

Abstract
The non‐canonical inflammasome mediates pyroptotic cell death in response to bacterial lipopolysaccharide (LPS) found in the cytosol. Understanding the mechanism and regulation of this system is of great interest, given its central role in mouse models of bacterial septic shock. In this issue of EMBO Reports, Benaoudia and colleagues sought to discover extra players in the human non‐canonical inflammasome using a CRISPR library screen; the only strongly positive hit apart from the known components caspase‐4 and gasdermin D was interferon regulatory factor‐2 (IRF2) [1]. IRF2 was found to be a transcriptional activator of caspase‐4, and in its absence, induction of IRF1 could substitute to maintain caspase‐4 expression.
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Immunology; Microbiology, Virology & Host Pathogen Interaction
The canonical inflammasomes are protein complexes that activate caspase‐1 in response to a wide variety of microbial products as well as endogenous danger signals 2. Caspase‐1 then cleaves gasdermin D and the precursor forms of proinflammatory cytokines IL‐1β and IL‐18, resulting in cell death and inflammation 2. The non‐canonical inflammasome was described more recently and involves direct recognition of cytosolic LPS by the caspase recruitment domains (CARD) of the other inflammatory caspases—caspase‐4 and caspase‐5 in humans and caspase‐11 in mice 3, 4. This promotes caspase dimerisation and activation followed by the cleavage of gasdermin D 2, 3, 4. The N‐terminal fragment of gasdermin D then oligomerises to form membrane pores, leading to pyroptosis—a rapid lytic cell death. The membrane damage inflicted by gasdermin D pores additionally activates the NLRP3 canonical inflammasome via K+ efflux 3, 4, 5, 6. The result is cell death to remove a possible replicative niche for a bacterium, as well as inflammation to promote clearance of infection.
Benaoudia et al 1 identified IRF2 as a regulator of caspase‐4 levels in human U937 promonocytic cells, using a genome‐wide CRISPR‐Cas9 screen for effects on pyroptosis in response to electroporated LPS. IRF2 knockout cell lines had dramatically reduced cell death in response to cytosolic LPS from F. novicidia or E. coli, and the pyroptosis was restored by reconstituting IRF2 expression. Caspase‐4, as well as caspase‐1, expression was induced by IRF2 at the mRNA and protein levels. Analysis of IRF2 ChIP‐Seq (chromatin immunoprecipitation coupled to high‐throughput sequencing) data from primary human monocytes identified IRF2 binding in the promoters of CASP4 and CASP1 genes, indicating direct regulation. In support of this, LPS‐dependent pyroptosis was restored in IRF2 knockout cells by expressing CASP4 under a constitutive promoter. These results indicate that under steady‐state conditions, IRF2 directly controls the expression of caspase‐4 which is required for the pyroptotic cell death response to electroporated LPS (Fig 1).
Figure 1. IRF1 and IRF2 regulation of inflammasome components in humans and mice.
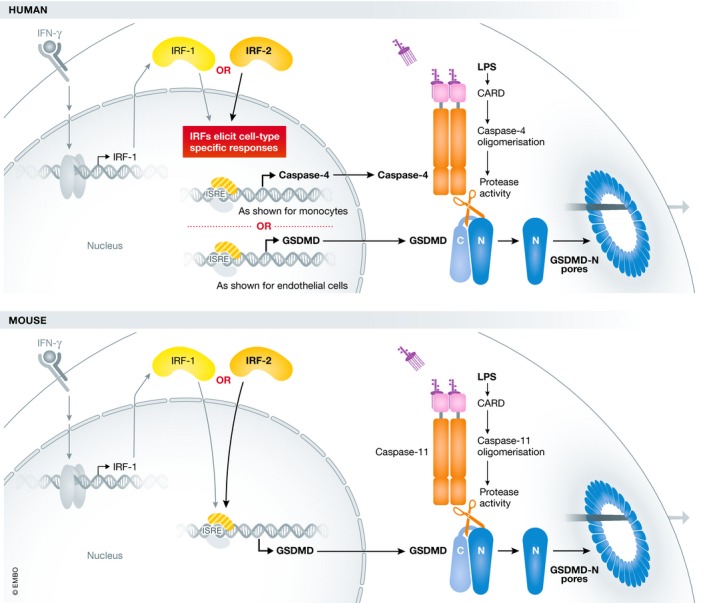
Cytosolic LPS, sensed by caspase‐4/‐5 in humans and caspase‐11 in mice, activates the non‐canonical inflammasome. This leads to the cleavage of gasdermin D and subsequent formation of pores in the plasma membrane. IRF2 transcription factor directly regulates the expression levels of gasdermin D in mouse macrophages and either gasdermin D or caspase‐4 in humans in a cell type‐specific manner. In the presence of IFN‐γ, IRF1 can compensate for IRF2 deficiency and induce caspase‐4 or gasdermin D expression.
In the presence of interferon‐γ (IFN‐γ), mimicking conditions of infection, IRF1 compensated for IRF2 deficiency in regulating the cell death response to LPS transfection in U937 monocytes and PMA‐differentiated U937 macrophages. Macrophage death in response to bacterial infections is more complex than when LPS is delivered by electroporation, and can involve multiple pathways. However, cell death and IL‐1β release in response to infection by E. coli and F. novicida was largely abolished in IRF1/2 double knockout cells 1. IRF2 was originally identified as a competitive inhibitor of IRF1 function and negative regulator of type I IFN 7. However, a cooperative role between these transcription factors is not unprecedented. IRF1 and IRF2 have been shown to both positively regulate Tlr3 expression, with IRF2 controlling basal expression levels and IRF1 upregulating expression upon IFN stimulation 8, similar to the situation for caspase‐4.
Interestingly, other recently published work found a role for IRF1 and IRF2 in the non‐canonical inflammasome in mouse and human, but via action on the gasdermin D promoter 9. Kayagaki et al 9 first used an ENU mutagenesis forward genetic screen to describe a role for IRF2 in regulating the mouse non‐canonical inflammasome. In mouse macrophages, IRF2 induced the expression of gasdermin D but not caspase‐11, which is inducibly expressed in response to type I IFN 2. Macrophages from Irf2 −/− mice exhibited defective responses to canonical and non‐canonical inflammasome stimuli, comparable to Gsdmd −/− cells 9. Again through ChIP‐Seq analysis, an IRF2 binding motif was identified in the Gsdmd promoter, and was essential for expression in macrophages. These results could explain the older observation that Irf2 −/− mice are protected from LPS‐induced septic shock 10.
Kayagaki et al 9 also examined the effect of IRF2 deficiency in the human EA.hy926 hybrid endothelial cell line. In contrast to the results of Benaoudia and colleagues, it was found that IRF2 induced gasdermin D but not caspase‐4. Both studies agree on the compensatory role of induced IRF1 when IRF2 is absent; however, deficiency of both IRF1 and IRF2 still did not affect caspase‐4 abundance in the EA.hy926 cells. The IRF2 binding motif identified in the promoter region of gasdermin D 9 is conserved in humans and mice, yet IRF2 was not required for gasdermin D expression in U937 cells 1. Benaoudia et al validated the role of IRF2 as a regulator of caspase‐4 expression in macrophages differentiated from human‐induced pluripotent cells. However, unlike the case with U937 cells, IRF2 knockout did not affect caspase‐1 expression levels. These convincing but contrasting results (Table 1) support potential cell type‐specific effects of IRF2 on different promoter elements. The explanation for the differences seen may lie in both promoter‐specific and cell type‐specific interactions between IRFs and other transcription factors.
Table 1.
Inflammasome gene induction by IRF1/2 varies with cell type
These studies demonstrate the key role of IRF2 and IRF1 as regulators of inflammasome protein expression and, subsequently, the function of these pathways in both humans and mice. Further investigation will be required to explain cell type or species variations in the specific non‐canonical inflammasome components induced by IRF2. The fact that this screen found no other strong hits means that this remains a surprisingly simple system, in which caspase‐4 acts both as LPS receptor and effector protein.
Acknowledgement
KJS is supported by NHMRC Senior Research Fellowship 1059729.
EMBO Reports (2019) 20: e48891
See also: S Benaoudia et al (September 2019)
References
- 1. Benaoudia S, Martin A, Gamez MP et al (2019) EMBO Rep 20: e48235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rathinam VAK, Zhao Y, Shao F (2019) Nat Immunol 20: 527–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kayagaki N, Stowe IB, Lee BL et al (2015) Nature 526: 666–671 [DOI] [PubMed] [Google Scholar]
- 4. Shi J, Zhao Y, Wang K et al (2015) Nature 526: 660–665 [DOI] [PubMed] [Google Scholar]
- 5. Ruhl S, Broz P (2015) Eur J Immunol 45: 2927–3296 [DOI] [PubMed] [Google Scholar]
- 6. Schmid‐Burgk JL, Gaidt MM, Schmidt T et al (2015) Eur J Immunol 45: 2911–2917 [DOI] [PubMed] [Google Scholar]
- 7. Harada H, Fujita T, Miyamoto M et al (1989) Cell 58: 729–739 [DOI] [PubMed] [Google Scholar]
- 8. Ren G, Cui K, Zhang Z et al (2015) Cell Biosci 5: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kayagaki N, Lee BL, Stowe IB et al (2019) Sci Signal 12: eaax4917 [DOI] [PubMed] [Google Scholar]
- 10. Cuesta N, Salkowski CA, Thomas KE et al (2003) J Immunol 170: 5739–5747 [DOI] [PubMed] [Google Scholar]