

Abstract
Intrinsic apoptosis requires mitochondrial outer membrane disruption triggered by recruitment, activation, and oligomerization of the Bcl‐2 homology protein Bax. Following oxidative stress, we demonstrated that the transcriptional regulator cyclin C is released into the cytosol where it directs mitochondrial fragmentation and efficient apoptotic induction. This study reveals that cytoplasmic cyclin C is required for both normal Bax activation and its efficient mitochondrial localization. This activity appears direct as cyclin C co‐immunoprecipitates with active Bax in stressed cells and binds recombinant Bax in vitro. In addition, stable cyclin C–Bax association requires the fission complex. Pharmacologically stimulating cyclin C nuclear release is sufficient for Bax association and their mitochondrial localization in the absence of any stress signals. However, these cells do not undergo cell death as Bax fails to oligomerize. These data support a model that cyclin C association defines an initial step in Bax mitochondrial recruitment and provides a physical connection between the fission and apoptotic factors. This strategy allows the cell to discriminate stress‐induced fission able to recruit Bax from other types of mitochondrial divisions.
Keywords: apoptosis, Bcl‐2 homology, Cdk8, cyclin C, mitochondria
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport
Introduction
Mitochondria undergo constant fission and fusion cycles depending on the stress and energy requirements of the cell 1. For example, following cellular damage, the mitochondria undergo extensive fission that is dependent on recruitment of the dynamin‐like GTPase Drp1 by one of the several mitochondrial outer membrane (MOM) receptors (Mff, Mid49, Mid51, hFis1) 2. Hyper‐fission is associated with MOM permeabilization (MOMP) and release of pro‐apoptotic factors (e.g., cytochrome c) 3, 4, 5. Consistent with this model, Drp1 knockdown cells are resistant to apoptosis/programmed cell death (PCD) 6, 7. MOMP requires mitochondrial recruitment and oligomerization of two pro‐apoptotic proteins (Bax and Bak) 8. Connections between fission and the PCD machinery have been suggested by the finding that Bax is recruited to sites of fission 9. However, additional reports indicate that fission itself is not necessary for the timely release of some pro‐apoptotic proteins 5, 10. Therefore, the connection between the fission machinery and PCD still remains to be clarified. In addition, it was not clear why Bax is only recruited to fission sites following stress and not by other instances of Drp1‐dependent fragmentation (e.g., mitosis, mitophagy). This paradox argued for the existence of an additional stress‐activated factor that differentiates one type of mitochondrial division from another.
Cyclin C, Cdk8, Med12, and Med13 are components of the Cdk8 kinase module (CKM) that associate with the Mediator complex of RNA polymerase II to both positively and negatively regulate gene transcription 11. In yeast, cyclin C–Cdk8 function primarily as repressors of genes involved in environmental sensing, stress response, cell type control, and meiosis 12, 13, 14, 15. In response to several stressors including pro‐oxidants, we previously demonstrated that cyclin C (but not Cdk8) translocates from the nucleus to the cytoplasm where it stimulates mitochondrial fission in both yeast 16, 17 and mammalian 18 cells. The role of cyclin C in directing stress‐induced fission appears direct as cyclin C co‐immunoprecipitates with Drp1 in mammalian cells 18 and recombinant cyclin C and Drp1 interact in pull‐down experiments 19. In budding yeast, cyclin C also co‐immunoprecipitates with the fission adaptor Mdv1 16. In addition, cyclin C is required for efficient execution of the intrinsic PCD pathway by promoting MOMP 18. These findings suggested a central role for cyclin C in coordinating mitochondrial fission and PCD in response to cellular stress. The present study focuses on the role cyclin C plays in directing intrinsic apoptosis. We find that cyclin C directly binds Bax and is both necessary and sufficient to efficiently recruit Bax to the mitochondria. In addition, Bax–cyclin C association requires an intact fission complex biochemically connecting the two processes. Finally, our studies reveal a role for mitochondrial‐derived reactive oxygen species (ROS) in mediating cyclin C‐induced Bax activation. These findings connect the fission and apoptotic machinery through cyclin C and provide a mechanism for how the cell identifies a stress‐activated fission complex able to recruit Bax to the mitochondria.
Results and Discussion
Cyclin C is required for Bax activation
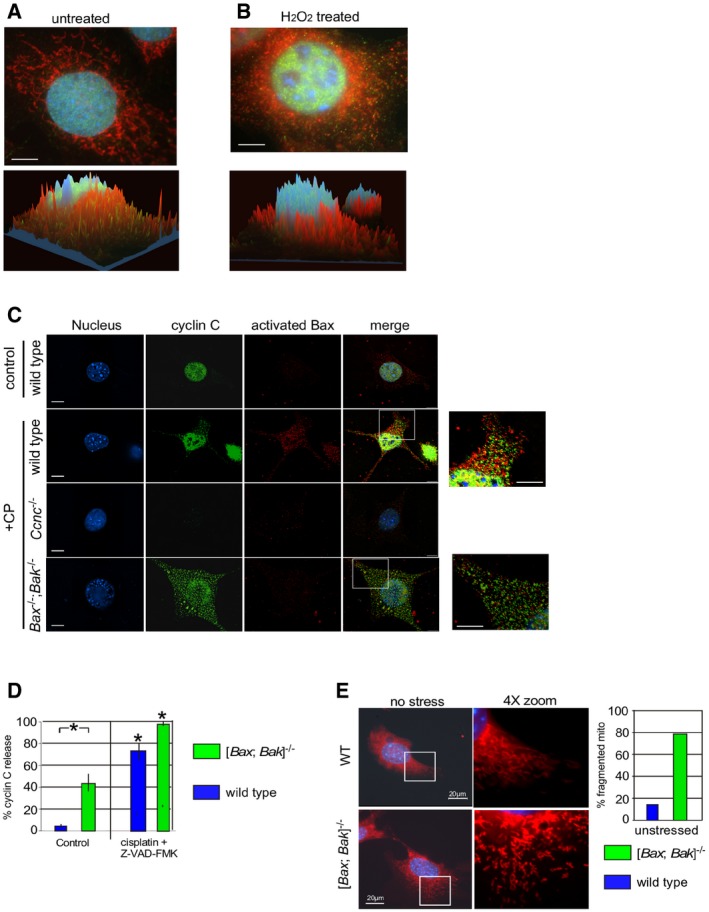
We previously reported that the nuclear transcriptional regulator cyclin C translocates from the nucleus to the mitochondria in response to H2O2 treatment (Ref. 18, see Fig EV1A and B). Bax‐induced MOMP requires its conformational change, MOM insertion, and oligomerization 20. Therefore, we first examined whether cyclin C played a role in promoting Bax activation. Wild‐type and Ccnc −/− MEF cultures were treated with H2O2 for 4 h and protein extracts were immunoprecipitated with antibodies directed against an amino‐terminal epitope exposed upon Bax conformational change 21. This assay is commonly used to infer that Bax is in its open or “active” configuration. Western blot analysis of the immunoprecipitates with a conformation specific Bax antibody revealed that, as expected, activated Bax was only detected in wild‐type extracts prepared from cells exposed to H2O2 (Fig 1A). Repeating these experiments with H2O2‐stressed Ccnc −/− cultures failed to identify significant Bax activation. This result was not due to changes in overall Bax levels (bottom panels). To assay Bax activation in a different way, indirect immunofluorescence (IF) was used with the activation antibody in wild‐type and Ccnc −/− MEF cultures exposed to the anti‐cancer drug cisplatin. These studies also demonstrated a reduction in Bax activation in Ccnc −/− treated cells versus the wild‐type control (Fig EV1C). Taken together, these results indicate that cyclin C is required for normal Bax activation. However, Bak and Bax are not required for cyclin C nuclear release (Fig EV1C, quantitated in Fig EV1D). Interestingly, the double‐mutant cells exhibit elevated cyclin C nuclear release in the absence of stress (Fig EV1D) that resulted in partial mitochondrial fragmentation (Fig EV1E). Taken together, these results suggest that the mitochondrial fission activity of cyclin C is largely independent of Bax or Bak.
Figure EV1. Cyclin C is required for normal Bax activation.
-
A, BWild‐type MEF culture was stained for the nuclei (DAPI), cyclin C (indirect immunofluorescence), and mitochondria (MitoTracker Red) before (A) and after (B) H2O2 treatment (0.4 mM, 4 h). Confocal images were collected and analyzed for signal localization within the cell with the z‐axis indicating intensity. Red and green peaks indicate co‐localization between cyclin C and mitochondria. Note the cyclin C shading is normally on the side of the peak indicating association with the end of the mitochondrion indicative of a role in fission. Bars = 10 μm.
-
CCyclin C is required for normal Bax activation. MEF cultures with the indicated genotypes were treated with cisplatin (CP) (30 mM for 16 h) and then analyzed by fluorescence microscopy visualizing the nucleus (DAPI), cyclin C (IF), activated Bax (IF). The images were merged in the fourth column. Image exposures were identical for each component examined. Bar indicates 20 μm. Zoom images (4×) are provided on the far right (bars = 10 μm).
-
DCyclin C nuclear release was quantitated for MEF cultures with the indicated genotype. A positive nuclear release score was given when > 15% of the cyclin C total signal was cytoplasmic. The caspase inhibitor Z‐VAD‐FMK was added to prevent apoptosis initiation allowing the entire populations to be counted. Three independent cultures were measured, 100 cells per culture. Error bars indicate SD, and asterisks indicate P values < 0.05 using the Student's t‐test. Note the increased cyclin C nuclear release in unstressed [Bax; Bak]−/− cells.
-
EUnstressed WT and [Bak; Bax]−/− MEF cells were stained for nuclei (DAPI) and mitochondria (MitoTracker). Mitochondrial morphology in the WT cell displays normal reticular configuration. The double‐mutant cells exhibited an intermediate phenotype displaying a portion of fragmented mitochondria but not to the extent observed in stressed cells (see Fig EV1B).
Figure 1. Oxidative stress‐induced Bax activation requires cyclin C and the fission machinery.
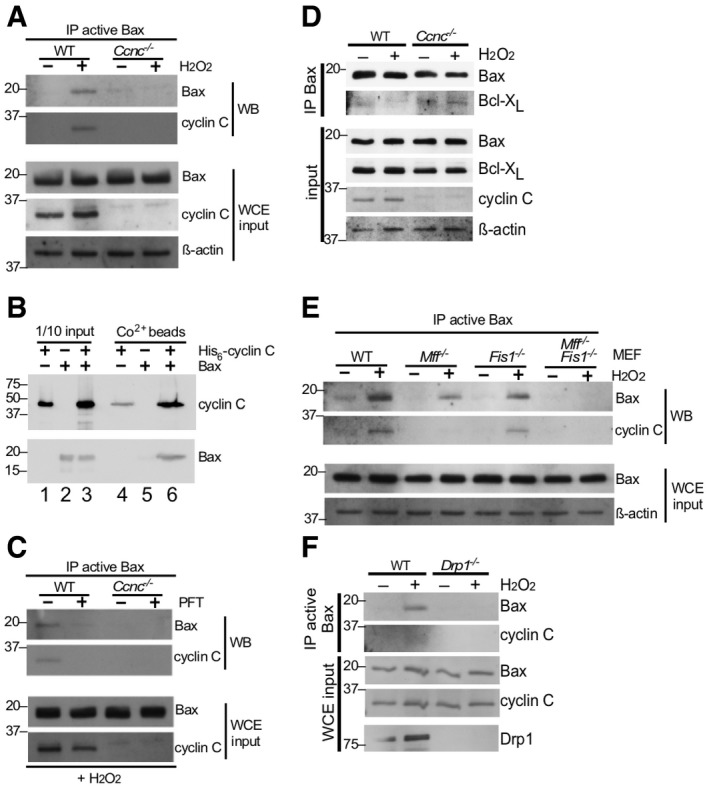
- WT and Ccnc −/− MEF cultures were treated with 0.4 mM H2O2 as indicated and extracts prepared and immunoprecipitated with antibody recognizing the active conformation of Bax. The immunoprecipitates were subjected to Western blot analysis and probed for total Bax or cyclin C. Whole cell extracts (WCE) were subjected to Western blot analysis as indicated to control for protein concentrations in the extracts. Molecular weight markers (kDa) are indicated on the left.
- Cyclin C and Bax directly interact. Recombinant Bax and SUMO‐His6‐cyclin C were either incubated together or separately at room temperature and then passed over Co2+ resin. The elutions were subjected to Western blot analysis probing for Bax and cyclin C (lanes 4–6). Input controls for each sample (1/10) are included in lanes 1–3. Molecular weight markers (kDa) are indicated on the left.
- Mitochondrial relocalization of cyclin C is required for Bax activation. The experiment described in (A) was repeated except that H2O2‐treated cells were treated with 1 μM pifithrin‐μ (PFT) for 24 h as indicated. Molecular weight markers (kDa) are indicated on the left.
- Cyclin C is required for efficient Bax‐Bcl‐XL dissociation. Extracts prepared from MEF cells with the indicated genotypes following treatment with H2O2 (0.4 mM, 4 h) were immunoprecipitated with general Bax antibodies. The immunoprecipitates were subjected to Western blot analysis probing with Bcl‐XL antibodies. Western blot analysis of WCE was performed as input concentration controls. Molecular weight markers (kDa) are indicated on the left.
- The mitochondrial fission complex is required for cyclin C‐dependent Bax activation. Extracts were prepared from MEF cultures with the indicated genotypes treated with H2O2 (0.4 mM, 4 h). The extracts were treated as in (A). Molecular weight markers (kDa) are indicated on the left.
- Drp1 is required for cyclin C–Bax interaction. Extracts were prepared from wild‐type or Drp1 −/− MEF cultures treated or not with H2O2 and analyzed for the presence of Bax and cyclin C as described in (A). Molecular weight markers (kDa) are indicated on the left.
We next examined the relationship between cyclin C and Bax following stress application. First, the blot described in Fig 1A was stripped and reprobed for cyclin C. These results identified cyclin C in the Bax immunoprecipitate suggesting the possibility that its role in Bax activation is direct. To test this possibility, recombinant Bax and His6‐cyclin C were purified from Escherichia coli and a pull‐down experiment was performed (see Materials and Methods). His6‐cyclin C was immobilized on Co2+ beads; then, purified Bax was added. His6‐cyclin C and Bax were added independently to the resin as well. After incubation, the beads were washed extensively, collected, and the remaining proteins removed by boiling in 2× sample buffer. The eluted proteins were separated by SDS–PAGE then blotted and probed for the two proteins. These results revealed that Bax was included in the His6‐cyclin C bound beads but not in the resin alone (Fig 1B, compare lanes 5 and 6). Compared to the input control, approximately 10% of Bax in the reaction associated with cyclin C (compare Bax signals in lanes 3 and 6). These results indicate that cyclin C directly associates with Bax.
Cyclin C regulates Bax activity at the mitochondria
To further explore the relationship between cyclin C and Bax, we next asked whether mitochondrial cyclin C relocalization is required for normal Bax activation. These experiments were therefore repeated with the chaperone inhibitor pifithrin‐μ (PFT) 22. We previously found that PFT treatment inhibits cyclin C translocation to the mitochondria and prevents stress‐induced hyper‐fission 18. Cells were incubated with PFT prior to H2O2 addition and Bax activation monitored as just described. In the absence of PFT, Bax activation and cyclin C association were observed as before (Fig 1C). However, PFT suppressed Bax activation supporting the model that cyclin C relocalization to the mitochondria contributes to Bax activation under oxidative stress conditions.
Bax insertion into the MOM is inhibited by the presence of the Bcl‐2 homology pro‐survival proteins Bcl‐2 or Bcl‐XL. Therefore, we next asked whether cyclin C‐dependent Bax activation was accompanied by reduction in Bcl‐XL interaction. In wild‐type MEFs, Bcl‐XL co‐immunoprecipitated with Bax but this association was reduced following H2O2 treatment (Fig 1D). However, Bax‐Bcl‐XL association was not reduced in Ccnc −/− cells following oxidative stress application. These results suggest that the presence of cyclin C helps restrict the association of Bcl‐XL to Bax. However, whether cyclin C disrupts Bcl‐XL association directly or indirectly is not known.
An intact fission complex is required for cyclin C–Bax interaction
We previously demonstrated that cyclin C directly binds Drp1 19 and is required for stress‐induced fission 18. Therefore, we next examined whether cyclin C association with Bax requires an intact fission complex. To examine this question, cyclin C–Bax activation experiments were repeated with MEF cells deleted for the Drp1 MOM receptors Mff and/or hFis1. These cultures were treated with H2O2, and Bax activation and cyclin C co‐immunoprecipitation were monitored as before. As reported previously 23, deleting Mff or hFis1 reduced, but did not eliminate, Bax activation (Fig 1E). Similarly, cyclin C association was reduced approximately corresponding to the level of active Bax signal observed. Interestingly, active Bax or cyclin C association was not observed in the Mff −/− hFis1 −/− double mutant. A similar result was obtained in Drp1 knockout MEF cells (Fig 1F). These results mirrored our previous findings that the mitochondrial fragmentation observed when cyclin C was exogenously added to permeabilized cells required Mff and hFis1 18. Interestingly, Drp1 −/− MEF cells displayed a reduced ability to execute the cell death pathway (Fig EV2). These findings suggest that only a limited amount of Bax activation is required for partial apoptotic induction or that Bak, or other cell death pathways, are involved (see Discussion). Taken together, the studies indicate that cyclin C association with Bax requires the presence of an intact fission complex.
Figure EV2. Drp1 is required for normal apoptotic efficiency following cisplatin treatment.
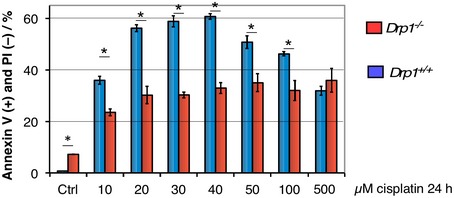
Wild‐type and Drp1 −/− MEF cells were treated with several concentrations of cisplatin, and apoptotic efficiency was determined as the percentage of the population Annexin V positive, propidium iodide (PI) negative. Error bars represent SD, n = 3 independent cultures. Asterisks indicate P < 0.01 using Student's t‐test.
Cyclin C is sufficient to recruit active Bax to the mitochondria
We next asked whether cyclin C relocalization to the cytoplasm is sufficient to induce mitochondrial fission and conversion of Bax to its active configuration or whether an additional stress signal(s) is required. Our previous yeast studies revealed that disrupting the interaction between cyclin C and its nuclear anchor, Med13, allows cyclin C release and subsequent mitochondrial fragmentation without additional stress signals 16, 17. We identified a short alpha helical region we termed the holoenzyme association domain (HAD) that is required for Med13 association 24. We developed a cell‐penetrating peptide mimetic (S‐HAD) of this region that rapidly induced cyclin C nuclear release and mitochondrial fragmentation (Fig 2A, quantified in Fig 2B). S‐HAD‐induced mitochondrial fragmentation required cyclin C as this response was not observed in Ccnc −/− MEF cells (Fig 2C). In addition, fluorescence microscopy revealed association between cyclin C and the mitochondria (arrows, Fig 2A). To biochemically test for cyclin C–mitochondrial association following peptide treatment, subcellular fractionation studies were performed. Nuclear and heavy membrane mitochondrial enriched fractions were prepared from wild‐type and Ccnc −/− MEF cultures with or without S‐HAD treatment. As expected from Fig 2A, S‐HAD treatment increased cyclin C concentration in the mitochondrial fraction (Fig 2D) while a significant fraction remained nuclear. No accumulation of cyclin C was observed in the soluble cytoplasmic fraction derived from these preparations (Fig EV3). Probing this blot for total Bax revealed an elevated signal in the Ccnc +/+ mitochondrial fraction following S‐HAD treatment but not in the Ccnc −/− cells. These findings indicate that cyclin C nuclear release into the cytoplasm is sufficient to induce mitochondrial fragmentation and recruit active Bax to the mitochondria.
Figure 2. Ectopic nuclear release of cyclin C is sufficient to induce mitochondrial fragmentation and Bax recruitment in its active confirmation.
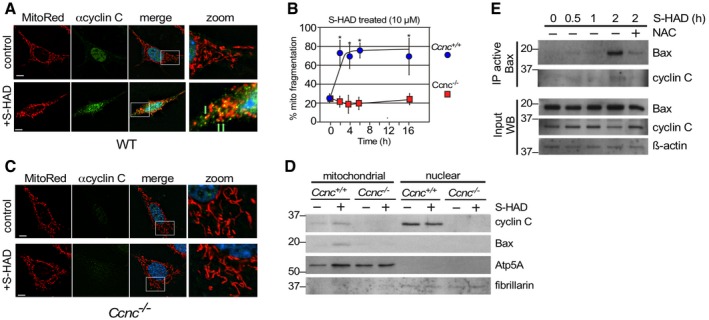
- Wild‐type MEF cells were fixed before (control) and following S‐HAD treatment (10 μM, 4 h); then, nuclei (DAPI), cyclin C (indirect immunofluorescence), and mitochondria (MitoTracker Red) were visualized by fluorescence microscopy. Merge panels present DAPI staining (blue) only. Zoom panels (4×) are indicated by the boxes in the merge panels. Green arrows indicate cyclin C and mitochondrial co‐localization. Bar indicates 10 μM.
- Timecourse experiment with wild‐type and Ccnc −/− MEF cells treated with S‐HAD (10 μM) for the times indicated (n = 3). Error bars indicate SD. Asterisks indicate P values < 0.05 from pretreatment control (Student's t‐test).
- S‐HAD peptide activity requires cyclin C. The experiment described in (A) was repeated with Ccnc −/− MEF cultures. Bar indicates 10 μm. Results quantitated in (B).
- S‐HAD treatment increases Bax mitochondrial localization. Western blot analysis of the indicated proteins in nuclear and mitochondrial fractions before and following S‐HAD treatment (10 μM, 4 h). Atp5A and fibrillarin levels controlled for mitochondrial and nuclear loadings, respectively. Molecular weight markers (kDa) are indicated on the left.
- Bax activation was monitored in wild‐type MEF cultures treated with S‐HAD (10 μM) for the times indicated. The anti‐oxidant N‐acetyl cysteine (NAC) was added (1 mM) 2 h prior to S‐HAD treatment. Active Bax immunoprecipitates were subjected to Western blot analysis for Bax and cyclin C as indicated. Bax, cyclin C, and β‐actin levels were monitored in extract preparations as input controls. Molecular weight markers (kDa) are indicated on the left.
Figure EV3. Cyclin C nuclear release promotes Bax mitochondrial association.
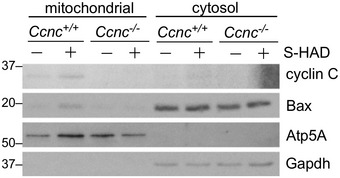
Mitochondrial‐heavy membrane and cytoplasmic subcellular fractions were prepared from the indicated MEF cultures after treatment with vehicle control (−) or S‐HAD (+) peptide (10 μM, 2 h). These fractions were probed for the indicated proteins. Atp5A and Gapdh were used to control for mitochondria and cytoplasmic fractions, respectively.
Next, we asked whether cyclin C nuclear release was sufficient to stimulate the conformational change of Bax to its active form in the absence of any additional stress signals. To test this possibility, a wild‐type MEF culture was treated with S‐HAD and samples taken over a 2‐h timecourse experiment. This timeframe coincides with maximal mitochondrial fragmentation observed for S‐HAD‐treated cells (Fig 2B). The timepoints were assayed for the presence of activated Bax as described in Fig 1. The results indicated that Bax activation was observed after a 2‐h peptide treatment (Fig 2E). Interestingly, addition of the free radical scavenger N‐acetyl cysteine (NAC) reduced peptide response, suggesting a role for endogenous ROS in S‐HAD stimulation of Bax activation (see below). Taken together, these studies indicate that introducing cyclin C into the cytoplasm is sufficient to induce Bax recruitment to the mitochondria and its conversion to an active conformation.
Cyclin C‐induced Bax activation sensitizes cells to apoptosis
Previous studies have associated Bax recruitment to the mitochondria and transition to an active conformation with MOMP and the initiation of the apoptotic pathway 25. To determine if cyclin C nuclear release induces cell death independently of additional stress signals, MEF cells were treated with S‐HAD for 16 h and several markers of apoptosis were examined. These experiments revealed a modest increase in caspase activation between the peptide‐treated culture and the control cells (Fig 3A). However, this effect was significantly less than that observed with cisplatin‐treated cells. Two additional assays for cell death, PARP cleavage (Fig 3B) and the rounding up of cultured cells (Fig 3C, quantified in Fig EV4A), exhibited little or no elevation in cell death markers between S‐HAD‐treated cells and vehicle control. As we reported previously 18, cell death was still observed over background with the Ccnc −/− cisplatin‐treated cells but still below the wild‐type control. These studies indicate that the presence of cytoplasmic cyclin C is not sufficient to induce PCD.
Figure 3. Cyclin C nuclear release alone is insufficient to induce PCD.
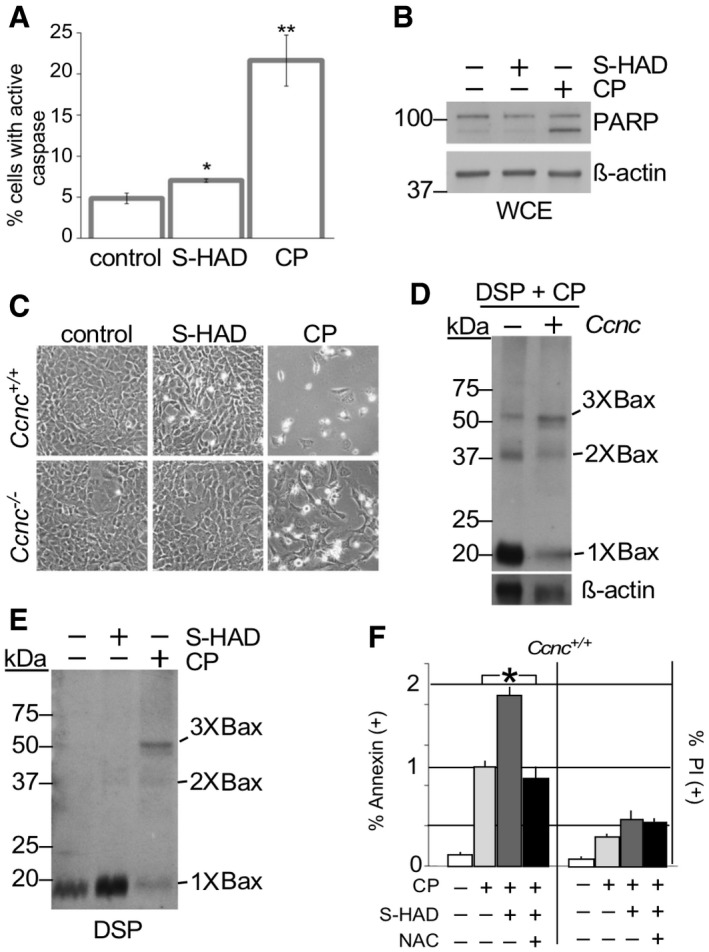
- Caspase activation was measured by fluorescent cell analysis in wild‐type MEF cells treated with S‐HAD (10 μM, 24 h) or cisplatin (CP, 30 μM, 16 h). n = 3. Error bars indicate SD. * and ** indicate P < 0.05 and P < 0.01, respectively, using Student's t‐test.
- PARP cleavage was not induced by S‐HAD treatment. PARP cleavage was monitored by Western blot analysis in HeLa cells treated with S‐HAD or cisplatin (CP) as described in (A). The blot was stripped and reprobed with β‐actin antibodies as a loading control. Molecular weight markers (kDa) are indicated on the left.
- Wild‐type and Ccnc −/− MEF cultures were treated with S‐HAD and cisplatin (CP) as just described. Cells were imaged and examined for a “rounding up” phenotype diagnostic for death (see Fig EV4A for quantitation).
- Cyclin C is required for normal Bax oligomerization in response to cisplatin. Whole cell extracts prepared from DSP crosslinked wild‐type and CCNC −/− MEF cells exposed to cisplatin (30 μM, 16 h) were probed for total Bax. Bax multimers are indicated. Molecular weight markers (kDa) are indicated on the left.
- S‐HAD does not induce Bax oligomerization. Experiments described in (D) were repeated with cultures treated with S‐HAD (10 μM, 24 h). Bax multimers are indicated. Molecular weight markers (kDa) are indicated on the left.
- S‐HAD treatment sensitizes Hela cells to cisplatin. Hela cultures were treated with cisplatin (30 μM), S‐HAD (10 μM), and N‐acetyl cysteine (NAC, 1 mM) as indicated. Fluorescent cell analysis was used to quantitate the apoptotic (Annexin V positive) and necrotic (PI positive) population percentages, respectively. n = 3 independent cultures. Error bars indicate SD. Asterisk indicates P < 0.05 (Student's t‐test).
Figure EV4. S‐HAD treatment does not significantly increase the population percentage of rounded, detached cells.
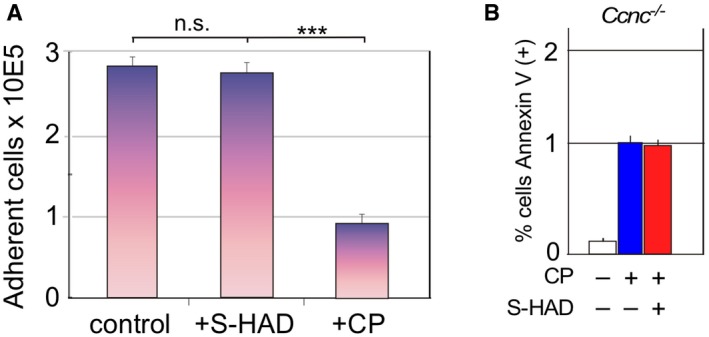
- Wild‐type MEF cultures were treated with vehicle (control), S‐HAD (10 μM, 24 h), or cisplatin (CP, 30 μM, 16 h), and the percent of the cells still adhered to the dish counted. Results are shown from three independent cultures, error bars indicate SD, n.s. = not statistically different; ***P < 0.001 as calculated by Student's t‐test.
- Cyclin C is required for S‐HAD‐enhanced apoptosis. Apoptotic efficiency was measured using Annexin V staining in Ccnc −/− MEF cultures treated with cisplatin (30 μM, 24 h) with and without 4 h S‐HAD addition (n = 3). Error bars indicate SD. The percentage of Annexin V‐positive cells in the cisplatin alone treated culture was set to one.
Bax oligomerization and subsequent pore formation are required for MOMP and cell death. To test the requirement of cyclin C for Bax oligomerization, cisplatin‐treated wild‐type and Ccnc −/− MEF cells were crosslinked with dithiobis(succinimidyl propionate; DSP), cellular membranes were solubilized with a detergent buffer (digitonin) and extracts prepared for Western blot analysis. In wild‐type cells, Bax multimers were readily detected with a corresponding loss in monomers (Fig 3D). In the Ccnc −/− MEF extracts, oligomerization was detected but at a reduced levels. In addition, an elevated pool of monomeric Bax was observed in the mutant extracts compared to wild type. These studies indicate that cyclin C stimulates Bax oligomerization but only in the presence of an additional stress signal. Therefore, we next sought to understand how cyclin C nuclear release induces the active conformation of Bax and mitochondrial recruitment without causing significant cell death. First, to determine if Bax oligomerization occurred following cyclin C release, S‐HAD peptide‐treated wild‐type MEF cells were treated with DSP as before. These results indicated that cisplatin addition resulted in readily detectable Bax multimers with a corresponding reduction in monomer concentration (Fig 3E). However, multimerization of Bax was not observed in the S‐HAD‐treated samples. These results indicate that nuclear release of cyclin C alone is not sufficient to induce Bax oligomerization providing an explanation why S‐HAD‐treated cells do not exhibit elevated cell death. These results are consistent with a model that cyclin C is sufficient to induce a conformational change in Bax allowing MOM retention but not for the oligomerization step required for MOMP.
Our results indicate that cyclin C nuclear release is able to “stage” Bax on the MOM in its active configuration but not oligomerize. Therefore, we next tested whether Bax mitochondrial staging made cells hypersensitive to apoptotic stimuli. HeLa cells were treated with S‐HAD, the DNA alkylating agent cisplatin, or both and examined for Annexin V and propidium iodide (PI) staining. We considered Annexin V positive, PI negative as an indicator of cells in early apoptosis. These studies revealed that ectopic nuclear cyclin C release stimulated cisplatin‐induced PCD approximately twofold (Fig 3F). No significant increase in propidium iodide permeable cells was observed, indicating that necrotic cell death was not enhanced in these studies. The S‐HAD‐induced elevation in cell death required cyclin C as no stimulation was observed in Ccnc −/− cells (Fig EV4B). These findings indicate that staging Bax at the mitochondria renders cells hypersensitive to the anti‐cancer drug cisplatin.
Cyclin C activity is stimulated by mitochondrial‐derived ROS
In the experiments described in Fig 2E, we found that cyclin C‐induced Bax conversion to the active configuration was negated by addition of the general free radical scavenger N‐acetyl cysteine (NAC). These results suggested a potential role for ROS in cyclin C function. Consistent with this possibility, cysteine 62 oxidation is reported to stimulate Bax oligomerization 26. Previous studies reported that extensive mitochondrial fragmentation elevates endogenous ROS levels (e.g., Ref. 27). Using a mitochondrial‐targeted redox sensor (MitoSOX), we found that S‐HAD treatment stimulated mitochondrial ROS in a cyclin C‐dependent manner (Fig 4A). To control for changes in mitochondrial ROS production via electron transport chain (ETC) defects, wild‐type and Ccnc −/− cells were treated with the ETC complex 1 inhibitor rotenone that produces extensive free radical release 28. Mitochondrial ROS release was similar in both wild‐type and Ccnc −/− delete cells. These results indicate that the loss of elevated mitochondrial ROS in S‐HAD‐treated Ccnc −/− cells is most likely due to the lack of fragmentation and not other mitochondrial defects. However, there still exists the possibility that the S‐HAD peptide or deleting Ccnc has undiscovered activities that could produce this result.
Figure 4. Endogenous ROS enhances cyclin C–Bax interaction.
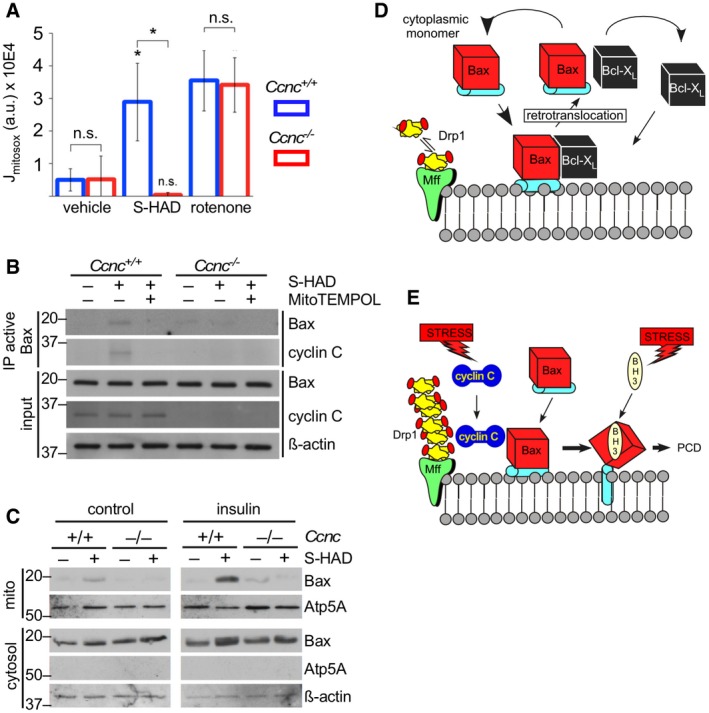
-
AS‐HAD treatment induces mitochondrial ROS. Wild‐type and Ccnc −/− MEF cultures were treated with S‐HAD (10 μM, 24 h), rotenone, or vehicle control as indicated. Mitochondrial ROS was measured by MitoSOX staining (4 μM, 15 min) and quantitative confocal imaging. Error bars indicate SD. n = 3, asterisk indicates P < 0.05 (Student's t‐test), n.s., not significantly different.
-
BQuenching mitochondrial ROS reduces cyclin C–Bax interaction. Wild‐type and Ccnc −/− MEF cultures were treated with S‐HAD (10 μM, 16 h) and 100 nM mitochondrial‐specific reactive oxygen scavenger MitoTEMPO. Western blot analysis was conducted as described in Fig 1. Molecular weight markers (kDa) are indicated on the left.
-
CInsulin treatment stimulates S‐HAD‐induced Bax activation. Wild‐type and Ccnc −/− MEF cultures were grown with or without insulin (10 μg/ml) and then treated with S‐HAD (10 μM, 4 h). Mitochondrial and cytosolic fractions were prepared and probed for the indicated proteins. Atp5A and β‐actin levels control for mitochondrial and cytosolic loading, respectively. The two blots were prepared, processed, and developed identically. Molecular weight markers (kDa) are indicated on the left.
-
D, EModel for Bax mitochondrial association before (D) and after (E) PCD‐inducing stress. In unstressed cells, Bax continuously samples the MOM but does not insert into the MOM and is removed by pro‐survival proteins (Bcl‐2, Bcl‐XL) in a process termed retrotranslocation. In response to stress, cyclin C associates with Bax stabilizing its interaction with the MOM in a fission complex‐dependent manner. Increased dwell time makes Bax more efficiently targeted by BH3‐only proteins activated by stress. BH3‐only protein binding induces conformational changes promoting Bax MOM insertion, homodimer formation, and oligomerization ultimately leading to PCD (see text for details).
Next, we determined if cyclin C‐dependent Bax activation required mitochondrial‐derived ROS. Wild‐type and Ccnc −/− MEF cultures were treated with S‐HAD as before with and without MitoTEMPO, a mitochondrial‐specific anti‐oxidant 29. These experiments revealed that mitochondrial ROS is required for cyclin C‐dependent Bax activation (Fig 4B). Since reducing endogenous ROS restricted cyclin C‐dependent Bax activation, we next tested whether Bax mitochondrial recruitment was enhanced under conditions of metabolically elevated ROS. In this experiment, wild‐type and Ccnc −/− MEF cells were treated with S‐HAD with or without insulin, which stimulates H2O2 production by stimulating Nox4 activity 30. Insulin treatment enhanced Bax recruitment to the mitochondria in a cyclin C‐dependent manner (Fig 4C).
Where does cyclin C fit into the established Bax activation pathway?
Several studies revealed a multi‐step process of Bax‐induced cytochrome c release 31. Under non‐stress conditions, Bax continuously samples on and off the MOM in a process called retrotranslocation (Fig 4D) 32, 33. Removing Bax from the MOM requires the pro‐survival Bcl‐2 proteins Bcl‐2 or Bcl‐XL. Once released into the cytosol, the proteins are predicted to separate and recycle onto the MOM. In the presence of a stress signal (Fig 4E), this cycle is disrupted as Bax MOM retention increases through its helix 9 transmembrane domain. Association with a BH3‐only family member (e.g., tBid, Bim) 34 causes release of the “latch domain” composed of helixes 6, 7, and 8 35. This conformational switch exposes the BH3 grove promoting homodimer formation, oligomerization, and eventually pore formation. Our results suggest that cyclin C plays an early role in Bax activation and recruitment to the mitochondria before BH3‐only protein activation. We demonstrate that cyclin C nuclear release is sufficient to enhance mitochondrial Bax association and expose the amino‐terminal epitope. It is not clear whether the presence of cyclin C stimulates release of the Bax transmembrane domain to enhance mitochondrial dwell time. Our in vitro experiments revealed cyclin C–Bax association although Bax was not in an activated state. However, we were unable to detect cytosolic Bax–cyclin C association in cells. These findings suggest cyclin C–Bax interaction and mitochondrial association may require additional proteins in vivo. For example, we found that the fission complex is required for cyclin C–Bax interaction. Association of Bax to cyclin C may increase its dwell time on the mitochondria providing more opportunity for recognition and stimulation by the BH3‐only proteins. This model is consistent with our finding that staging Bax on the MOM using the S‐HAD peptide mimetic makes cells hypersensitive to cisplatin. Therefore, although Bax is observed in its “active” conformation, it may be associating with the mitochondria through the fission machinery components via cyclin C and not inserting into the MOM. Although this possibility has not been directly tested, we have discovered that recombinant cyclin C and Drp1 interact in vitro 19. In this model, cyclin C represents a biochemical bridge between the fission and PCD complexes. However, cyclin C is not sufficient to induce Bax oligomerization providing an explanation why S‐HAD‐treated cells do not exhibit elevated cell death.
The present study indicates that cyclin C and the fission machinery help establish a pro‐death initiating complex. The inclusion of cyclin C in this process allows the cell to distinguish damage‐activated fission from the fragmentation that occurs in response to other stimuli such as cell division. Our finding that Drp1, Fis1, and Mff are required for Bax activation and cyclin C–Bax association in response to H2O2 explains the resistance we observe to this pro‐oxidant in Cncc −/− MEF cells (this study and Ref. 18). However, this study and others still observe some level of apoptosis in the absence of a functional fission complex. One possibility is that the proposed retention of Bax via cyclin C association is not required for Bak activation. Alternative models for Bax or Bak activation through VDAC2 association have been reported 36. A functional connection between cyclin C, Bak, and VDAC2 waits to be explored. Our results may also address an ongoing question concerning the connection between fission and MOMP. Studies have found that the presence of dominant‐negative Drp1, or GTPase negative derivative, does not prevent Bax recruitment 9, 37. However, a reduction in MOMP is observed in Drp1 −/− knockout cells 38. Therefore, a cyclin C–Drp1 complex, with or without fission, may be sufficient to also stage Bax on the MOM. This may explain the reduced, but not eliminated, ability of a cell to undergo PCD in the absence of mitochondrial fragmentation. The mitochondrial ROS release associated with the scission process may then increase PCD efficiency but not the absolute ability to undergo cell death. For example, actively recruiting Bax to fission foci has been proposed to facilitate pore formation due to membrane stress at these sites 39. However, it is clear from our studies that cyclin C–mitochondrial association is not sufficient to induce cell death. This might be expected as the decision to initiate PCD is complex and requires the satisfaction of several checkpoints. For example, SUMO modification of Drp1 is required for PCD downstream of Bax/Bak oligomerization 40. Combined with this study, these results indicate that additional hurdles exist, both upstream and downstream of Bax multimerization, which have yet to be fully elaborated.
Several factors have been identified that promote nuclear to mitochondria communication 41. The new locations for these proteins are accompanied by new functions as well. For example, p53 translocates from the nucleus to the mitochondria where it promotes apoptosis by sequestering the pro‐survival proteins Bcl‐2 and Bcl‐XL 42, 43, 44, 45, 46. Although the roles of p53 and cyclin C are similar, there are several differences. Unlike p53, aberrant mitochondrial localization of cyclin C does not induce substantial lethality. In addition, p53 does not appear to have a role in mitochondrial fission. How, or if, these two factor cooperate to regulate PCD has yet to be elucidated. The use of transcription factor relocalization as the harbinger of cell damage could serve two roles. First, removing cyclin C from the nucleus (Cdk8 remains nuclear during stress) would inactivate Cdk8, thus altering transcription while simultaneously inducing mitochondrial fission and preparing the cell for PCD. Consistent with this model, we have identified cyclin C‐repressed genes that are induced following oxidative stress 47. This strategy allows coordination of the stress response between these two important organelles. Taken together, our results are consistent with a model that cyclin C nuclear release following oxidative stress allows the cell to differentiate between stress and non‐stress fission events. In addition, this mechanism would coordinate changes in gene expression with mitochondrial dynamics.
Materials and Methods
Chemicals and reagents
Cisplatin, glycerol, and H2O2 were from Thermo Fisher Scientific (Waltham, MA). SDS was from US Biological (Salem, MA). Bromophenol blue, NaCl, and Tris were from VWR (Radnor, PA). CHAPS, DTT, and HEPES were from Gold Biotechnology (St. Louis, MO). All other reagents were from Sigma‐Aldrich (St. Louis, MO), and all antibodies were from Abcam (Cambridge, MA), unless stated otherwise. Stapled peptide WILDKQ‐S5‐LLK‐S5‐RQKDLKF (Bio‐Synthesis, Lewisville, TX), hydrocarbon‐stapled via two (S)‐2‐(4‐pentenyl) alanine residues (S5), was reconstituted at 25 mM stock concentration in 4°C PBS (Corning, Corning, NY) and stored at −20°C.
Cell culture
MEF cell lines were generated from WT and conditional cyclin C‐knockout mice as previously described 18. MEF and HeLa (ATCC, Manassas, VA) cells were cultured in DMEM (Corning, 10‐013), 10% fetal bovine serum (FBS; Denville Scientific, Holliston, MA; FB5001), and 1% penicillin/streptomycin (PS). All incubations were conducted in this medium except for H2O2 treatment, which was performed in FBS‐ and PS‐free medium. For insulin treatment (Sigma I9278), the cells were grown in the presence of the hormone (10 μg/ml) for multiple passages prior to S‐HAD treatment.
Western blot and immunoprecipitation
Whole cell extracts (WCE) were prepared from cells harvested by trypsin‐EDTA treatment (Thermo Fisher Scientific). Adherent and floating fractions were combined, washed with 4°C PBS, and incubated with CHAPS lysis buffer (CLB; 150 mM NaCl, 10 mM HEPES, 1% CHAPS, pH 7.4) containing 1% protease inhibitor cocktail (PIC; Sigma, P8340) for 30 min at 4°C. WCEs were centrifuged at 14,000 × g for 15 min at 4°C to separate soluble proteins from cell debris. Soluble protein concentrations were determined by Bradford assay (Bio‐Rad, Hercules, CA). WCEs (0.25 or 0.5 mg) were pre‐cleared with protein G agarose (PGA) beads (Gold Biotechnology) for 1 h at 4°C and incubated overnight with a monoclonal antibody directed against the active conformation of Bax (E63 Abcam, ab32503) or total Bax antibody (Cell Signaling Technology, 2772S), respectively. Immunoprecipitates were collected with PGA beads (1 h at 4°C), washed five times in CLB, and eluted with sample buffer (SB; 100 mM Tris, 4% SDS, 20% glycerol, 2 mg/ml bromophenol blue, pH 6.8) at 42°C for 10 min. Immunoprecipitates and input WCEs (dissolved in SB) were supplemented with 100 mM DTT, boiled for 5 min, separated by SDS–PAGE, and resulting Western blots probed for Bax (Cell Signaling Technology, 2772S), Bcl‐XL (Abcam, ab77571), or cyclin C (Bethyl Laboratories, Montgomery, TX; A301‐989A). Western blots were visualized by film exposure or phosphorimaging using alkaline phosphatase‐conjugated rabbit (Abcam, ab97061) or mouse (Abcam, ab97027) secondary antibody and CDP‐Star (Thermo Fisher Scientific) as a substrate. Blots were stripped and reprobed between each primary antibody application. β‐Actin (Sigma‐Aldrich, A1978) was used as a loading control. Poly(ADP)‐ribose polymerase (PARP) cleavage was monitored as described 48 using antibody from Cell Signaling Technology (9542).
Cell fractionation/AMS assays
Subcellular fractions were prepared in the presence of 1% PIC (Sigma, P8340) as described (Abcam, ab109719). ATP synthase subunit alpha (ATP5A; Abcam, ab14748) and fibrillarin (Abcam, ab4566) served as mitochondrial and nuclear markers, respectively. Monitoring the redox state of cyclin C was conducted as previously described 26 using 4‐acetamido‐4′‐maleimidylstilbene‐2,2′‐disulfonic acid (AMS; Molecular Probes).
Cyclin C immunofluorescence and mitochondrial fragmentation assay
Cells were cultured on poly‐l‐lysine‐coated coverslips for 2 days, stained with 100 nM MitoTracker Red CMXRos (Thermo Fisher Scientific) for 30 min at 37°C, fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X‐100 for 10 min, blocked with 2% BSA for 30 min, and incubated with 4 mg/ml cyclin C antibody (Thermo Fisher Scientific, PA5‐16227) at 4°C overnight and 1 mg/ml Alexa Fluor 488‐conjugated secondary antibody (Thermo Fisher Scientific, A11008) for 1 h. Fixed cells were mounted with 4′,6‐diamidino‐2‐phenylindole (DAPI)‐containing medium (Vector Laboratories, Burlingame, CA) to stain the nuclei. Images were acquired with an Eclipse 90i microscope (Nikon, Tokyo, Japan) using the 100× objective. All images of a particular stain were collected with the identical exposure times. Imaging processing was limited to adjusting exposures to permit visualization of all components in merged images. All merged images were adjusted identically. Mitochondrial network containing 15 or more puncta was considered fragmented. NIS‐Elements software (Nikon) was used for image deconvolution and analysis.
Bax oligomerization
Whole cell extracts (WCE) were prepared in digitonin lysis buffer (150 mM NaCl, 10 mM HEPES, 1% digitonin, pH 7.4), crosslinked with 2 mM dithiobis(succinimidyl propionate) (DSP; Thermo Fisher Scientific) for 30 min at room temperature, separated by electrophoresis under non‐reducing or denaturing conditions (no DTT or boiling), and probed for Bax as described above.
Mitochondrial superoxide assay
Cells were grown on poly‐d‐lysine‐coated glass‐bottom Petri dishes (MatTek, Ashland, MA; P35GC‐1.5‐14‐C) for 1 day, treated as indicated, stained with 4 μM MitoSOX Red (Thermo Fisher Scientific) for 15 min at 37°C, washed twice with FluoroBrite DMEM (Thermo Fisher Scientific), and imaged using the 60× objective on an Eclipse Ti‐E confocal microscope (Nikon) equipped with a stage‐top incubator (Tokai Hit, Fujinomiya, Japan) for controlling the temperature (37°C) and CO2 (5%). Excitation was accomplished using the Sapphire 561‐nm laser (Coherent, Santa Clara, CA), and emission was collected through a 570‐nm‐long pass filter in 1‐min intervals (15 min total). Rates of superoxide release were quantified in mitochondrial regions of interest using Ellipse (ViDiTo, Kosice, Slovakia) as previously described 49.
Cell death assays
Cells were grown on 12‐well plates (VWR) for 2 days and treated with cisplatin (30 μM for 16 h) as indicated in the text. Annexin V (Clontech, 630110) assay was conducted as described by the manufacturer on an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). Annexin V+ and propidium iodide (PI)− cells were considered early apoptotic. Caspase activity was quantified using the CaspSCREEN apoptosis detection kit (BioVision, Milpitas, CA; K200) utilizing flow cytometry detection of (aspartyl)2‐rhodamine 110 (D2R). Cell adhesion following S‐HAD treatment was quantified by direct counting of all attached cells following PBS washing.
GST pull‐down assays
Recombinant SUMO‐His6‐cyclin C was purified from E. coli as previously described 19. Bax (without an affinity tag) was a generous gift from P. Czabotar that was purified as described 35 and incubated with His6‐cyclin C in 500 μl HCB150 (150 mM KCl, 50 mM HEPES, 5 mM tris(2‐carboxyethyl)phosphine‐HCl) at ambient temperature for 1 h. Equal amounts of Co2+ slurry were added to each reaction and incubated further for 15 min. The unbound fraction was removed by centrifugation and discarded. The beads were washed twice with 500 μl of HCB150. The bound fraction was eluted by boiling the beads in 50 μl SDS‐loading dye and resolved by SDS–PAGE. The gels were subjected to Western blot analysis.
Statistical analysis
P values were determined using a two‐tailed Student's t‐test. *P < 0.05, **P < 0.01, ***P < 0.001. The number of studies for each assay is given.
Author contributions
JJ conducted experiments, wrote the article, and prepared figures. K‐TC prepared figures and performed experiments. AMJ performed experiments. RS conceived the experiments, edited the article, and prepared figures.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank David Chan (California Institute of Technology, Pasadena, CA) for the generous gift of the hFis1, Mff, and Drp1 knockout MEF cultures. We thank Peter Czabotar (The Walter and Eliza Hall Institute, Parkville, Australia) for purified Bax protein. We thank Katrina Cooper and Vidyaramanan Ganesan for helpful comments on article preparation. This work was supported by a grant from the National Institutes of Health (GM113052) and the New Jersey Health Foundation to R.S.
EMBO Reports (2019) 20: e47425
References
- 1. Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148: 1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337: 1062–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC (2003) Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 160: 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frank S, Gaume B, Bergmann‐Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ (2001) The role of dynamin‐related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1: 515–525 [DOI] [PubMed] [Google Scholar]
- 5. Estaquier J, Arnoult D (2007) Inhibiting Drp1‐mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ 14: 1086–1094 [DOI] [PubMed] [Google Scholar]
- 6. Fannjiang Y, Cheng WC, Lee SJ, Qi B, Pevsner J, McCaffery JM, Hill RB, Basanez G, Hardwick JM (2004) Mitochondrial fission proteins regulate programmed cell death in yeast. Genes Dev 18: 2785–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y et al (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11: 958–966 [DOI] [PubMed] [Google Scholar]
- 8. Shamas‐Din A, Brahmbhatt H, Leber B, Andrews DW (2011) BH3‐only proteins: orchestrators of apoptosis. Biochem Biophys Acta 1813: 508–520 [DOI] [PubMed] [Google Scholar]
- 9. Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ (2002) Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol 159: 931–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, Martinou JC (2006) Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak‐dependent apoptosis. Mol Cell Biol 26: 7397–7408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bourbon HM (2008) Comparative genomics supports a deep evolutionary origin for the large, four‐module transcriptional mediator complex. Nucleic Acids Res 36: 3993–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Surosky RT, Strich R, Esposito RE (1994) The yeast UME5 gene regulates the stability of meiotic mRNAs in response to glucose. Mol Cell Biol 14: 3446–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wahi M, Johnson AD (1995) Identification of genes required for alpha 2 repression in Saccharomyces cerevisiae . Genetics 140: 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuchin S, Yeghiayan P, Carlson M (1995) Cyclin‐dependent protein kinase and cyclin homologs SSN3 and SSN8 contribute to transcriptional control in yeast. Proc Natl Acad Sci USA 92: 4006–4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cooper KF, Mallory MJ, Smith JB, Strich R (1997) Stress and developmental regulation of the yeast C‐type cyclin Ume3p (Srb11p/Ssn8p). EMBO J 16: 4665–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cooper KF, Khakhina S, Kim SK, Strich R (2014) Stress‐induced nuclear‐to‐cytoplasmic translocation of cyclin C promotes mitochondrial fission in yeast. Dev Cell 28: 161–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khakhina S, Cooper KF, Strich R (2014) Med13p prevents mitochondrial fission and programmed cell death in yeast through nuclear retention of cyclin C. Mol Biol Cell 25: 2807–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang K, Yan R, Cooper KF, Strich R (2015) Cyclin C mediates stress‐induced mitochondrial fission and apoptosis. Mol Biol Cell 26: 1030–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ganesan V, Willis SD, Chang KT, Beluch S, Cooper KF, Strich R (2019) Cyclin C directly stimulates Drp1 GTP affinity to mediate stress‐induced mitochondrial hyperfission. Mol Biol Cell 30: 302–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cosentino K, Garcia‐Saez AJ (2017) Bax and Bak pores: are we closing the circle? Trends Cell Biol 27: 266–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsu YT, Youle RJ (1997) Nonionic detergents induce dimerization among members of the Bcl‐2 family. J Biol Chem 272: 13829–13834 [DOI] [PubMed] [Google Scholar]
- 22. Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I, Bosykh DA, Burdelya LG, Macklis RM, Skaliter R et al (2006) Small‐molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol 2: 474–479 [DOI] [PubMed] [Google Scholar]
- 23. Loson OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24: 659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cooper KF, Strich R (1999) Functional analysis of the Ume3p/Srb11p‐RNA polymerase II holoenzyme interaction. Gene Expr 8: 43–57 [PMC free article] [PubMed] [Google Scholar]
- 25. Eskes R, Desagher S, Antonsson B, Martinou JC (2000) Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol 20: 929–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nie C, Tian C, Zhao L, Petit PX, Mehrpour M, Chen Q (2008) Cysteine 62 of Bax is critical for its conformational activation and its proapoptotic activity in response to H2O2‐induced apoptosis. J Biol Chem 283: 15359–15369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu T, Sheu SS, Robotham JL, Yoon Y (2008) Mitochondrial fission mediates high glucose‐induced cell death through elevated production of reactive oxygen species. Cardiovasc Res 79: 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP (2003) Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem 278: 8516–8525 [DOI] [PubMed] [Google Scholar]
- 29. Trnka J, Blaikie FH, Smith RA, Murphy MP (2008) A mitochondria‐targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic Biol Med 44: 1406–1419 [DOI] [PubMed] [Google Scholar]
- 30. Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H (2005) Role of insulin‐induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal 7: 1021–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Z, Subramaniam S, Kale J, Liao C, Huang B, Brahmbhatt H, Condon SG, Lapolla SM, Hays FA, Ding J et al (2016) BH3‐in‐groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J 35: 208–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, Neutzner A, Tjandra N, Youle RJ (2011) Bcl‐x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145: 104–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schellenberg B, Wang P, Keeble JA, Rodriguez‐Enriquez R, Walker S, Owens TW, Foster F, Tanianis‐Hughes J, Brennan K, Streuli CH et al (2013) Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol Cell 49: 959–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lovell JF, Billen LP, Bindner S, Shamas‐Din A, Fradin C, Leber B, Andrews DW (2008) Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135: 1074–1084 [DOI] [PubMed] [Google Scholar]
- 35. Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ et al (2013) Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152: 519–531 [DOI] [PubMed] [Google Scholar]
- 36. Ma SB, Nguyen TN, Tan I, Ninnis R, Iyer S, Stroud DA, Menard M, Kluck RM, Ryan MT, Dewson G (2014) Bax targets mitochondria by distinct mechanisms before or during apoptotic cell death: a requirement for VDAC2 or Bak for efficient Bax apoptotic function. Cell Death Differ 21: 1925–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Montessuit S, Somasekharan SP, Terrones O, Lucken‐Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy‐Wetzel E, Basanez G, Meda P et al (2010) Membrane remodeling induced by the dynamin‐related protein Drp1 stimulates Bax oligomerization. Cell 142: 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oettinghaus B, D'Alonzo D, Barbieri E, Restelli LM, Savoia C, Licci M, Tolnay M, Frank S, Scorrano L (2016) DRP1‐dependent apoptotic mitochondrial fission occurs independently of BAX, BAK and APAF1 to amplify cell death by BID and oxidative stress. Biochem Biophys Acta 1857: 1267–1276 [DOI] [PubMed] [Google Scholar]
- 39. Brooks C, Cho SG, Wang CY, Yang T, Dong Z (2011) Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am J Physiol Cell Physiol 300: C447–C455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM (2015) MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol Cell 59: 941–955 [DOI] [PubMed] [Google Scholar]
- 41. Buchner N, Altschmied J, Jakob S, Saretzki G, Haendeler J (2010) Well‐known signaling proteins exert new functions in the nucleus and mitochondria. Antioxid Redox Signal 13: 551–558 [DOI] [PubMed] [Google Scholar]
- 42. Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11: 577–590 [DOI] [PubMed] [Google Scholar]
- 43. Leu JI, Dumont P, Hafey M, Murphy ME, George DL (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak‐Mcl1 complex. Nat Cell Biol 6: 443–450 [DOI] [PubMed] [Google Scholar]
- 44. Jiang P, Du W, Heese K, Wu M (2006) The Bad guy cooperates with good cop p53: Bad is transcriptionally up‐regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol Cell Biol 26: 9071–9082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Park BS, Song YS, Yee SB, Lee BG, Seo SY, Park YC, Kim JM, Kim HM, Yoo YH (2005) Phospho‐ser 15‐p53 translocates into mitochondria and interacts with Bcl‐2 and Bcl‐xL in eugenol‐induced apoptosis. Apoptosis 10: 193–200 [DOI] [PubMed] [Google Scholar]
- 46. Green DR, Kroemer G (2009) Cytoplasmic functions of the tumour suppressor p53. Nature 458: 1127–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stieg DC, Chang KT, Cooper KF, Strich R (2019) Cyclin C regulated oxidative stress responsive transcriptome in Mus musculus embryonic fibroblasts.G3 (Bethesda) 9: 1901–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG (1993) Specific proteolytic cleavage of poly(ADP‐ribose) polymerase: an early marker of chemotherapy‐induced apoptosis. Cancer Res 53: 3976–3985 [PubMed] [Google Scholar]
- 49. Jezek J, Dlaskova A, Zelenka J, Jaburek M, Jezek P (2015) H(2)O(2)‐activated mitochondrial phospholipase iPLA(2)gamma prevents lipotoxic oxidative stress in synergy with UCP2, amplifies signaling via G‐protein‐coupled receptor GPR40, and regulates insulin secretion in pancreatic beta‐cells. Antioxid Redox Signal 23: 958–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File