2007;21:1745‐1755. [DOI] [PubMed] [Google Scholar]
- 8. Huffman DM, Grizzle WE, Bamman MM, et al. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612‐6618. [DOI] [PubMed] [Google Scholar]
- 9. Chen X, Sun K, Jiao S, et al. High levels of SIRT1 expression enhance tumorigenesis and associate with a poor prognosis of colorectal carcinoma patients. Sci Rep. 2014;4:7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin J, Chu Z, Ma P, Meng Y, Yang Y. SIRT1 promotes the proliferation and metastasis of human pancreatic cancer cells. Tumour Biol. 2017;39:1010428317691180. [DOI] [PubMed] [Google Scholar]
- 11. Ford J, Jiang M, Milner J. Cancer‐specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 2005;65:10457‐10463. [DOI] [PubMed] [Google Scholar]
- 12. Heltweg B, Gatbonton T, Schuler AD, et al. Antitumor activity of a small‐molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006;66:4368‐4377. [DOI] [PubMed] [Google Scholar]
- 13. Ota H, Tokunaga E, Chang K, et al. Sirt1 inhibitor, Sirtinol, induces senescence‐like growth arrest with attenuated Ras‐MAPK signaling in human cancer cells. Oncogene. 2006;25:176‐185. [DOI] [PubMed] [Google Scholar]
- 14. Lain S, Hollick JJ, Campbell J, et al. Discovery, in vivo activity, and mechanism of action of a small‐molecule p53 activator. Cancer Cell. 2008;13:454‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Firestein R, Blander G, Michan S, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE. 2008;3:e2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang RH, Zheng Y, Kim HS, et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1‐associated tumorigenesis. Mol Cell. 2008;32:11‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Salminen A, Kaarniranta K, Kauppinen A. Crosstalk between oxidative stress and SIRT1: impact on the aging process. Int J Mol Sci. 2013;14:3834‐3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ghosh S, Karin M. Missing pieces in the NF‐kappaB puzzle. Cell. 2002;109(Suppl):S81‐S96. [DOI] [PubMed] [Google Scholar]
- 19. Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF‐kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25:1939‐1948. [DOI] [PubMed] [Google Scholar]
- 20. Chen Z, Shentu TP, Wen L, Johnson DA, Shyy JY. Regulation of SIRT1 by oxidative stress‐responsive miRNAs and a systematic approach to identify its role in the endothelium. Antioxid Redox Signal. 2013;19:1522‐1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X, Gao Y, Tian N, et al. Astragaloside IV represses high glucose‐induced mesangial cells activation by enhancing autophagy via SIRT1 deacetylation of NF‐kappaB p65 subunit. Drug Des Devel Ther. 2018;12:2971‐2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pan S, Wu Y, Pei L, et al. BML‐111 reduces neuroinflammation and cognitive impairment in mice with sepsis via the SIRT1/NF‐kappaB signaling pathway. Front Cell Neurosci. 2018;12:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Park JY, Jeong AL, Joo HJ, et al. Development of suspension cell culture model to mimic circulating tumor cells. Oncotarget. 2018;9:622‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ferragina P, Manzini G. Opportunistic data structures with applications. In: Proc. 41st Annual Symposium on Foundations of Computer Science, IEEE Computer Society, 2000:390–398. 10.1109/SFCS.2000.892127 [DOI]
- 25. Heinz S, Benner C, Spann N, et al. Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chung S, Yao H, Caito S, Hwang JW, Arunachalam G, Rahman I. Regulation of SIRT1 in cellular functions: role of polyphenols. Arch Biochem Biophys. 2010;501:79‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suski JM, Lebiedzinska M, Bonora M, Pinton P, Duszynski J, Wieckowski MR. Relation between mitochondrial membrane potential and ROS formation. Methods Mol Biol. 2012;810:183‐205. [DOI] [PubMed] [Google Scholar]
- 28. Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria‐dependent and mitochondria‐independent pathways of apoptosis. Arch Toxicol. 2013;87:1157‐1180. [DOI] [PubMed] [Google Scholar]
- 29. Zitka O, Skalickova S, Gumulec J, et al. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett. 2012;4:1247‐1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dvoriantchikova G, Ivanov D. Tumor necrosis factor‐alpha mediates activation of NF‐kappaB and JNK signaling cascades in retinal ganglion cells and astrocytes in opposite ways. Eur J Neurosci. 2014;40:3171‐3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ito K. Impact of post‐translational modifications of proteins on the inflammatory process. Biochem Soc Trans. 2007;35:281‐283. [DOI] [PubMed] [Google Scholar]
- 32. Meng S, Tripathy D, Frenkel EP, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10:8152‐8162. [DOI] [PubMed] [Google Scholar]
- 33. Alix‐Panabieres C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014;14:623‐631. [DOI] [PubMed] [Google Scholar]
- 34. Yu XJ, Guo XZ, Li C, et al. SIRT1‐ZEB1‐positive feedback promotes epithelial‐mesenchymal transition process and metastasis of osteosarcoma. J Cell Biochem. 2019;120:3727‐3735. [DOI] [PubMed] [Google Scholar]
- 35. Qin T, Liu W, Huo J, et al. SIRT1 expression regulates the transformation of resistant esophageal cancer cells via the epithelial‐mesenchymal transition. Biomed Pharmacother. 2018;103:308‐316. [DOI] [PubMed] [Google Scholar]
- 36. Abdelmawgoud H, El Awady RR. Effect of Sirtuin 1 inhibition on matrix metalloproteinase 2 and Forkhead box O3a expression in breast cancer cells. Genes Dis. 2017;4:240‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang L, Wang X, Chen P. MiR‐204 down regulates SIRT1 and reverts SIRT1‐induced epithelial‐mesenchymal transition, anoikis resistance and invasion in gastric cancer cells. BMC Cancer. 2013;13:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karin M, Lin A. NF‐kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221‐227. [DOI] [PubMed] [Google Scholar]
- 39. Hayden MS, Ghosh S. Shared principles in NF‐kappaB signaling. Cell. 2008;132:344‐362. [DOI] [PubMed] [Google Scholar]
- 40. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF‐kappaB signaling. Cell Res. 2011;21:103‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials