

Abstract
A CGG trinucleotide repeat expansion in the 5′ UTR of FMR1 causes the neurodegenerative disorder Fragile X‐associated tremor/ataxia syndrome (FXTAS). This repeat supports a non‐canonical mode of protein synthesis known as repeat‐associated, non‐AUG (RAN) translation. The mechanism underlying RAN translation at CGG repeats remains unclear. To identify modifiers of RAN translation and potential therapeutic targets, we performed a candidate‐based screen of eukaryotic initiation factors and RNA helicases in cell‐based assays and a Drosophila melanogaster model of FXTAS. We identified multiple modifiers of toxicity and RAN translation from an expanded CGG repeat in the context of the FMR1 5′UTR. These include the DEAD‐box RNA helicase belle/DDX3X, the helicase accessory factors EIF4B/4H, and the start codon selectivity factors EIF1 and EIF5. Disrupting belle/DDX3X selectively inhibited FMR1 RAN translation in Drosophila in vivo and cultured human cells, and mitigated repeat‐induced toxicity in Drosophila and primary rodent neurons. These findings implicate RNA secondary structure and start codon fidelity as critical elements mediating FMR1 RAN translation and identify potential targets for treating repeat‐associated neurodegeneration.
Keywords: DDX3X, eIF, Fragile X‐associated tremor/ataxia syndrome, RAN translation, RNA helicase
Subject Categories: Neuroscience, Protein Biosynthesis & Quality Control, RNA Biology
Introduction
Over 30 different nucleotide repeat expansions (NREs) cause neurodegeneration in humans 1. NREs within consensus coding sequences (CCDS) cause disease predominantly via protein‐based gain‐of‐function mechanisms that depend on the intrinsic toxicity of homopolymeric peptides or dysfunction of the proteins in which they reside 2, 3, 4, 5. Alternatively, NREs can elicit toxicity via mRNA‐based mechanisms where expanded repeats sequester essential RNA‐binding proteins, leading to transcriptome dysregulation (e.g., myotonic dystrophy I and II) and gelation of RNA‐protein complexes into RNA containing foci 6, 7, 8, 9. More recently, NREs were found to support translational initiation in the absence of an AUG start codon through a process known as repeat‐associated, non‐AUG (RAN) translation 10. Proteins generated through RAN translation accumulate in patient tissues 10, 11, 12, 13, 14, 15 and are toxic in animal and cellular models of disease 10, 14, 16, 17, 18. Since its discovery, RAN translation has been implicated in several NRE‐associated neurodegenerative disorders 19, including Fragile X‐associated tremor/ataxia syndrome (FXTAS), C9orf72‐associated amyotrophic lateral sclerosis and frontotemporal dementia (C9ALS/FTD), and Huntington's disease 20.
FXTAS is an adult‐onset neurodegenerative disorder caused by a CGG NRE in the 5′ UTR of FMR1 from approximately 30 repeats to 55–200 repeats 21. The NRE is transcribed into mRNA, which can bind to and sequester specific RNA‐binding proteins 22, 23, 24, 25, 26, 27. In addition, expanded CGG repeats are translated via RAN translation into toxic proteins, which accumulate in ubiquitinated aggregates in tissue of both FXTAS patients and animal models of FXTAS 14, 17, 18. Synthesis of RAN products is necessary for CGG repeats to elicit toxicity in over‐expression systems, including Drosophila melanogaster, cultured human cells, and transgenic mice 14, 17, 28. FXTAS shares its causative locus with the neurodevelopmental disorder Fragile X syndrome, but it is clinically and mechanistically distinct: Fragile X syndrome results from larger (> 200) CGG NREs that transcriptionally silence the Fragile X locus, resulting in loss of FMR1 mRNA, no expression of expanded CGG repeats as RNA, and loss of FMRP 29.
The mechanism of RAN translation, and how it differs from canonical translation, remains unclear. Early reports demonstrated that, at least under some circumstances, RAN translation's initial stages resemble canonical translation 30, 31, 32: The 43S pre‐initiation complex (PIC)—composed of the 40S ribosomal subunit, tRNAiMet, and a number of essential eukaryotic initiation factors (eIFs)—binds to the 5′ methyl‐7‐guanosine (m7G) cap on mRNA and scans through the 5′ untranslated region (UTR) 33. In canonical translation, the 43S PIC scans until it encounters an AUG start codon, which triggers a cascade of structural rearrangements that ends with binding of the 60S ribosomal subunit and initiation of translation. In RAN translation, initiation occurs at non‐AUG codons, either upstream of or within the NRE 10, 30, 31, 32, 34. At CGG repeats and possibly GGGGCC repeats, this failure of codon fidelity is thought to result from impairment of 43S PIC scanning by stable RNA secondary structures formed by the expanded repeats, since such structures facilitate initiation at non‐AUG sites 35, 36, 37, 38. At other repeats and cellular contexts, RAN translation may utilize cap‐independent initiation mechanisms and/or initiator tRNAs other than tRNAiMet 10, 39, 40. Which mechanisms occur in the context of each human disease is unclear and could vary based on gene context, repeat content, and cell type 19, 41, 42.
Discerning how RAN translation initiates, and how that process diverges from canonical translation, might reveal new therapeutic strategies for FXTAS, C9ALS/FTD, and other NRE‐associated disorders. Two features distinguish RAN translation from canonical translation: the presence of highly stable RNA secondary structures composed of NREs 43, 44, 45 and the use of non‐AUG start codons. 43S PIC scanning is known to require several RNA helicases in order to resolve mRNA structure within 5′ UTRs, including Ded1/Belle/DDX3X 46, 47, 48, 49, 50, 51, 52, eIF4A and its cofactors eIF4B and eIF4H 50, 53, 54, 55, and DHX29 56, 57. In addition, start codon fidelity in yeast is regulated by a series of eIFs—including eIF1, eIF1A, eIF2, and eIF5—and upstream signaling pathways 58, 59, 60, 61, 62, 63, 64, 65.
With these features in mind, we conducted a candidate‐based screen of eIFs, RNA helicases, and other RNA‐binding proteins to identify regulators of RAN translation, using both cell‐based assays and a Drosophila melanogaster model of FXTAS 66. In FXTAS, RAN translation from the sense strand of FMR1 yields at least two RAN products from the CGG repeat: a polyglycine peptide (FMRpolyG), reflecting initiation in the GGC (+1, relative to the reading frame of the downstream CCDS product, FMRP) frame, and a polyalanine peptide (FMRpolyA), reflecting initiation in the GCG (+2) frame 14, 31. Antisense FMR1 transcripts also support RAN translation, yielding three distinct RAN products 67. In Drosophila, we previously demonstrated that CGG repeat‐associated toxicity is largely dependent on RAN translation 14, 68. Our screen identified multiple factors that were necessary for RAN translation from the FMR1 5′ UTR, disruption of which suppressed CGG repeat‐associated toxicity in Drosophila. Disruption of one in particular, the DEAD‐box RNA helicase DDX3X (belle in Drosophila), selectively inhibited FMR1 RAN translation in human cell‐based systems and suppressed repeat‐induced neurodegeneration in rodent neurons. Our findings implicate RNA secondary structure and start codon fidelity in FMR1 RAN translation and suggest specific targets for future therapeutic development.
Results
A Drosophila screen for modifiers of CGG repeat toxicity
To identify regulators of FMR1 RAN translation, we conducted a candidate‐based screen using a Drosophila melanogaster model of FXTAS 66. This model carries an EGFP transgene 3′ to an upstream activation sequence (UAS) and the 5′ UTR of human FMR1 with 90 CGG repeats, with EGFP in the GGC (+1, FMRpolyG) reading frame [(CGG)90‐EGFP)]. The expression in the eye via a GMR‐GAL4 driver manifests in a significant rough‐eye phenotype observable at eclosion, with ubiquitin‐positive aggregates of the RAN product FMRpolyG‐EGFP accumulating in retinal neurons 14. For the screen, females expressing (CGG)90‐EGFP under a GMR‐GAL4 driver were crossed to males carrying germline mutations in, UAS‐driven transgenes of, and UAS‐driven shRNA constructs to 10 canonical eIFs, 4 RNA helicases, a ribosomal protein associated with non‐canonical translation initiation 69, 70, and an RNA‐binding protein implicated in ALS/FTD 71 (Appendix Fig S1). We selected these candidates in a hypothesis‐driven fashion based on their known functions in non‐canonical translation initiation and regulation of start codon fidelity, as well their potential to modulate GC‐rich secondary structures in RNA. By design, this candidate list was non‐exhaustive. Because previous work has demonstrated the importance of eIF4E‐m7G binding and 43S PIC scanning to RAN translation 30, 31, 32 and because eIF4E and eIF4G are necessary for canonical translation, we did not evaluate them in this screen.
We examined the rough‐eye phenotype of the F1 flies at eclosion to identify suppressors and enhancers of (CGG)90‐driven toxicity. Of the 57 candidate lines tested, 21 acted as suppressors of toxicity, and 17 acted as enhancers (Fig 1, Appendix Fig S1). All lines were subsequently crossed to flies carrying GMR‐GAL4 alone to control for toxic effects independent of (CGG)90‐based toxicity (Appendix Fig S1). Six of the 17 toxicity‐enhancing lines had no effect in the absence of (CGG)90‐EGFP, suggesting this enhancement is specific to the presence of expanded CGG repeats. We selected three suppressors for further analysis—belle (bel)/DDX3X, eIF4B, and eIF4H1—based on their individual functions in translation initiation.
Figure 1. A candidate‐based screen reveals modifiers of repeat‐associated toxicity in Drosophila .
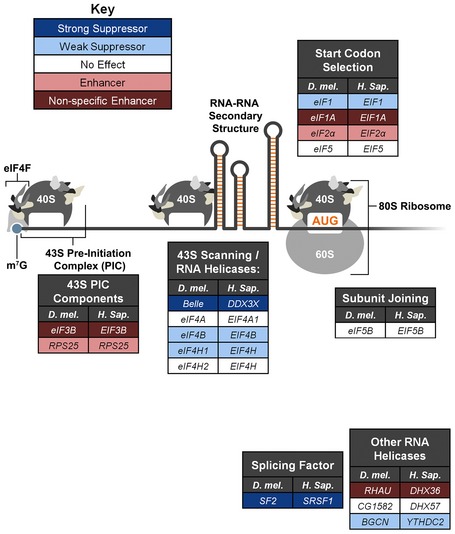
Candidate modifiers are categorized here based on their known functions in gene expression. Fly genes are listed in the left columns, while their human homologs are listed in the right columns. Disruption of genes highlighted in dark blue strongly suppressed (CGG)90‐EGFP toxicity. Disruption of genes highlighted in light blue weakly suppressed (CGG)90‐EGFP toxicity. Disruption of genes highlighted in light red enhanced (CGG)90‐EGFP toxicity selectively. Disruption of genes highlighted in dark red enhanced the toxicity of both (CGG)90‐EGFP and GMR‐GAL4 (these were toxic independent of the repeat.) All other genes are displayed in white. The methyl‐7‐guanosine (m7G) cap, eIF4F complex, 43S pre‐initiation complex (PIC), and ribosomes are indicated.
Bel/DDX3X selectively modulates FMR1 RAN translation in Drosophila
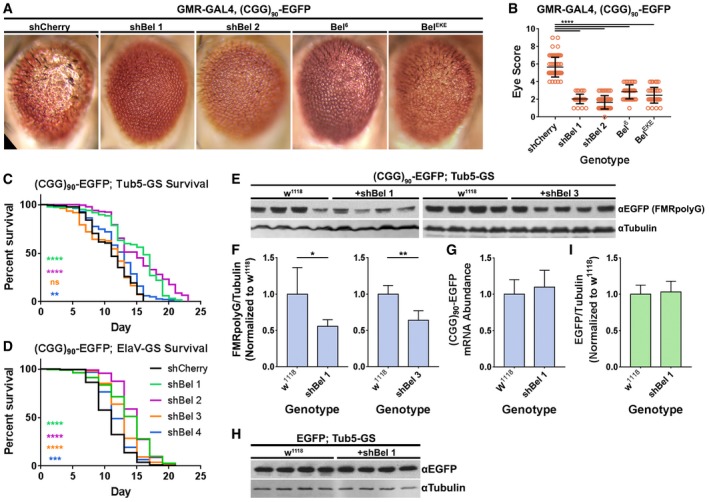
In our candidate‐based Drosophila screen, bel disruption by multiple genetic means suppressed CGG100‐elicited toxicity. Four shRNAs against bel and five heterozygous loss‐of‐function bel mutants significantly suppressed the rough‐eye phenotype in (CGG)90‐EGFP‐coexpressing flies (Fig 2A and B; Appendix Fig S2A and B). Bel mutants tested included nonsense mutations and P[lacW] and P[PZ] element insertions 72, 73, 74, 75. The bel shRNA lines generally suppressed (CGG)90‐EGFP toxicity more effectively than the heterozygous, loss‐of‐function bel mutants, potentially because the shRNAs had stronger effects on the abundance of bel protein. None of the bel shRNAs had phenotypic effects in flies expressing an AUG‐initiated EGFP transgene under a GMR‐GAL4 driver (Appendix Fig S2C and D). Modulation of (CGG)90‐driven toxicity was not limited to the eye, since three bel shRNAs increased the lifespan of adult flies expressing (CGG)90‐EGFP ubiquitously post‐eclosion under an inducible Tub5 Geneswitch driver (Fig 2C) 76. Similarly, four bel shRNAs increased lifespan when (CGG)90‐EGFP was expressed pan‐neuronally in adult flies under an inducible Geneswitch ElaV driver (Fig 2D).
Figure 2. Knockdown of belle mitigates repeat‐associated toxicity by inhibiting RAN translation in Drosophila .
-
ARepresentative photographs of fly eyes expressing (CGG)90‐EGFP under a GMR‐GAL4 driver, with various belle disruptions.
-
BQuantitation of GMR‐GAL4 and (CGG)90‐EGFP eye phenotypes with belle disruptions (Mann–Whitney U‐test with Bonferroni corrections for multiple comparisons; n = 35–77/genotype).
-
C, DLongevity assays of (CGG)90‐EGFP; Tub5‐GS (log‐rank Mantel–Cox test with Bonferroni corrections for multiple comparisons; n = 110–219/genotype) and (CGG)90‐EGFP; ElaV‐GS (n = 147–299/genotype) flies with belle knockdown.
-
EWestern blots of the FMRpolyG‐EGFP RAN product in (CGG)90‐EGFP; Tub5‐GS flies with and without belle knockdown by two independent shRNAs.
-
FQuantitation of FMRpolyG‐EGFP band density, normalized to β‐tubulin band density, from blots in (E) (Student's t‐test; n = 4–5/genotype).
-
GAbundance of (CGG)90‐EGFP mRNA normalized to RPL32 mRNA, following belle knockdown, determined by qRT–PCR (n = 8/genotype).
-
HWestern blot of AUG‐driven EGFP in EGFP; Tub5‐GS flies with and without belle knockdown.
-
IQuantitation of EGFP band density, normalized to β‐tubulin band density, from blot in (H) (n = 4/genotype).
Bel and its homologs in yeast (Ded1) and humans (DDX3X) are important for translation of specific mRNAs, particularly those with long or structured 5′ UTRs 46, 47, 48, 49, 50, 51, 52, 77. Given the role that secondary structure is hypothesized to play in the initiation of RAN translation, we asked whether knockdown of bel suppressed the (CGG)90‐EGFP phenotype by suppressing RAN translation. Knockdown of bel by two independent shRNAs reduced the expression of the RAN product FMRpolyG‐EGFP in (CGG)90‐expressing flies (Fig 2E and F), supporting the hypothesis that suppression of (CGG)90 toxicity is driven by inhibition of RAN translation. In contrast, bel knockdown had no effect on the abundance of (CGG)90‐EGFP transcripts (Fig 2G). Finally, knockdown of bel had no effect on the expression of an AUG‐initiated EGFP reporter lacking the FMR1 5′ UTR (Fig 2H and I), suggesting that the decrease in FMRpolyG‐EGFP we observed reflects a selective effect on RAN translation, rather than a global decline in translation.
Bel/DDX3X selectively modulates FMR1 RAN translation in human cells
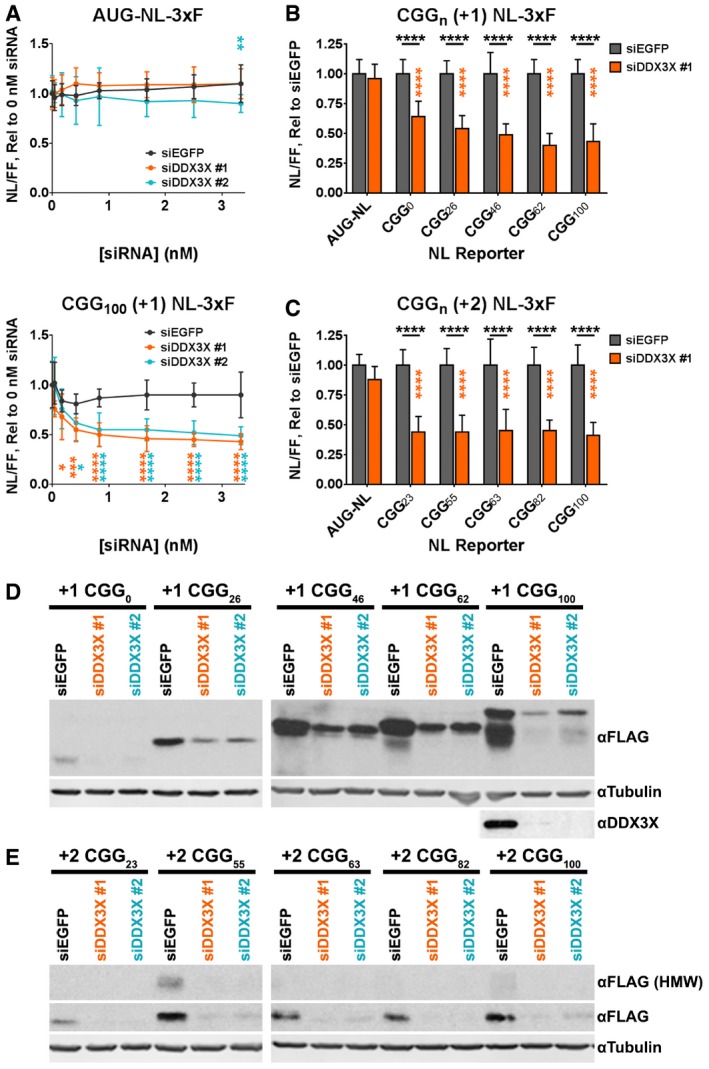
We next asked whether DDX3X, the human homolog of bel, might play a similar role in facilitating RAN translation of CGG repeats in human cells. We previously generated transfectable luciferase‐based reporters consisting of a 3xFLAG‐tagged nanoluciferase (NL‐3xF) downstream of the 5′UTR of human FMR1, with multiple repeat sizes (0–100 repeats) and with the NL‐3xF in both the GGC (+1, FMRpolyG) and GCG (+2, FMRpolyA) reading frames (Appendix Fig S3) 31. These reporters enable quantitative and qualitative detection of RAN product expression by luminescence assays and Western blotting, respectively. Knockdown of DDX3X by five independent siRNAs reduced the expression of plasmid‐based +1 (CGG)100 NL‐3xF reporters in a dose‐dependent manner (Fig 3A; Appendix Fig S4). As in our Drosophila experiments, these siRNAs had minimal effect on the expression of an AUG‐initiated NL‐3xF reporter (AUG‐NL‐3xF). To further test whether DDX3X knockdown inhibits protein synthesis across mRNAs, we tested the effects of two DDX3X siRNAs on NL‐3xF reporters bearing the short, minimally structured 5′ UTRs of β‐actin and β‐globin. siDDX3X #1 had no effect on Actin‐NL‐3xF and increased the expression of Globin‐NL‐3xF but decreased the expression of +1 (CGG)100 NL‐3xF (Appendix Fig S5A). Though siDDX3X #2 decreased both Actin‐ and Globin‐NL‐3xF, it did so significantly less than it inhibited +1 (CGG)100 NL‐3xF. Finally, to assess the effects of DDX3X knockdown on global protein synthesis of endogenous mRNAs, we performed polysome fractionation on HeLa cells transfected with siDDX3X or siEGFP (Appendix Fig S6A and B). Consistent with our NL‐3xF reporter data, knockdown of DDX3X (Appendix Fig S6C) did not result in a reproducible shift in the relative monosome and polysome fractions. This indicates that DDX3X knockdown does not lead to global inhibition of mRNA translation, again highlighting the role of DDX3X in the expression of select genes. This finding is consistent with previous work demonstrating that the expression of only a specific subset of mRNAs—those with long and/or secondary‐structured 5′ UTRs—is reduced following DDX3X/Ded1 disruption 50, 51, 77, 78.
Figure 3. Knockdown of DDX3X inhibits RAN translation in cultured human cells.
-
ADose–response curves showing the effects of two independent anti‐DDX3X siRNAs on the expression of AUG‐NL‐3xF (top) and (CGG)100 +1 NL‐3xF (bottom) reporters. Plasmid‐based reporters were transfected into HeLa cells 24 h after knockdown, and reporter expression was quantified by luminescence. Nanoluciferase (NL) luminescence has been normalized to luminescence from firefly luciferase (FF), which was co‐transfected, in order to control for transfection variability. Asterisks refer to comparisons between anti‐DDX3X siRNAs and siRNAs against EGFP (siEGFP; two‐way ANOVA with Dunnett's multiple comparisons test; n = 12/condition).
-
B, C(CGG)n +1 and (CGG)n +2 NL‐3xF expression (normalized to FF) with and without DDX3X knockdown across a range of CGG repeat sizes. Black asterisks refer to comparisons between siDDX3X‐ and siEGFP‐treated cells; orange asterisks refer to comparisons between siDDX3X‐treated cells expressing AUG‐NL‐3xF and those expressing a different reporter (two‐way ANOVA with Tukey's multiple comparisons test; n = 17–30/condition).
-
D, EWestern blots of FMRpolyG‐NL‐3xF and FMRpolyA‐NL‐3xF products with and without DDX3X knockdown across a range of repeat sizes.
We next asked whether DDX3X regulates RAN translation on FMR1 transcripts in other reading frames. The expression of the +2 (CGG)100 NL‐3xF RAN product (FMRpolyA100), which likely derives from initiation within the NRE 31, was reduced to a similar degree as the FMRpolyG100 product in luminescence assays (Fig 3B and C; Appendix Fig S5B and C). We also observed these effects following detection of the FMRpolyGn and FMRpolyAn RAN products by Western blotting (Fig 3D and E). These data indicate that the function of DDX3X in promoting RAN translation is not limited to a single reading frame.
DDX3X and its homologs in yeast and Drosophila function in several aspects of RNA metabolism. We therefore asked whether DDX3X functions in RAN translation directly or whether its effects might instead be mediated by modulating RNA stability or transcription. Knockdown of DDX3X reduces the expression of +1 and +2 (CGG)100 NL‐3xF, regardless of whether the reporters are transfected as plasmids or in vitro‐transcribed RNAs (Fig 4A; Appendix Fig S5D and E), suggesting that DDX3X acts post‐transcriptionally. We next asked whether DDX3X knockdown impacted the abundance of reporter mRNAs following plasmid transfection. We observed that the mRNA abundance of both AUG‐NL‐3xF and (CGG)100‐NL‐3xF in the +1 and +2 frames was not consistently affected after transfection with siDDX3X #1 and siDDX3X #2 (Fig 4B). In order to isolate translational effects per se from other, concurrent effects on gene expression, we conducted in vitro translation assays using transcribed reporter RNAs and cytoplasmic extracts generated from cells depleted of DDX3X by two independent siRNAs. DDX3X‐depleted extracts yielded reduced translation of +1 (CGG)100 NL‐3xF, while having either no effect on or increasing the synthesis of an AUG‐NL‐3xF reporter (Fig 4C). This effect was consistent across independently prepared in vitro translation extract replicates (four extracts per siRNA; two anti‐DDX3X siRNAs, Appendix Fig S7A and B). These in vitro experiments point specifically to a direct translational function of DDX3X, while leaving open the possibility that changes in mRNA stability and abundance may further impact the expression of RAN products.
Figure 4. DDX3X facilitates expression of RAN products at the level of translation.
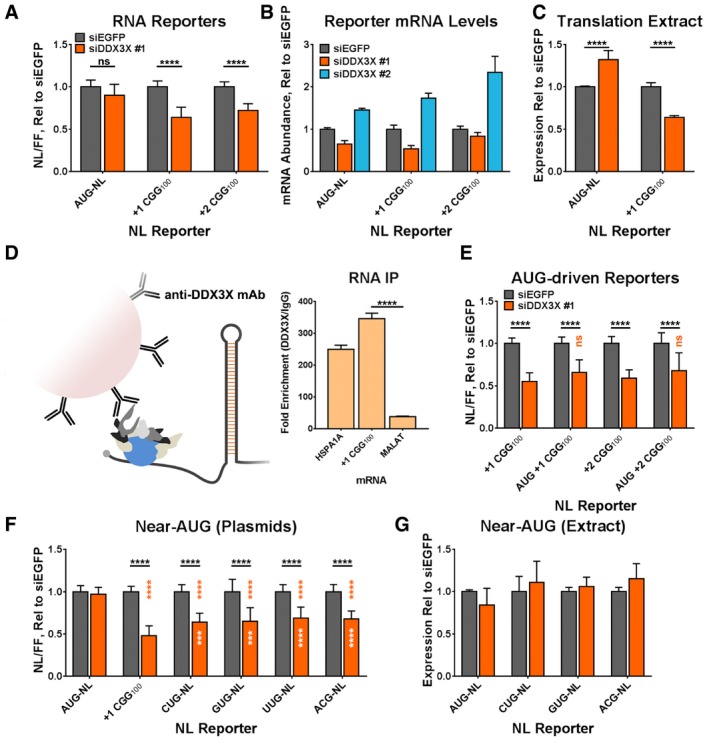
- The expression of in vitro‐transcribed AUG, +1 (CGG)100, and +2 (CGG)100 NL‐3xF RNAs following DDX3X knockdown in HeLa cells, expressed as NL luminescence normalized to FF luminescence. (Student's t‐test with Bonferroni corrections for multiple comparisons; n = 21/condition).
- Abundance of reporter mRNAs following DDX3X knockdown and plasmid‐reporter transfection, determined by qRT–PCR (n = 7/condition). This panel depicts data as means ± SEM.
- The expression of AUG‐NL‐3xF and +1 (CGG)100 NL‐3xF in in vitro translation extracts, collected from HeLa cells treated with siRNAs against EGFP or DDX3X (two‐way ANOVA with Tukey's multiple comparisons test; n = 4/condition).
- Enrichment of HSPA1A and +1 (CGG)100 NL‐3xF mRNA following anti‐DDX3X RIP, relative to incubation with isotype control IgG. MALAT RNA, in contrast, is not enriched (Student's t‐test, n = 3). Data from the additional replicate are presented in Appendix Fig S8A.
- The expression of +1 and +2 (CGG)100 NL‐3xF plasmid reporters with and without an AUG inserted 5′ to the CGG repeat, with and without DDX3X knockdown. Black asterisks refer to comparisons between siDDX3X‐ and siEGFP‐treated cells; orange asterisks refer to comparisons between siDDX3X‐treated cells expressing either +1 or +2 (CGG)100 NL‐3xF and those expressing the respective AUG‐driven variant (two‐way ANOVA with Tukey's multiple comparisons test; n = 11–12/condition).
- The expression of NL‐3xF plasmids with initiator AUG codons mutated to near‐AUG codons, with and without DDX3X knockdown (two‐way ANOVA with Dunnett's multiple comparisons test; n = 18–24/condition). Black asterisks refer to comparisons between siEGFP‐treated and siDDX3X‐treated cells; orange asterisks refer to comparisons between siDDX3X‐treated cells expressing AUG‐NL‐3xF and those expressing a different reporter; white asterisks refer to comparisons between siDDX3X‐treated cells expressing +1 (CGG)100 NL‐3xF and those expressing a different reporter.
- The expression of in vitro‐transcribed near‐AUG reporter RNAs in in vitro translation extracts, collected from HeLa cells treated with siRNAs against EGFP or DDX3X. Experiments with independent, replicate lysates are presented in Appendix Fig S7C (n = 4/group).
We next asked how DDX3X regulates FMR1 RAN translation. To determine whether DDX3X can directly interact with CGG reporter mRNAs, we performed photo‐crosslinking RNA immunoprecipitation (RIP) assays to probe for an interaction between DDX3X and our FMR1 (CGG)100 reporters in cultured cells. As we anticipated, significantly more HSPA1A mRNA (translation of which requires DDX3X) 52 was co‐purified using antibodies against DDX3X than isotype control IgG (Fig 4D). Similarly, significantly more +1 (CGG)100 NL‐3xF mRNA was co‐purified using antibodies against DDX3X than antibodies against EGFP or isotype control IgG (Fig 4D; Appendix Fig S8A). +1 (CGG)100 NL‐3xF mRNA was also enriched by DDX3X RIP in comparison with endogenous MALAT RNA, indicating a transcript‐selective interaction between DDX3X and +1 (CGG)100 NL‐3xF mRNA. To determine whether that interaction is an artifact of the NL‐3xF tag on +1 (CGG)100 NL‐3xF mRNA, we repeated the experiment using a 3′‐truncated “tagless” construct in which the NL‐3xF tag had been deleted (leaving the FMR1 5′ UTR and minimal vector sequence intact). In two independent replicates, tagless (CGG)100 mRNA also co‐precipitated with DDX3X, near or above the levels of HSPA1A mRNA as a positive control (Appendix Fig S8B). Finally, to determine whether that interaction depends on the expanded CGG repeats in the tagless (CGG)100 construct, we repeated this experiment using a modified tagless construct bearing 0 CGG repeats [tagless (CGG)0]. (CGG)0 mRNAs co‐precipitated with DDX3X at levels comparable to (CGG)100 and HSPA1A mRNA, indicating that expanded CGG repeats are unnecessary for this interaction. This is not surprising, however, as the 5′UTR of FMR1 is highly GC‐rich (76%) even excluding the CGG NRE, and previously published work has demonstrated that Ded1 (the yeast homolog of DDX3X) preferentially binds 5′ to secondary structures within 5′ UTRs 77.
To determine which features of the FMR1 5′UTR enable DDX3X to modulate RAN translation, we first varied the size of the CGG NRE in our reporters. Using plasmid‐based reporters, we observed no significant effect of CGG repeat size on the impact of DDX3X knockdown in either the GGC (+1) frame or the GCG (+2) frame (Fig 3B–E; Appendix Fig S5B and C). All exhibited decreased expression as measured by luminescence assays and Western blotting. We observed similar results when we transfected reporters as in vitro‐transcribed RNA (Appendix Fig S5D and E). These results indicate that expanded CGG repeats are unnecessary for DDX3X to modulate RAN translation initiating within the FMR1 5′ UTR.
One of the unique features of RAN translation is its use of non‐AUG codons for initiation. We therefore asked whether DDX3X facilitated RAN translation by regulating start codon selection. We first tested the effect of DDX3X knockdown on a modified +1 (CGG)100 reporter, in which the major near‐AUG codon (ACG) utilized for GGC (+1)‐frame RAN translation had been removed and replaced with a nearby AUG (Appendix Fig S9) 31. This change enhanced basal expression of the construct, but DDX3X knockdown still impaired AUG‐(CGG)100 (+1) NL‐3xF expression (Fig 4E). Similarly, insertion of an AUG codon in a strong Kozak context 5′ to the NRE in the GCG (+2) frame enhanced basal expression, but the expression of this AUG‐(CGG)100 (+2) NL‐3xF reporter remained DDX3X‐dependent (Fig 4E). In a complementary experiment, we evaluated whether DDX3X knockdown affected translation of NL‐3xF reporters that initiate with the near‐cognate codons CUG, GUG, UUG, or ACG absent any NRE or FMR1 sequence. As expected, the expression of CUG‐, GUG‐, UUG‐, and ACG‐NL‐3xF reporter plasmids was lower than the expression of AUG‐NL‐3xF 30, 37. When these plasmids were transfected into cells, DDX3X knockdown reduced their expression compared to AUG‐NL‐3xF but significantly less than +1 (CGG)100 NL‐3xF (Fig 4F). In contrast, the expression of in vitro‐transcribed near‐AUG‐initiated NL‐3xF reporters was unaffected by DDX3X depletion in in vitro translation assays (Fig 4G; Appendix Fig S7C). In total, our results indicate that altered start codon fidelity is unlikely to be the sole factor mediating the effects of DDX3X knockdown on FMR1 RAN translation.
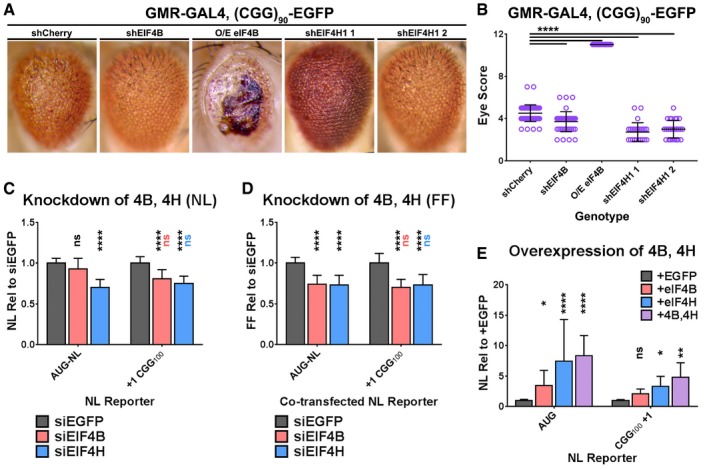
eIF4B and eIF4H modulate RAN Translation at CGG repeats
eIF4B and eIF4H are co‐stimulatory factors for the RNA helicase eIF4A, and like Belle/DDX3X, they are required for translation of mRNAs with long or structured 5′ UTRs 50, 53, 54, 55. Previous work demonstrated that eIF4A is specifically required for RAN translation at both CGG repeats and GGGGCC repeats associated with ALS/FTD 30, 31, 32. We therefore asked whether these co‐stimulatory factors might play a similar, specific function in RAN translation like DDX3X. In our initial Drosophila screen, three shRNAs against eIF4H1, one shRNA against eIF4H2, and one shRNA against eIF4B suppressed the rough‐eye phenotype induced by (CGG)90‐EGFP (Fig 5A and B; Appendix Fig S10A and B). In addition, eIF4B over‐expression exacerbated this phenotype, while having no effect in the absence of (CGG)90‐EGFP (Appendix Fig S10C and D). As with belle shRNAs, these effects were not limited to the eye, as eIF4B shRNA increased and over‐expression decreased the lifespan of flies expressing (CGG)90‐EGFP under an inducible Tub5 driver (Appendix Fig S10E). These experiments suggest that, like Belle/DDX3X, eIF4B and eIF4H are capable of modulating RAN translation.
Figure 5. EIF4B and EIF4H modulate RAN translation and general translation in Drosophila and cultured human cells.
-
ARepresentative photographs of GMR‐GAL4; (CGG)90‐EGFP fly eyes expressing manipulations of eIF4B and eIF4H.
-
BQuantitation of GMR‐GAL4, (CGG)90‐EGFP eye phenotypes with eIF4B/H manipulations (Mann–Whitney U‐test with Bonferroni corrections for multiple comparisons; n = 26–55/genotype).
-
C, DThe expression of plasmid‐based AUG‐NL and +1 (CGG)100 NL‐3xF reporters (C), or co‐transfected AUG‐FF reporters (D), following knockdown of EIF4B or EIF4H. Black asterisks refer to comparisons between siEGFP‐ and siEIF4B/H‐treated cells; pink and blue asterisks refer to comparisons between siEIF4B‐ (pink) or siEIF4H‐ (blue) treated cells expressing AUG‐NL‐3xF and those expressing +1 (CGG)100 (two‐way ANOVA with Tukey's multiple comparisons test, n = 9/condition).
-
EThe expression of plasmid‐based AUG‐NL‐3xF and (CGG)100 +1 NL‐3xF reporters with and without over‐expression of EIF4B, EIF4H, or both (two‐way ANOVA with Dunnett's multiple comparisons test; n = 20/condition). Asterisks refer to comparisons between cells over‐expressing either EGFP or EIF4B, EIF4H, or EIF4B and EIF4H and expressing the same reporter.
In contrast to Belle/DDX3X, the impact of modulating eIF4B or eIF4H expression was not specific to RAN translation. In cultured HeLa cells, knockdown of either EIF4B or EIF4H (Appendix Fig S10F) similarly decreased the expression of +1 CGG100 NL‐3xF and a co‐transfected AUG‐driven firefly luciferase (AUG‐FF) reporter (Fig 5C and D), suggesting that we cannot separate the role of eIF4B/H in RAN translation from their functions in translation generally. In support of this interpretation, over‐expression of EIF4H alone, or both EIF4B and EIF4H together, significantly increased the expression of +1 CGG100 NL‐3xF and AUG‐NL‐3xF (Fig 5E). These data suggest that eIF4B and eIF4H both regulate RAN translation but do so in a manner that is not specific to transcripts that are RAN translated.
eIF1 and eIF5 modulate RAN translation in human cells via start codon selectivity
By definition, RAN translation follows a failure in start codon fidelity. Therefore, we predict that eIFs that enhance start codon fidelity would suppress RAN translation, while eIFs that reduce start codon fidelity would enhance RAN translation. We and others have pursued this concept in the context of the integrated stress response 30, 32, 39: Cellular stressors trigger phosphorylation of eIF2α, which suppresses global protein translation by reducing ternary complex (eIF2α‐GTP‐tRNAiMet) recycling and availability. Cellular stressors and mutations in multiple eIF2 subunits have both been shown to enhance initiation at non‐AUG codons in yeast and mammalian cells 79, 80. Consistent with this, we and others observed that exogenous stressors or eIF2α phosphorylation selectively enhances RAN translation at both CGG repeats and GGGGCC repeats associated with ALS 30, 32, 39 in a near‐AUG codon‐dependent manner. Several other eIFs are known to modulate start codon fidelity 33, 81. eIF1 maintains the fidelity of scanning 43S PICs for AUG start codons by antagonizing the structural reconfigurations that follow AUG recognition 58, 60, 61, 62, 63. We therefore asked whether EIF1 over‐expression might impact RAN translation. Over‐expression of EIF1 in cultured human cells decreased the expression of (CGG)100 NL‐3xF in the +1 and +2 frames (Fig 6A). Notably, inserting an AUG codon upstream of the CGG NRE in the +1 frame abolished this effect, indicating that the identity of the start codon is essential for modulation by eIF1. This demonstrates that manipulation of the molecular machinery that determines start codon fidelity can modulate RAN translation at CGG repeats in human cells.
Figure 6. EIF1 and EIF5 modulate RAN translation by determining AUG start codon specificity.
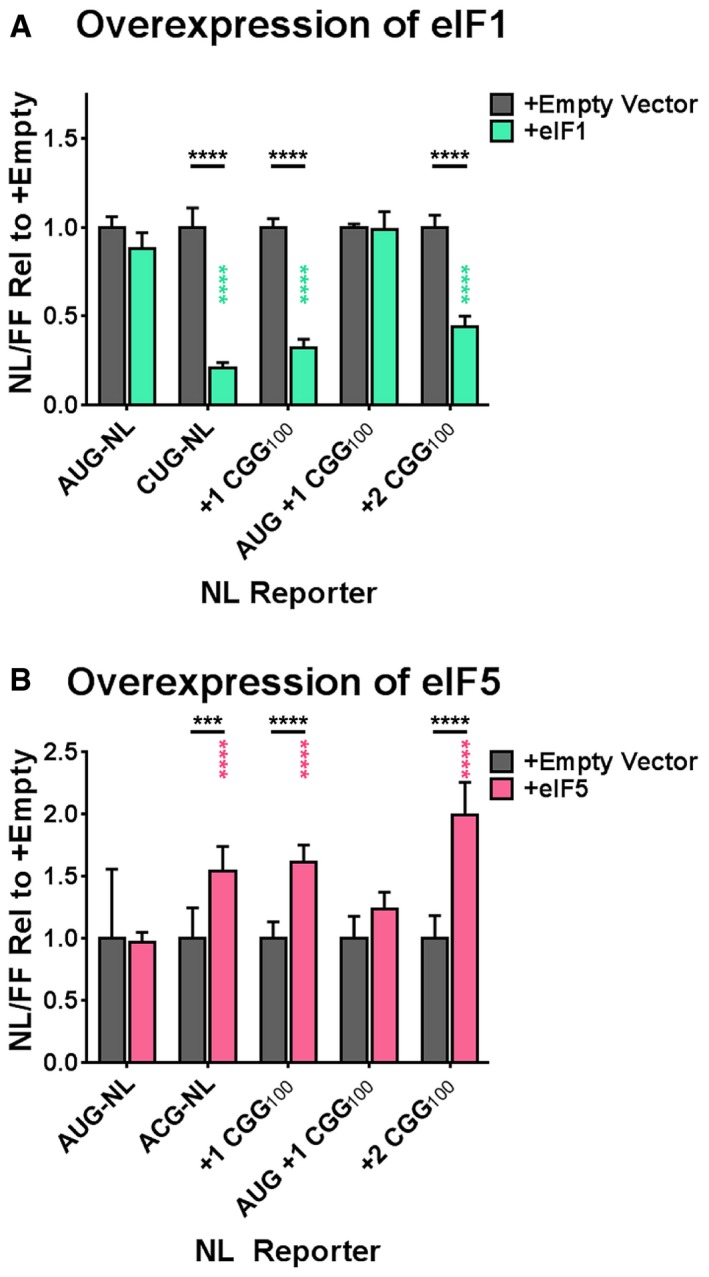
- The expression of plasmid‐based NL‐3xF reporters in HEK293 cells with and without over‐expression of EIF1 (two‐way ANOVA with Sidak's multiple comparisons test; n = 9–12/condition). Black asterisks refer to comparisons between empty vector‐transfected and EIF1‐transfected cells; green asterisks refer to comparisons between EIF1‐transfected cells expressing AUG‐NL‐3xF and those expressing a different reporter.
- The expression of plasmid‐based NL‐3xF reporters in HEK293 cells with and without over‐expression of EIF5 (two‐way ANOVA with Sidak's multiple comparisons test; n = 9–12/condition). Black asterisks refer to comparisons between empty vector‐transfected and EIF5‐transfected cells; pink asterisks refer to comparisons between EIF5‐transfected cells expressing AUG‐NL‐3xF and those expressing a different reporter.
We next asked whether siRNA‐mediated EIF1 knockdown would modulate RAN translation, and we observed that EIF1 knockdown resulted in significant inhibition of all transfected reporters (Appendix Fig S11A). This finding is consistent with the known scanning‐promoting functions of eIF1 during general translation initiation 61, 62 and potentially explains the toxicity we observed with some EIF1‐disrupting Drosophila lines in the absence of (CGG)90‐EGFP (Appendix Fig S1), as well as the reduced (CGG)90‐elicited toxicity we observed with EIF1‐disrupting lines that were not, on their own, toxic.
We next asked whether enhancing expression of eIF5 would affect RAN translation. eIF5 is an eIF2γ‐specific GTPase‐activating protein (GAP) 82, 83. Once a 43S PIC recognizes a start codon, eIF1 dissociates from the PIC, and eIF5 promotes the hydrolysis of eIF2γ‐bound ATP 84, 85, 86, 87, a critical step in the dissociation of other eIFs that must precede ribosomal subunit joining. Furthermore, Loughran et al 88 demonstrated that higher eIF5 abundance leads to increased initiation at non‐AUG codons. Consistent with these results, we observed that EIF5 over‐expression in cultured human cells led to higher expression of ACG‐initiated NL‐3xF and +1 and +2 (CGG)100 NL‐3xF reporter plasmids, but not AUG‐NL‐3xF or AUG‐initiated +1 (CGG)100 NL‐3xF reporters (Fig 6B). Moreover, we confirmed by Western blot that transfection of EIF1 and EIF5 cDNA‐containing plasmids resulted in a higher level of eIF1 and eIF5 expression (Appendix Fig S11B). In total, these experiments demonstrate that manipulation of factors that influence start codon fidelity can up‐ or down‐regulate RAN translation at CGG repeats.
Knockdown of DDX3X suppresses toxicity in (CGG)100‐expressing primary neurons
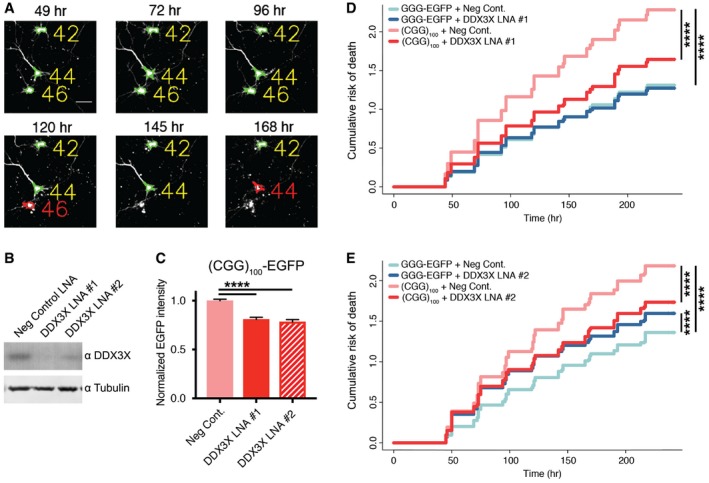
The toxicity of the CGG NRE in FMR1 is driven at least in part by the products of RAN translation 14, 17, 18. Having demonstrated that DDX3X regulates the abundance of RAN products in mammalian cells, we asked whether knockdown of DDX3X can mitigate the toxicity of (CGG)100 repeats in mammalian neurons. We transfected primary rat neurons with locked nucleic acids (LNAs) against DDX3X or non‐targeting controls, along with plasmids containing the human FMR1 5′ UTR with 100 CGG repeats upstream of an EGFP reporter in the +1 frame [+1 (CGG)100 EGFP], along with an AUG‐driven mApple construct to highlight transfected cells. Over the following 10 days, we used automated longitudinal fluorescence microscopy to track the survival of transfected cells (Fig 7A) 89, 90, 91. The expression of +1 (CGG)100 EGFP markedly reduced neuronal survival compared to neurons expressing either EGFP (Appendix Fig S12) or an EGFP reporter in which the AUG start codon has been replaced with a GGG codon (GGG‐EGFP). Knockdown of DDX3X (Fig 7B) by two independent LNAs reduced the expression of FMRpolyG100‐EGFP in these neurons (Fig 7C) and significantly improved the survival of (CGG)100‐expressing neurons relative to transfection of non‐targeting control LNAs (Fig 7D and E). We observed some neurotoxicity with one of the LNA‐targeted DDX3X (#2) but not the other, potentially indicative of adverse off‐target effects from LNA #2. These findings suggest that, like in Drosophila, knockdown of DDX3X suppresses CGG NRE‐elicited toxicity by inhibiting RAN translation.
Figure 7. Knockdown of DDX3X mitigates (CGG)100 toxicity in primary rodent neurons.
-
ASample micrographs collected by automated longitudinal fluorescence microscopy, demonstrating the automated determination of cell death.
-
BAnti‐DDX3X Western blot of B35 cells transfected with either of two independent anti‐DDX3X LNAs or a control LNA.
-
CThe expression of EGFP in primary rat neurons transfected with (CGG)100 (+1) EGFP and either anti‐DDX3X LNAs or a control (one‐way ANOVA with Tukey's multiple comparisons test; n = 2,408–5,689 cells/condition). All graphs depict pooled data, normalized first within the replicate.
-
D, ETransfection of anti‐DDX3X LNA #1 (D) or #2 (E) reduced the cumulative risk of death in (CGG)100 (+1) EGFP‐expressing neurons (Cox proportional hazard analysis; n = 2,408–3,676 cells/condition).
Discussion
We performed a screen of eIFs and RNA helicases to identify modifiers of FMR1 NRE‐associated RAN translation and toxicity in Drosophila and human cells, and found both selective (bel/DDX3X, EIF1, and EIF5) and non‐selective (EIF4B and EIF4H) modifiers. Manipulation of these genes both reduced the expression of the RAN products FMRpolyG and FMRpolyA and mitigated CGG repeat‐associated toxicity. This work both extends our understanding of RAN translation mechanisms and identifies potential therapeutic targets for FXTAS and potentially other RAN translation‐associated disorders.
RAN translation occurs in association with expanded, GC‐rich, secondary structure‐forming repeats that promote initiation in the absence of an AUG codon 10, 14, 31. We originally hypothesized that disruption of RNA helicases, which resolve RNA–RNA secondary structures, would enhance RAN translation at CGG repeats. Our results demonstrated the opposite: Knockdown of bel/DDX3X, EIF4B, or EIF4H inhibited FMR1 RAN translation. How can we account for this discrepancy? DDX3X and its yeast homolog Ded1 are DEAD‐box RNA helicases that are required for resolution of RNA–RNA structures in long, GC‐rich 5′ UTRs of particular genes 46, 47, 48, 49, 50, 51, 52, 77, 92, 93. The FMR1 5′ UTR is GC‐rich (76%) independent of the CGG repeats and is predicted to form highly stable secondary structures 94, 95, 96 capable of stalling scanning 43S PICs 35, 36. Our results, in which DDX3X is required for initiation within the FMR1 5′ UTR but not for expression of AUG‐NL‐3xF, are most consistent with a model in which DDX3X interacts with and resolves RNA–RNA secondary structures within the FMR1 5′ UTR 5′ to and within the CGG repeat, which allows access of scanning PICs to sites of initiation. Without DDX3X, scanning 43S PICs are unable to access the initiation sites for RAN translation, leading to a selective decrease in their use. In contrast, EIF4B and EIF4H are stimulatory factors 53, 54, 55, 97, 98 for the DEAD‐box RNA helicase eIF4A, which is critical for 43S PIC‐mRNA binding and PIC scanning 50. Our data, in which eIF4B and eIF4H regulate not only RAN translation but also general translation, are best explained with a model in which eIF4B and eIF4H facilitate 43S PIC attachment and basal scanning in the initial stages of RAN translation in a similar fashion to that for canonical translation. This model is consistent with previous observations that FMR1 RAN translation resembles canonical translation during these early stages of initiation 30, 31, 32.
A key feature that distinguishes RAN translation from canonical translation is its use of non‐AUG codons for initiation 10, 30, 31, 32. The specificity for AUG start codons is simultaneously (and paradoxically) central to the integrity of eukaryotic and prokaryotic proteomes and an essential point of regulation for determining which, when, and how much protein is synthesized from a given mRNA transcript 81, 99, 100, 101, 102. RNA secondary structure‐forming elements (including GC‐rich NREs) are predicted to slow or stall PICs during scanning, enhancing the dwell time of codon‐anticodon interactions at non‐AUG codons and increasing the frequency of initiation events at non‐AUG sites 37. RAN translation might initiate in a similar manner. If correct, this model predicts that increasing the abundance of eIFs that boost start codon fidelity would inhibit RAN translation, while increasing abundance of eIFs that reduce start codon fidelity would enhance RAN translation. Multiple factors and regulatory pathways converge to govern start codon selection, including EIF1 and EIF5. eIF1 (originally identified as Sui1) is known to increase the specificity of scanning 43S PICs for AUG start codons 58, 103, while higher availability and abundance of eIF5 (originally Sui5) have been shown to decrease start codon fidelity 88, 104, 105.
Consistent with our prediction, we observed that EIF1 over‐expression decreased RAN translation in a start codon‐dependent manner, while over‐expression of EIF5 did the opposite. However, as factors such as EIF1 have multiple roles in translational initiation, suppression of their expression led to global decreases in translation that impacted both FMR1 RAN translation and canonical translation, which elicited intrinsic toxicity in Drosophila. These results align with previous work demonstrating that RAN translation at both CGG and GGGGCC NREs and across multiple reading frames is induced by activation of the ISR in a start codon‐dependent manner via a mechanism that impinges on the AUG selectivity of the 43S PIC 30, 34, 39. This line of research supports a model in which RAN translation represents a failure in start codon fidelity, suggesting that factors that regulate start codon fidelity also regulate this pathologic process.
A central goal of understanding how RAN translation occurs, and how it is distinct from canonical translation, is to identify potential therapeutic targets. Prevention of FMR1 RAN translation is sufficient to suppress toxicity/neurodegeneration in human cells in vitro and Drosophila and mice in vivo 14, 17, 18. Here, we demonstrate that disruption of bel/DDX3X not only inhibits FMR1 RAN translation selectively in vitro and in vivo, but significantly mitigates repeat‐induced toxicity across model systems. We suggest that targeting factors critical for resolving RNA secondary structures and/or enhancing start codon fidelity could represent viable therapeutic strategies for FXTAS and related neurodegenerative disorders. DDX3X in particular is currently the target of multiple lines of pharmacological research aimed at treating various cancers and viruses 106, 107, 108. By targeting a proximal event in the pathophysiology of RAN translation‐associated neurodegenerative disorders, this strategy has the potential to be more efficacious than targeting the toxic effects of each RAN product.
Materials and Methods
Drosophila lines
All fly lines used here and their sources are listed in Appendix Table S1.
Antibodies
For Western blotting, all primary antibodies were used at 1:1,000 in 5% non‐fat dairy milk (wt/vol), 0.02% NaN3 (wt/vol), and 0.1% Tween‐20 (vol/vol) in TBS. Monoclonal mouse anti‐EGFP antibody was acquired from Sigma (clones 7.1 and 13.1, catalog #11814460001). Monoclonal mouse anti‐β‐tubulin antibody, developed by Michael Klymkowsky, was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. Monoclonal mouse anti‐FLAG antibody was acquired from Sigma (clone M2, catalog #F1804). Polyclonal rabbit anti‐DDX3X antibody (catalog #2635), anti‐eIF4B antibody (catalog #3592), anti‐eIF4H antibody (catalog #2444), anti‐eIF1 antibody (catalog #12496), and anti‐eIF5 antibody (catalog #13894) were acquired from Cell Signaling Technology. For information on the anti‐DDX3X and isotype control antibodies used for RNA immunoprecipitation, see the relevant section below.
Drosophila phenotyping
All flies were raised and crossed at 25°C on SY10 food unless otherwise stated. For the screen, virgin female flies carrying the UAS‐FMR1 (CGG)90‐EGFP reporter 66 and a GMR‐GAL4 driver [Bloomington Drosophila Stock Center (BDSC) 8605] were crossed to males carrying UAS‐driven shRNA constructs against, UAS‐driven transgenes of, or germline mutations in candidate genes. Rough‐eye phenotypes in F1 progeny were photographed and scored at 0–1 day post‐eclosion according to a rubric adapted from Pandey et al 109. One point was given for each of the following morphological aberrations: supernumerary inter‐ommatidial bristles, abnormal orientation of inter‐ommatidial bristles, disorganization of the ommatidial array, ommatidial fusion, and total loss of the ommatidial array over 10% of the eye surface. Two points were given for each of the following: the presence of necrotic lesions, collapse of the eye's convex surface, and shrinkage of the eye's surface area by 25%. Individual flies could therefore score between 0 and 11, with higher scores indicating a more severe phenotype. Eye images were captured using a Leica M125 stereomicroscope and a Leica DFC425 digital camera.
For longevity assays, flies carrying (CGG)90‐EGFP and either a Tub5‐GAL4 GeneSwitch or ElaV‐GAL4 GeneSwitch driver (both RU486‐inducible) 76 were placed on SY10 food supplemented with 200 μM RU486 and flipped onto fresh RU486‐supplemented SY10 every 24 (Tub5) or 48 (ElaV) h, and kept at 29°C until expiration. Deaths were counted and dead flies removed every 24 or 48 h.
Western blotting and quantitative reverse‐transcription–PCR (qRT–PCR) in Drosophila
For Western blotting and qRT–PCR of fly material, < 2 days post‐eclosion flies carrying (CGG)90‐EGFP and the Tub5‐GAL4 GeneSwitch driver were placed on 200 μM RU486‐supplemented SY10 for 72 h, with fresh RU486‐supplemented food provided every 24 h, at 29°C. For Western blotting, flies were homogenized at 4°C in ice‐cold radioimmunoprecipitation assay (RIPA) buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 0.1% SDS (wt/vol), 0.5% sodium deoxycholate (wt/vol), 1% IGEPAL CA‐630 (vol/vol), and complete mini protease inhibitor (Roche)] and then centrifuged at 13,300 g for 2 min at 4°C to pellet cuticle and wing debris. The supernatant was removed and the chromatin sheared by 10 strokes through a 28.5G syringe. Lysates were subsequently mixed with 6× reducing Laemmli buffer, separated by SDS–PAGE, and transferred to PVDF membranes (Bio‐Rad).
For qRT–PCR assays, flies were homogenized in TRIzol (Thermo Fisher Scientific) and total RNA was subsequently extracted. Ten micrograms of RNA per sample was twice incubated with 2 U of TURBO DNase (Thermo Fisher Scientific) in reaction volumes of 50 μl at 37°C for 30 min, per manufacturer's instructions, and then recovered using RNA Clean and Concentrator‐25 kits (Zymo Research). Five hundred nanograms of DNase‐treated RNA per sample was used to generate cDNAs using a mixture of oligo(dT) and random hexamer primers (iScript cDNA Synthesis Kit, Bio‐Rad). cDNA abundance was measured using iQ SYBR Green Supermix (Bio‐Rad), appropriate primers at 300 nM (Appendix Table S2), and an iQ5 qPCR system (Bio‐Rad).
Plasmids
All reporter constructs used here were generated and described by Kearse et al 31 and/or Green et al 30. In brief, all nanoluciferase reporters were developed by site‐directed mutagenesis or digestion/ligation from pcDNA3.1(+)/AUG‐NL‐3xF. All pcDNA3.1(+)/FMR1 (CGG)n NL‐3xF constructs bear the full human 5′ UTR of FMR1 upstream of the CGG repeats. In addition, in every FMR1 (CGG)n constructs used here, the initiator ATG of NL‐3xF has been mutated to GGG to abolish initiation at this site. The firefly luciferase construct pGL4.13 was acquired from Promega. For in vitro transcription of firefly luciferase RNA, the firefly luciferase construct was digested and ligated into a pcDNA3.1(+) vector using 5′ HindIII and 3′ XbaI restriction sites (Rapid DNA Dephos and Ligation Kit, Sigma). All plasmids used for transfection and in vitro transcription were prepared from Escherichia coli cultures using ZymoPURE Plasmid Midiprep Kits (Zymo Research).
pCMV6‐XL5/EIF4B and pCMV5‐XL5/EIF4H, which drive expression of human EIF4B and EIF4H, respectively, were acquired from OriGene. pcDNA3.1(+)/EIF1 and pcDNA3.1D/EIF5‐V5‐His, which drive expression of human EIF1 and EIF5, respectively, were acquired from J. Schofield.
Cell culture and transfection
HeLa (CCL‐2, ATCC), HEK293 (CRL‐1573, ATCC), HEK293T (CRL‐3216), and B35 (CRL‐2754, ATCC) cells were cultured and passaged at 37°C, 5% CO2, with HeLa cells in Dulbecco's modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% non‐essential amino acids, and HEK293, HEK293T, and B35 cells in DMEM supplemented with 10% FBS.
For luciferase assays, HeLa cells were plated in 96‐well plates at 1.0 × 104 cells/well in 100 μl media and reverse transfected with Stealth siRNAs against human DDX3X (DDX3XHSS102712 and DDX3XHSS176054, Thermo Fisher Scientific) or EGFP at 1.67 nM, unless otherwise noted, using Lipofectamine RNAiMAX (Thermo Fisher Scientific). Alternatively, they were transfected with ON‐TARGETplus SMARTpool siRNAs against human EIF4B (L‐020179‐00, Dharmacon), EIF4H (L‐013054‐00), or EIF1 (L‐015804‐02) or a non‐targeting pool (D‐001810‐10). In brief, siRNA and RNAiMAX were diluted in Opti‐MEM, combined, incubated for 10 min at room temperature, and then mixed with cells. For subsequent plasmid transfection, 24 h after plating cells were transfected with 25 ng/well pcDNA3.1(+)/NL‐3xF plasmid and 25 ng/well firefly luciferase transfection control plasmid (pGL4.13) using jetPRIME (Polyplus). The transfection media was removed and replaced 4 h post‐transfection. For RNA reporter transfection, 24 h after plating cells were transfected with 25 ng/well in vitro‐transcribed nanoluciferase RNA and 25 ng/well firefly luciferase RNA using TransIT mRNA (Mirus Bio). Luciferase assays were performed 24 h after plasmid transfection, as described by Kearse et al 31 and Green et al 30.
For over‐expression experiments, cells were plated in 96‐well plates at 1.0 × 104 cells/well (HeLa) or 2.0 × 104 cells/well (HEK293T) in 100 μl media. Twenty‐four hours after plating, HeLa cells were transfected with 5 ng/well pcDNA3.1(+)/NL‐3xF, 5 ng/well pGL4.13, and 40 ng/well pCMV6‐XL5/EIF4B, pCMV5‐XL5/EIF4H, pEGFP N1, or a combination thereof, using jetPRIME (Polyplus). HEK293T cells were transfected with 25 ng/well pcDNA3.1(+)/NL‐3xF, 25 ng/well pGL4.13, and 250 ng/well pcDNA3.1(+)/EIF1, empty pcDNA3.1(+), pcDNA3.1D/V5‐His‐EIF5, or empty pcDNA3.1D/V5‐His using FuGENE HD (Promega). Luciferase assays were performed as above.
For Western blotting experiments, HeLa cells were plated in 12‐well plates at 1.5 × 105 cells/well in 1 ml media and reverse transfected, as above, with Stealth siRNAs against DDX3X or EGFP at 1.67 nM using Lipofectamine RNAiMAX. Alternatively, they were transfected with ON‐TARGETplus SMARTpool siRNAs against EIF4B, EIF4H, or a non‐targeting pool at 15 nM. Twenty‐four hours after plating, cells were transfected with 500 ng/well pcDNA3.1(+)/NL‐3xF using jetPRIME (Polyplus). The transfection media was removed and replaced 4 h post‐transfection. Twenty‐four hours after plasmid transfection, cells were lysed on‐plate in RIPA buffer. The lysate was homogenized by 10 strokes through a 28.5G syringe (without centrifugation), mixed with 6× reducing Laemmli buffer, heated at 90°C for 10 min, resolved by SDS–PAGE, and transferred to a PVDF membrane before incubation in primary antibody.
For qRT–PCR experiments, HeLa cells were plated in 6‐well plates at 2.5 × 105 cells/well in 2.5 ml media and reverse transfected with Stealth siRNAs against DDX3X or EGFP at 1.67 nM using Lipofectamine RNAiMAX. Twenty‐four hours after plating cells were transfected with 625 ng/well pcDNA3.1(+)/NL‐3xF and 625 ng/well pGL4.13 using jetPRIME (Polyplus). The transfection media was removed and replaced 4 h post‐transfection. Twenty‐four hours after plasmid transfection, cells were lysed and total cellular RNA collected using Quick‐RNA MiniPrep Kit (Zymo Research). Five micrograms of RNA per sample was incubated twice with 2 U of TURBO DNase (Thermo Fisher Scientific) for 30 min at 37°C to remove contaminating genomic and plasmid DNA, and then recovered using the RNA Clean and Concentrator‐25 Kit (Zymo Research). cDNA was generated from 500 ng of DNase‐treated RNA per sample and a mixture of oligo(dT) and random hexamer primers (iScript cDNA Synthesis Kit, Bio‐Rad). cDNA abundance was measured using iQ SYBR Green Supermix (Bio‐Rad), an iQ5 qPCR system (Bio‐Rad), and the appropriate primers at 300 nM. cDNA abundance was quantified using a modified ΔΔC t method recommended by the manufacturer and was presented as normalized to spiked‐in in vitro‐transcribed RNAs to account for differences in RT efficiency.
For confirmation of anti‐DDX3X locked nucleic acid (LNA) efficacy, B35 cells were plated in 12‐well plates at 2.0 × 105 cells/well in 1 ml media and reverse transfected with anti‐DDX3X Silencer Select LNAs (s165214 and s165216, Thermo Fisher Scientific) or a non‐targeting control (4390843, Thermo Fisher Scientific) at 40 nM using Lipofectamine RNAiMAX. Forty‐eight hours after plating, cells were lysed as above for Western blot analysis.
Crosslinking and RNA immunoprecipitation
HEK293 cells were plated in poly‐l‐lysine‐coated 10‐cm plates at 3.0 × 106 cells/plate. Forty‐eight hours after plating, cells were transfected with 5 μg of pcDNA3.1+/CGG100 (+1) NL‐3xF and 5 μg of either pGL4.13 or pEGFP N1 using ViaFect (Promega). Twenty‐four hours post‐transfection, the media was aspirated and replaced with fresh media supplemented with 6‐thioguanine (6SG) 110 at 100 μM and allowed to incubate for 12 h.
Cells were rinsed 3× in PBS (pH 7.4), the PBS was aspirated, and the cells were irradiated uncovered with 0.6 J/cm2 of 365 nm UV light using a Stratalinker 2400 (Stratagene). Cells were then harvested using trypsin (0.25%)‐EDTA and rubber policemen, collected by centrifugation, rinsed 2× in PBS, flash‐frozen in a dry ice/EtOH bath, and stored at −80°C. For processing, cell pellets were lysed in NP‐40 lysis buffer [50 mM Tris (pH 7.5), 150 mM NaCl, 1 mM DTT, 0.5% IGEPAL CA‐630 (wt/vol)] supplemented with complete mini EDTA‐free protease inhibitors (Roche), PhosSTOP phosphatase inhibitors (Roche), 1 U/μl recombinant RNAsin (Promega), and 200 U/ml SuperaseIN (Thermo Fisher Scientific) for 25 min on ice, incubated with 42 U/ml RQ1 DNase (Promega) at 37°C for 10 min, and then centrifuged at 10,000 g, 4°C for 10 min.
Protein G Dynabeads (Thermo Fisher Scientific) were prepared and incubated with mouse anti‐DDX3X (clone 2253C5a, Santa Cruz sc‐81247), mouse anti‐EGFP (clones 7.1/13.1, Sigma 11814460001), or mouse isotype control IgG (Thermo Fisher Scientific 10400C) as per the manufacturer's instructions. Antibody‐conjugated beads were rinsed 3× in NP‐40 lysis buffer and incubated with the cleared lysate for 16 h with inversion at 4°C. Only those cells transfected with pEGFP N1 were subjected to anti‐EGFP RIP. The lysate was removed, and the beads were washed 3× with NP‐40 lysis buffer and 3× with 5× PBS (pH 7.4) supplemented with 0.5% IGEPAL CA‐630. RNA was eluted by incubation with 2 mg/ml proteinase K at 55°C for 1 h in 50 mM Tris–HCl (pH 7.0), 75 mM NaCl, 6 mM EDTA, and 2% SDS (wt/vol), extracted using TRIzol (Thermo Fisher Scientific) and GlycoBlue co‐precipitant (Thermo Fisher Scientific), treated twice with 40 U/ml TURBO DNase at 37°C for 30 min, and purified using the RNA Clean and Concentrator‐5 Kit (Zymo Research). cDNAs were generated using the iScript cDNA Synthesis Kit, as described above, with each reaction spiked with equal amounts of in vitro‐transcribed AUG‐FF or EGFP RNA (depending on which plasmid had not been co‐transfected) as a reverse‐transcription control. qPCR was performed as described above, with cDNA abundance normalized to FF or EGFP cDNA abundance.
In vitro transcription and translation reactions
pcDNA3.1(+)/NL‐3xF and pcDNA3.1(+)/FF were linearized by PspOMI and XbaI restriction enzymes (NEB), respectively, and recovered using DNA Clean and Concentrator‐25 kits (Zymo Research). m7G‐capped and poly‐adenylated RNAs were transcribed in vitro from these plasmids using HiScribe T7 ARCA mRNA Kit (with tailing; NEB) as per the manufacturer's instructions and recovered using RNA Clean and Concentrator‐25 kits (Zymo Research). The integrity and size of all transcribed RNAs were confirmed by denaturing agarose gel electrophoresis with formaldehyde/formamide.
For preparation of translation‐competent extracts, HeLa cells were plated in 14.5‐cm dishes at 8 × 106 cells/plate. Twenty‐four hours later, they were forward transfected with Stealth siRNAs against DDX3X or EGFP at 1.67 nM using RNAiMAX, as described above (adapted from Rakotondrafara & Heintze 111). The transfection media was removed 5 h post‐transfection and replaced with fresh media. Two days post‐transfection, cells were harvested using trypsin (0.25%)‐EDTA, centrifuged, and rinsed 3× with PBS (pH 7.4). Cells were allowed to swell on ice in a volume of hypotonic lysis buffer [10 mM HEPES‐KOH (pH 7.6), 10 mM KOAc, 0.5 mM Mg2OAc, 5 mM DTT, supplemented with complete mini, EDTA‐free protease inhibitor] equal to the cell pellet volume for 30 min. Cells were mechanically disrupted at 4°C using 20 strokes in a 27G syringe and then allowed to incubate on ice for an additional 20 min. Lysis was confirmed visually in > 95% of cells by trypan blue inclusion. The lysate was centrifuged at 10,000 g for 10 min at 4°C. The supernatant was then collected, diluted in lysis buffer to 8.0 μg/μl using a modified Bradford protein quantification assay (Bio‐Rad), flash‐frozen in liquid N2, and stored at −80°C.
For in vitro translation reactions, lysates were brought to final concentrations of 20 mM HEPES‐KOH (pH 7.6), 44 mM KOAc, 2.2 mM Mg2AOc, 2 mM DTT, 20 mM creatine phosphate (Roche), 0.1 μg/μl creatine kinase (Roche), 0.1 mM spermidine, and on average 0.1 mM of each amino acid (with relative amounts approximating those in eukaryotes) 112. To this, in vitro‐transcribed RNAs were added to 4 nM in a final volume of 10 μl per reaction. After incubation at 30°C for 30 min, 25 μl room temperature Glo Lysis Buffer (Promega) was added to halt the reaction and allowed to incubate for 5 min at room temperature. To 25 μl of this mixture was added 25 μl of Nano‐Glo substrate freshly diluted in Nano‐Glo buffer (Promega). This mixture was allowed to incubate in opaque 96‐well plates on a rocking shaker in the dark for 5 min before the luminescence detection and quantification using a GloMax microplate luminometer (Promega).
Polysome fractionation
HeLa cells were seeded in four to eight 10‐cm dishes per condition. Twenty‐four hours after plating, cells were transfected with siRNAs against DDX3X or EGFP at 1.6 nM using RNAiMAX, as above, with the media exchanged at 5 h post‐transfection. When cells reached 70–90% confluent, 24–36 h post‐knockdown, they were treated with 100 μg/ml cycloheximide (CHX) for 5 min at 37°C. Cells were then transferred to ice and washed with 2.5 ml ice‐cold PBS containing 100 μg/ml CHX, collected by scraping in 2.5 ml cold PBS + CHX, and pelleted at 234 g and 4°C for 5 min. PBS was aspirated and pellets re‐suspended in polysome‐profiling lysis buffer (20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 15 mM MgCl2, 8% (vol/vol) glycerol, 20 U/ml SUPERase, 80 U/ml murine RNase inhibitor, 0.1 mg/ml heparin, 100 μg/ml CHX, 1 mM DTT, 1× EDTA‐free protease inhibitor cocktail, 20 U/ml Turbo DNase, 1% Triton X‐100) 113. Lysates were passed through a 20G needle 10× and incubated on ice for 5 min. Cellular debris was pelleted at 14,000 g and 4°C for 5 min, and supernatant transferred to a fresh tube. Total lysate RNA was estimated by NanoDrop. Lysates were flash‐frozen in liquid N2 and stored at −80°C until fractionation.
Sucrose gradients were prepared by successively freezing equal volumes of 50, 36.7, 23.3, and 10% sucrose (wt/vol) in 12‐ml Seton tubes. Sucrose‐gradient buffer consisted of 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 15 mM MgCl2, 10 U/ml SUPERase, 20 U/ml murine RNase inhibitor, 100 μg/ml CHX, and 1 mM DTT (Simsek et al 113). Prior to use, gradients were allowed to thaw and linearize overnight at 4°C (Luthe, Analytical Biochemistry, 1983). For fractionation, approximately 90 (trial 1 with four 10‐cm dishes), 220, and 250 μg (trials 2 and 3, respectively, with eight 10‐cm dishes) total RNA was applied to the top of the sucrose gradient. Gradients were spun at 151,263 g and 4°C for 3 h using a Beckman Coulter Optima L‐90K ultracentrifuge and SW 41 Ti swinging‐bucket rotor.
Gradients were fractionated with Brandel's Gradient Fractionation System, measuring absorbance at 254 nm. The detector was base‐lined with 60% sucrose chase solution, and its sensitivity set to 0.5 for trial 1, and 1.0 for trials 2 and 3. For fractionation, 60% sucrose was pumped at a rate of 1.5 ml/min. Brandel's PeakChart software was used to collect data, overlay profiles, and calculate the area under the curve for monosome and polysome fractions.
Primary neuronal cultures and automated fluorescence microscopy
Embryonic day (E) 19–20 Long‐Evans rat (Rattus norvegicus) cortices were harvested and the neurons dissociated and plated in 96‐well plates at 6.0 × 106 cells/ml as previously described 114. On in vitro day (DIV) 4, neurons were transfected using Lipofectamine 2000 (Thermo Fisher Scientific) 115 with 100 ng/well pGW/(CGG)100 +1 EGFP or pGW/GGG‐EGFP, 50 ng/well pGW/mApple, and LNAs to a final concentration of 40 nM. Following transfection, neurons were maintained in NEUMO photostable media (Cell Guidance Systems) for the length of the experiment.
Neurons were imaged using a Nikon Eclipse Ti inverted microscope with PerfectFocus3 and Nikon Plan Fluor 20× objective lens 90. Cells were illuminated with a Lambda XL Xenon lamp (Sutter Instrument) and detected using an Andor iXon3 897 EMCCD or Andor Zyla4.2 (+) sCMOS camera. Stage, filter, and shutter movements were controlled with scripts written in BeanShell for use in μManager. Separate ImageJ/Fiji macros and Python scripts were employed for automated identification of transfected neurons and the drawing of regions of interest (ROIs) around each neuron 91. Cell death was indicated by rounding of the cell body, deterioration of neuronal processes, and loss of mApple fluorescence intensity.
Data analysis
Band intensity on Western blots was quantified using ImageJ (NIH; anti‐EGFP) or Odyssey Image Studio (LI‐COR; anti‐tubulin) software. Primary neuron survival analysis and determination of hazard ratios through Cox proportional hazard analysis were conducted using the publicly available survival package in R. All other data were analyzed using GraphPad Prism 7.00.
Author contributions
AEL conceived the project, designed and performed experiments, analyzed and interpreted data, and wrote the article. FH helped conceive the project and design and perform experiments. AMM, MRG, BNF, AK, HCA, and KMG designed/performed experiments and analyzed/interpreted data. SN and SJF performed experiments. SJB provided necessary equipment, designed experiments, and interpreted data. PKT provided funding, conceived the project, designed experiments, interpreted data, and wrote the article.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
We thank Scott Pletcher's lab at the University of Michigan for providing Tub5‐GS and ElaV‐GS lines and all fly food. We thank Peng Jin's lab at Emory University for providing the (CGG)90‐EGFP line. We thank Wu‐Min Deng's lab at Florida State University for providing mutant belle lines. We thank Ting Xie's lab at the University of Kansas for providing the UAS‐eIF4A line. We thank J. Schofield for pcDNA3.1+/EIF1 and pcDNA3.1D/EIF5‐V5‐His plasmids. We thank David Turner for technical assistance with photo‐crosslinking and RIP experiments. We thank Michael Kearse and everyone in the Todd lab for many thoughtful conversations and their collective wisdom. This work was funded by grants from the VA BLRD (1I21BX001841 and 1I01BX003231), the NIH (R01NS099280 and R01NS086810), and the Michigan Alzheimer's Disease Center and Protein Folding Disease Initiative to PKT. KMG, BNF, and AEL were supported by NIH T32GM007315. KMG was further supported by NIH F31NS100302 and AEL by NIH F30NS098571. MRG was supported by NIH T32NS007222‐35S1. SJB and BNF were supported by the NIH (R01‐NS097542, 1P30AG053760‐01) and Ann Arbor Active Against ALS.
EMBO Reports (2019) 20: e47498
References
- 1. Nelson DL, Orr HT, Warren ST (2013) The unstable repeats–three evolving faces of neurological disease. Neuron 77: 825–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. La Spada AR, Paulson HL, Fischbeck KH (1994) Trinucleotide repeat expansion in neurological disease. Ann Neurol 36: 814–822 [DOI] [PubMed] [Google Scholar]
- 3. Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN (1997) Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19: 333–344 [DOI] [PubMed] [Google Scholar]
- 4. Paulson HL, Shakkottai VG, Clark HB, Orr HT (2017) Polyglutamine spinocerebellar ataxias – from genes to potential treatments. Nat Rev Neurosci 18: 613–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Warrick JM, Paulson HL, Gray‐Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM (1998) Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila . Cell 93: 939–949 [DOI] [PubMed] [Google Scholar]
- 6. Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA (2001) Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet 10: 2165–2170 [DOI] [PubMed] [Google Scholar]
- 7. Miller JW, Urbinati CR, Teng‐Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 19: 4439–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ranum LP, Cooper TA (2006) RNA‐mediated neuromuscular disorders. Annu Rev Neurosci 29: 259–277 [DOI] [PubMed] [Google Scholar]
- 9. Jain A, Vale RD (2017) RNA phase transitions in repeat expansion disorders. Nature 546: 243–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zu T, Gibbens B, Doty NS, Gomes‐Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA et al (2011) Non‐ATG‐initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA 108: 260–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus‐Hernandez M, van Blitterswijk MM, Jansen‐West K, Paul JW III, Rademakers R et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77: 639–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gendron TF, Bieniek KF, Zhang YJ, Jansen‐West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes‐Casey M, Chew J et al (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat‐associated non‐ATG translation in c9FTD/ALS. Acta Neuropathol 126: 829–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide‐repeat proteins in FTLD/ALS. Science 339: 1335–1338 [DOI] [PubMed] [Google Scholar]
- 14. Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V et al (2013) CGG repeat‐associated translation mediates neurodegeneration in Fragile X tremor ataxia syndrome. Neuron 78: 440–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zu T, Liu Y, Banez‐Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH et al (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA 110: E4968–E4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IOC, Pietrzyk J et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine‐rich proteins. Science 345: 1192–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sellier C, Buijsen RAM, He F, Natla S, Jung L, Tropel P, Gaucherot A, Jacobs H, Meziane H, Vincent A et al (2017) Translation of expanded CGG repeats into FMRpolyG is pathogenic and may contribute to Fragile X tremor ataxia syndrome. Neuron 93: 331–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buijsen RA, Visser JA, Kramer P, Severijnen EA, Gearing M, Charlet‐Berguerand N, Sherman SL, Berman RF, Willemsen R, Hukema RK (2016) Presence of inclusions positive for polyglycine containing protein, FMRpolyG, indicates that repeat‐associated non‐AUG translation plays a role in Fragile X‐associated primary ovarian insufficiency. Hum Reprod 31: 158–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zu T, Pattamatta A, Ranum LPW (2018) Repeat‐associated Non‐ATG translation in neurological diseases. Cold Spring Harb Perspect Biol 10: a033019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Banez‐Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, Pletnikova O, Borchelt DR, Ross CA, Margolis RL et al (2015) RAN translation in huntington disease. Neuron 88: 667–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hagerman PJ, Hagerman RJ (2015) Fragile X‐associated tremor/ataxia syndrome. Ann N Y Acad Sci 1338: 58–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, Babineau B, Lebrilla CB, Hagerman RJ, Hagerman PJ (2006) Protein composition of the intranuclear inclusions of FXTAS. Brain 129: 256–271 [DOI] [PubMed] [Google Scholar]
- 23. Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST (2007) Pur alpha binds to rCGG repeats and modulates repeat‐mediated neurodegeneration in a Drosophila model of Fragile X tremor/ataxia syndrome. Neuron 55: 556–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muslimov IA, Patel MV, Rose A, Tiedge H (2011) Spatial code recognition in neuronal RNA targeting: role of RNA‐hnRNP A2 interactions. J Cell Biol 194: 441–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, Alunni V, Moine H, Thibault C, Page A et al (2013) Sequestration of DROSHA and DGCR25 by expanded CGG RNA repeats alters microRNA processing in fragile X‐associated tremor/ataxia syndrome. Cell Rep 3: 869–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ et al (2010) Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J 29: 1248–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J (2007) RNA‐binding proteins hnRNP A2/B1 and CUGBP1 suppress Fragile X CGG premutation repeat‐induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55: 565–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Glineburg MR, Todd PK, Charlet‐Berguerand N, Sellier C (2018) Repeat‐associated non‐AUG (RAN) translation and other molecular mechanisms in Fragile X tremor ataxia syndrome. Brain Res 1693: 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hagerman RJ, Berry‐Kravis E, Hazlett HC, Bailey DB Jr, Moine H, Kooy RF, Tassone F, Gantois I, Sonenberg N, Mandel JL et al (2017) Fragile X syndrome. Nat Rev Dis Primers 3: 17065 [DOI] [PubMed] [Google Scholar]
- 30. Green KM, Glineburg MR, Kearse MG, Flores BN, Linsalata AE, Fedak SJ, Goldstrohm AC, Barmada SJ, Todd PK (2017) RAN translation at C9orf72‐associated repeat expansions is selectively enhanced by the integrated stress response. Nat Commun 8: 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kearse MG, Green KM, Krans A, Rodriguez CM, Linsalata AE, Goldstrohm AC, Todd PK (2016) CGG repeat‐associated non‐AUG translation utilizes a cap‐dependent scanning mechanism of initiation to produce toxic proteins. Mol Cell 62: 314–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tabet R, Schaeffer L, Freyermuth F, Jambeau M, Workman M, Lee CZ, Lin CC, Jiang J, Jansen‐West K, Abou‐Hamdan H et al (2018) CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nat Commun 9: 152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jackson RJ, Hellen CU, Pestova TV (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sonobe Y, Ghadge G, Masaki K, Sendoel A, Fuchs E, Roos RP (2018) Translation of dipeptide repeat proteins from the C9ORF72 expanded repeat is associated with cellular stress. Neurobiol Dis 116: 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kozak M (1986) Influences of mRNA secondary structure on initiation by eukaryotic ribosomes. Proc Natl Acad Sci USA 83: 2850–2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kozak M (1989) Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol Cell Biol 9: 5134–5142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kozak M (1989) Context effects and inefficient initiation at non‐AUG codons in eucaryotic cell‐free translation systems. Mol Cell Biol 9: 5073–5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leppek K, Das R, Barna M (2018) Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol 19: 158–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cheng W, Wang S, Mestre AA, Fu C, Makarem A, Xian F, Hayes LR, Lopez‐Gonzalez R, Drenner K, Jiang J et al (2018) C9ORF72 GGGGCC repeat‐associated non‐AUG translation is upregulated by stress through eIF2alpha phosphorylation. Nat Commun 9: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jan E, Thompson SR, Wilson JE, Pestova TV, Hellen CU, Sarnow P (2001) Initiator Met‐tRNA‐independent translation mediated by an internal ribosome entry site element in cricket paralysis virus‐like insect viruses. Cold Spring Harb Symp Quant Biol 66: 285–292 [DOI] [PubMed] [Google Scholar]
- 41. Gao FB, Richter JD, Cleveland DW (2017) Rethinking unconventional translation in neurodegeneration. Cell 171: 994–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Green KM, Linsalata AE, Todd PK (2016) RAN translation‐What makes it run? Brain Res 1647: 30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EM, Parkinson G, Isaacs AM (2012) C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G‐quadruplexes. Sci Rep 2: 1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sobczak K, Michlewski G, de Mezer M, Kierzek E, Krol J, Olejniczak M, Kierzek R, Krzyzosiak WJ (2010) Structural diversity of triplet repeat RNAs. J Biol Chem 285: 12755–12764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zumwalt M, Ludwig A, Hagerman PJ, Dieckmann T (2007) Secondary structure and dynamics of the r(CGG) repeat in the mRNA of the Fragile X mental retardation 1 (FMR1) gene. RNA Biol 4: 93–100 [DOI] [PubMed] [Google Scholar]
- 46. Chuang RY, Weaver PL, Liu Z, Chang TH (1997) Requirement of the DEAD‐Box protein ded1p for messenger RNA translation. Science 275: 1468–1471 [DOI] [PubMed] [Google Scholar]
- 47. Hilliker A, Gao Z, Jankowsky E, Parker R (2011) The DEAD‐box protein Ded1 modulates translation by the formation and resolution of an eIF4F‐mRNA complex. Mol Cell 43: 962–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ihry RJ, Sapiro AL, Bashirullah A (2012) Translational control by the DEAD box RNA helicase belle regulates ecdysone‐triggered transcriptional cascades. PLoS Genet 8: e1003085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marsden S, Nardelli M, Linder P, McCarthy JE (2006) Unwinding single RNA molecules using helicases involved in eukaryotic translation initiation. J Mol Biol 361: 327–335 [DOI] [PubMed] [Google Scholar]
- 50. Sen ND, Zhou F, Harris MS, Ingolia NT, Hinnebusch AG (2016) eIF4B stimulates translation of long mRNAs with structured 5′ UTRs and low closed‐loop potential but weak dependence on eIF4G. Proc Natl Acad Sci USA 113: 10464–10472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sen ND, Zhou F, Ingolia NT, Hinnebusch AG (2015) Genome‐wide analysis of translational efficiency reveals distinct but overlapping functions of yeast DEAD‐box RNA helicases Ded1 and eIF4A. Genome Res 25: 1196–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Soto‐Rifo R, Rubilar PS, Limousin T, de Breyne S, Decimo D, Ohlmann T (2012) DEAD‐box protein DDX3 associates with eIF4F to promote translation of selected mRNAs. EMBO J 31: 3745–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rogers GW Jr, Richter NJ, Lima WF, Merrick WC (2001) Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. J Biol Chem 276: 30914–30922 [DOI] [PubMed] [Google Scholar]
- 54. Rogers GW Jr, Richter NJ, Merrick WC (1999) Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J Biol Chem 274: 12236–12244 [DOI] [PubMed] [Google Scholar]
- 55. Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell G, Belsham GJ, Sonenberg N (2001) The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA 7: 382–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dhote V, Sweeney TR, Kim N, Hellen CU, Pestova TV (2012) Roles of individual domains in the function of DHX29, an essential factor required for translation of structured mammalian mRNAs. Proc Natl Acad Sci USA 109: E3150–E3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pisareva VP, Pisarev AV, Komar AA, Hellen CU, Pestova TV (2008) Translation initiation on mammalian mRNAs with structured 5′UTRs requires DExH‐box protein DHX29. Cell 135: 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Castilho‐Valavicius B, Yoon H, Donahue TF (1990) Genetic characterization of the Saccharomyces cerevisiae translational initiation suppressors SUI1, SUI2 and SUI3 and their effects on HIS4 expression. Genetics 124: 483–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maag D, Algire MA, Lorsch JR (2006) Communication between eukaryotic translation initiation factors 5 and 1A within the ribosomal pre‐initiation complex plays a role in start site selection. J Mol Biol 356: 724–737 [DOI] [PubMed] [Google Scholar]
- 60. Passmore LA, Schmeing TM, Maag D, Applefield DJ, Acker MG, Algire MA, Lorsch JR, Ramakrishnan V (2007) The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol Cell 26: 41–50 [DOI] [PubMed] [Google Scholar]
- 61. Pestova TV, Borukhov SI, Hellen CU (1998) Eukaryotic ribosomes require initiation factors 1 and 1A to locate initiation codons. Nature 394: 854–859 [DOI] [PubMed] [Google Scholar]
- 62. Pestova TV, Kolupaeva VG (2002) The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev 16: 2906–2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pisarev AV, Kolupaeva VG, Pisareva VP, Merrick WC, Hellen CU, Pestova TV (2006) Specific functional interactions of nucleotides at key −3 and +4 positions flanking the initiation codon with components of the mammalian 48S translation initiation complex. Genes Dev 20: 624–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Singh CR, Curtis C, Yamamoto Y, Hall NS, Kruse DS, He H, Hannig EM, Asano K (2005) Eukaryotic translation initiation factor 5 is critical for integrity of the scanning preinitiation complex and accurate control of GCN4 translation. Mol Cell Biol 25: 5480–5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wek RC (2018) Role of eIF2alpha kinases in translational control and adaptation to cellular stress. Cold Spring Harb Perspect Biol 10: a032870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jin P, Zarnescu DC, Zhang F, Pearson CE, Lucchesi JC, Moses K, Warren ST (2003) RNA‐mediated neurodegeneration caused by the Fragile X premutation rCGG repeats in Drosophila . Neuron 39: 739–747 [DOI] [PubMed] [Google Scholar]
- 67. Krans A, Kearse MG, Todd PK (2016) Repeat‐associated non‐AUG translation from antisense CCG repeats in Fragile X tremor/ataxia syndrome. Ann Neurol 80: 871–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Oh SY, He F, Krans A, Frazer M, Taylor JP, Paulson HL, Todd PK (2015) RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X‐associated tremor ataxia syndrome. Hum Mol Genet 24: 4317–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Carvajal F, Vallejos M, Walters B, Contreras N, Hertz MI, Olivares E, Caceres CJ, Pino K, Letelier A, Thompson SR et al (2016) Structural domains within the HIV‐1 mRNA and the ribosomal protein S25 influence cap‐independent translation initiation. FEBS J 283: 2508–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Landry DM, Hertz MI, Thompson SR (2009) RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev 23: 2753–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hautbergue GM, Castelli LM, Ferraiuolo L, Sanchez‐Martinez A, Cooper‐Knock J, Higginbottom A, Lin YH, Bauer CS, Dodd JE, Myszczynska MA et al (2017) SRSF1‐dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat Commun 8: 16063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Castrillon DH, Gonczy P, Alexander S, Rawson R, Eberhart CG, Viswanathan S, DiNardo S, Wasserman SA (1993) Toward a molecular genetic analysis of spermatogenesis in Drosophila melanogaster: characterization of male‐sterile mutants generated by single P element mutagenesis. Genetics 135: 489–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Johnstone O, Deuring R, Bock R, Linder P, Fuller MT, Lasko P (2005) Belle is a Drosophila DEAD‐box protein required for viability and in the germ line. Dev Biol 277: 92–101 [DOI] [PubMed] [Google Scholar]
- 74. Poulton JS, Huang YC, Smith L, Sun J, Leake N, Schleede J, Stevens LM, Deng WM (2011) The microRNA pathway regulates the temporal pattern of Notch signaling in Drosophila follicle cells. Development 138: 1737–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Spradling AC, Stern D, Beaton A, Rhem EJ, Laverty T, Mozden N, Misra S, Rubin GM (1999) The Berkeley Drosophila Genome Project gene disruption project: single P‐element insertions mutating 25% of vital Drosophila genes. Genetics 153: 135–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Osterwalder T, Yoon KS, White BH, Keshishian H (2001) A conditional tissue‐specific transgene expression system using inducible GAL4. Proc Natl Acad Sci USA 98: 12596–12601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Guenther UP, Weinberg DE, Zubradt MM, Tedeschi FA, Stawicki BN, Zagore LL, Brar GA, Licatalosi DD, Bartel DP, Weissman JS et al (2018) The helicase Ded1p controls use of near‐cognate translation initiation codons in 5′ UTRs. Nature 559: 130–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Valentin‐Vega YA, Wang YD, Parker M, Patmore DM, Kanagaraj A, Moore J, Rusch M, Finkelstein D, Ellison DW, Gilbertson RJ et al (2016) Cancer‐associated DDX3X mutations drive stress granule assembly and impair global translation. Sci Rep 6: 25996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hinnebusch AG, Ivanov IP, Sonenberg N (2016) Translational control by 5′‐untranslated regions of eukaryotic mRNAs. Science 352: 1413–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Young SK, Wek RC (2016) Upstream open reading frames differentially regulate gene‐specific translation in the integrated stress response. J Biol Chem 291: 16927–16935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kearse MG, Wilusz JE (2017) Non‐AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev 31: 1717–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chakrabarti A, Maitra U (1991) Function of eukaryotic initiation factor 5 in the formation of an 80 S ribosomal polypeptide chain initiation complex. J Biol Chem 266: 14039–14045 [PubMed] [Google Scholar]
- 83. Das S, Maiti T, Das K, Maitra U (1997) Specific interaction of eukaryotic translation initiation factor 5 (eIF5) with the beta‐subunit of eIF2. J Biol Chem 272: 31712–31718 [DOI] [PubMed] [Google Scholar]
- 84. Nanda JS, Saini AK, Munoz AM, Hinnebusch AG, Lorsch JR (2013) Coordinated movements of eukaryotic translation initiation factors eIF1, eIF1A, and eIF5 trigger phosphate release from eIF2 in response to start codon recognition by the ribosomal preinitiation complex. J Biol Chem 288: 5316–5329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nanda JS, Cheung YN, Takacs JE, Martin‐Marcos P, Saini AK, Hinnebusch AG, Lorsch JR (2009) eIF1 controls multiple steps in start codon recognition during eukaryotic translation initiation. J Mol Biol 394: 268–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Saini AK, Nanda JS, Martin‐Marcos P, Dong J, Zhang F, Bhardwaj M, Lorsch JR, Hinnebusch AG (2014) Eukaryotic translation initiation factor eIF5 promotes the accuracy of start codon recognition by regulating Pi release and conformational transitions of the preinitiation complex. Nucleic Acids Res 42: 9623–9640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Pisareva VP, Pisarev AV (2014) eIF5 and eIF5B together stimulate 48S initiation complex formation during ribosomal scanning. Nucleic Acids Res 42: 12052–12069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Loughran G, Sachs MS, Atkins JF, Ivanov IP (2012) Stringency of start codon selection modulates autoregulation of translation initiation factor eIF5. Nucleic Acids Res 40: 2898–2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Archbold HC, Jackson KL, Arora A, Weskamp K, Tank EM, Li X, Miguez R, Dayton RD, Tamir S, Klein RL et al (2018) TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci Rep 8: 4606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Barmada SJ, Ju S, Arjun A, Batarse A, Archbold HC, Peisach D, Li X, Zhang Y, Tank EM, Qiu H et al (2015) Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc Natl Acad Sci USA 112: 7821–7826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Malik AM, Miguez RA, Li X, Ho YS, Feldman EL, Barmada SJ (2018) Matrin 3‐dependent neurotoxicity is modified by nucleic acid binding and nucleocytoplasmic localization. Elife 7: e35977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Berthelot K, Muldoon M, Rajkowitsch L, Hughes J, McCarthy JE (2004) Dynamics and processivity of 40S ribosome scanning on mRNA in yeast. Mol Microbiol 51: 987–1001 [DOI] [PubMed] [Google Scholar]
- 93. Lai MC, Chang WC, Shieh SY, Tarn WY (2010) DDX3 regulates cell growth through translational control of cyclin E1. Mol Cell Biol 30: 5444–5453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL (2008) The Vienna RNA websuite. Nucleic Acids Res 36: W70–W74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL (2011) ViennaRNA package 2.0. Algorithms Mol Biol 6: 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Grifo JA, Tahara SM, Leis JP, Morgan MA, Shatkin AJ, Merrick WC (1982) Characterization of eukaryotic initiation factor 4A, a protein involved in ATP‐dependent binding of globin mRNA. J Biol Chem 257: 5246–5252 [PubMed] [Google Scholar]
- 98. Ray BK, Lawson TG, Kramer JC, Cladaras MH, Grifo JA, Abramson RD, Merrick WC, Thach RE (1985) ATP‐dependent unwinding of messenger RNA structure by eukaryotic initiation factors. J Biol Chem 260: 7651–7658 [PubMed] [Google Scholar]
- 99. Hann SR (1994) Regulation and function of non‐AUG‐initiated proto‐oncogenes. Biochimie 76: 880–886 [DOI] [PubMed] [Google Scholar]
- 100. Ingolia NT, Brar GA, Stern‐Ginossar N, Harris MS, Talhouarne GJ, Jackson SE, Wills MR, Weissman JS (2014) Ribosome profiling reveals pervasive translation outside of annotated protein‐coding genes. Cell Rep 8: 1365–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Meijer HA, Thomas AA (2002) Control of eukaryotic protein synthesis by upstream open reading frames in the 5′‐untranslated region of an mRNA. Biochem J 367: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Touriol C, Bornes S, Bonnal S, Audigier S, Prats H, Prats AC, Vagner S (2003) Generation of protein isoform diversity by alternative initiation of translation at non‐AUG codons. Biol Cell 95: 169–178 [DOI] [PubMed] [Google Scholar]
- 103. Ivanov IP, Loughran G, Sachs MS, Atkins JF (2010) Initiation context modulates autoregulation of eukaryotic translation initiation factor 1 (eIF1). Proc Natl Acad Sci USA 107: 18056–18060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kozel C, Thompson B, Hustak S, Moore C, Nakashima A, Singh CR, Reid M, Cox C, Papadopoulos E, Luna RE et al (2016) Overexpression of eIF5 or its protein mimic 5MP perturbs eIF2 function and induces ATF4 translation through delayed re‐initiation. Nucleic Acids Res 44: 8704–8713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tang L, Morris J, Wan J, Moore C, Fujita Y, Gillaspie S, Aube E, Nanda J, Marques M, Jangal M et al (2017) Competition between translation initiation factor eIF5 and its mimic protein 5MP determines non‐AUG initiation rate genome‐wide. Nucleic Acids Res 45: 11941–11953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bol GM, Raman V, van der Groep P, Vermeulen JF, Patel AH, van der Wall E, van Diest PJ (2013) Expression of the RNA helicase DDX3 and the hypoxia response in breast cancer. PLoS One 8: e63548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Brai A, Fazi R, Tintori C, Zamperini C, Bugli F, Sanguinetti M, Stigliano E, Este J, Badia R, Franco S et al (2016) Human DDX3 protein is a valuable target to develop broad spectrum antiviral agents. Proc Natl Acad Sci USA 113: 5388–5393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Samal SK, Routray S, Veeramachaneni GK, Dash R, Botlagunta M (2015) Ketorolac salt is a newly discovered DDX3 inhibitor to treat oral cancer. Sci Rep 5: 9982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O et al (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447: 859–863 [DOI] [PubMed] [Google Scholar]
- 110. Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M et al (2010) Transcriptome‐wide identification of RNA‐binding protein and microRNA target sites by PAR‐CLIP. Cell 141: 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rakotondrafara AM, Hentze MW (2011) An efficient factor‐depleted mammalian in vitro translation system. Nat Protoc 6: 563–571 [DOI] [PubMed] [Google Scholar]
- 112. Caschera F, Noireaux V (2015) Preparation of amino acid mixtures for cell‐free expression systems. Biotechniques 58: 40–43 [DOI] [PubMed] [Google Scholar]
- 113. Simsek D, Tiu GC, Flynn RA, Byeon GW, Leppek K, Xu AF, Chang HY, Barna M (2017) The mammalian ribo‐interactome reveals ribosome functional diversity and heterogeneity. Cell 169: 1051–1065 e1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95: 55–66 [DOI] [PubMed] [Google Scholar]
- 115. Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S (2010) Cytoplasmic mislocalization of TDP‐43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 30: 639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File