

Abstract
Upon activation, lymphocytes exit quiescence and undergo substantial increases in cell size, accompanied by activation of energy‐producing and anabolic pathways, widespread chromatin decompaction, and elevated transcriptional activity. These changes depend upon prior induction of the Myc transcription factor, but how Myc controls them remains unclear. We addressed this issue by profiling the response to LPS stimulation in wild‐type and c‐myc‐deleted primary mouse B‐cells. Myc is rapidly induced, becomes detectable on virtually all active promoters and enhancers, but has no direct impact on global transcriptional activity. Instead, Myc contributes to the swift up‐ and down‐regulation of several hundred genes, including many known regulators of the aforementioned cellular processes. Myc‐activated promoters are enriched for E‐box consensus motifs, bind Myc at the highest levels, and show enhanced RNA Polymerase II recruitment, the opposite being true at down‐regulated loci. Remarkably, the Myc‐dependent signature identified in activated B‐cells is also enriched in Myc‐driven B‐cell lymphomas: hence, besides modulation of new cancer‐specific programs, the oncogenic action of Myc may largely rely on sustained deregulation of its normal physiological targets.
Keywords: B‐cell, Myc, transcription
Subject Categories: Immunology, Transcription
Introduction
Mature splenic B‐cells can be activated ex vivo to re‐enter the cell cycle and differentiate into antibody‐producing cells, accompanied by massive increases in cell size and RNA content 1, 2, 3, 4, 5. This implies a concomitant intensification of the metabolic pathways needed to provide energy and building blocks for macromolecular biosynthesis and cell growth and, in turn, the necessity for the cells to adapt their transcriptional and translational outputs to the augmented cell size and metabolic activity 6. A key regulator in this overall process is the Myc transcription factor, encoded by the c‐myc proto‐oncogene: indeed, Myc is directly induced by mitogenic signals and, in turn, is thought to orchestrate the plethora of transcriptional changes that foster cell growth and proliferation, as exemplified in cultured mouse fibroblasts 7, 8. In either B or T lymphocytes, c‐myc serves as a direct sensor of activating signals 3, 9, 10, 11, 12, 13 and is required for multiple facets of cellular activation, including metabolic reprogramming, ATP production, ATP‐dependent chromatin decompaction, RNA and biomass accumulation, and cell growth 3, 4, 5, 11, 13, 14, 15, 16, 17, 18. However, how Myc activity impacts on those diverse cellular features remains largely unclear.
Myc binds DNA and activates transcription as a dimer with its partner protein Max 19, 20, 21, but its precise contribution to transcriptional programs in cells has been subject of an intense debate in the field in recent years: while multiple studies indicated that Myc can either activate or repress select target genes 8, 20, 21, 22, 23, 24, others concluded that it acts instead as a general activator—or amplifier—of all expressed genes 3, 25, 26, 27. However, careful scrutiny of the available data 3, 4, 5, 8, 25, 26, 27, 28, 29, 30, 31 lends no formal support to this model, suggesting instead that RNA amplification—when present—is one of the consequences of the metabolic and genomic changes that occur during cellular activation and/or transformation 30, 32. Hence, understanding the precise contribution of Myc to global changes in RNA biology will require a fine mapping of direct, Myc‐dependent transcriptional programs.
The B‐cell system, with its global increase in RNA transcription during cell activation 1, offers a valuable tool to assess the order of events that lead from the triggering of a signaling event to cell cycle entry, cell growth, and RNA amplification, and to understand how these events depend upon prior activation of Myc. Toward this aim, we profiled gene expression along with the genomic distribution of Myc and RNA polymerase II (RNAPII) during B‐cell activation in vitro in wild‐type and c‐myc knockout cells. Our data led to the identification of a specific Myc‐dependent transcriptional program occurring within the first few hours upon cell activation, pre‐setting the stage for the subsequent global increase in metabolic and biosynthetic activities.
Results and Discussion
In order to characterize the contribution of Myc to B‐cell activation, we took advantage of mice homozygous for a conditional c‐myc knockout allele (c‐myc f/f) 33. Freshly purified c‐myc f/f and control c‐myc wt/wt splenic B‐cells were treated with a preparation of cell‐permeable Tat‐Cre recombinase, deleting c‐myc f/f with 70‐80% efficiency (henceforth c‐myc ∆/∆) (Fig EV1A) and preventing the LPS‐induced accumulation of the c‐myc mRNA and protein (Fig EV1B and C, Appendix Fig S1). Chromatin immunoprecipitation (ChIP) analysis confirmed rapid binding of Myc to a known target locus (Ncl) in c‐myc wt/wt cells (Fig EV1D). Ncl and Pus7, another Myc‐dependent mRNAs previously identified in fibroblasts 8, responded to LPS in B‐cells and required Myc for maximal accumulation, from 4 h onward (Fig 1A). While global RNA levels also increased in a Myc‐dependent manner 3, 4, this occurred later (24 h, Fig EV1E) concomitant with increases in bulk RNA synthesis and nuclear size (Fig 1B), overall cell size 2, 4, 5, and S‐phase entry (Fig EV1F) 4. As expected 4, 5, 14, 15, all of the above effects were lost in c‐myc ∆/∆ cells, accompanied by a reduced proliferative response (Fig EV1F and G), residual expansion at 48–72 h resulting from the selection of non‐deleted c‐myc f/f cells (Fig EV1H). Finally, the apoptotic response observable at late time‐points (72 h onwards) was also reduced in c‐myc ∆/∆ cells (Fig EV1I). Hence, c‐myc f/f B‐cells provide a reliable system to address the role of Myc within the first cell division cycle after LPS stimulation.
Figure EV1. Characterization of c‐myc wt/wt and c‐myc ∆/∆ cells.
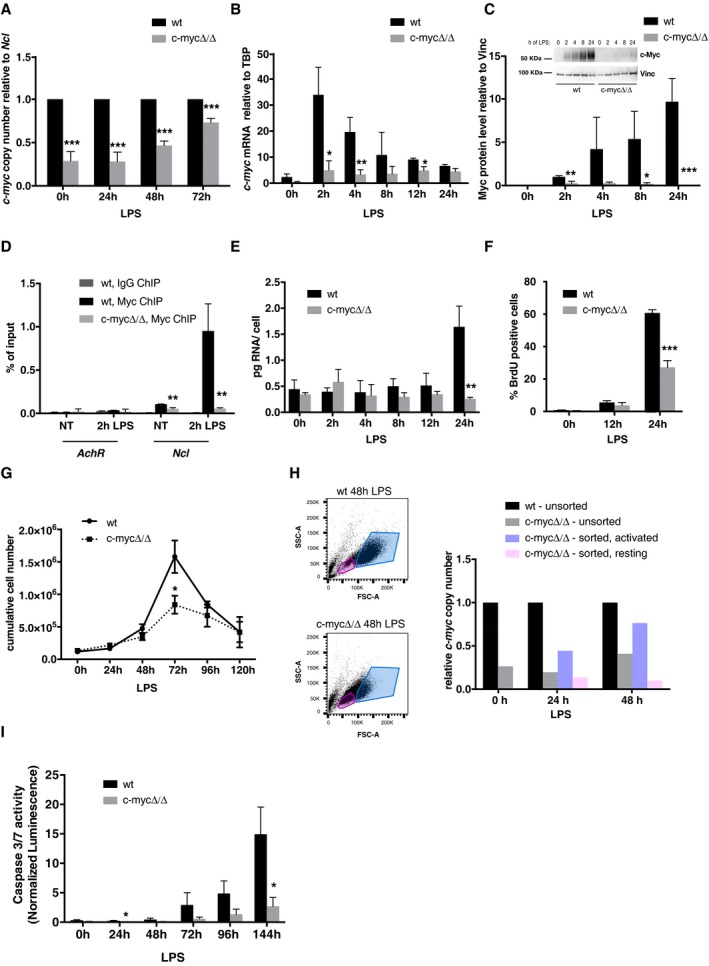
- c‐myc copy number relative to a reference amplicon on the Ncl gene at different time‐points after LPS stimulation in c‐myc wt/wt and c‐myc ∆/∆ cells, as indicated. n = 6 for the t0 and 24 h samples, n = 4 for the 48‐h and 72‐h samples.
- c‐myc mRNA expression (normalized to TBP) at different time‐points after LPS stimulation in c‐myc wt/wt and c‐myc ∆/∆ cells. n = 3.
- Myc protein levels at different time‐points after LPS stimulation in c‐myc wt/wt and c‐myc ∆/∆ cells, based on quantification of four independent immunoblots (each produced with B‐cell lysates obtained from a pool of 3 mice per genotype): a representative blot is shown above the plot. Note that in wild‐type cells, c‐myc mRNA levels peaked 2 h after LPS stimulation 9, while the protein steadily accumulated over time, consistent with post‐transcriptional regulation of its synthesis and/or stability 63, 64: as expected, both mRNA and protein accumulation were blunted in c‐myc ∆/∆ cells.
- Myc and IgG ChIP, as indicated, in non‐treated (NT) or LPS‐treated (2 h) c‐myc wt/wt and c‐myc ∆/∆ B‐cells. PCR primers in the AchR promoter (as a non‐bound control) and in Ncl intron 1 (as a known Myc target with 5 E‐boxes) were used for quantification (% if input) as previously described 39, 65. n = 3.
- Quantification of total RNA levels per cell along the LPS time‐course in c‐myc wt/wt and c‐myc ∆/∆ cells. n = 3.
- Percentages of BrdU‐positive cells at 0, 12, and 24 h, as indicated. n = 3.
- Growth curve of c‐myc wt/wt and c‐myc ∆/∆ cells, as indicated. n = 3.
- c‐myc wt/wt and c‐myc ∆/∆ cells were sorted according to their size and shape (FSC and SSC, respectively) after 24 or 48 h of LPS stimulation: cells with low and high FSC + SSC were defined as resting and activated, respectively, and were purified by FACS sorting prior to qPCR quantification of c‐myc copy number, alongside unsorted c‐myc ∆/∆ and c‐myc wt/wt control samples. Results from a representative experiment are shown. The experiment was repeated twice with similar results.
- Caspase‐3/7 activity normalized on cell numbers along the LPS time‐course in c‐myc wt/wt and c‐myc ∆/∆ cells. n = 3.
Figure 1. Myc is required for the regulation of a subset of LPS‐responsive genes during B‐cells activation.
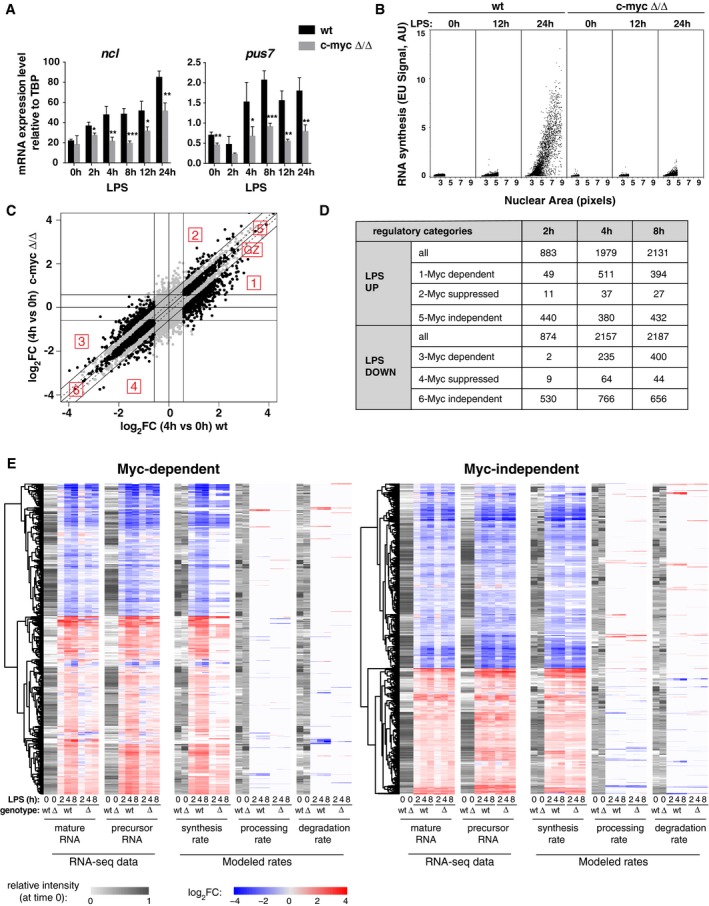
- Ncl and Pus7 mRNA levels (normalized to TBP) at different time‐points after LPS stimulation in c‐myc wt/wt and c‐myc ∆/∆ cells. Data are presented as mean ± SD; n = 3.
- To quantify RNA synthesis, we exposed our cultures to a brief pulse of ethyl‐uridine (EU) and measured its incorporation into RNA by light microscopy. The scatter plots show the Nuclear Area as detected in the 2D plane (x‐axis, in pixels) versus the EU signal (y‐axis, as arbitrary units, AU) for each single cell identified by DAPI staining in c‐myc wt/wt and c‐myc ∆/∆ populations at the indicated time‐points after LPS treatment. One representative experiment out of 5 is shown.
- Variation of each expressed mRNA at 4 h of LPS treatment (log2FC, relative to time 0) for c‐myc wt/wt (x‐axis) and c‐myc ∆/∆ cells (y‐axis), as measured by RNA‐Seq. Regulatory groups 1–6 and gray zone (GZ) are as defined in the text.
- Numbers of genes classified in the different regulatory categories at the indicated time‐points after LPS stimulation.
- Variations (log2FC) in mature and precursor RNAs (as measured by RNA‐Seq) and in the modeled rates (RNA synthesis, processing, and degradation) for Myc‐dependent and Myc‐independent genes. The gray scale represents the starting level for each parameter in unstimulated cells.
We used RNA‐Seq to profile the Myc‐dependent transcriptional response shortly after LPS stimulation (2, 4, and 8 h): Based on a ≥ 1.5 fold‐change to call differentially expressed genes (DEGs: log2FC ≥ 0.58; qval ≤ 0.05), LPS induced the rapid up‐ and down‐regulation of several thousand mRNAs (Fig EV2A). We defined Myc‐regulated genes as those for which the magnitude of the LPS response was reduced by at least 1.5‐fold in c‐myc ∆/∆ relative to c‐myc wt/wt cells (groups 1–4, Fig 1C and D, Dataset EV1 and EV2): Among these, the most abundant were Myc‐dependent LPS‐induced and repressed genes, both showing dampened responses in c‐myc ∆/∆ cells (groups 1 and 3), while much fewer mRNAs showed reinforced responses (groups 2 and 4). On the other hand, significant fractions of all mRNAs showed Myc‐independent up‐ or down‐regulation by LPS (altered ≤ 1.15‐fold in c‐myc ∆/∆ relative to c‐myc wt/wt cells; groups 5, 6; Fig 1C and D, Dataset EV1 and EV2). As expected, other genes fell in what we qualify as the “gray zone”, with intermediate levels of Myc dependency (Fig 1C). In line with the kinetics of Myc accumulation, Myc‐dependent responses were rare at 2 h but increased at later time‐points (Figs 1D and EV2B).
Figure EV2. Time‐course analysis of the transcriptional regulatory categories.
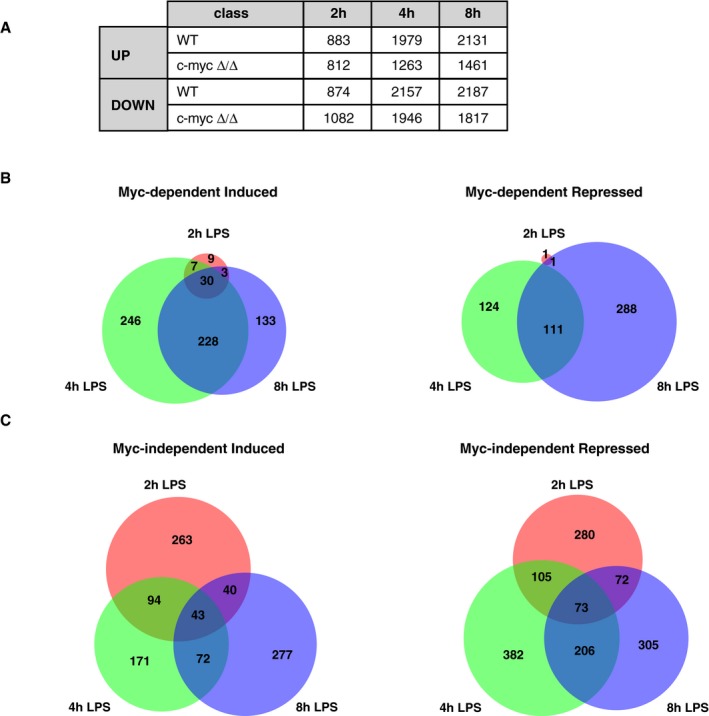
-
ANumbers of genes classified in the different regulatory categories on the basis of the RNA‐Seq data at each time‐point after LPS stimulation.
-
B, CVenn diagrams representing the overlap between genes identified as Myc‐dependent induced (left) or repressed (right) (B), or Myc‐independent LPS‐regulated (C), at the different time‐points after LPS treatment.
Besides mature mRNAs, we sought to measure the accumulation of unspliced precursors along the time‐course and to use these data for computational modeling of RNA synthesis, processing, and degradation rates: While originally based on metabolic labeling of newly synthesized RNA 34, this can readily be achieved with intronic and exonic reads in total RNA‐Seq data (Fig 1E) [preprint: 35]. Remarkably, the changes in pre‐mRNA and mRNA levels elicited by either LPS or c‐myc deletion at either Myc‐dependent or independent genes were largely explained (93% and 91%, respectively) by variations in RNA synthesis rates, with no widespread contribution of either processing or degradation, indicating that regulation occurred mainly at the transcriptional level. Altogether, Myc was rapidly induced by LPS and modulated the transcriptional response of select groups of genes in early G1 (4–8 h), preceding general effects on biomass accumulation and cell size 4.
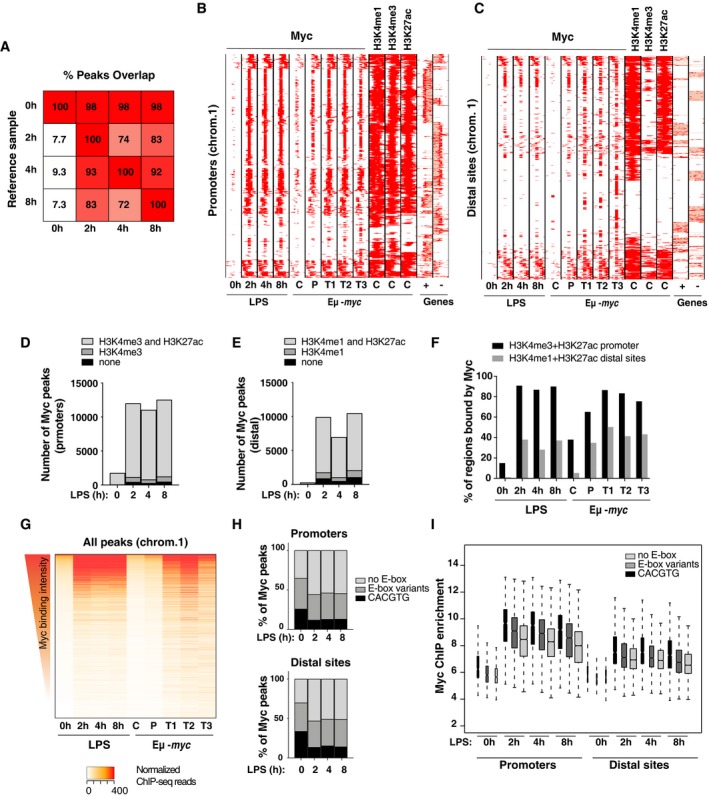
We then used ChIP‐Seq to profile Myc along the genome: Approximately 2,000 binding sites were detected in resting wild‐type B‐cells, rising to ca. 22000 after LPS stimulation (either 2, 4, or 8 h), with consistent overlaps along the time‐course (Fig 2A). Most Myc‐binding sites were proximal (−2 to +1 kb) to an annotated transcription start site (TSS), albeit the fraction of distal sites increased upon stimulation (from 12% at 0 h to 45% at 8 h). Remarkably, the progression of Myc‐binding profiles upon LPS stimulation was virtually overlapping with that seen in vivo when comparing control c‐myc wt/wt splenic B‐cells (C) with Eμ‐myc transgenic pre‐tumoral B‐cells (P) and lymphomas (tumor: T) (Fig 2B and C), consistent with parallel increases in Myc levels 4. Further comparison with histone mark profiles in c‐myc wt/wt B‐cells 4 showed that Myc‐binding sites pre‐existed in an active state characterized by elevated H3K4me3 and H3K4me1 at proximal and distal sites, respectively, with H3K27ac at both (Fig 2B–E), indicative of active promoters and enhancers: while few of these regulatory regions (< 20% and < 5%, respectively) were bound by Myc in naïve B‐cells (0 h or C), most were targeted upon either LPS stimulation or Eμ‐myc expression (Fig 2F). Importantly, those regions bound in resting conditions corresponded to high‐affinity sites that were also the most efficiently bound upon Myc activation (Fig 2G) and showed the highest enrichments for E‐box consensus motifs (Fig 2H). In line with these observations, E‐box‐containing sites were the most efficiently bound by Myc in all conditions, with intermediate levels at regions with variant E‐boxes 8, 36, 37 and lowest in the absence of any E‐box motifs (Fig 2I); note however that these categories showed large ranges and significant overlaps in binding intensities, consistent with the involvement of additional chromatin‐ and protein‐based determinants 38. Altogether, in line with previous observations in either B‐cells 3, 4, 25, 39 or other cell types 24, 25, 27, 28, 29, 30, 40, 41, acute accumulation of Myc upon LPS treatment led to its widespread association with pre‐existing promoters and enhancers: this phenomenon, also termed “invasion” 3, 4, 25, most probably occurs through low‐affinity, non‐sequence‐specific interactions with genomic DNA 38. Together with the findings that Myc deletion only impacts a subset of the genes regulated by either LPS (this work) or serum 8, we infer that chromatin association cannot be systematically equated with productive regulatory engagement of the transcription factor onto genomic DNA 24, 31, 34.
Figure 2. Myc widely associates with active promoters and enhancers upon LPS stimulation.
-
AOverlap between Myc ChIP‐Seq peaks. For each time‐point indicated at the bottom, the percentage of peaks overlapping over ≥ 1 bp with any other time‐point (Reference sample) is reported. Peaks showed consistent distributions along the time‐course, almost all those called in untreated cells being included in LPS‐stimulated samples
-
B, CDistribution of Myc at promoters (B) and distal sited (C) in LPS‐treated B‐cells (0, 2, 4 8 h) and in B‐cells in vivo, in either control c‐myc wt/wt (C), pre‐tumoral Eμ‐myc mice (P), or tumors (T) 4. Each row represents a promoter‐associated Myc peak within a 6‐kb genomic interval (centered on the midpoint of the peak). The heatmap includes every annotated promoter in chromosome 1 (as representative of the whole genome) that was called as Myc‐associated by ChIP‐Seq in at least one sample. For the same intervals, we also show the distribution of H3K4me1, H3K4me3, and H3K27ac in the in vivo control sample, and gene annotations (exons in red, introns in pink; + sense, − antisense strand).
-
D, ENumbers of Myc peaks at promoters (−2 kb, +1 kb) (D) and distal sites (E) at different time‐points after LPS stimulation. The shadings mark the subsets of peaks associated with H3K4me3 (D) or H3K4me1 (E), with or without H3K27ac.
-
FPercentage of active promoters (positive for H3K4me3 and H3K27ac) or distal sites (H3K4me1 and H3K27ac) that overlap with at least a Myc peak at the different time‐points after LPS stimulation (0, 2, 4, 8 h) or in vivo (C, P, T1, T2, T3).
-
GQuantitative heatmap showing Myc signal intensities (read counts normalized by library size) at each bound region in chromosome 1, ranked based on the signal in the control sample (0 h LPS).
- H
-
IMyc enrichment by ChIP‐Seq at different time‐points after LPS treatment for peaks located either in promoters or distal sites and containing the indicated DNA motifs. In the boxplot, the central tendency marks the median, the box ranges mark the upper and lower quartiles, and the whiskers extend to the upper and lower extreme datapoints, which are no more than 1.5 times the interquartile range from the box. The Myc ChIP‐Seq time‐course was performed on a unique pool of cells, obtained from > 10 mice.
One parameter predicting transcriptional responses in other cell types was the relative efficiency in Myc binding at promoters 29, 30, 31, 34, which we expressed as the “Myc share” (obtained by normalizing the signal at each promoter by the total amount of promoter‐bound Myc in the genome) 34: indeed, those genes for which the RNA synthesis rate increased upon LPS stimulation also showed the highest gains in Myc binding (i.e., increasing share), while those with a reduced synthesis showed the lowest gains (decreasing share) (Fig 3A and B). This correlation was the strongest for Myc‐dependent genes, although still present at Myc‐independent loci, as also observed in Myc‐driven liver cancer 30, 34: albeit paradoxical at first sight, this observation is consistent with the notion that promoter activity and Myc binding are mutually dependent, owing to the interaction of Myc with other chromatin‐associated factors, RNAPII, and components of the basal transcription machinery 39, 42, 43. In other words, changes in transcriptional activity at Myc‐independent genes are likely to impact in return on recruitment of Myc itself. In proliferating U2OS cells, high‐affinity E‐box‐containing loci were deemed to be already saturated by Myc at the population level, thus showing no—or negligible—increases in either Myc binding or transcriptional activity upon Myc overexpression 29: in our experiments instead, virtually all promoters—regardless of initial binding intensities—showed increased Myc binding upon stimulation (Fig 3C), implying that these were far from saturation to start with. Accordingly, Myc‐dependent LPS‐induced genes showed not only the highest increase in Myc binding (Fig 3A) but also the highest frequency of E‐boxes (Fig 3D). Hence, upon B‐cell stimulation and the concomitant transition from very low to high Myc levels, Myc drove rapid and selective activation of high‐affinity promoters, most frequently associated with the presence of the E‐box binding motif.
Figure 3. Changes in Myc share are predictive of gene regulation.
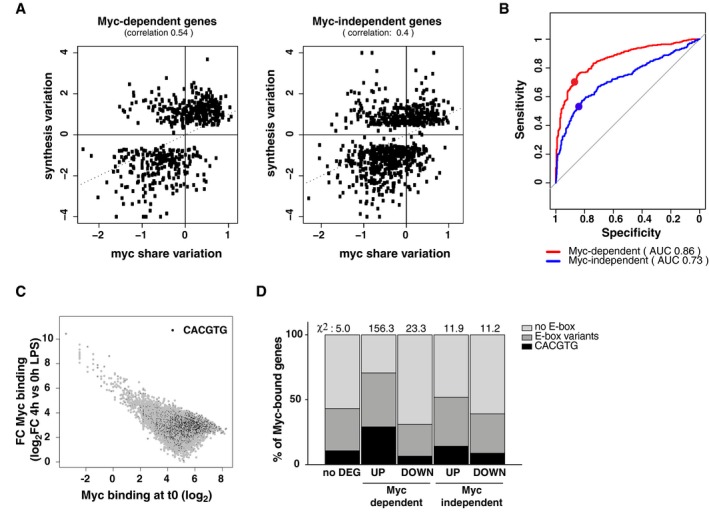
- Scatter plots correlating the variations in Myc share at promoters (averaged between the 2‐, 4‐, and 8‐h time‐points) and in RNA synthesis (at 8 h), relative to untreated cells, for Myc‐dependent and Myc‐independent genes. The Spearman correlation between the two parameters is reported.
- The ability of the Myc share in predicting the transcriptional outcome can be represented in terms of a receiver operating characteristic (ROC) curve, which represents the Sensitivity (true‐positive rate) and Specificity (true‐negative rate) of a predictor (i.e., Myc share) in discriminating between two classes (i.e., up‐ and down‐regulated genes), using different thresholds. The largest the area under the curve (AUC), the largest is the predictive power, in a scale from 0.5 (random classification: gray line) to 1 (perfect predictor). For each system, the dot corresponds to the variation of Myc at which promoters begin increasing their share of Myc binding.
- Fold‐change in Myc levels at each bound promoter (at 4 h LPS, relative to time zero) as a function of the initial binding intensity at time zero (expressed as log2 of the coverage in a 200‐bp window around the summit of the peak). Peaks containing a canonical E‐box are highlighted in black.
- Percentage of Myc‐bound promoters within the indicated transcriptionally regulatory categories that contains a canonical or variant E‐box. The overall chi‐square is 207.7 with a P‐value < 0.00001. The contribution to the chi‐square of each category is reported above the corresponding bar in the plot.
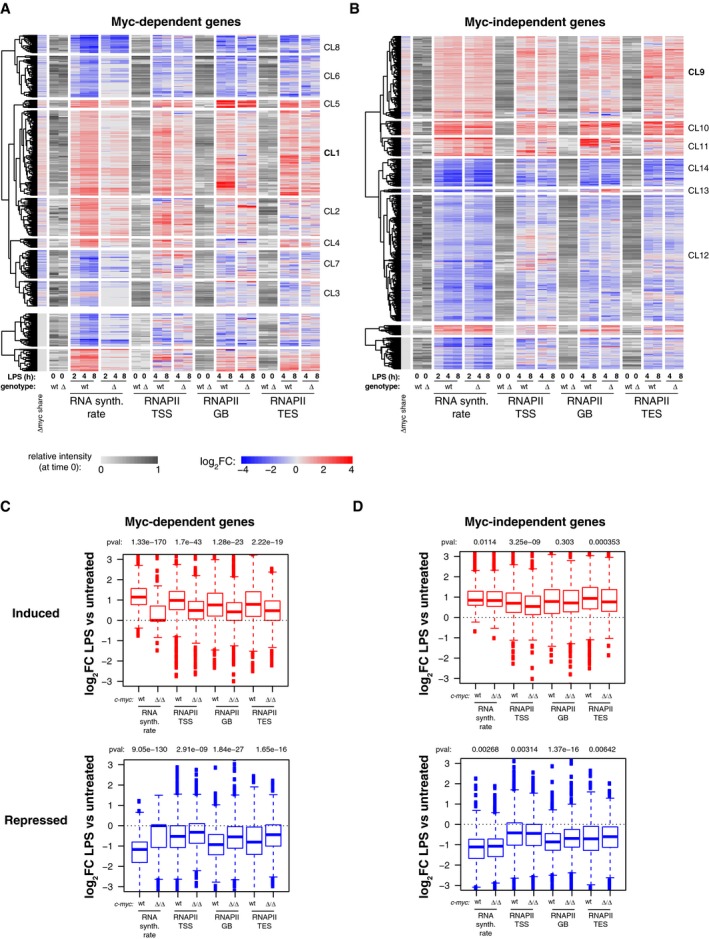
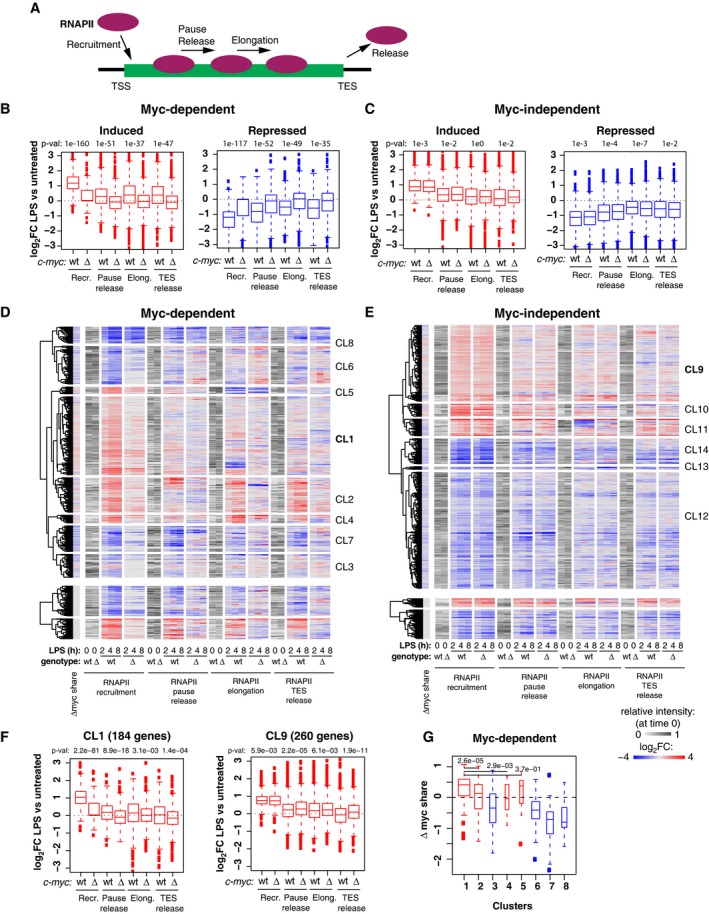
In order to address the mechanisms of transcriptional regulation by Myc, we profiled RNA polymerase II (RNAPII) by ChIP‐Seq, computed its quantitative changes in the promoter (TSS), gene‐body (GB), and termination site (TES) of regulated genes, and confronted those to the changes in RNA synthesis rates (Fig EV3A and B). As expected, LPS‐induced genes showed consistent increases in RNAPII densities in all of their domains, the opposite being true for repressed genes: most importantly, these effects of LPS on RNAPII were suppressed by c‐myc deletion at Myc‐dependent, but not Myc‐independent loci (Fig EV3A–D). We then used a dedicated algorithm 34 to model the kinetic rates governing the RNAPII cycle at each locus, including promoter recruitment, pause‐release, elongation, and release from the transcription end site (TES) (Fig 4A). Remarkably, these steps were up‐ or down‐modulated by LPS consistently with mRNA levels, and these changes were dampened in c‐myc ∆/∆ cells at Myc‐dependent, but not Myc‐independent genes (Fig 4B and C): however, as seen after MycERT2 activation in fibroblasts 34, RNAPII recruitment was the most significantly affected upon either LPS stimulation or Myc deletion.
Figure EV3. RNAPII ChIP‐Seq data quantification.
-
A, BHeatmap showing the variations in RNAPII ChIP‐Seq read density (log2FC) in different regions (TSS, gene‐body, TES) for Myc‐dependent (A) and Myc‐independent (B) genes along the LPS time‐course, as indicated. The gray scale represents the starting level for each parameter in unstimulated cells. Columns on the left show the changes in Myc share and RNA synthesis rates, as indicated, for the same genes.
-
C, DBoxplots reporting LPS‐induced changes (log2FC) for each of the four RNAPII kinetic rates in c‐myc wt/wt (wt) and c‐myc ∆/∆ cells for Myc‐dependent (C) and Myc‐independent (D) induced and repressed genes, as indicated. All boxplots are as defined in Fig 2I, and RNAPII replicates as defined in Fig 4B and C.
Figure 4. Myc regulates all steps of the RNAPII transcriptional cycle.
-
AScheme of the different steps of RNAPII regulation for which the corresponding rates were modeled 34.
-
B, CBoxplots reporting LPS‐induced changes (log2FC) for each of the four RNAPII kinetic rates in c‐myc wt/wt (wt) and c‐myc ∆/∆ cells for Myc‐dependent (B) and Myc‐independent (C) induced and repressed genes. P‐values obtained with two‐sample Wilcoxon tests for the comparison between c‐myc wt/wt and c‐myc ∆/∆ cells are reported. The RNAPII ChIP‐Seq time‐course was performed on two biological replicates (each on pools of cells from > 10 mice), and reads were merged after sequencing. All boxplots are as defined in Fig 2I.
-
D, EHeatmaps showing the variations in RNAPII kinetic rates (log2FC) on Myc‐dependent (D) and Myc‐independent (E) genes during the LPS time‐course. The gray scale represents the starting level for each parameter in unstimulated cells. The first column on the left reports the changes in Myc share for the same genes. Genes were grouped using hierarchical clustering.
-
FBoxplots reporting LPS‐induced changes (log2FC) for each of the four RNAPII kinetic rates in c‐myc wt/wt (wt) and c‐myc ∆/∆ cells for genes of clusters 1 and 9. P‐values obtained with two‐sample Wilcoxon tests for the comparison between c‐myc wt/wt and c‐myc ∆/∆ cells are reported.
-
GChanges in Myc share in LPS‐treated versus LPS‐untreated cells for the different clusters of panel d. P‐values obtained with two‐sample Wilcoxon tests for the comparison between c‐myc wt/wt and c‐myc ∆/∆ cells are reported.
To increase the resolution of our analysis and unravel possible regulatory subsets, we clustered LPS‐regulated genes on the basis of RNA synthesis rate and RNAPII dynamics (Fig EV3A and B, Dataset EV3). Remarkably, the largest sets of LPS‐activated genes (CL1: Myc‐dependent; CL9: Myc‐independent) were almost exclusively regulated through RNAPII loading (Fig 4D–F), while others showed additional effects on other regulatory steps (Appendix Fig S2 and S3). Among the Myc‐dependent induced genes, those of CL1 also showed the highest increases in Myc share (Fig 4G). Myc‐dependent repressed genes, instead (CL3, 6, 7, 8), were characterized by a relative loss of Myc binding (decreased share) and by decreased RNAPII recruitment (Fig 4B and G, Appendix Fig S2), consistent with the concept that Myc‐dependent repression may largely be a passive process 34—albeit not excluding the existence of active repressive mechanisms at select loci 28, 44. Altogether, Myc appears to primarily drive RNAPII recruitment at activated loci 34, with differential contributions of other regulatory steps, in particular pause‐release 45 or elongation 46. Together with recent studies, our work shows that a careful integrative analysis of RNAPII and RNA dynamics 34, 47 ideally complemented by dedicated assays to monitor elongation (e.g., Pro‐Seq or 4sU‐FP‐seq) 46, 48 is needed to unravel the hierarchical contribution of distinct regulatory steps.
To discern the biological pathways directly modulated by Myc, we analyzed the gene ontology (GO) terms enriched in the various subgroups of LPS‐induced genes: Myc‐dependent (or Group 1, Fig 1C, Dataset EV2: 647 genes), Myc‐independent (Group 5: 697 genes), and the intervening “gray zone” (GZ: 1,129 genes) as the latter was likely to comprise genes modulated by Myc with low—albeit possibly biologically meaningful—amplitude. We also analyzed Cluster 1, characterized by the highest increase in Myc share (CL1, Fig 4D, 184 genes). Group 1, GZ, and CL1 most significantly enriched for common GO terms pertaining to RNA and amino acid metabolism, as well as mitochondrial biogenesis (Dataset EV4), with additional terms enriched only in the GZ, such as translation, ribosome biogenesis, RNAPI transcription, or nucleotide biosynthesis. Importantly, Myc‐independent genes showed little overlap with any of the above, enriching for distinct functional categories such as lymphocyte activation, cell cycle, apoptosis, and DNA metabolism (repair, recombination). Thus, biological processes that depend upon Myc activity (e.g., cell cycle, DNA replication) are regulated—to a large extent—by genes that show Myc‐independent expression, suggesting that the requirement for Myc in these processes is due to the regulation of genes involved in associated essential events (e.g., protein and nucleic acid biosynthesis). Most importantly in this context, the differential occurrence of consensus binding motifs suggests that other transcription factors, such as E2F or NFY, may have predominant roles—or be redundant with Myc 49—in regulating Myc‐independent genes (Fig EV4).
Figure EV4. DNA Motifs analysis of Myc‐dependent and Myc‐independent LPS‐induced genes.
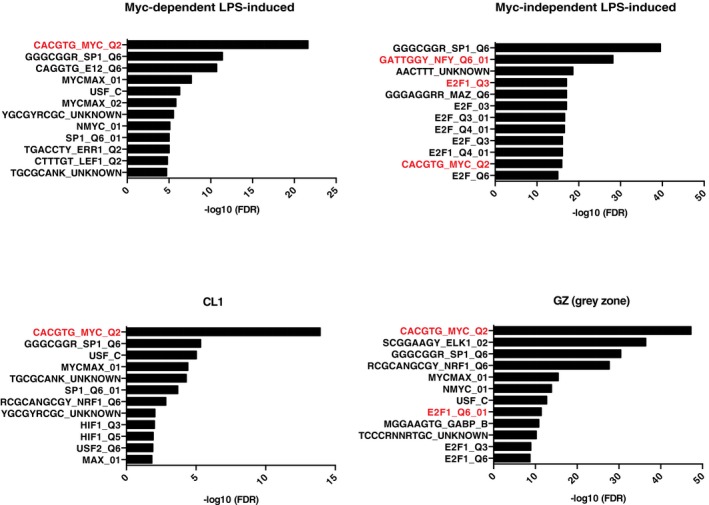
Bar plots showing the −log10 (FDR) of the top 12 cis‐regulatory motifs enriched in the promoters of the genes belonging to the indicated regulatory groups.
It is noteworthy here that several of the Myc‐dependent ontological categories (e.g., RNA metabolism, ribosome biogenesis, nucleotide biosynthesis) were also enriched among Myc‐regulated genes in different cell types 8, 29, 31, in germinal center B‐cells 10, or in primary B‐cells at longer time‐points of LPS stimulation (72 h) 16. Yet, the overlaps between these and our gene lists were only modest (Fig EV5A, Dataset EV5). Hence, while different genes may show Myc‐dependent expression in different contexts, the biological processes that rely on Myc activity appear to be generally conserved. Remarkably, larger fractions of the Myc‐dependent genes identified in this work were induced during lymphomagenesis in Eμ‐myc transgenic mice 4 (Fig EV5B), emphasizing the need to confront normal and pathological Myc‐regulated programs in the corresponding cellular contexts.
Figure EV5. Overlap between Myc‐dependent LPS‐induced genes and other datasets.
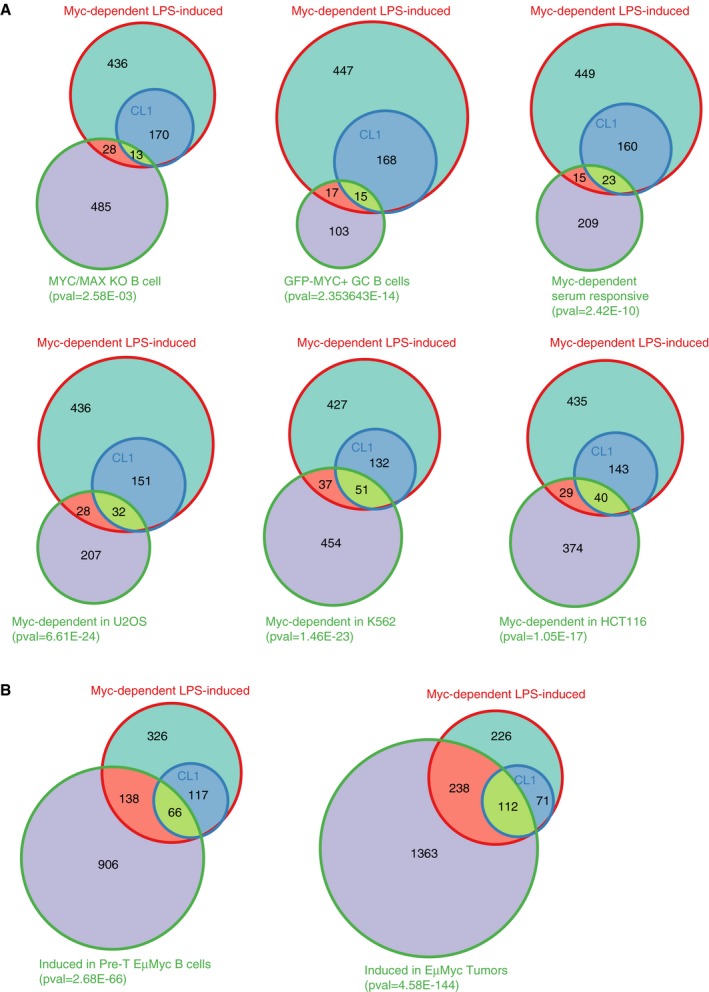
- Venn diagrams representing the overlap between Myc‐dependent LPS‐induced genes defined in this study (either all genes, or only those in CL1) with other lists of Myc‐regulated genes. From left to right, and top to bottom: genes less expressed in Myc/Max KO B‐cells activated with LPS for 72 h, compared to wt cells 16; genes more expressed in Myc‐positive versus negative germinal center B‐cells 10 Myc‐dependent serum‐responsive (MDSR) genes in fibroblasts 8; genes down‐regulated in Myc‐depleted U2OS 29, K562 and HCT116 cells 31.
- Venn diagrams representing the overlap between Myc‐dependent LPS‐induced genes defined in this study with genes induced in Eμ‐myc B‐cells at the pre‐tumoral (left) or tumoral stage (right).
Altogether, we identified approximately 650 genes that were induced in a Myc‐dependent manner within 4–8 h following B‐cell stimulation by LPS, and an additional group of ca. 1,100 additional genes regulated by Myc with modest quantitative effects: while the latter were below threshold for being reliably called as Myc‐dependent, both groups enriched for highly consistent functional categories. By and large, Myc‐dependent LPS‐responsive genes encode proteins involved in RNA biology, energy production, and anabolic pathways 11, 13, 23: these, in turn, may provide the building blocks (nucleotides, amino acids) required to sustain the large accumulation of biomass—including RNA—characteristic of activated B‐cells 3, 4, 5. In an alternative model, Myc was deemed to act as a direct activator—or amplifier—of all expressed genes 3, 25, 26, 27. However, our results and others 4, 8, 28, 31 provide no formal support for this model: instead, when observed, RNA amplification is most consistently interpretable as a late, indirect consequence of Myc action, mediated by a selective—yet complex—set of target genes 24, 32. In stimulated primary B‐cells in particular, Myc was needed for ATP production, which in turn supported widespread chromatin remodeling and decompaction 5, in line with the delayed increase in bulk RNA synthesis reported here. Finally, the Myc‐dependent program that we identified in activated B‐cells was constitutively deregulated during lymphomagenesis in Eμ‐myc transgenic mice 4. Hence, besides aberrant regulation of novel, tumor‐specific targets 4, 28, 29, the oncogenic action of Myc may largely rely on the uncontrolled activation of genes regulated during mitogenic stimulation in normal cells.
Materials and Methods
Mouse strains and primary B‐cells
C57BL/6 c‐myc f/f mice were obtained from Andreas Trumpp 33. In all experiments, we used gender‐ and age‐matched mice (both males and females) without randomization or blinding. Naïve mouse B‐cells were isolated from the spleen of 7‐ to 10‐week‐old wild‐type or c‐myc f/f mice with the B‐cell isolation kit (MACS Miltenyi Biotec Cat. no. 130‐090‐862). After purification, naïve B‐cells were incubated for 1 h at 37°C with recombinant Tat‐Cre protein (50 μg/ml) in optimem + 1% fetal bovine serum 50 in order to induce deletion of the c‐myc f/f allele. Cells were washed with PBS and then grown in B‐cell medium composed of DMEM medium (Dulbecco's modified Eagle's medium) and IMDM medium (Iscove's modified Dulbecco's medium) in a 1:1 ratio and additionated of 10% fetal calf serum (FCS) (Globefarm Ltd, Cranleigh, UK), 2 mM l‐glutamine (Invitrogen Life Technologies, Paisley, UK), 1% non‐essential amino acids (NEAA), 1% penicillin/streptomycin, and 25 μM β‐mercaptoethanol 51. 12 h after seeding, B‐cells were stimulated with lipopolysaccharide LPS (50 μg/ml; SIGMA L6237) to induce cell activation. The Tat‐Cre protein was produced and purified as previously described starting from pTriEx‐His‐TAT‐NLS‐Cre expressing bacteria 50. As Tat‐Cre can be contaminated with bacterially derived molecules, including LPS, we controlled for possible effects of Tat‐Cre treatment on wild‐type B‐cell activation or proliferation, as shown in Appendix Fig S1.
Experiments involving animals were carried out in accordance with the Italian Law D.lgs. 26/2014, which enforces Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes.
Proliferation, cell cycle analysis, and sorting
For proliferation analysis, cells were counted with Trypan blue to exclude dead cells every 24 h. For cell cycle analysis, cells were incubated with 33 μM BrdU for a pulse labeling of 30 min. Cells were then harvested, washed with PBS, and fixed in ice‐cold ethanol. Upon DNA denaturation using 2N HCl, cells were stained with an anti‐BrdU primary antibody (BD Biosciences) and anti‐mouse FITC‐conjugated secondary antibody (Jackson Immunoresearch). DNA was stained by resuspending the cells in 2.5 μg/ml Propidium Iodide (Sigma) overnight at 4°C before FACS analysis. All samples were acquired on a FACS Canto II (BD Biosciences) flow cytometer. At least 15,000 events were acquired, and the analysis was performed using the FlowJo X software. For cell sorting, cells were resuspended in cold Macs Buffer (0.5% BSA, 2 mM EDTA in PBS) and sorted on the basis of the FSC/SSC parameters with a Facs‐Aria II machine (BD Biosciences).
Caspase‐3/7 assay
In order to evaluate apoptosis, caspase activity was measured using the Caspase‐Glo 3/7 luminescence kit (Promega G8090) following manufacturer's instruction. The luminescent signal obtained was normalized to cell numbers counted by Trypan blue.
Immunoblot analysis
5 × 106 B‐cells were lysed with RIPA Buffer (20 mM HEPES at pH 7.5, 300 mM NaCl, 5 mM EDTA, 10% Glycerol, 1% Triton X‐100), supplemented with protease inhibitors (Roche) and phosphatase inhibitors (0.4 mM Orthovanadate, 10 mM NaF), and briefly sonicated. Cleared lysates were electrophoresed and immunoblotted with the indicated primary antibodies: c‐Myc Y69 (ab32072) from Abcam, Vinculin (V9264) from Sigma. Chemiluminescent detection, after incubation of the membranes with appropriate secondary antibodies, was done through a CCD camera using the ChemiDoc System (Bio‐Rad). Quantification of protein levels was done using the Image Lab Software (Bio‐Rad, version 4.0).
EU staining and data analysis
In order to label newly synthesized RNA, B‐cells were plated at a cell density of 8 × 105/ml and incubated with the alkyne‐modified nucleoside, 5‐ethynyl uridine (EU), 1 mM for 1 h before fixation in 4% PFA for 10 min at RT. Fixed cells were washed in PBS and resuspended in PBS+BSA 3%. Cells were then cytospinned on polylysine‐coated slides, permeabilized with Triton X‐100 0.5% in PBS, and treated with the Click‐iT reaction cocktail for 30 min at RT as indicated by manufacturer's instruction (Click‐iT RNA Imaging Kit—Invitrogen, C10329). DNA was than stained with DAPI.
The image analysis was performed using a custom pipeline developed and executed in the Acapella software development/run‐time environment (Perkin Elmer). Nuclei were detected on the basis of DAPI staining using a PerkinElmer proprietary algorithm, and each nucleus was associated with its planar area (in pixels) and its integrated EU signal.
Isolation of genomic DNA
Cell pellets (1.5 × 106) were collected at different time‐points after LPS stimulation, and DNA was extracted with the Nucleospin tissue kit (Macherey‐Nagel, 740952). The genomic DNA was eluted in 50 μl of BE buffer (5 mM Tris–HCl, pH 8.5). The analysis of c‐myc deletion efficiency was performed by real‐time PCR using 10 ng of genomic DNA as template and the primers reported in Appendix Table S1 33.
RNA extraction and analysis
Total RNA (at least from 2.5 × 106 cells) was purified onto RNeasy columns (Qiagen) and treated on‐column with DNase (Qiagen). Complementary DNA (cDNA) was prepared using ImProm‐II™ reverse transcription kit (Promega, A3800), and 10 ng of cDNA was used as template for each real‐time PCR. cDNA was detected by fast SyberGreen Master Mix (Applied Biosystems, 4385614) on CFX96 Touch™ Real‐Time PCR Detection System (Bio‐Rad). Sequences of the used PCR primers were reported in Appendix Table S1.
For RNA‐Seq experiments, total RNA from 8 × 106 B‐cells was purified as above, and then, 0.5 μg was treated with Ribozero rRNA removal kit (Epicentre) and EtOH precipitated. RNA quality and removal of rRNA were checked with the Agilent 2100 Bioanalyser (Agilent Technologies). Libraries for RNA‐Seq were then prepared with the TruSeq RNA Sample Prep Kits v2 (Illumina) following manufacturer instruction (except for skipping the first step of mRNA purification with poly‐T oligo‐attached magnetic beads). RNA‐Seq libraries were then run on the Agilent 2100 Bioanalyser (Agilent Technologies) for quantification and quality control and then sequenced on Illumina HiSeq2000.
Chromatin immunoprecipitation
Purified splenic B‐cells were resuspended in PBS at room temperature and fixed for 10 min by addition of formaldehyde to a final concentration of 1%. Fixation was stopped by addition of glycine to a final concentration of 0.125 M. Cells were washed in PBS, resuspended in SDS buffer (50 mM Tris at pH 8.1, 0.5% SDS, 100 mM NaCl, 5 mM EDTA, and protease inhibitors), and stored at −80°C before further processing for ChIP as described in 4. For ChIP‐Seq analysis, lysates obtained from 30 to 50 × 106 B‐cells were immunoprecipitated with 10 μg of Myc (Santa Cruz, sc‐764) or RNAPII (Santa Cruz, sc‐899) antibodies. Immunoprecipitated DNA was eluted in TE‐2% SDS, and crosslinks were reversed by incubation overnight at 65°C. DNA was then purified by Qiaquick columns (Qiagen) and quantified using Qubit™ dsDNA HS Assay kits (Invitrogen). 1.5–2 ng of ChIP DNA was end‐repaired, A‐tailed, ligated to the sequencing adapters, and amplified by 17‐cycles of PCR, size selected (200–300 bp) according with TruSeq ChIP Sample Prep Kit (Illumina). ChIP‐Seq libraries were then run on the Agilent 2100 Bioanalyser (Agilent Technologies) for quantification and quality control and then sequenced on the Illumina HiSeq2000. Sequences of the primers used in qPCR were reported in Appendix Table S1.
Next generation sequencing data filtering and quality assessment
ChIP‐Seq and RNA‐Seq reads were filtered using the fastq_quality_trimmer and fastq_masker tools of the FASTX‐Toolkit suite (http://hannonlab.cshl.edu/fastx_toolkit/). Their quality was evaluated and confirmed using the FastQC application: (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Pipelines for primary analysis (filtering and alignment to the reference genome of the raw reads) and secondary analysis (expression quantification, differential gene expression, and peak calling) have been integrated in the HTS‐flow system 52. Bioinformatic and statistical analysis were performed using R with Bioconductor and comEpiTools packages 53, 54.
ChIP‐Seq data analysis
ChIP‐Seq NGS reads were aligned to the mm9 genome through the BWA aligner 55 using default settings. Peaks were called using the MACS software (v2.0.9) 56 with the option “– mfold = 7,30 ‐p 0.00001”, thus outputting only enriched regions with P‐value < 10−5. Normalized read counts within a genomic region were determined as the number of reads per million of library reads (total number of aligned reads in the sequencing library). Peak enrichment was determined as log2(Peakw/Nc—inputw/Ni), where Peakw is the read count on the enriched region in the ChIP sample, inputw is the read count on the same region in the corresponding input sample, Nc is the total number of aligned reads in the ChIP sample, and Ni is the total number of aligned reads in the input sample. Promoter peaks were defined as all peaks with at least one base pair overlapping with the interval between −2 kb to +1 kb from the nearest TSS. For Myc share calculation, promoters were defined as −2 kb to +2 kb from the nearest TSS. For RNAPII ChIP‐seq analysis, the following genomic regions were considered: promoters (−50, +700 from TSS); TES (−1 kb,+4 kb from TES), gene‐body (from promoter end to TES start). The presence of canonical and variant E‐boxes (CACGCG, CATGCG, CACGAG, CATGTG) 8, 36, 37 in the Myc ChIP peaks was scored in a region of 200 bp around the peak summit.
When comparing Myc ChIP‐Seq with H3K4me3, H3K4me1, or H3K27ac histone marks to define peaks in active promoter or enhancers 57, 58, we considered two peaks as overlapping when sharing at least one base pair (subsetByOverlaps tool of the comEpiTools R package).
Fold‐change of Myc binding at promoters was calculated as the log2 ratio of the coverage in a 200‐bp window around the summit of the peaks identified at 4 h of LPS in stimulated versus not stimulated conditions. To avoid regions with 0 coverage in the unstimulated situation, all those regions were set to the minimum coverage identified.
RNA‐Seq data analysis
RNA‐Seq NGS reads were aligned to the mm9 mouse reference genome using the TopHat aligner (version 2.0.8) 59 with default parameters. In case of duplicated reads, only one read was kept. Read counts were associated to each gene (based on UCSC‐derived mm9 GTF gene annotations), using the featureCounts software (http://bioinf.wehi.edu.au/featureCounts/) 60 setting the options ‐T 2 ‐p ‐P. Absolute gene expression was defined determining reads per kilobase per million mapped reads defining total library size as the number of reads mapping to exons only (eRPKM). After removing very low expressed genes (below 1 eRPKM in all samples), we obtained a set of 12,690 expressed genes that were used for further analysis. Differentially expressed genes (DEGs) were identified using the Bioconductor Deseq2 package 61 as genes whose q‐value is lower than 0.05 and |FoldChange|> 1.5. The different categories of Myc‐dependent LPS response and Myc‐independent LPS response were defined at each time‐point (ti) relative to time zero (t0), as follows:
px = qval ti vs. t0 (c‐myc wt/wt)
py = qval ti vs. t0 (c‐myc ∆/∆)
x = log2FC ti vs. t0 (c‐myc wt/wt)
y = log2FC ti vs. t0 (c‐myc ∆/∆)
NO DEG: (px > 0.05 & py > 0.05) OR ((|x| ≤ log2(1.5)) & (|y| ≤ log2(1.5)))
Class 5 (Myc‐independent UP): (px ≤ 0.05 & py ≤ 0.05) & (x > log2(1.5) & y > log2(1.5)) & (|y−x| ≤ log2(1.15))
Class 6 (Myc‐independent DOWN): (px ≤ 0.05 & py ≤ 0.05) & (x < −log2(1.5) & y < −log2(1.5)) & (|y−x| ≤ log2(1.15))
Class 1 (Myc‐dependent UP): (px ≤ 0.05) & (x > log2(1.5)) & (y−x <−log2(1.5))
Class 3 (Myc‐dependent DOWN): (px ≤ 0.05) & (x < −log2(1.5)) & (y−x > log2(1.5))
Class 2 (Myc‐suppressed UP): (px ≤ 0.05 & py ≤ 0.05) & (x > log2(1.5)) & (y−x > log2(1.5))
Class 4 (Myc‐suppressed DOWN): (px ≤ 0.05 & py ≤ 0.05) & (x < −log2(1.5)) & (y−x < −log2(1.5))
Functional annotation analysis to determine enriched Gene Ontology was performed using the ClueGo v2.5.3 Application of CytoScape v3.7.1 with the following parameters:
Statistical Test Used = Enrichment/Depletion (Two‐sided hypergeometric test)
Correction Method Used = Bonferroni step down
Min GO Level = 6
Max GO Level = 8
GO Fusion = true
Show only pathways with P‐value ≤ 0.05
GO Group = true
Kappa Score Threshold = 0.4
Group By Kappa Statistics = true
Initial Group Size = 1
Sharing Group Percentage = 50.0
Merge redundant groups with > 50.0% overlap
GO categories enriched in at least one gene list were reported in Dataset EV4 and manually grouped on the basis of the ClueGo assignation to different groups.
Analysis of cis‐regulatory motif enrichment in the promoter of the different classes of genes has been performed with the online tool available at http://software.broadinstitute.org/gsea/msigdb/index.jsp.
For the calculation of a significant overlap between Myc‐dependent LPS‐induced genes and the datasets of interest, a hypergeometric distribution was assumed. The external data were converted in the mouse orthologous when required and filtered keeping only the genes expressed in the reference dataset (B‐cells, this study). Gene lists are reported in Dataset EV5 and obtained from the following sources:
Column D: MYC/MAX KO B‐cells 16
Column E: GFP‐MYC+ GC B‐cells 10
Column F: Myc‐dependent serum responsive 8
Column G: Myc‐dependent in U2OS cells 29 (q‐val < 0.01; log2FC < −1)
Column H: Myc‐dependent in K562 cells 31 (q‐val < 0.05; log2FC < −1)
Column I: Myc‐dependent in HCT116 cells 31 (q‐val < 0.05; log2FC < −0.5)
Column J: Induced in Pre‐tumoral Eμ‐myc B‐cells 4 (q‐val < 0.05; log2FC > 0.585)
Column K: Induced in Eμ‐myc tumors 4 (q‐val < 0.05; log2FC > 0.585)
Estimation of synthesis, processing, and degradation rates
Our previous R/Bioconductor package INSPEcT 62 was designed to estimate the rates of RNA synthesis, processing, and degradation following metabolic labeling and quantification of total and newly synthesized RNA: We recently extended this package to pursue the same goal in the absence of experimental data on newly synthesized RNA, taking advantage of intronic reads in total RNA‐Seq experiments (INSPEcT2) [preprint: 35]. Myc‐dependent and Myc‐independent genes at either 4 of 8 h following LPS stimulation (Dataset EV2) were used for the modeling after removal of genes without introns or with scarce intronic signal. Out of the 925 Myc‐dependent genes modeled, 674 were identified as modulated in their synthesis rate in the c‐myc wt/wt condition and kept for subsequent analysis. Similarly, 1,005 Myc‐independent genes out of the 1,660 modeled were identified as modulated in their synthesis rates both in the c‐myc wt/wt and c‐myc ∆/∆ conditions and kept for subsequent analysis. The quality of the models was evaluated through the fraction of variance explained (FVE), by using the formula:
where the squared error (SE = (model ‐ <experiment>)2) is normalized to the experimental variance (Var), evaluated from replicated experiments, and subtracted to 1. The selected 674 Myc‐dependent gene models had a median FVE of 0.94 and 0.86 in WT and FLOX experiments, respectively, while the 1,005 Myc‐independent gene models had a median FVE of 0.92 in both WT and FLOX. Overall, the models obtained from INSPEcT showed to explain the largest part of the dataset variation.
Estimation of RNAPII recruitment, pause‐release, elongation, and termination rates
Similarly to what done in previous work 34, we modeled RNAPII progression on each gene as a dynamic system composed by four steps: recruitment (p 1), pause‐release (p 2), elongation (p 3), and the release from termination end sites (release, p 4). This system relates to RNAPII quantification at promoters (R tss), gene‐body (R gb), and termination end sites (R tes) as follows:
In practical terms, this model assumes that:
RNAPII at transcriptional start site (R tss) is the balance between the amount of recruited RNAPII (p 1(t)) and the amount of RNAPII that enters the gene‐body due to pause‐release (R tss(t)·p 2(t));
RNAPII at the gene‐body (R gb) is the balance between the amount of RNAPII that enters the gene‐body due to pause‐release and the amount that enters into the termination sites due to elongation (R gb(t)·p 3(t));
RNAPII at the termination sites (R tes) is the balance between the amount of RNAPII that enters into the termination sites due to elongation and the amount that is released to nucleoplasm (R tes(t)·p 4(t)).
Additionally, we assumed that the RNAPII that enters in termination sites after elongation has synthesized an RNA molecule:
Estimated values of p 1(t), p 2(t), p 3(t), and p 4(t) are obtained at each experimental time‐point (0 h, 2 h, 4 h, 8 h) directly by the solution of the system of the four equations above, where dR tss(t)/dt, dR gb(t)/dt, and dR tes(t)/dt are estimated from the time‐course of R tss, R gb, and R tes, respectively, and k 1(t) is the synthesis rate calculated by INSPEcT at the time t.
The relative error of each modeled gene is calculated as:
where , , , and are the modeled values of R tss(t), R gb(t), R tes(t), and k 1(t) obtained by integrating the estimated rates p 1(t), p 2(t), p 3(t), and p 4(t) into the system of differential equations, assuming linear behavior of the estimated rates between the experimental time‐points. We considered valid only those models with relative error lower than 1 and no missing‐values in any of the rates at any time‐point (618/674 Myc‐dependent genes and 891/1005 Myc‐independent genes).
Statistical analysis
All the experiments, except for ChIP, were performed at least in biological triplicates. Sample size was not predetermined, but is reported in the respective figure legends. Two‐tailed Student's t‐test was used to compare between two groups and expressed as P‐values. In the figures: *P < 0.05, **P < 0.01, ***P < 0.001.
Data and code availability
RNA‐Seq and ChIP‐Seq data have been deposited in NCBI's Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GSE126340 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126340).
All R scripts used in data analysis and generation of figures are available upon request.
Author contributions
AS and BA designed experiments, supervised the work, and wrote the manuscript. AT performed the experiments. AV and MD provided technical support. AA analyzed the EU staining. SP, MFu, MFi, MJM, MP, and AS were involved in bioinformatic data analysis.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Review Process File
Acknowledgements
We thank P. Nicoli, A. Gobbi, and M. Capillo for their help with the management of mouse colonies; S. Bianchi, L. Rotta, and T. Capra for assistance with the Illumina HiSeq; S. Barozzi and D. Parazzoli for assistance with imaging technologies; A. Bisso, S. Campaner, O. Croci, G. Natoli, and J. Zuber for discussions, comments, and advice on data analysis. A.S. and B.A. acknowledge the support from the Istituto Italiano di Tecnologia in the early phases of the work. This study was supported by funding from the European Research Council (grant agreement no. 268671‐MYCNEXT), the Italian Health Ministry (RF‐2011‐02346976), and the Italian Association for Cancer Research (2012‐13182 and 2015‐16768) to B.A., and from Worldwide Cancer Research (15‐1260) to A.S.
EMBO Reports (2019) 20: e47987
Contributor Information
Bruno Amati, Email: bruno.amati@ieo.it.
Arianna Sabò, Email: arianna.sabo@ieo.it.
References
- 1. Pogo BG, Allfrey VG, Mirsky AE (1966) RNA synthesis and histone acetylation during the course of gene activation in lymphocytes. Proc Natl Acad Sci USA 55: 805–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kouzine F, Wojtowicz D, Yamane A, Resch W, Kieffer‐Kwon KR, Bandle R, Nelson S, Nakahashi H, Awasthi P, Feigenbaum L et al (2013) Global regulation of promoter melting in naive lymphocytes. Cell 153: 988–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R et al (2012) c‐Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151: 68–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sabò A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A et al (2014) Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511: 488–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kieffer‐Kwon KR, Nimura K, Rao SSP, Xu J, Jung S, Pekowska A, Dose M, Stevens E, Mathe E, Dong P et al (2017) Myc regulates chromatin decompaction and nuclear architecture during B cell activation. Mol Cell 67: 566–578.e510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marguerat S, Schmidt A, Codlin S, Chen W, Aebersold R, Bahler J (2012) Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 151: 671–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winkles JA (1998) Serum‐ and polypeptide growth factor‐inducible gene expression in mouse fibroblasts. Prog Nucleic Acid Res Mol Biol 58: 41–78 [DOI] [PubMed] [Google Scholar]
- 8. Perna D, Faga G, Verrecchia A, Gorski MM, Barozzi I, Narang V, Khng J, Lim KC, Sung WK, Sanges R et al (2012) Genome‐wide mapping of Myc binding and gene regulation in serum‐stimulated fibroblasts. Oncogene 31: 1695–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kelly K, Cochran BH, Stiles CD, Leder P (1983) Cell‐specific regulation of the c‐myc gene by lymphocyte mitogens and platelet‐derived growth factor. Cell 35: 603–610 [DOI] [PubMed] [Google Scholar]
- 10. Dominguez‐Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, Dalla‐Favera R (2012) The proto‐oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol 13: 1083–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caro‐Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, Gavin AL, Abel ED, Kelsoe G, Green DR et al (2014) Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF‐exposed B cells. J Immunol 192: 3626–3636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luo W, Weisel F, Shlomchik MJ (2018) B cell receptor and CD40 signaling are rewired for synergistic induction of the c‐Myc transcription factor in germinal center B cells. Immunity 48: 313–326.e315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J et al (2011) The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35: 871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Alboran IM, O'Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, Alt FW (2001) Analysis of C‐MYC function in normal cells via conditional gene‐targeted mutation. Immunity 14: 45–55 [DOI] [PubMed] [Google Scholar]
- 15. Murn J, Mlinaric‐Rascan I, Vaigot P, Alibert O, Frouin V, Gidrol X (2009) A Myc‐regulated transcriptional network controls B‐cell fate in response to BCR triggering. BMC Genom 10: 323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perez‐Olivares M, Trento A, Rodriguez‐Acebes S, Gonzalez‐Acosta D, Fernandez‐Antoran D, Roman‐Garcia S, Martinez D, Lopez‐Briones T, Torroja C, Carrasco YR et al (2018) Functional interplay between c‐Myc and Max in B lymphocyte differentiation. EMBO Rep 19: e45770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Silva NS, Klein U (2015) Dynamics of B cells in germinal centres. Nat Rev Immunol 15: 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Link JM, Hurlin PJ (2015) The activities of MYC, MNT and the MAX‐interactome in lymphocyte proliferation and oncogenesis. Biochim Biophys Acta 1849: 554–562 [DOI] [PubMed] [Google Scholar]
- 19. Blackwood EM, Eisenman RN (1991) Max: a helix‐loop‐helix zipper protein that forms a sequence‐specific DNA‐binding complex with Myc. Science 251: 1211–1217 [DOI] [PubMed] [Google Scholar]
- 20. Kretzner L, Blackwood EM, Eisenman RN (1992) Myc and Max proteins possess distinct transcriptional activities. Nature 359: 426–429 [DOI] [PubMed] [Google Scholar]
- 21. Amati B, Dalton S, Brooks MW, Littlewood TD, Evan GI, Land H (1992) Transcriptional activation by the human c‐Myc oncoprotein in yeast requires interaction with Max. Nature 359: 423–426 [DOI] [PubMed] [Google Scholar]
- 22. Eilers M, Eisenman RN (2008) Myc's broad reach. Genes Dev 22: 2755–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dang CV (2013) MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med 3: a014217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kress TR, Sabò A, Amati B (2015) MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 15: 593–607 [DOI] [PubMed] [Google Scholar]
- 25. Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA (2012) Transcriptional amplification in tumor cells with elevated c‐Myc. Cell 151: 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Porter JR, Fisher BE, Baranello L, Liu JC, Kambach DM, Nie Z, Koh WS, Luo J, Stommel JM, Levens D et al (2017) Global inhibition with specific activation: how p53 and MYC redistribute the transcriptome in the DNA double‐strand break response. Mol Cell 67: 1013–1025.e1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zeid R, Lawlor MA, Poon E, Reyes JM, Fulciniti M, Lopez MA, Scott TG, Nabet B, Erb MA, Winter GE et al (2018) Enhancer invasion shapes MYCN‐dependent transcriptional amplification in neuroblastoma. Nat Genet 50: 515–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay‐Odelot H, Karim S, Bartkuhn M et al (2014) Activation and repression by oncogenic MYC shape tumour‐specific gene expression profiles. Nature 511: 483–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lorenzin F, Benary U, Baluapuri A, Walz S, Jung LA, von Eyss B, Kisker C, Wolf J, Eilers M, Wolf E (2016) Different promoter affinities account for specificity in MYC‐dependent gene regulation. Elife 5: e15161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kress TR, Pellanda P, Pellegrinet L, Bianchi V, Nicoli P, Doni M, Recordati C, Bianchi S, Rotta L, Capra T et al (2016) Identification of MYC‐dependent transcriptional programs in oncogene‐addicted liver tumors. Can Res 76: 3463–3472 [DOI] [PubMed] [Google Scholar]
- 31. Muhar M, Ebert A, Neumann T, Umkehrer C, Jude J, Wieshofer C, Rescheneder P, Lipp JJ, Herzog VA, Reichholf B et al (2018) SLAM‐seq defines direct gene‐regulatory functions of the BRD4‐MYC axis. Science 360: 800–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sabò A, Amati B (2018) BRD4 and MYC‐clarifying regulatory specificity. Science 360: 713–714 [DOI] [PubMed] [Google Scholar]
- 33. Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, Bishop JM (2001) c‐Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414: 768–773 [DOI] [PubMed] [Google Scholar]
- 34. de Pretis S, Kress TR, Morelli MJ, Sabò A, Locarno C, Verrecchia A, Doni M, Campaner S, Amati B, Pelizzola M (2017) Integrative analysis of RNA polymerase II and transcriptional dynamics upon MYC activation. Genome Res 27: 1658–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Furlan M, de Pretis S, Galeota E, Caselle M, Pelizzola M (2019) Dynamics of transcriptional regulation from total RNA‐seq experiments. bioRxiv 10.1101/520155 [PREPRINT] [DOI] [Google Scholar]
- 36. Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, Weintraub H (1990) Sequence‐specific DNA binding by the c‐Myc protein. Science 250: 1149–1151 [DOI] [PubMed] [Google Scholar]
- 37. Grandori C, Mac J, Siebelt F, Ayer DE, Eisenman RN (1996) Myc‐Max heterodimers activate a DEAD box gene and interact with multiple E box‐related sites in vivo . EMBO J 15: 4344–4357 [PMC free article] [PubMed] [Google Scholar]
- 38. Sabò A, Amati B (2014) Genome recognition by MYC. Cold Spring Harb Perspect Med 4: a014191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall’ Olio V, Zardo G, Nervi C, Bernard L, Amati B (2006) Myc‐binding‐site recognition in the human genome is determined by chromatin context. Nat Cell Biol 8: 764–770 [DOI] [PubMed] [Google Scholar]
- 40. Soufi A, Donahue G, Zaret KS (2012) Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151: 994–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guo J, Li T, Schipper J, Nilson KA, Fordjour FK, Cooper JJ, Gordan R, Price DH (2014) Sequence specificity incompletely defines the genome‐wide occupancy of Myc. Genome Biol 15: 482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, Olejniczak ET, Clark T, Dey S, Lorey S et al (2015) Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell 58: 440–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Richart L, Carrillo‐de Santa Pau E, Rio‐Machin A, de Andres MP, Cigudosa JC, Lobo VJ, Real FX (2016) BPTF is required for c‐MYC transcriptional activity and in vivo tumorigenesis. Nat Commun 7: 10153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tu WB, Shiah YJ, Lourenco C, Mullen PJ, Dingar D, Redel C, Tamachi A, Ba‐Alawi W, Aman A, Al‐Awar R et al (2018) MYC interacts with the G9a histone methyltransferase to drive transcriptional repression and tumorigenesis. Cancer Cell 34: 579–595.e578 [DOI] [PubMed] [Google Scholar]
- 45. Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA (2010) c‐Myc regulates transcriptional pause release. Cell 141: 432–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baluapuri A, Hofstetter J, Dudvarski Stankovic N, Endres T, Bhandare P, Vos SM, Adhikari B, Schwarz JD, Narain A, Vogt M et al (2019) MYC recruits SPT5 to RNA polymerase II to promote processive transcription elongation. Mol Cell 74: 674–687.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Davari K, Lichti J, Gallus C, Greulich F, Uhlenhaut NH, Heinig M, Friedel CC, Glasmacher E (2017) Rapid genome‐wide recruitment of RNA polymerase II drives transcription, splicing, and translation events during T cell responses. Cell Rep 19: 643–654 [DOI] [PubMed] [Google Scholar]
- 48. Liang K, Smith ER, Aoi Y, Stoltz KL, Katagi H, Woodfin AR, Rendleman EJ, Marshall SA, Murray DC, Wang L et al (2018) Targeting Processive transcription elongation via SEC disruption for MYC‐induced cancer therapy. Cell 175: 766–779.e717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu H, Tang X, Srivastava A, Pecot T, Daniel P, Hemmelgarn B, Reyes S, Fackler N, Bajwa A, Kladney R et al (2015) Redeployment of Myc and E2f1‐3 drives Rb‐deficient cell cycles. Nat Cell Biol 17: 1036–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peitz M, Pfannkuche K, Rajewsky K, Edenhofer F (2002) Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: a tool for efficient genetic engineering of mammalian genomes. Proc Natl Acad Sci USA 99: 4489–4494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gerondakis S, Grumont RJ, Banerjee A (2007) Regulating B‐cell activation and survival in response to TLR signals. Immunol Cell Biol 85: 471–475 [DOI] [PubMed] [Google Scholar]
- 52. Bianchi V, Ceol A, Ogier AG, de Pretis S, Galeota E, Kishore K, Bora P, Croci O, Campaner S, Amati B et al (2016) Integrated systems for NGS data management and analysis: open issues and available solutions. Front Genet 7: 75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J et al (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kishore K, de Pretis S, Lister R, Morelli MJ, Bianchi V, Amati B, Ecker JR, Pelizzola M (2015) methylPipe and compEpiTools: a suite of R packages for the integrative analysis of epigenomics data. BMC Bioinformatics 16: 313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou VW, Goren A, Bernstein BE (2011) Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 12: 7–18 [DOI] [PubMed] [Google Scholar]
- 58. Calo E, Wysocka J (2013) Modification of enhancer chromatin: what, how, and why? Mol Cell 49: 825–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- 61. Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. de Pretis S, Kress T, Morelli MJ, Melloni GE, Riva L, Amati B, Pelizzola M (2015) INSPEcT: a computational tool to infer mRNA synthesis, processing and degradation dynamics from RNA‐ and 4sU‐seq time course experiments. Bioinformatics 31: 2829–2835 [DOI] [PubMed] [Google Scholar]
- 63. Ehninger A, Boch T, Uckelmann H, Essers MA, Mudder K, Sleckman BP , Trumpp A (2014) Posttranscriptional regulation of c‐Myc expression in adult murine HSCs during homeostasis and interferon‐alpha‐induced stress response. Blood 123: 3909–3913 [DOI] [PubMed] [Google Scholar]
- 64. Farrell AS, Sears RC (2014) MYC degradation. Cold Spring Harb Perspect Med 4: a014365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B (2001) Binding of c‐Myc to chromatin mediates mitogen‐induced acetylation of histone H4 and gene activation. Genes Dev 15: 2069–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Review Process File
Data Availability Statement
RNA‐Seq and ChIP‐Seq data have been deposited in NCBI's Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GSE126340 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126340).
All R scripts used in data analysis and generation of figures are available upon request.