

Abstract
Long noncoding RNAs (lncRNAs) are emerging as key regulators in cancer initiation and progression. TP53TG1 is a recently identified lncRNA and several studies have shown that TP53TG1 may play the role of tumor suppressor gene or oncogene in different tumors. Nevertheless, the involvement of TP53TG1 in carcinogenesis of pancreatic ductal adenocarcinoma (PDAC) has not been characterized. In our studies, we identified that TP53TG1 was highly expressed in PDAC and was a novel regulator of PDAC development. Knockdown of TP53TG1 inhibited proliferation, induced apoptosis, and decreased migration and invasion in PDAC cells, whereas enhanced expression of TP53TG1 had the opposite effects. Mechanistically, TP53TG1 could directly bind to microRNA (miR)‐96 and effectively function as a sponge for miR‐96, thus antagonizing the functions of miR‐96 and leading to derepression of its endogenous target KRAS, which is a core oncogene in the initiation and maintenance of PDAC. Taken together, these observations imply that TP53TG1 contributes to the growth and progression of PDAC by acting as a competing endogenous RNA (ceRNA) to competitively bind to miR‐96 and regulate KRAS expression, which highlights the importance of the complicated miRNA‐lncRNA network in modulating the progression of PDAC.
Keywords: ceRNA, KRAS, lncRNA TP53TG1, miR‐96, pancreatic ductal adenocarcinoma
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive malignancies with a dismal 8% 5‐year survival rate and is currently the third leading cause of cancer‐related death in the USA. In contrast to the steady increase in survival for most cancers in recent decades, disease‐specific survival has only improved marginally for PDAC.1, 2 The high mortality rate of PDAC stems primarily from the lack of early diagnosis and ineffective treatment for advanced tumors. It is of paramount importance to understand the underlying pathogenesis and molecular alterations causing PDAC in order to develop novel diagnostic biomarkers and effective therapeutic strategies and improve the prognosis of PDAC.
Development of high‐throughput transcriptome analyses and genome‐wide surveys have shown that >90% of the total mammalian genome can be transcribed into various short or long noncoding RNAs (lncRNAs).3, 4 MicroRNAs (miRNAs), a class of short noncoding (nc)RNA molecules that range in size from 19 to 25 nucleotides, regulate the expression of protein‐coding genes by targeting mRNAs, leading to translational inhibition or RNA degradation.5 LncRNAs are transcripts that are longer than 200 nucleotides in length and do not encode protein.6 Although only a small portion of lncRNAs have been characterized in detail, many lncRNAs have emerged as regulators of various biological processes, such as epigenetic regulation, alternative splicing, RNA decay, cell cycle control, and cell fate determination.7, 8, 9 Furthermore, multiple lines of evidence suggest that the abnormal expression of lncRNAs is associated with the development of diverse human diseases, and specific lncRNAs have also been shown to play a critical role in tumor initiation and progression, including PDAC.10, 11, 12, 13 For example, LINC00673 is able to promote protein tyrosine phosphatase PTPN11 degradation through ubiquitination, resulting in decreased proliferation of pancreatic cancer cells.14 Another characterized lncRNA, lincRNA‐ROR, acts as an important regulator of ZEB1, promotes invasion and metastasis in pancreatic cancer, and may represent a novel therapeutic target.15 LncRNA ENST00000480739 suppresses PDAC cell invasion by regulating osteosarcoma amplified‐9 (OS‐9) and hypoxia‐inducible factor‐1α (HIF‐1α).16 Our previous studies have also shown that lncRNA MIR31HG is upregulated in PDAC and shows oncogenic property in PDAC development.17
Recently, a new regulatory mechanism has been reported in which crosstalk between lncRNAs and mRNAs occurs by competing for shared miRNAs response elements. In this case, lncRNAs could interact with specific miRNAs and function as competing endogenous RNAs (ceRNA) to prevent targeted transcripts of these miRNAs from being degraded. An example of this type of regulation is exemplified by lncRNA MIR31HG, which binds miR‐193b and regulates the expression level of its downstream miRNA‐target genes in PDAC.17 Similarly, lncRNA HOTAIR may function as a ceRNA to regulate HER2 expression through competition for miR‐331‐3p, thus playing an oncogenic role in gastric pathogenesis.18 Other studies also showed that ceRNAs were involved in the tumorigenesis of multiple cancer types, including prostate, breast, liver and lung cancers and so on.19, 20, 21, 22 Although ceRNA research is still in its initial stages, exploration of ceRNA interplay in cancer will provide new insight into mechanisms underlying the pathogenesis of cancers.
Long non‐coding RNA TP53TG1 (NCBI Reference Sequence: NR_015381.1), located on chromosome 12, is transcribed as an approximately 0.7‐kb lncRNA molecule. It has been demonstrated that the expression of TP53TG1 could be induced under conditions of cellular stress in a wild‐type TP53‐dependent way.23 Diaz‐Lagares et al found that TP53TG1 shows tumor‐suppressor features and that DNA methylation‐associated silencing of TP53TG1 produces aggressive tumors that are resistant to cellular death in gastrointestinal tumors.24 However, as in glioma, the expression of TP53TG1 was significantly upregulated, and overexpression of TP53TG1 promoted cell proliferation and migration but inhibited cell apoptosis under glucose deprivation.25 TP53TG1 was also highly expressed in nasopharyngeal carcinoma and functionally promoted nasopharyngeal carcinoma malignant phenotypes.26 Although several studies showed that TP53TG1 may play the role of tumor suppressor gene or oncogene in different tumors, the involvement of TP53TG1 in tumorigenesis of PDAC remains poorly understood.
In the present study, we identified that TP53TG1 was significantly upregulated in PDAC tissues compared with paired adjacent tissues. After downregulating TP53TG1, the proliferation and invasion of PDAC cells were significantly inhibited, whereas upregulation of TP53TG1 had the opposite effects. Dual‐luciferase reporter assay validated that TP53TG1 and 3′UTR of KRAS mRNA competitively bind with miR‐96. Our data further indicate that TP53TG1 could effectively function as a sponge for miR‐96 to modulate the derepression of KRAS.
2. MATERIALS AND METHODS
2.1. Cell culture and tissue collection
The PDAC cell lines PANC‐1 and MIA PaCa‐2 were purchased from ATCC. BxPC‐3 cells were kindly provided by Cell Bank, Chinese Academy of Sciences. These cell lines were subjected to morphological examination, growth curve assay, and Mycoplasma detection according to the ATCC cell line verification test recommendations 1 month before the study. PDAC tissues and matched adjacent normal tissues were obtained from patients who had undergone surgery at The Second Hospital of Hebei Medical University between 2014 and 2017. No local or systemic treatment had been conducted in these patients prior to surgery. All cases were histologically confirmed as PDAC. All the tissue samples were immediately snap frozen in liquid nitrogen, and stored at −80°C until RNA extraction. The study was approved by the Research Ethics Committee of The Second Hospital of Hebei Medical University. Informed consent was obtained from all patients.
2.2. Rapid amplification of cDNA ends
The rapid amplification of cDNA ends (RACE) program was carried out using Smarter RACE cDNA Amplification kit (Clontech, Mountain View, CA, USA). Gene‐specific primers for 5′‐RACE (GSP1), 3′‐RACE (GSP2), and each nested PCR NGSP1 and NGSP2 were designed in accordance with the NCBI sequence (NR_015381.1) and are listed in Table 1. RACE PCR products were separated on a 1% agarose gel. Individual bands were gel purified and cloned into PGEM‐T vector (Promega, Madison, WI, USA) for sequencing.
Table 1.
Primers for rapid amplification of cDNA ends (RACE)
| 5′‐RACE | GSP1 | 5′‐CGCCAGCCTGGGAAATGACTTTGGGT‐3′ |
| NGSP1 | 5′‐TCAGCCCTGCCACTCTCTGCTGTCA‐3′ | |
| 3′‐RACE | GSP2 | 5′‐CTCGCCGGGTGCCAAATGAGCTGTC‐3′ |
| NGSP2 | 5′‐CTGCATGATGCTGGGGAGCTTGGCG‐3′ |
2.3. RNA extraction and RT‐qPCR detection
Human PDAC cell and tissue RNA was extracted using TRIzol reagents (Invitrogen, Carlsbad, CA, USA). Total RNA was reverse transcribed to cDNA using a ReverTra Ace qPCR RT Kit (Toyobo, Osaka, Japan). cDNA was then diluted for RT‐qPCR analysis using THUNDERBIRD SYBR qPCR mix (Toyobo) with specific primers. All reactions were run on Step One Plus Real Time PCR System (Applied Biosystems, Foster City, CA, USA). Primers used for RT‐qPCR analysis are listed in Table 2.
Table 2.
Primers for RT‐qPCR
| Gene | Primer direction | Sequence |
|---|---|---|
| TP53TG1 | PCR primer F | 5′‐CAGTGAGCCGCTTTTTGCAG‐3′ |
| PCR primer R | 5′‐TCTCAGAGTCCTTGGTGGTTA‐3′ | |
| KRAS | PCR primer F | 5′‐AGGTGCGGGAGAGAGGCCTG‐3′ |
| PCR primer R | 5′‐ACTGTACTCCTCTTGACCTGCTGTG‐3′ | |
| GAPDH | PCR primer F | 5′‐GCACCGTCAAGGCTGAGAAC‐3′ |
| PCR primer R | 5′‐GCCTTCTCCATGGTGGTGAA‐3′ | |
| miR‐96 | RT primer | 5′‐GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAgcaaa‐3′ |
| PCR primer F | 5′‐TTTGGCACTAGCACATT‐3′ | |
| PCR primer R | 5′‐GTGCAGGGTCCGAGGT‐3′ | |
| U6 | RT primer | 5′‐CGCTTCACGAATTTGCGTGTCAT‐3′ |
| PCR primer F | 5′‐GCTTCGGCAGCACATATACTAAAAT‐3′ | |
| PCR primer R | 5′‐CGCTTCACGAATTTGCGTGTCAT‐3′ |
miR, microRNA.
2.4. siRNA and miRNA transfection
For RNA interference (RNAi)‐mediated knockdown of TP53TG1, two different siRNAs were generated by GenePharma (Suzhou, Jiangsu, China). miRNA mimics were also synthesized and purified by GenePharma. Cells were transfected with 20 nmol/L siRNAs or 50 nmol/L miRNA mimics using the Lipofectamine RNAiMAX Reagent (Invitrogen). Sequences of the RNA used in transfections are described in Table 3.
Table 3.
Sequences of siRNAs, miRNA mimics and inhibitors
| Name | Nucleotide sequence | |
|---|---|---|
| siTP53TG1‐1 | Sense | 5′‐CUGGUAACAAUUCUCUUCATT‐3′ |
| Antisense | 5′‐UGAAGAGAAUUGUUACCAGTT‐3′ | |
| siTP53TG1‐2 | Sense | 5′‐CUUCCCUCUUAAUGAAUAATT‐3′ |
| Antisense | 5′‐UUAUUCAUUAAGAGGGAAGTT‐3′ | |
| siKRAS‐1 | Sense | 5′‐CAGCUAAUUCAGAAUCAUU‐3′ |
| Antisense | 5′‐AAUGAUUCUGAAUUAGCUG‐3′ | |
| siKRAS‐2 | Sense | 5′‐AAAGACUCCUAAUAGCTT‐3′ |
| Antisense | 5′‐GCUAUUAGGAGUCUUUTT‐3′ | |
| hsa‐miR‐96‐5p‐mimic | Sense | 5′‐UUUGGCACUAGCACAUUUUUGCU‐3′ |
| Antisense | 5′‐CAAAAAUGUGCUAGUGCCAAAUU‐3′ | |
| Negative control (NC) | Sense | 5′‐GUACCUGACUAGUCGCAGATT‐3′ |
| Antisense | 5′‐UCUGCGACUAGUCAGGUACTT‐3′ | |
miR, microRNA.
2.5. Plasmid construction and transfection
The full length of TP53TG1 was synthesized and subcloned into the pCDNA3.1 (Invitrogen) vector, generating the pCDNA3.1‐TP53TG1 plasmid for overexpression of this lncRNA in PDAC cells. To investigate the direct binding between TP53TG1, KRAS and miR‐96, the TP53TG1 sequence or 3′UTR of KRAS was inserted into the pmirGLO vector (Promega) downstream of the luc2 reporter gene, generating the wild‐type plasmid. The mutated vector was constructed by site‐directed mutagenesis of the putative miR‐96 binding site in TP53TG1 or 3′UTR of the KRAS sequence. All plasmids were verified by DNA sequencing. Lipofectamine 2000 Reagent (Invitrogen) was used for plasmid transfection.
2.6. Cell proliferation, apoptosis, migration and invasion analysis
Cell viability was measured using CCK‐8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) and the Cell‐Light EdU Apollo 567 In Vitro Imaging Kit (RiboBio, Guangzhou, China). Apoptosis was assessed using the FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen, San Diego, CA, USA) by Accuri C6 Flow Cytometer (BD Biosciences, Franklin Lakes, NJ, USA) analysis. Wound‐healing assays were carried out to assess cell migration ability. Cell invasion assays were evaluated using Transwell Permeable Supports (8.0 μm; Costar 3422; Corning, Corning, NY, USA). Soft agar colony formation assays were used to measure the anchorage‐independent growth ability.
2.7. In situ hybridization
RNA probes were prepared for in situ hybridization assays using the DIG RNA Labeling Kit (Roche, Basel, Switzerland). A digoxigenin‐UTP labeled antisense RNA probe was derived from 183 to 607nt of TP53TG1 for detecting TP53TG1 in PDAC cells. The digoxigenin‐UTP labeled sense RNA probe derived from nucleotides 183 to 607 of TP53TG1 was used as a negative control.
2.8. Cell cytoplasm/nucleus fraction isolation
Separation of nuclear and cytosolic fractions was done using the NE‐PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Waltham, MA, USA). RNAs were extracted from each fraction using TRIzol reagent (Invitrogen) and subjected to RT‐qPCR analysis.
2.9. Dual luciferase reporter assay
Luciferase reporter assays were carried out using the Dual‐Luciferase Reporter Assay System (Promega). PmirGLO‐TP53TG1 or pmirGLO‐KRAS plasmids were cotransfected with miR‐96 mimic into PANC‐1 cells by using Lipofectamine 2000 (Invitrogen). Relative luciferase activity was normalized to Renilla luciferase activity 24 hours after transfection.
2.10. RNA binding protein immunoprecipitation assay
RNA binding protein immunoprecipitation (RIP) experiments were carried out using the Magna RIP RNA‐Binding Protein Immunoprecipitation Kit (Millipore, Burlington, MA, USA) according to the manufacturer's protocol. The Ago2 antibody used in RIP assays was obtained from Abcam (Cambridge, UK). Purified RNA was subjected to RT‐qPCR to determine the presence of the binding targets using gene‐specific primers.
2.11. Western blot analysis
Cells were harvested and lysed in RIPA buffer (Pierce Biotechnology, Waltham, MA, USA) for protein collection. Samples were separated on 12% SDS‐PAGE, transferred to PVDF membranes (Millipore), and incubated with antibodies. The following antibodies were used in this study: Ago2 (ab57113; Abcam), KRAS (ab55391; Abcam), and ACTB (sc‐47778; Santa Cruz Biotechnology, Dallas, TX, USA).
2.12. Statistical analysis
Two‐tailed Student's t‐tests and one‐way ANOVAs were used to analyze data with SPSS 22.0 software (IBM). Each experiment was carried out at least three times. Results of experiments are shown as mean ± SE. P‐values <.05 were considered to be statistically significant.
3. RESULTS
3.1. TP53TG1 is upregulated in PDAC
A 5′‐ and 3′‐RACE analysis was carried out to determine the full‐length transcript of TP53TG1 (Figure S1). As presented in the genome browser view of Figure 1A, TP53TG1 is a 694‐bp transcript present on chr7 (q21,12), and contains three exons and a poly‐A tail. Compared with the NCBI sequence (NR_015381.1), the RACE data showed that there are 12 more bases at the 5′ ends of TP53TG1 (Figure 1B). To confirm that TP53TG1 is indeed a noncoding RNA, we assessed the protein‐coding potential of TP53TG1 using the Coding‐Potential Assessment Tool (CPAT, https://lilab.research.bcm.edu/cpat/index),27 which showed that the coding probability of TP53TG1 is very low (P < .05) and TP53TG1 has no coding label (Table 4).
Figure 1.
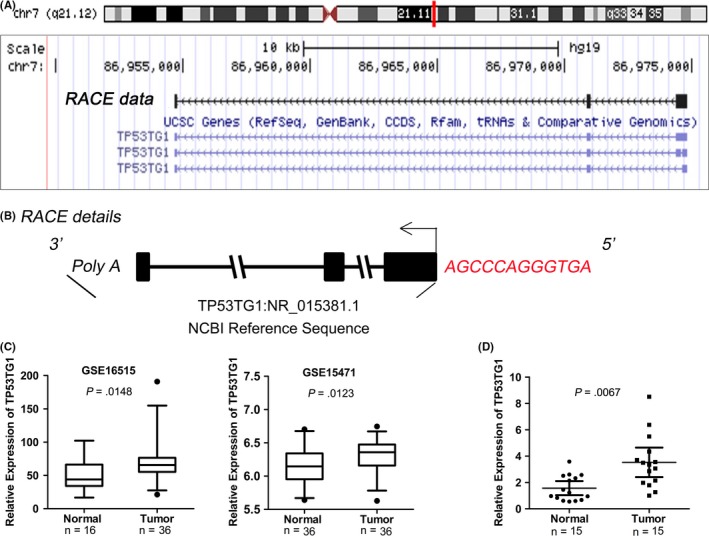
TP53TG1 is a novel long noncoding RNA and is upregulated in pancreatic ductal adenocarcinoma (PDAC). A, Genome browser representation of TP53TG1. Current gene annotations from our rapid amplification of cDNA ends (RACE) data and The University of California Santa Cruz (UCSC) genes are shown. B, The full sequence of TP53TG1 extending from 5′ and 3′ ends RACE technique is compared with the NCBI sequence (NR_015381.1). Red letters represent the 12 extra bases at the 5′ end of TP53TG1. C, TP53TG1 expression in normal pancreatic tissues and PDAC tissues, according to the array data (NCBI/GEO/GSE16515, GSE15471). Values are median with 95% CI. D, TP53TG1 expression in normal pancreatic tissues and PDAC tissues, as determined by RT‐qPCR
Table 4.
Coding potential of TP53TG1 was calculated by CPAT software
| Sequence name | RNA size (bp) | ORF size (bp) | Fickett score | Hexamer score | Coding probability | Coding label |
|---|---|---|---|---|---|---|
| TP53TG1 (NR_015381.1) | 751 | 273 | 0.5716 | −0.0549 | 0.0228 | No |
CPAT, Coding‐Potential Assessment Tool.27
Data mining of publicly available gene profiling analysis results (GSE16515 and GSE15471) showed that TP53TG1 was expressed at higher levels in PDAC tissues than in nontumor pancreatic tissues (Figure 1C). To further validate this result, RT‐qPCR analysis was done to determine the expression level of TP53TG1 in 15 paired PDAC and adjacent pancreatic tissue samples, and the results also showed that TP53TG1 is significantly upregulated in PDAC tumor tissues (Figure 1D).
3.2. siRNA‐mediated knockdown of TP53TG1 inhibits PDAC cell proliferation and invasion
We then tested whether TP53TG1 was functionally involved in PDAC tumorigenesis. For this purpose, we developed a TP53TG1 knockdown model by transfecting PDAC cell lines with two discrete anti‐TP53TG1 siRNAs (Figure 2A). By carrying out CCK‐8 assays, we found that knockdown of endogenous TP53TG1 expression dramatically inhibited the proliferative capacity of PANC‐1 and MIA PaCa‐2 cells compared with the control group (Figure 2B). EdU assay was also used to evaluate the effects of TP53TG1 on cell proliferation. Consistent with the results of CCK‐8, EdU incorporation was drastically reduced following TP53TG1 downregulation in both cell types (Figure 2C). Next, the effect of TP53TG1 knockdown on apoptosis was investigated. As expected, few early apoptotic cells were detected in the negative control (NC)‐treated cells, whereas treatment of siTP53TG1 significantly increased the percentage of early apoptotic cells as detected by Annexin V staining (Figure 2D). Taken together, these results provide evidence that TP53TG1 downregulation inhibited cell proliferation in PDAC cell lines.
Figure 2.
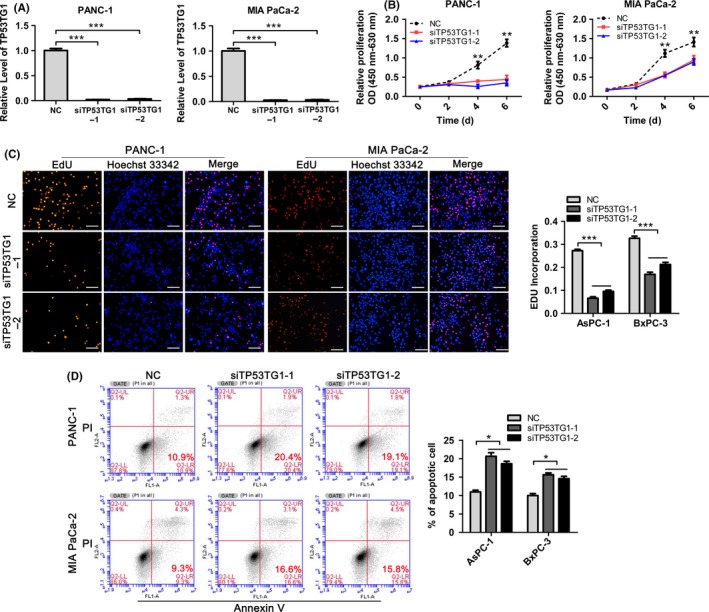
Downregulation of TP53TG1 inhibits pancreatic ductal adenocarcinoma cell proliferation. A, TP53TG1 expression was measured by RT‐qPCR following treatment of PANC‐1 and MIA PaCa‐2 cells with negative control (NC) or with TP53TG1 siRNAs. ***P < .001. B, CCK‐8 assay was carried out to determine the growth curves of si‐TP53TG1‐transfected PANC‐1 and MIA PaCa‐2 cells. **P < .01. C, Proliferation in PANC‐1 and MIA PaCa‐2 cells after TP53TG1 knockdown was detected through EdU‐incorporation assays. Proliferating cells were labeled with EdU. Scale bars, 100 μm. ***P < .001. D, Apoptosis was determined following treatment of PANC‐1 and MIA PaCa‐2 cells with NC or si‐TP53TG1 by flow cytometric analysis. *P < .05
To explore whether TP53TG1 could influence progression of PDAC, we analyzed the effect of TP53TG1 expression on the migratory and invasive function of PANC‐1 and MIA PaCa‐2 cells. Wound‐healing assays showed that TP53TG1 downregulation caused a marked reduction of cell migration during closure of an artificial wound created in a confluent cell monolayer (Figure 3A). The assay was carried out in the presence of mitomycin C to block cell proliferation. Using Transwell experiments, we determined the invasion ability of PDAC cells following treatment with siTP53TG1. Upon downregulation of TP53TG1, the invasive abilities of PANC‐1 and MIA PaCa‐2 cells were significantly decreased (Figure 3B). Furthermore, soft agar colony formation assays revealed that TP53TG1 knockdown cells showed significantly decreased anchorage‐independent growth compared with the control group in PANC‐1 and MIA PaCa‐2 cells, as they displayed fewer and smaller colonies formed in soft agar (Figure 3C). Overall, it was concluded that TP53TG1 might have important roles in PDAC development and progression.
Figure 3.
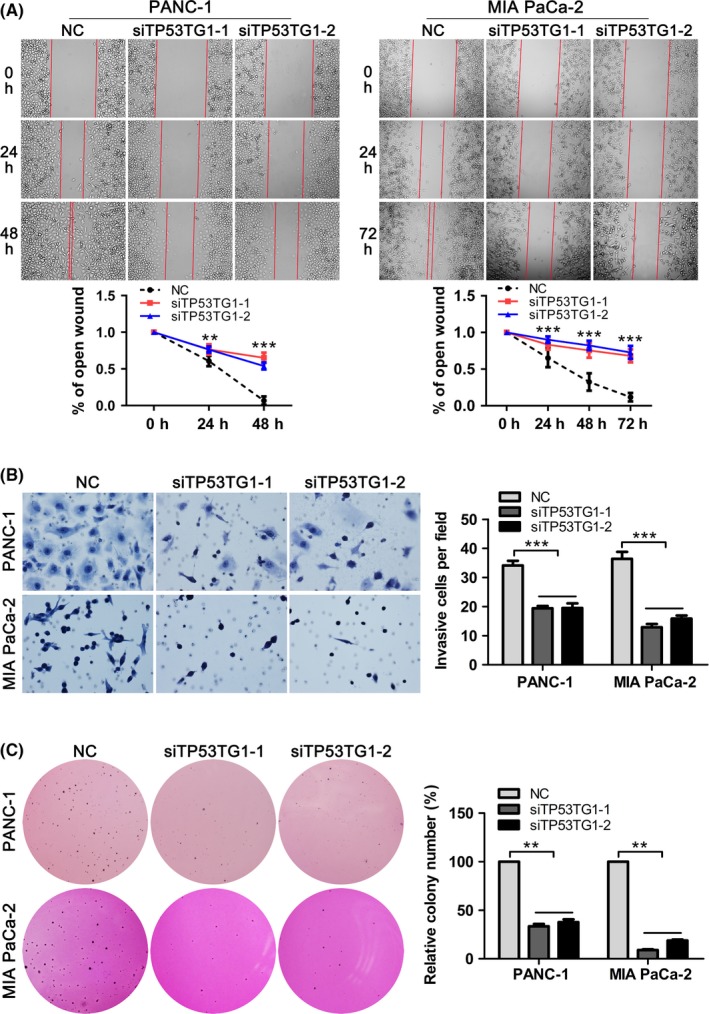
Downregulation of TP53TG1 inhibits pancreatic ductal adenocarcinoma (PDAC) cell migration and invasion. A, Wound‐healing assays were carried out to determine the migratory ability of si‐TP53TG1‐transfected PANC‐1 and MIA PaCa‐2 cells. **P < .01, ***P < .001. B, Effect of TP53TG1 downregulation on the invasive abilities of PANC‐1 and MIA PaCa‐2 cells was measured using Transwell assays. ***P < .001. C, Colony formation assays were carried out to determine the anchorage‐independent growth abilities of PANC‐1 and MIA PaCa‐2 cells after TP53TG1 knockdown. **P < .001. NC, negative control
3.3. Upregulation of TP53TG1 promotes PDAC cell proliferation and invasion
To further investigate the biological significance of TP53TG1 in PDAC, we carried out gain‐of‐function studies in PDAC cells using the pcDNA3.1‐TP53TG1 expression plasmid. PANC‐1 and MIA PaCa‐2 cells were transfected with pcDNA3.1‐TP53TG1 vector or pcDNA3.1 empty vector and TP53TG1 expression was drastically increased after 48 hours of transfection in comparison with the control group (Figure 4A). CCK‐8 assay showed that TP53TG1 overexpression markedly increased the proliferative capacity of PANC‐1 and MIA PaCa‐2 cells (Figure 4B). Edu incorporation to measure cell proliferation yielded similar results in that the percentage of EdU‐incorporating cells in the pcDNA3.1‐TP53TG1‐treated group was significantly increased as compared to the empty vector group (Figure 4C). Moreover, soft agar colony formation analysis showed that overexpression of TP53TG1 also enhanced the non‐anchored growing ability of PDAC cells. The number of colonies formed in pcDNA3.1‐TP53TG1 transfected groups was significantly increased compared with that of the control groups after culturing for 3 weeks (Figure 4D). Furthermore, Transwell assays showed that enhanced expression of TP53TG1 clearly increased the invasive capacity of PDAC cells (Figure 4E). These results showed that TP53TG1 can function as a tumor promoter to enhance the proliferation and progression of PDAC.
Figure 4.
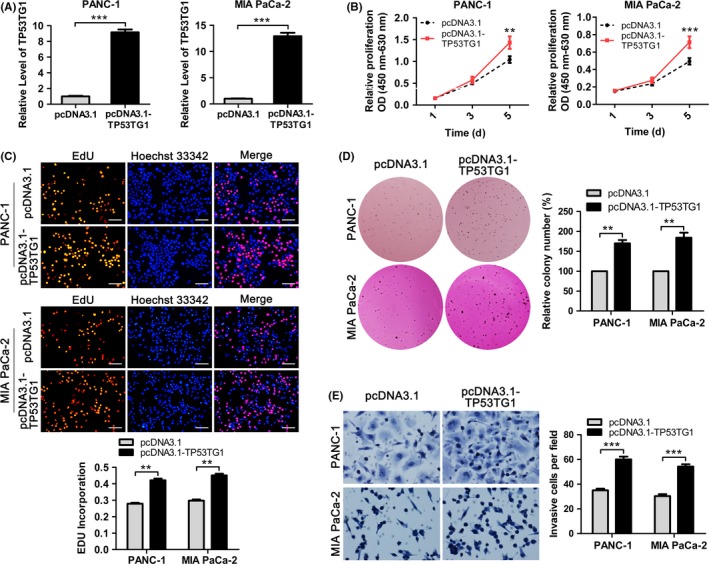
Enforced expression of TP53TG1 promotes the proliferation and invasion of pancreatic ductal adenocarcinoma (PDAC) cells. A, TP53TG1 expression in PANC‐1 and MIA PaCa‐2 cells transfected with pcDNA3.1‐TP53TG1 or empty pcDNA3.1 vector was measured by RT‐qPCR. ***P < .001. B, CCK‐8 assay was carried out to determine the growth curves of PANC‐1 and MIA PaCa‐2 cells after transfection with pcDNA3.1‐TP53TG1 or empty pcDNA3.1 vector. **P < .01, ***P < .001. C, Proliferation in PANC‐1 and MIA PaCa‐2 cells after TP53TG1 overexpression was detected through EdU‐incorporation assays. Proliferating cells were labeled with EdU. Scale bars, 100 μm. **P < .01. D, Effect of TP53TG1 upregulation on the anchorage‐independent growth capacity of PANC‐1 and MIA PaCa‐2 cells was determined using colony formation assays. **P < .01. E, Transwell assays were carried out to determine the invasive ability of PANC‐1 and MIA PaCa‐2 cells after upregulation of TP53TG1. ***P < .001
3.4. TP53TG1 is negatively regulated by miR‐96
Recent studies have shown that lncRNAs may function through binding specific miRNAs. To examine whether TP53TG1 has a similar mechanism, we used DIANA‐LncBase and miRcode to invest TP53TG1‐miRNA interactions,28, 29 and several miRNAs were predicted to be bound to TP53TG1 with high scores. We transfected PDAC cells with mimics of these miRNAs, and miR‐96, a tumor suppressor gene in various types of cancer,30, 31 was chosen for further studies given its most significant effect on TP53TG1 expression (Figure S2). The predicted binding sites of miR‐96 to the TP53TG1 sequence are illustrated in Figure 5A. RT‐qPCR showed that overexpression of miR‐96 significantly reduced TP53TG1 expression (>50%) in PANC‐1 and MIA PaCa‐2 cell lines (Figure 5B), showing the negative regulation of TP53TG1 by miR‐96. Next, we examined the cellular localization of TP53TG1 in PDAC cell lines by in situ hybridization and the result showed that the majority of TP53TG1 signal was located in the cell cytoplasm, while a small amount of punctate staining was detected in the nucleus. Reduction of in situ hybridization signal was observed in PDAC cells transfected with TP53TG1‐targeted siRNA, which supported that the probe was specifically detecting the TP53TG1 transcript (Figure 5C). Moreover, TP53TG1 and miR‐96 expression in nuclear and cytosolic fractions from PANC‐1 and MIA PaCa‐2 cells were measured by RT‐qPCR. Differential enrichments of GAPDH and MALAT1 RNA were used as fractionation indicators. We observed a considerable increase in TP53TG1 and miR‐96 expression in the cytoplasm versus the nucleus (Figure 5D), thus further confirming that TP53TG1 and miR‐96 were both mainly localized in the cytoplasm. This provided a prerequisite for reciprocal interaction between TP53TG1 and miR‐96.
Figure 5.
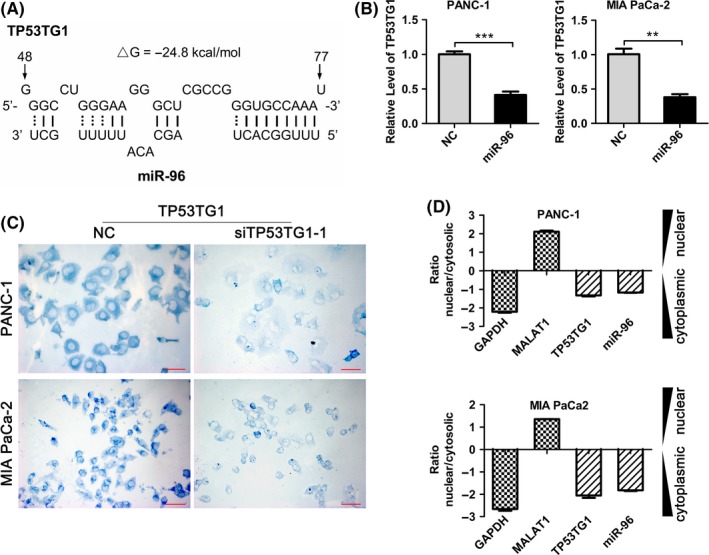
Long noncoding RNA (lncRNA) TP53TG1 is negatively regulated by microRNA (miR)‐96. A, Predicted binding sites for miR‐96 in TP53TG1 sequences. Numbers show the nucleotides relative to the transcriptional start site of TP53TG1. B, PANC‐1 and MIA PaCa‐2 cells were transfected with miR‐96 mimic and, 48 h later, total RNA was isolated and the expression of TP53TG1 was analyzed by RT‐qPCR. **P < .01, ***P < .001. C, Localization of TP53TG1 in PANC‐1 and MIA PaCa‐2 cells was determined by RNA‐in situ hybridization assays. The signal of TP53TG1 is stained blue. Scale bars, 50 μm. D, RT‐qPCR detection of the percentage of TP53TG1, miR‐96, GAPDH and MALAT1 in the cytoplasmic and nuclear fractions of PANC‐1 and MIA PaCa‐2 cells. GAPDH and MALAT1 serve as cytoplasmic and nuclear localization indicators, respectively
3.5. miR‐96 competitively binds to TP53TG1 and KRAS
Our previous study showed that miR‐96 functions as a tumor‐suppressing miRNA in PDAC and directly targets the KRAS oncogene.31 According to the prediction results, there are putative conserved binding sites for miR‐96 on both TP53TG1 and the 3′UTR of KRAS mRNA (Figure 6A). To test whether miR‐96 directly binds to TP53TG1 and KRAS, the full‐length of TP53TG1 or 3′UTR of KRAS were inserted into the pmirGLO dual luciferase reporter vector, which contains wild‐type (WT) or mutated‐type (MUT) miR‐96 binding sites (Figure 6A). Results of luciferase reporter assays demonstrated that miR‐96 mimics significantly reduced the luciferase activities of wild‐type TP53TG1 and KRAS reporter vector, respectively. However, luciferase activities in cells transfected with mutated‐type TP53TG1 and KRAS vector were almost comparable to that of control cells (Figure 6B). These data suggested that miR‐96 is able to directly bind to TP53TG1 and KRAS. Previous studies showed that miRNAs exert their gene‐silencing functions through the RNA induced silencing complex (RISC).32 Ago2 is an essential component of RISC complex necessary for siRNA or miRNA‐mediated gene silencing. We carried out RNA immunoprecipitation (RIP) assays with Ago2 antibody and observed a significant enrichment of miR‐96, TP53TG1 and KRAS by Ago2 antibody compared with the IgG control (Figure 6C), indicating that miR‐96 may recruit TP53TG1 and KRAS to the miR‐96‐RISC complex. Furthermore, RT‐qPCR analysis showed that KRAS is significantly more highly expressed in PDAC tissues compared with adjacent normal pancreatic tissues, which is consistent with the expression trend of TP53TG1, and there is a significant positive correlation between TP53TG1 and KRAS expression (Figure 6D). Moreover, RT‐qPCR analysis showed that, whereas TP53TG1 and KRAS were both upregulated in PDAC tissues, miR‐96 was downregulated in the same tumor specimens, resulting in a significant inverse correlation between miR‐96 and TP53TG1, and between miR‐96 and KRAS expression (Figure 6E). Collectively, these results suggested that miR‐96 induces post‐transcriptional silencing of KRAS or TP53TG1 by directly binding to KRAS mRNA 3′UTR or TP53TG1 specific sites. Further, the results also suggested that there is competition for miR‐96 between KRAS and TP53TG1.
Figure 6.
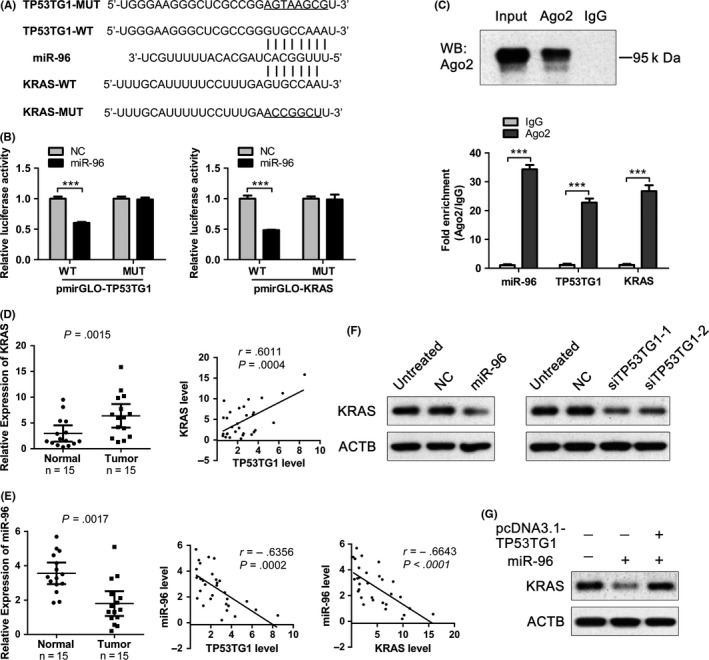
TP53TG1 acts as a competing endogenous RNA to regulate KRAS expression by sponging microRNA (miR)‐96. A, Bioinformatics analysis shows that there are putative binding sites for miR‐96 on both TP53TG1 and the 3′UTR of KRAS mRNA and mutation was generated in the complementary site for the seed region of miR‐96. B, Luciferase activity in PANC‐1 cells cotransfected with miR‐96 mimics and wild type (WT) or mutant (MUT) TP53TG1 or KRAS luciferase reporters was measured using a dual‐luciferase reporter gene assay system. ***P < .001. C, Upper panel: Immunoprecipitation of Ago2 protein by Ago2 or IgG antibody was detected by western blot assay. Lower panel: RIP followed by RT‐qPCR was used to detect miR‐96, TP53TG1 and KRAS endogenously associated with Ago2. Expression level of miR‐96, TP53TG1 and KRAS was normalized to that in the IgG antibody group. ***P < .001. D‐E, Expression of KRAS and miR‐96 in pancreatic ductal adenocarcinoma tissues and corresponding normal pancreatic tissues was detected by RT‐qPCR, and the associations between expression levels of KRAS, miR‐96 and TP53TG1 were determined by Pearson correlation analysis. F, Effect of miR‐96 mimic or TP53TG1‐siRNAs on the expression of KRAS in PANC‐1 cells was detected by western blot. G, Effect of cotransfected PANC‐1 cells with TP53TG1 expression plasmid and miR‐96 mimic on the expression of KRAS was detected by western blot
3.6. TP53TG1 functions as a ceRNA to regulate KRAS expression by sponging miR‐96
Mutated KRAS was proposed to be a hallmark of PDAC as this protein is mutated in more than 90% of PDAC cases. KRAS mutation was also found in almost all PDAC cell lines, but BxPC‐3 was determined to be wild‐type.33 Knockdown of TP53TG1 in KRAS mutant (PANC‐1, MIA PaCa‐2) and wild type (BxPC‐3) cell lines showed the reducing proliferative and invasive ability of these cells (Figures 2, 3 and S3). Moreover, the effect of KRAS knockdown in KRAS mutant (PANC‐1) and wild type (BxPC‐3) cell lines has been analyzed, and the results showed that KRAS downregulation significantly inhibited the proliferative and invasive capacity in both cell lines (Figure S4), which is consistent with the antitumor effect of TP53TG1 knockdown in the two cell lines. miR‐96 has been reported as a tumor suppressor gene in PDAC by directly targeting oncogene KRAS. We examined KRAS expression after transfection with miR‐96 mimic and western blot analysis showed that overexpression of miR‐96 resulted in a significant reduction in KRAS expression, confirming that KRAS is a target of miR‐96 (Figure 6F). Then we transfected PANC‐1 cells with TP53TG1 siRNAs and found that knockdown of TP53TG1 significantly suppressed KRAS expression, which was consistent with the results of forced expression of miR‐96 (Figure 6F). In contrast, we cotransfected PANC‐1 cells with TP53TG1 expression plasmid and miR‐96 mimic and the expression of KRAS was partly rescued by overexpression of TP53TG1 compared with the miR‐96 group (Figure 6G), suggesting that TP53TG1 could partly abolish the silencing effect of miR‐96 on KRAS. Collectively, these results indicated that TP53TG1 could upregulate the miRNA‐96 target gene KRAS expression by competitively ‘sponging’ miRNA‐96.
4. DISCUSSION
Recent reports showed that TP53TG1 was either overexpressed or underexpressed, exerting oncogenic or tumor‐suppressing functions in different tumors.24, 25, 26 In the present study, we identified the function of TP53TG1 in PDAC by applying loss‐of‐function and gain‐of‐function approaches. We found that knockdown of TP53TG1 led to decreased proliferation, migration and invasion of PDAC cells, whereas ectopic expression of TP53TG1 promoted malignant activity. These findings suggest that TP53TG1 may play a critical role in the promotion of multiple oncogenic properties in PDAC during initial development and progression.
Results of RNA‐seq (BioProject: PRJEB4337), which was carried out on tissue samples from 95 human individuals representing 27 different tissues, showed that the expression of TP53TG1 was second highest in normal colon and lowest in normal pancreas tissues (Figure S5). We speculated that the different functions of TP53TG1 in gastrointestinal cancer and PDAC may be partly attributable to its tissue‐specific expression pattern. However, further study is required to elucidate the multiple roles of this lncRNA in tumors.
Previous studies have shown that the mechanism of lncRNAs in human disease is associated with their cellular localization. lncRNAs located in the nucleus could interact with chromatin modifiers and specific genomic loci, whereas lncRNAs located in the cytoplasm can serve as miRNA sponges to regulate the expression of miRNA target genes through competition for shared miRNA response elements.34 In the present study, subcellular localization analysis of TP53TG1 by RNA fluorescence in situ hybridization assay showed that TP53TG1 was mainly localized in the cytoplasm, so we hypothesized that TP53TG1 may also serve as a ceRNA.
The target prediction algorithm indicated the potential existence of specific crosstalk between TP53TG1 and KRAS through competitive binding with miR‐96. Our previous reports have shown that miR‐96 targets KRAS in PDAC. The present study confirmed that miR‐96 can bind KRAS and TP53TG1 directly at the “seed site” though dual luciferase reporter assays. Moreover, RIP assays with Ago2 antibody indicated that miR‐96 may recruit TP53TG1 and KRAS to the miR‐96‐RISC complex. Additionally, western blot analysis showed that knockdown of TP53TG1 was sufficient to suppress KRAS expression, which was consistent with the results of forced expression of miR‐96. In contrast, ectopic expression of TP53TG1 could partly abolish the silencing effect of miR‐96 on KRAS. Collectively, our results provide a ceRNA model including TP53TG1, miR‐96 and KRAS in PDAC. TP53TG1, which shared miR‐96 response elements with KRAS, regulated the expression of KRAS though sponging miR‐96.
Activating mutations in the KRAS oncogene are found in a broad range of human cancers including pancreatic, colorectal, lung and urachal carcinoma, which is widely considered essential for carcinogenesis.35, 36, 37, 38, 39 Although research shows that KRAS mutations occur in approximately 90%‐95% of PDAC cases,40 which act as a key event in the initiation and maintenance of PDAC,41 some results further imply that KRAS overexpression is also valuable in the process of tumorigenesis. Zeng et al42 demonstrated that siRNA‐directed KRAS silencing caused defective abilities of proliferation and invasion of pancreatic cancer cells. Moreover, miR‐217 and miR‐193b were found as inhibitors in the development of PDAC by suppressing KRAS expression. In the present study, we further provide evidence that TP53TG1 can function as ceRNA to upregulate KRAS expression through competitive combination with miR‐96, thus promoting PDAC cell proliferation and invasion.
Although ceRNA research is in its initial stages, accumulating data indicate that this is a new dimension of post‐transcriptional gene regulation in which RNA transcripts control miRNA activity. This effect has profound implications for cancer initiation and progression. For example, lncRNA DANCR promotes osteosarcoma development by acting as a molecular sponge of miR‐335‐5p and miR‐1972.43 Similarly, lncRNA‐UCA1 exerts oncogenic functions in non‐small cell lung cancer, acting mechanistically by upregulating ERBB4 in part through sponging miR‐193a‐3p.44 Additionally, lncRNA SNHG6‐003 could upregulate TAK1 expression by sequestering miR‐26a/b and promoting the progression of hepatocellular carcinoma.45 Our present study showed that ceRNA‐associated KRAS modulation is involved in the carcinogenesis of PDAC.
In summary, our study identifies that lncRNA TP53TG1 is upregulated in PDAC and contributes to the growth and progression of PDAC. TP53TG1 operates as a sponge for miR‐96 to weaken the suppressive effect of miR‐96 on KRAS, and thus increases the expression of KRAS. Modulation of KRAS activity by ceRNA will provide new insights into the mechanisms underlying PDAC tumorigenesis.
DISCLOSURE
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This study was supported by the Natural Science Foundation of Hebei province, China (H2017206022 to Haiyan Yang), the National Nature Science Foundations of China (81702299 to Haiyan Yang, 81472326 to Jie Chen) and the Scientific Research Foundation from Public Health Department of Hebei province, China (20170556 to Yufeng Zhang).
Zhang Y, Yang H, Du Y, et al. Long noncoding RNA TP53TG1 promotes pancreatic ductal adenocarcinoma development by acting as a molecular sponge of microRNA‐96. Cancer Sci. 2019;110:2760‐2772. 10.1111/cas.14136
Zhang and Yang contributed equally to this study.
Contributor Information
Xiaoying Xue, Email: xxy0636@163.com.
Jie Chen, Email: xhblk@163.com.
Xianghong Zhang, Email: zhangxianghong2008@163.com.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Yeo TP, Lowenfels AB. Demographics and epidemiology of pancreatic cancer. Cancer J. 2012;18:477‐484. [DOI] [PubMed] [Google Scholar]
- 3. Elgar G, Vavouri T. Tuning in to the signals: noncoding sequence conservation in vertebrate genomes. Trends Genet. 2008;24:344‐352. [DOI] [PubMed] [Google Scholar]
- 4. Guttman M, Amit I, Garber M, et al. Chromatin signature reveals over a thousand highly conserved large non‐coding RNAs in mammals. Nature. 2009;458:223‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904‐914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mercer TR, Dinger ME, Mattick JS. Long non‐coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155‐159. [DOI] [PubMed] [Google Scholar]
- 8. Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guttman M, Donaghey J, Carey BW, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang JL, Cao SW, Ou QS, et al. The long non‐coding RNA PTTG3P promotes cell growth and metastasis via up‐regulating PTTG1 and activating PI3K/AKT signaling in hepatocellular carcinoma. Mol Cancer. 2018;17:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li W, Zheng J, Deng J, et al. Increased levels of the long intergenic non‐protein coding RNA POU3F3 promote DNA methylation in esophageal squamous cell carcinoma cells. Gastroenterology. 2014;146:1714‐1726. e1715. [DOI] [PubMed] [Google Scholar]
- 12. Huang K, Geng J, Wang J. Long non‐coding RNA RP11‐552M11.4 promotes cells proliferation, migration and invasion by targeting BRCA2 in ovarian cancer. Cancer Sci. 2018;109:1428‐1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zheng S, Chen H, Wang Y, et al. Long non‐coding RNA LOC389641 promotes progression of pancreatic ductal adenocarcinoma and increases cell invasion by regulating E‐cadherin in a TNFRSF10A‐related manner. Cancer Lett. 2016;371:354‐365. [DOI] [PubMed] [Google Scholar]
- 14. Zheng J, Huang X, Tan W, et al. Pancreatic cancer risk variant in LINC00673 creates a miR‐1231 binding site and interferes with PTPN11 degradation. Nat Genet. 2016;48:747‐757. [DOI] [PubMed] [Google Scholar]
- 15. Zhan HX, Wang Y, Li C, et al. LincRNA‐ROR promotes invasion, metastasis and tumor growth in pancreatic cancer through activating ZEB1 pathway. Cancer Lett. 2016;374:261‐271. [DOI] [PubMed] [Google Scholar]
- 16. Sun YW, Chen YF, Li J, et al. A novel long non‐coding RNA ENST00000480739 suppresses tumour cell invasion by regulating OS‐9 and HIF‐1alpha in pancreatic ductal adenocarcinoma. Br J Cancer. 2014;111:2131‐2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang H, Liu P, Zhang J, et al. Long noncoding RNA MIR31HG exhibits oncogenic property in pancreatic ductal adenocarcinoma and is negatively regulated by miR‐193b. Oncogene. 2016;35:3647‐3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu XH, Sun M, Nie FQ, et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR‐331‐3p in gastric cancer. Mol Cancer. 2014;13:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao W, Lin S, Cheng C, et al. Long non‐coding RNA CASC2 regulates Sprouty2 via functioning as a competing endogenous RNA for miR‐183 to modulate the sensitivity of prostate cancer cells to docetaxel. Arch Biochem Biophys. 2019;665:69‐78. 10.1016/j.abb.2018.01.013 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 20. Jiang X, Zhou Y, Sun AJ, Xue JL. NEAT1 contributes to breast cancer progression through modulating miR‐448 and ZEB1. J Cell Physiol. 2018;233:8558‐8566. [DOI] [PubMed] [Google Scholar]
- 21. Xie CR, Wang F, Zhang S, et al. Long noncoding RNA HCAL facilitates the growth and metastasis of hepatocellular carcinoma by acting as a ceRNA of LAPTM4B. Mol Ther Nucleic Acids. 2017;9:440‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu H, Zhou C. Long non‐coding RNA UCA1 promotes lung cancer cell proliferation and migration via microRNA‐193a/HMGB1 axis. Biochem Biophys Res Commun. 2018;496:738‐745. [DOI] [PubMed] [Google Scholar]
- 23. Takei Y, Ishikawa S, Tokino T, Muto T, Nakamura Y. Isolation of a novel TP53 target gene from a colon cancer cell line carrying a highly regulated wild‐type TP53 expression system. Genes Chromosom Cancer. 1998;23:1‐9. [PubMed] [Google Scholar]
- 24. Diaz‐Lagares A, Crujeiras AB, Lopez‐Serra P, et al. Epigenetic inactivation of the p53‐induced long noncoding RNA TP53 target 1 in human cancer. Proc Natl Acad Sci USA. 2016;113:E7535‐E7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen X, Gao Y, Li D, Cao Y, Hao B. LncRNA‐TP53TG1 participated in the stress response under glucose deprivation in glioma. J Cell Biochem. 2017;118:4897‐4904. [DOI] [PubMed] [Google Scholar]
- 26. Yuan J, Jiang YY, Mayakonda A, et al. Super‐enhancers promote transcriptional dysregulation in nasopharyngeal carcinoma. Cancer Res. 2017;77:6614‐6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W. CPAT: Coding‐Potential Assessment Tool using an alignment‐free logistic regression model. Nucleic Acids Res. 2013;41:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paraskevopoulou MD, Georgakilas G, Kostoulas N, et al. DIANA‐LncBase: experimentally verified and computationally predicted microRNA targets on long non‐coding RNAs. Nucleic Acids Res. 2013;41:D239‐D245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeggari A, Marks DS, Larsson E. miRcode: a map of putative microRNA target sites in the long non‐coding transcriptome. Bioinformatics. 2012;28:2062‐2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yao Q, Pei Y, Zhang X, Xie B. microRNA‐96 acts as a tumor suppressor gene in human osteosarcoma via target regulation of EZRIN. Life Sci. 2018;203:1‐11. [DOI] [PubMed] [Google Scholar]
- 31. Yu S, Lu Z, Liu C, et al. miRNA‐96 suppresses KRAS and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res. 2010;70:6015‐6025. [DOI] [PubMed] [Google Scholar]
- 32. Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631‐640. [DOI] [PubMed] [Google Scholar]
- 33. Deer EL, Gonzalez‐Hernandez J, Coursen JD, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bournet B, Muscari F, Buscail C, et al. KRAS G12D mutation subtype is a prognostic factor for advanced pancreatic adenocarcinoma. Clin Transl Gastroenterol. 2016;7:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Won DD, Lee JI, Lee IK, Oh ST, Jung ES, Lee SH. The prognostic significance of KRAS and BRAF mutation status in Korean colorectal cancer patients. BMC Cancer. 2017;17:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dietz S, Harms A, Endris V, et al. Spatial distribution of EGFR and KRAS mutation frequencies correlates with histological growth patterns of lung adenocarcinomas. Int J Cancer. 2017;141:1841‐1848. [DOI] [PubMed] [Google Scholar]
- 38. Hang JF, Pan CC. Response to: Absence of GNAS and BRAF mutations but presence of KRAS mutation in urachal adenocarcinoma: author reply. Pathology. 2017;49:562‐563. [DOI] [PubMed] [Google Scholar]
- 39. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457‐2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zeng L, Li J, Wang Y, et al. Combination of siRNA‐directed Kras oncogene silencing and arsenic‐induced apoptosis using a nanomedicine strategy for the effective treatment of pancreatic cancer. Nanomedicine. 2014;10:463‐472. [DOI] [PubMed] [Google Scholar]
- 43. Wang Y, Zeng X, Wang N, et al. Long noncoding RNA DANCR, working as a competitive endogenous RNA, promotes ROCK1‐mediated proliferation and metastasis via decoying of miR‐335‐5p and miR‐1972 in osteosarcoma. Mol Cancer. 2018;17:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nie W, Ge HJ, Yang XQ, et al. LncRNA‐UCA1 exerts oncogenic functions in non‐small cell lung cancer by targeting miR‐193a‐3p. Cancer Lett. 2016;371:99‐106. [DOI] [PubMed] [Google Scholar]
- 45. Cao C, Zhang T, Zhang D, et al. The long non‐coding RNA, SNHG6‐003, functions as a competing endogenous RNA to promote the progression of hepatocellular carcinoma. Oncogene. 2017;36:1112‐1122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials