

Abstract
Despite playing physiological roles in specific situations, DNA–RNA hybrids threat genome integrity. To investigate how cells do counteract spontaneous DNA–RNA hybrids, here we screen an siRNA library covering 240 human DNA damage response (DDR) genes and select siRNAs causing DNA–RNA hybrid accumulation and a significant increase in hybrid‐dependent DNA breakage. We identify post‐replicative repair and DNA damage checkpoint factors, including those of the ATM/CHK2 and ATR/CHK1 pathways. Thus, spontaneous DNA–RNA hybrids are likely a major source of replication stress, but they can also accumulate and menace genome integrity as a consequence of unrepaired DSBs and post‐replicative ssDNA gaps in normal cells. We show that DNA–RNA hybrid accumulation correlates with increased DNA damage and chromatin compaction marks. Our results suggest that different mechanisms can lead to DNA–RNA hybrids with distinct consequences for replication and DNA dynamics at each cell cycle stage and support the conclusion that DNA–RNA hybrids are a common source of spontaneous DNA damage that remains unsolved under a deficient DDR.
Keywords: ATM, ATR, DNA damage response, post‐replicative repair, R loop
Subject Categories: DNA Replication, Repair & Recombination
Introduction
R loops, nucleic acid structures composed of a DNA–RNA hybrid and the displaced non‐template DNA strand, are central to a number of cellular processes, including mitochondrial DNA replication and immunoglobulin diversification as well as some cases of transcription regulation 1. Despite their positive role in cell physiology, an increasing number of reports have shown that R loops can be pathological 2, 3.
Given the potential deleterious consequences of R loops, cells bear a number of factors to prevent and resolve DNA–RNA hybrids 2, 4. As first discovered in yeast mutants of the THO complex 5, the optimal co‐transcriptional assembly of the messenger ribonucleoprotein particle (mRNP) is crucial to prevent that the nascent RNA hybridizes back with the DNA template. The accumulation of R loops has been subsequently demonstrated in human cells depleted of the splicing factor SRSF1 6, the THO complex 7, and several other mRNP processing factors, including THSC/TREX‐2 8, the mRNA cleavage and polyadenylation protein FIP1L1 9, or the XRN2 exoribonuclease 10. Hyper‐acetylated chromatin also facilitates DNA–RNA hybrid accumulation 11. Although R loops can be removed by the action of RNase H, which specifically cleaves the RNA moiety of the DNA–RNA hybrid, emerging evidence suggests the existence of a varied number of different helicases that can unwind DNA–RNA hybrids, including senataxin (SETX) 12, aquarius (AQR) 13, DHX9 14, DDX1 15, DDX19 16, DDX23 17, and DDX21 18.
A number of reports indicate that replication impairment is a major mechanism of DNA–RNA hybrid‐mediated genetic instability 11, 19, 20, 21, 22, 23, 24. Along this line, R loop and R loop‐mediated DNA damage accumulation was reported in cells depleted of the double‐strand break (DSB) repair and tumor suppressor genes BRCA1 and BRCA2, the Fanconi anemia (FA) factors involved in inter‐strand crosslink (ICL) repair or the FACT chromatin reorganizer complex, required for the progression of replication fork (RF) through chromatin 25, 26, 27, 28, 29. Since all these factors are involved in replication fork progression, these findings also support that R loops constitute a potential obstacle to replication 30.
Interestingly, R loops seem not to be harmful by themselves. Instead, certain chromatin modifications may be required for R loop‐induced genome instability, as recently shown in budding yeast for histone H3 Ser10 phosphorylation (H3S10‐P) 31. The connection between DNA–RNA hybrids and specific chromatin marks, such as H3S10‐P or H3K9me2, has been observed also in human cells and C. elegans depleted of the THO complex or SETX or at specific fragile sites, like those of Friedreich ataxia (FRDA) and Fragile X syndrome (FXS) 32, 33, 34. Also, genome‐wide mapping has shown a correlation between spontaneous R loops and a set of histone modifications 35, 36. Although the cause–effect relationship between these chromatin marks and R loops is yet to be understood, the accumulated evidence suggests that DNA–RNA hybrids can modulate chromatin remodeling and vice versa 4, 30.
To prevent the potential harmful effect of DNA damage and genetic instability in cell physiology and development, cells have evolved a coordinated cellular response, the DNA damage response (DDR), which is an intrinsic barrier to the early phases of human tumorigenesis 37, 38. The DDR includes a varied number of mechanisms that detect and enable the repair of different types of DNA damage 39, 40. Among them, DSBs activate the ATM/CHK2 DNA damage checkpoint (DDC) pathway 39, 40. By contrast, the RPA‐coated single‐stranded DNA (ssDNA) generated by stalled replication forks triggers the activation of the 9‐1‐1/ATR/CHK1 DDC pathway, which would mainly protect stalled forks from breakage. In addition, replication can restart downstream of the DNA lesion, leaving ssDNA gaps behind the fork that would be subsequently repaired by post‐replicative repair (PRR).
Under the premise that R loops may be a natural source of DNA damage, we wondered whether DDR factors might be important for spontaneous R loop detection and/or dissolution to protect genome integrity. To search for DDR factors involved in R loop homeostasis, we screened a 240 siRNA library of DDR genes using immunofluorescence (IF) with the S9.6 anti‐DNA–RNA antibody. After a first selection of candidates based on the IF‐positive signals, the presence of DNA–RNA hybrids was confirmed by RNase H‐sensitive IF and DNA–RNA immunoprecipitation (DRIP). These included the DDC sensors RAD17 and the 9‐1‐1 complex as well as the DDC kinases, ATR, ATM, CHK1, and CHK2 and the PRR factors UBE2B and RAD18. Cells depleted of each of these DDR factors increased H3S10‐P and H3K9me2 chromatin marks and DNA damage partially in a DNA–RNA hybrid‐dependent manner. However, R loop‐dependent replication fork stalling was detected in cells depleted of ATR or CHK1 but not in cells depleted of ATM, CHK2, or PRR factors. We propose a model in which in the absence of ATR/CHK1, harmful DNA–RNA hybrids accumulate leading to fork stalling, but in the absence of the ATM/CHK2 and PRR machineries they accumulate in association with unrepaired DSBs and post‐replicative ssDNA gaps, respectively. Altogether, these results show that, in addition to the previously reported role of replication‐associated repair factors such as FACT or the Fanconi anemia pathway 25, 26, 27, 28, 41, the 9‐1‐1/ATR/CHK1 and ATM/CHK2 checkpoints and the post‐replicative repair pathways are a safeguard against the accumulation of DNA–RNA hybrids as a common source of DNA damage.
Results
Screening for DDR factors involved in R loop homeostasis
To search for DDR factors that could be involved in the protection against potentially harmful R loops, we performed an siRNA screening in HeLa cells using an arrayed collection of siRNAs targeting 240 human genes involved in DDR (Dharmacon Human ON‐TARGETplus DDR siRNA library). We prepared 96‐well plates containing a duplicate of each four‐siRNA pool for every DDR gene, and a non‐targeted siRNA used as negative control (Fig 1A). Since THOC1 depletion was previously shown to cause accumulation of DNA–RNA hybrids 7, an siRNA pool targeting THOC1 was included as a positive control. For the screening, cells were immunostained with the S9.6 monoclonal antibody that recognizes DNA–RNA hybrids and with the anti‐nucleolin antibody to exclude any nucleolar signal 42. Experiments were repeated twice and all S9.6 nuclear intensity data analyzed excluding the nucleolar signal. We selected factors whose depletion led to a 10% higher S9.6 average nuclear intensity with respect to the negative control in both experiments as those to be considered further. For those siRNAs for which only one of the experiments was positive, the analysis was repeated up to 6 times to obtain a more reliable median value. With these criteria, we selected 16 candidate factors including four genes involved in the DNA damage checkpoint (DDC) (RAD1, RAD9A, TOPBP1, and MDC1), two post‐replicative repair (PRR) factors (UBE2B and RAD18), two nucleotide excision repair (NER) factors (GTF2H5 and DDB2), the tyrosyl‐DNA phosphodiesterase 2 (TDP2), the APEX1 gene of base excision repair (BER), the PMS1 gene of mismatch repair (MMR), the superoxide dismutase (SOD1) gene, and two Fanconi anemia (FA) genes, FANCD2 and FANCA (Fig 1A). In agreement with the reported role for FA in R loop resolution 25, 28, all FA siRNAs present in the library, except FANCB and FANCJ, conferred an S9.6 signal above the control (Fig 1A). Given that RAD18 is involved in FANCD2 ubiquitination in addition to PRR 43, 44, we assayed the possibility that the observed accumulation of hybrids in PRR‐deficient cells was due to a lack of FA function. We observed a significant decrease in the percentage of cells with FANCD2 foci when RAD18 was depleted (Fig EV1A). By contrast, siATM, siATR, and siUBE2B cells had no significant defect in FANCD2 foci formation. This is likely due to the redundant role of UBE2A, with which UBE2B shares 95% identity, both proteins being the homologs of the yeast PRR protein Rad6 45, since the double depletion of UBE2B and UBE2A is reported to cause a defect in FANCD2 ubiquitination 46. By contrast, depletion of UBE2B alone is reported to be impaired in PRR 47. Therefore, we support that the DNA–RNA hybrid accumulation phenotype observed in RAD18 and UBE2B‐depleted cells was caused by a defect in PRR rather than in FA.
Figure 1. Screening for DDR factors involved in R loop homeostasis.

- Top‐hit selected candidates are listed. The plot shows relative S9.6 nuclear intensity values for cells transformed with each of the indicated pool of siRNAs.
- Representative images of HeLa cells immunostained with S9.6 and nucleolin antibodies after transfection with at least a pool of two siRNAs of each original pool.
- Relative S9.6 signal intensity per nucleus after nucleolus signal removal in HeLa cells transfected with the indicated siRNAs. More than 250 total cells from three independent experiments were considered. The median of each population is shown. Boxes and whiskers indicate 25–75 and 10–90 percentiles, respectively. ***P < 0.001 (Mann–Whitney U‐test).
Figure EV1. Validation of the top‐hit candidates.
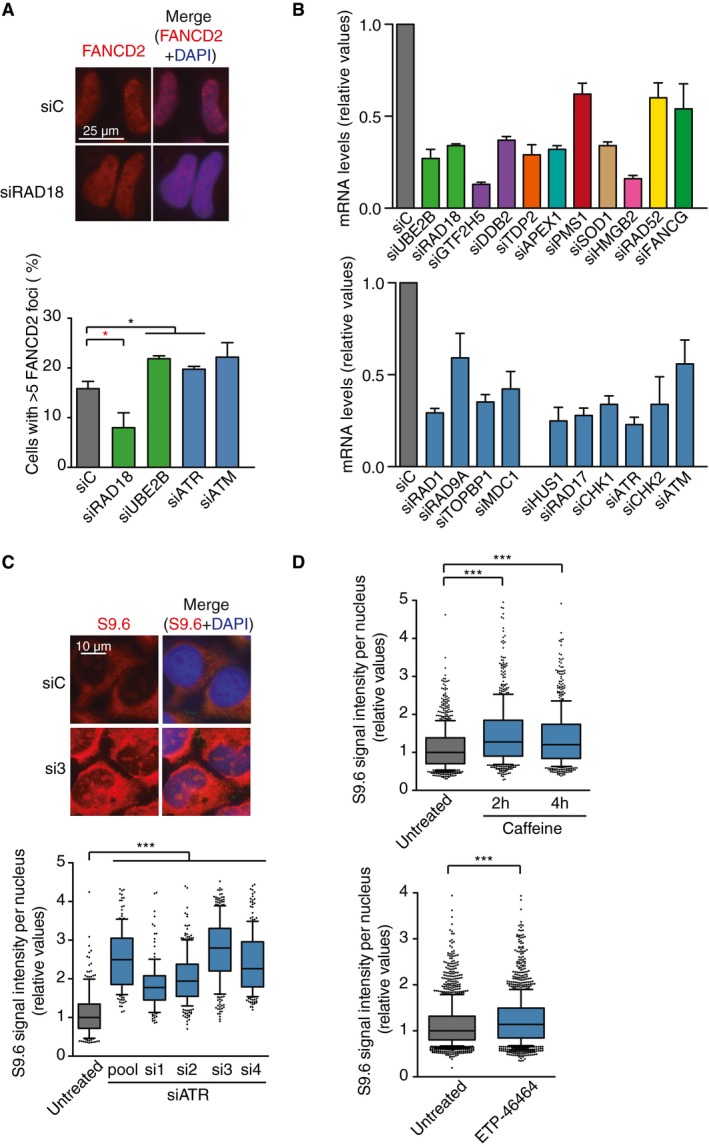
- FANCD2 foci in HeLa cells depleted of the selected candidates. The mean of three independent experiments with at least 100 cells each was considered. *P < 0.05 (Mann–Whitney U‐test). Red stars denote significant decreases. Representative images are shown on top.
- Relative mRNA levels of the indicated candidates as measured by RT–qPCR after siRNA depletion. Error bars represent relative target quantity (RQ) minimum and maximum from three technicals replicates.
- Relative S9.6 signal intensity per nucleus after nucleolus signal removal in HeLa cells transfected with the indicated siRNAs. More than 130 total cells from three independent experiments were considered. The median of each population is shown. Boxes and whiskers indicate 25–75 and 10–90 percentiles, respectively. ***P < 0.001 (Mann–Whitney U‐test).
- Relative S9.6 signal intensity per nucleus after nucleolus signal removal in HeLa cells transfected with the indicated siRNAs and treated with caffeine (10 mM, 2 or 4 h) or the ATR inhibitor ETP‐46464 (5 mM, 2 h). More than 600 total cells from five independent experiments were considered. The median of each population is shown. Boxes and whiskers indicate 25–75 and 10–90 percentiles, respectively. ***P < 0.001 (Mann–Whitney U‐test).
To validate the top‐hit candidates, we performed S9.6 IF in coverslips with the 14 selected candidates not involved in FA and with FANCG, as a representative of the FA pathway (Fig 1B and C). For this purpose, we used in each case a pool of two to four siRNAs from the original pool, which we previously validated to confer RNA silencing by RT–qPCR (Fig EV1B). As shown in Fig 1B and C, 6 out of the 14 candidates showed a significant increase in S9.6 nuclear intensity. The most representative functional group was the DNA damage checkpoint (DDC), with three genes: the RAD1 and RAD9A components of the 9‐1‐1 complex and the MDC1 mediator. Consequently, we extended our analysis to other relevant DDC genes such as the remaining 9‐1‐1 complex member HUS1, the clamp loader RAD17, and the main DDC kinases CHK1, ATR, CHK2, and ATM. Although these additional candidates, with the exception of siCHK2, had scored above the siC control in our screening (Fig 1A), a significant increase in S9.6 signal was only validated after depletion of CHK1, ATR, and CHK2. We next confirmed the effect of ATR depletion on S9.6 signal accumulation with four different siRNAs (Fig EV1C). Furthermore, treating cells with the phosphatidylinositol 3‐kinase‐like kinases (PIKK) inhibitors caffeine and ETP‐46464 had a similar effect (Fig EV1D), which indicates that the increase in DNA–RNA hybrids is unlikely due to siRNA off‐targets. We therefore decided to focus on DDC and PRR pathways as potentially involved in the protection against DNA–RNA hybrids.
DNA–RNA hybrids accumulate in DDC‐ and PRR‐deficient cells
To confirm further that the DDC‐ and PRR‐deficient cells accumulate DNA–RNA hybrids, we performed DNA–RNA immunoprecipitation (DRIP) with or without in vitro RNase H treatment, followed by qPCR at APOE and RPL13A genes, previously identified as R loop‐prone regions and used as model human genes for these studies 8, 25, 26, 35. The SNRPN gene was used as a negative control region at which low levels of detection correspond to background (Fig EV2A). As shown in Fig 2A, depletion of most of the DDC‐ and PRR‐selected genes, including both ATM/CHK2 and ATR/CHK1 branches, increased the DRIP signal in the RPL13A and APOE genes to similar levels than FANCD2‐depleted cells, which were used as positive control 25. Importantly, the DRIP signals were significantly reduced by RNase H treatment, implying that DNA–RNA hybrids do indeed accumulate in DDC‐ or PRR‐defective conditions. This is unlikely related to altered gene expression since, although slightly increased in siATM cells, the RNA levels of RPL13A were not significantly changed in siATR or siUBE2B cells (Fig EV2B). We next confirmed DNA–RNA hybrids at two other genes, MIB2 and RHOT2, when each of the three selected pathways was depleted (siATM, siATR, and siUBE2B, Fig 2A).
Figure EV2. Transcription dependency of the DNA–RNA hybrid accumulation and DNA breaks after DDR depletion.
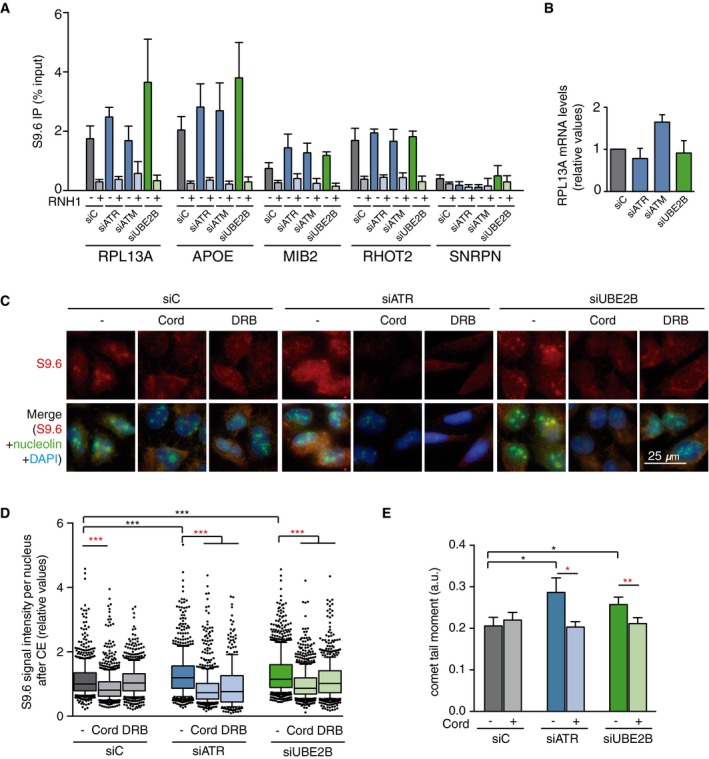
- DRIP–qPCR signal values at RPL13A, APOE, MIB2, RHOT2, and SNRPN genes in HeLa cells transfected with the indicated siRNAs and treated in vitro with RNase H pre‐immunoprecipitation where indicated. The mean ± SEM from at least three independent experiments is shown.
- Relative mRNA levels from the RPL13A gene in HeLa cells after transfection with the indicated siRNAs. The mean ± SEM from at least two independent experiments is shown.
- Representative images of HeLa cells immunostained with S9.6 and nucleolin antibodies after transfection with the indicated siRNAs and after cytoplasm pre‐extraction (CE).
- Relative S9.6 signal intensity per nucleus in HeLa cells transfected with the indicated siRNAs and treated with the transcription inhibitors 5,6‐dichloro‐1‐β‐D‐ribofuranosylbenzimidazole (DRB) or cordycepin (Cord). The median of the S9.6 signal intensity per nucleus relative to siC. Boxes and whiskers indicate 25–75 and 10–90 percentiles, respectively. More than 300 total cells from four independent experiments were considered. Values were normalized to the median of siC. ***P < 0.001 (Mann–Whitney U‐test). Black stars denote significant increases, whereas red stars denote significant decreases.
- Tail moment from single‐cell alkaline gel electrophoresis (comet assay) of HeLa cells transfected with the indicated siRNAs and treated with the transcription inhibitor cordycepin (Cord). More than 250 total cells were considered. The mean ± SEM of the median from five independent experiments is shown. *P < 0.05, **P < 0.01 (one‐tailed unpaired t‐test). Black stars denote significant increases, whereas red stars denote significant decreases.
Figure 2. R loop accumulation after depletion of the selected DNA Damage Checkpoint (DDC) and Post‐replicative Repair (PRR) candidates.
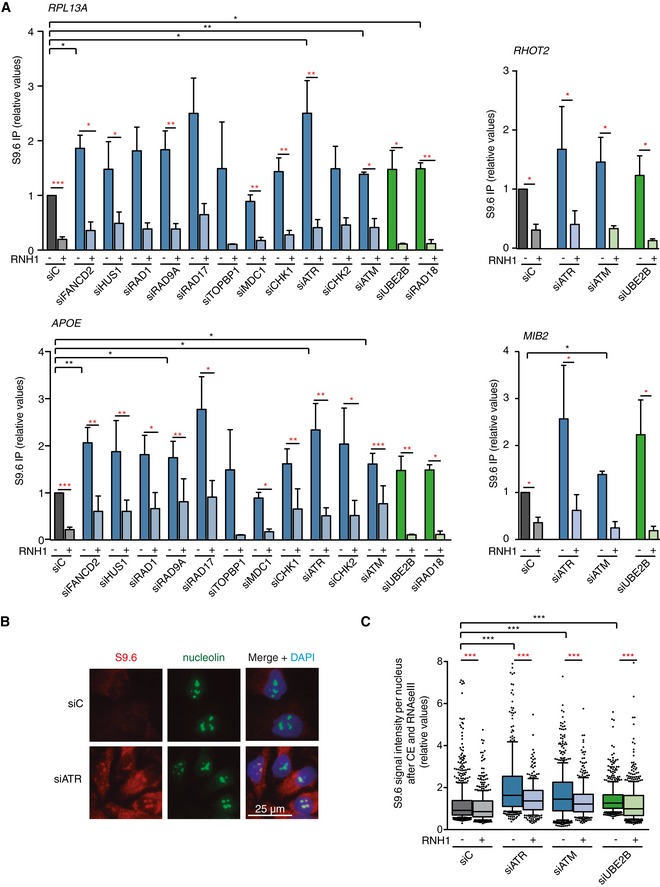
- Relative DRIP–qPCR signal values at RPL13A, APOE, MIB2, and RHOT2 genes in HeLa cells transfected with the indicated siRNAs and treated in vitro with RNase H pre‐immunoprecipitation where indicated. The mean ± SEM from at least three independent experiments is shown. *P < 0.05, **P < 0.01, ***P < 0.001 (one‐tailed paired t‐test).
- Representative images of immunostaining with S9.6 and anti‐nucleolin antibodies in HeLa cells transfected with the indicated siRNAs.
- Relative S9.6 signal intensity per nucleus after nucleolus signal removal in HeLa cells after cytoplasm pre‐extraction (CE) and treated in vitro with RNase III and RNase H where indicated. More than 500 total cells from three independent experiments were considered. The median of each population is shown. Boxes and whiskers indicate 25–75 and 10–90 percentiles, respectively. ***P < 0.001 (Mann–Whitney U‐test).
Interestingly, some depletions (such as siATM) increased the RNase H‐sensitive DRIP signal at some of the genes studied, but showed no effect in the S9.6 IFs used for validation of the screening results. However, despite the high reactivity with DNA–RNA hybrids, the S9.6 antibody also recognizes dsRNA 48, 49. Whereas DRIP is a highly specific method of detection of DNA–RNA hybrids, given that putative dsRNA molecules are not amplified by qPCR and signals are considered positive when sensitive to in vitro treatment with RNase H, which only removes RNA–DNA hybrids 50, dsRNA could be masking our initial validation by IF. Consequently, we repeated the IF analysis after in vitro treatment with RNase III, which degrades dsRNA and after pre‐extraction of the cytoplasm, to avoid any cytoplasmic interference 50. As shown in Fig 2B and C, we observed an increased S9.6 signal when each of the three selected DDR pathways was inactivated (siATR, siATM, and siUBE2B). These signals were sensitive to RNase H (Fig 2C), further confirming that they correspond to DNA–RNA hybrids. To assess whether these hybrids were transcription‐dependent, we performed S9.6 IF in cytoplasm pre‐extracted cells treated with the transcription inhibitors 5,6‐dichloro‐1‐β‐D‐ribofuranosylbenzimidazole (DRB) and cordycepin (Cord). As shown in Fig EV2C and D, both compounds significantly reduced the S9.6 signal observed after depletion of ATR or UBE2B. Since the 9‐1‐1/ATR/CHK1 DDC branch responds to RPA‐coated single‐stranded DNA (ssDNA) such as that generated by stalled forks, the PRR machinery acts after replication, and ATM is activated by DSBs 39, 40, these results suggest that DNA–RNA hybrids are a common source for both replication stress and DSB‐mediated cellular responses. We conclude that DNA–RNA hybrids accumulate in the absence of a proper response to either replication stress or DSBs.
DNA–RNA hybrids are a source of DNA breaks in specific DDR‐deficient cells
Next, we reasoned that if DNA–RNA hybrids are a frequent source of DNA damage, part of the spontaneous damage potentially accumulated in these DDC‐ and PRR‐defective cells should be suppressed by RNase H overexpression. To directly analyze DNA damage, we performed single‐cell alkaline gel electrophoresis (comet assay) in cells depleted of the DDR‐selected factors with or without overexpression of RNase H (Fig 3A). The comet tail moment (a measurement of DNA breaks) in cells depleted of all the DDC and PRR factors tested increased with respect to cells transfected with a non‐targeting siC. This damage was likely a consequence of both spontaneous metabolism and the stress induced by the double transfection since single siATR‐ and siUBE2B‐transfected cells showed a much modest increase (Fig EV2E). Importantly, this damage was partially dependent on transcription as it was reduced by cordycepin treatment (Fig EV2E). Furthermore, although RNase H overexpression caused DNA damage by itself in siC‐treated cells, in agreement with previous reports 11, it caused no further damage in ATR‐depleted cells. By contrast, RNase H overexpression caused a slight decrease in the occurrence of DNA breaks in cells depleted of specific factors of the 9‐1‐1/ATR/CHK1 pathway, such as siRAD9A‐, siRAD17‐, and siCHK1‐treated cells (Fig 3B), even though they did not show statistical significance. These results suggest that, whereas the DNA breaks accumulated in the absence of the ATM/CHK2 pathway arise independently on DNA–RNA hybrids, part of the DNA breaks observed in the absence of the 9‐1‐1/ATR/CHK1 pathway are due to DNA–RNA hybrids. Notably, the tail moment increase observed in siUBE2B and siRAD18 cells was significantly reduced by RNase H overexpression, implying that a defect in the PRR machinery causes DNA damage due to the formation of DNA–RNA hybrids.
Figure 3. DNA–RNA hybrids are a source of DNA breaks in DDR‐deficient cells.

- Representative images of single‐cell alkaline gel electrophoresis (comet assay) of HeLa cells transfected with the indicated siRNAs and either pEGFP‐C1 (RNH1−) or pEGFP‐M27 (RNH1+).
- Comet tail moment from single‐cell alkaline gel electrophoresis (comet assay) of HeLa cells transfected with the indicated siRNAs and either pEGFP‐C1 (RNH1−) or pEGFP‐M27 (RNH1+). More than 250 total cells were considered. The mean ± SEM of the median from at least three independent experiments is shown, except for RAD1, RAD17, and UBE2B (n = 2). *P < 0.05, **P < 0.01, ***P < 0.001 (one‐tailed unpaired t‐test). Black stars denote significant increases, whereas red stars denote significant decreases.
DDR inactivation causes R loop‐dependent H3S10‐P and H3K9me2
Given the emerging connection between DNA–RNA hybrids and specific chromatin marks such as H3S10‐P and H3K9me2 31, 32, 33, 34 and the recent observation in yeast that certain chromatin alterations are required for R loop‐driven genetic instability 31, we determined by IF whether DNA–RNA hybrids accumulated in DDR‐deficient cells were also accompanied by these chromatin marks. As can be seen in Fig 4A, the proportion of cells with elevated H3S10‐P foci significantly increased after depletion of most of the factors analyzed. Importantly, such an increase was reversed by RNase H overexpression in at least five of them (siRAD9A, siRAD17, siCHK2, siATM, and siUBE2B), indicating a tight link between DNA–RNA hybrids and H3S10‐P. An increase in H3K9me2 nuclear intensity that was significantly reduced by RNH1 overexpression was also observed in cells depleted of the three selected DDR pathways (siATR, siATM, and siUBE2B) (Fig 4B). Importantly, no major changes were observed in the cell cycle distribution in cells depleted of the selected DDR factors (Fig EV3A). These results indicate that the DNA–RNA hybrid accumulation observed is linked to chromatin alterations.
Figure 4. DDR deficiencies lead to DNA–RNA hybrid‐dependent accumulation of histone H3S10‐P and H3K9me2 chromatin marks.
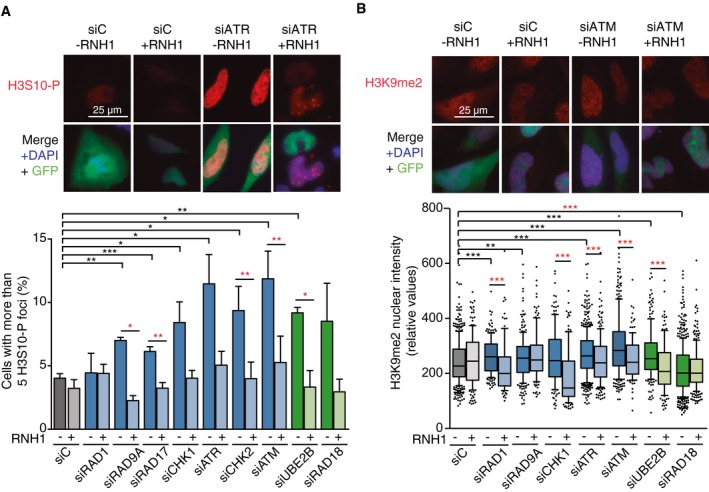
- Representative images and percentage of HeLa cells with more than 5 H3S10‐P foci after transfection with the indicated siRNAs and either pEGFP‐C1 (RNH1−) or pEGFP‐M27 (RNH1+). Mitotic cells were excluded for the analysis by DAPI staining. More than 300 total cells were considered. Data represent mean ± SEM from three independent experiments.
- Representative images and H3K9me2 nuclear signal intensity of HeLa cells transfected with the indicated siRNAs and either pEGFP‐C1 (RNH1−) or pEGFP‐M27 (RNH1+). At least three experiments were performed. The median of each population in a representative experiment with at least 100 cells per condition is shown. Boxes and whiskers indicate 25–75 and 10–90 percentiles, respectively.
Figure EV3. Representative cell cycle profiles of HeLa cells depleted of the selected DDC and PRR factors.
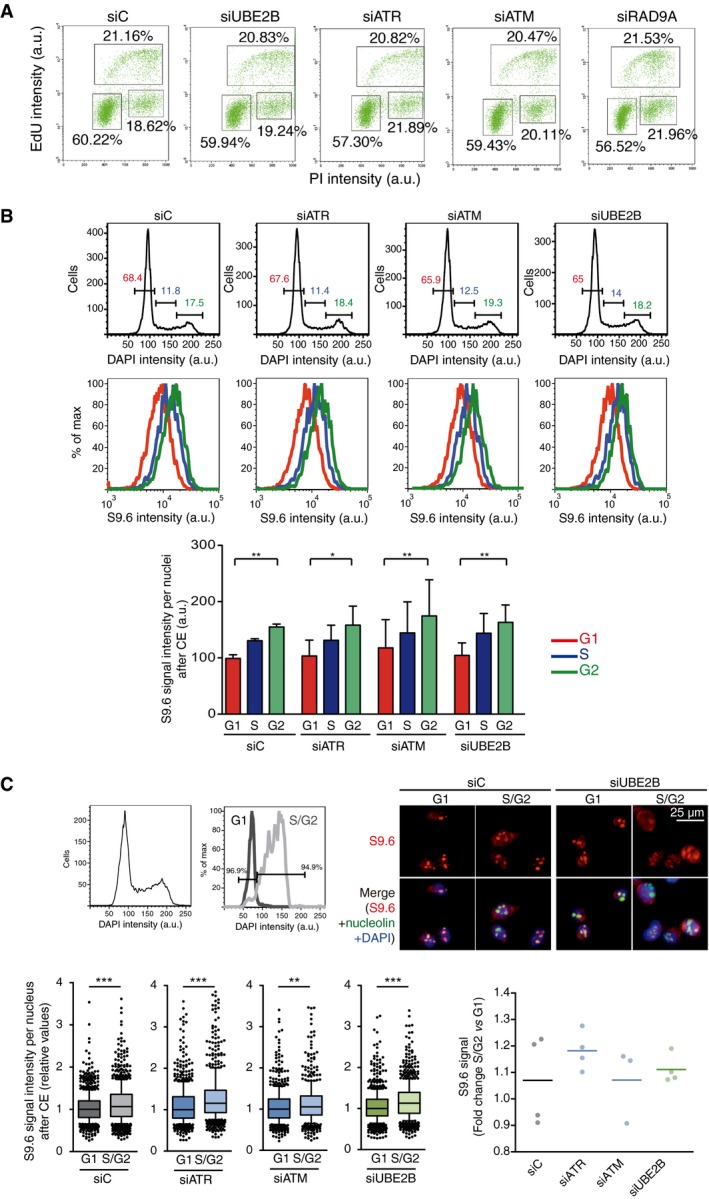
- Flow cytometry profiles showing EdU incorporation versus DNA content in the indicated HeLa cells. The percentage of cells in S (upper box), G0/1 (lower left box), and G2/M (lower right box) are indicated.
- Top row: flow cytometry histograms displaying the DNA content of the indicated HeLa cells after cytoplasm pre‐extraction (CE). G1 (red), S (blue), and G2 (green) phases are calculated from the profile. Bottom row: flow cytometry histograms depicting intensity of S9.6 signal in each phase of the cell cycle. Quantification is shown in panel below representing the mean ± SD of four experiments. *P < 0.05, **P < 0.01 (ANOVA test with Bonferroni's post‐test).
- Top left panel: flow cytometry histogram showing the DNA content of cells before (left) and after (right) sorting by DAPI signal to obtain samples enriched in G1 and S/G2 populations. A representative experiment of control cells is shown. Top right panel: representative images of S9.6 staining of G1 and S/G2 sorted fractions in control and UBE2B‐depleted cells after cytoplasm pre‐extraction (CE). Bottom left panel: relative S9.6 intensity in G1 and S/G2‐sorted cells. Median values are indicated. Boxes and whiskers indicate 25–75 and 10–90 percentiles, respectively. At least three experiments, each with more than 150 cells per condition, were considered. Bottom right panel: fold change of S9.6 signal median values in S/G2 with respect to G1 sorted cells. **P < 0.01, ***P < 0.001 (Mann–Whitney U‐test).
Differential effects on replication fork progression upon depletion of DDR factors
Compelling evidence supports that R loop accumulation perturbs DNA replication and that this is a major cause of R loop‐induced genetic instability 2, 51, 52, 53. Thus, we analyzed replication fork dynamics by DNA combing in the selected DDC‐ and PRR‐deficient cells. In agreement with the reported role for ATR/CHK1 in fork progression 54, a decrease in fork velocity and track length was observed after depletion of factors of the ATR/CHK1 pathway but not after depletion of ATM/CHK2 or the PRR factors (Figs 5A and EV4). However, this decrease was not suppressed by RNase H overexpression. Therefore, we analyzed fork asymmetry as a measurement for R loop‐mediated replication stalling. As shown in Fig 5B, we observed a significant increase in fork asymmetry after depletion of RAD1, RAD9A, RAD17, and CHK1. Although not significant, a similar tendency was observed after ATR depletion in agreement with increased fork stalling in the absence of a proper ATR/CHK1 checkpoint response. This increase was lost after RNase H overexpression in siRAD1‐, siRAD9A‐, and siRAD17‐treated cells, supporting that although DNA–RNA hybrids are obstacles to replication fork progression, they are not the only kind of spontaneously occurring obstacles, consistent with our actual knowledge 55. By contrast, the fork asymmetry observed after depletion of ATM, CHK2, or the PRR factors RAD18 or UBE2B was similar to that of the control cells, which implies an important difference between the impact on replication of the DNA–RNA hybrids accumulated by depletion of the two groups of DDR factors, ATR/CHK1 and 9‐1‐1, on the one hand and ATM/CHK2 and PRR on the other. Our results suggest that DNA–RNA hybrids formed before replication in the absence of DDC factors ATR/CHK1 and the 9‐1‐1 complex, thus potentially causing replication fork stalling. However, DNA–RNA hybridization might also be promoted by unrepaired DSBs (siATM and siCHK2 cells), in which case no additional effect is expected regarding replication fork stalling, or at post‐replicative ssDNA gaps (those accumulated in siUBE2B and siRAD18) after replication fork passage.
Figure 5. Differential effects on replication fork progression upon depletion of DDR factors.
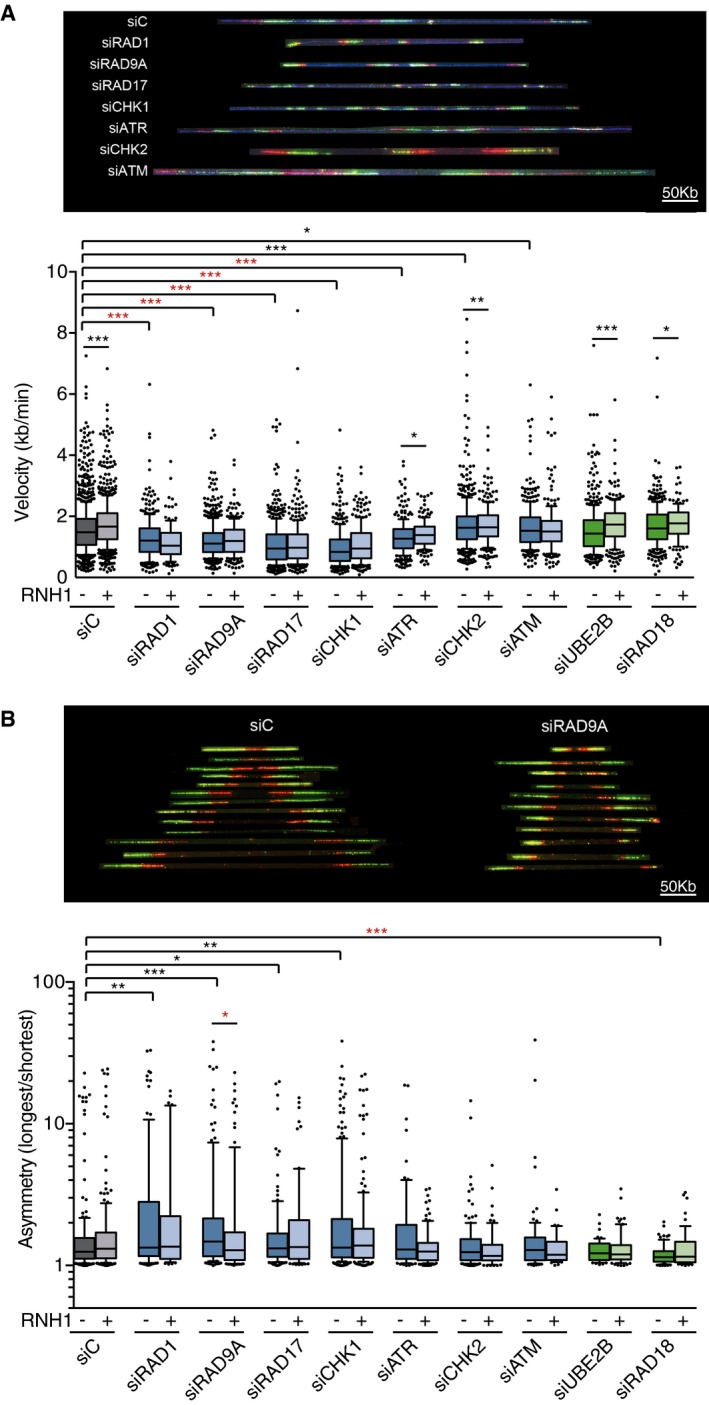
- Fork velocity as measured by DNA combing assay in HeLa cells transfected with the indicated siRNAs and either pEGFP‐C1 (RNH1−) or pEGFP‐M27 (RNH1+). More than 200 tracks were considered except for RAD1 + RNH1 (n = 125), ATR +RNH1 (n = 182), and RAD18 + RNH1 (n = 146).
- Fork asymmetry as measured by DNA combing assay in HeLa cells transfected with the indicated siRNAs and either pEGFP‐C1 (RNH1−) or pEGFP‐M27 (RNH1+). From 40 to 200 total measurements were considered for each candidate.
Figure EV4. Replication fork progression in HeLa cells depleted of DDC candidates.
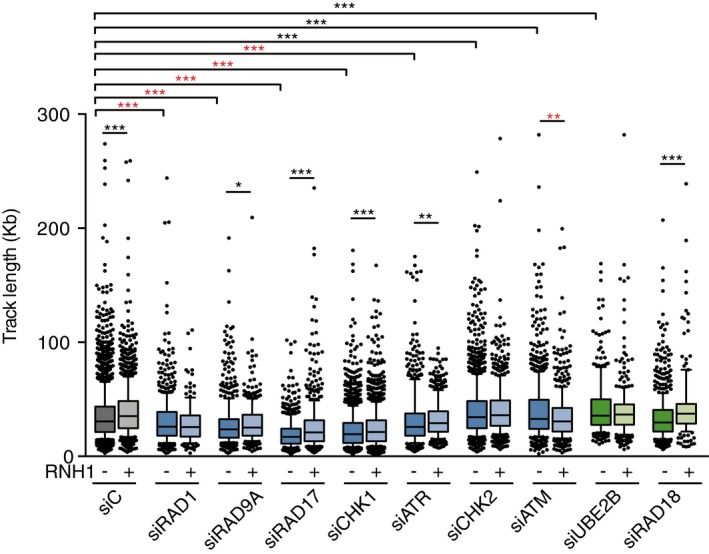
Track length as measured by DNA combing assay in HeLa cells transfected with the indicated siRNAs and either pEGFP‐C1 (RNH1−) or pEGFP‐M27 (RNH1+). More than 300 tracks were considered except for siRAD1 + RNH1 (156) and RAD18 + RNH1 (179). Median values are indicated. Boxes and whiskers indicate 25–75 and 10–90 percentiles, respectively. *P < 0.05, **P < 0.01, ***P < 0.001 (Mann–Whitney U‐test). Black stars denote significant increases, whereas red stars denote significant decreases.
During replication, spontaneous DNA obstacles including DNA–RNA hybrids should be frequently encountered by replisomes, potentially causing fork stalling or even breakage. In principle, if DNA–RNA hybrids are stimulated by breaks 56 they could be enriched at such broken forks. However, fork stalling can also lead to post‐replicative DNA gaps, which are potential substrates for RNA hybridization. Therefore, we hypothesized that DNA–RNA hybrids might be differently formed along the cell cycle. To test this hypothesis, we analyzed DNA–RNA hybrid accumulation in whole cells in the different cell cycle phases. We measured the S9.6 IF intensity of whole cells treated with RNase III by flow cytometry and observed a significant increase from G1 to S and from S to G2 phases in all samples (siC‐, siATR‐, siATM‐, and siUBE2B‐treated cells) (Fig 6A and B). This tendency was more noticeable when DDC or PRR pathways were compromised (Fig 6C). Similar results were obtained with cytoplasm pre‐extracted cells (Fig EV3B) as well as after sorting these pre‐extracted cells in two populations (before and after replication) and subjecting them to S9.6 IF (Fig EV3C). These results agree with spontaneous DNA–RNA hybrids being stimulated after DNA replication, thus creating a specific target for the ATR and PRR‐mediated DDR response, and are consistent with the idea that ssDNA gaps could efficiently hybridize with RNA.
Figure 6. DNA–RNA hybrid accumulation in different phases of the cell cycle.
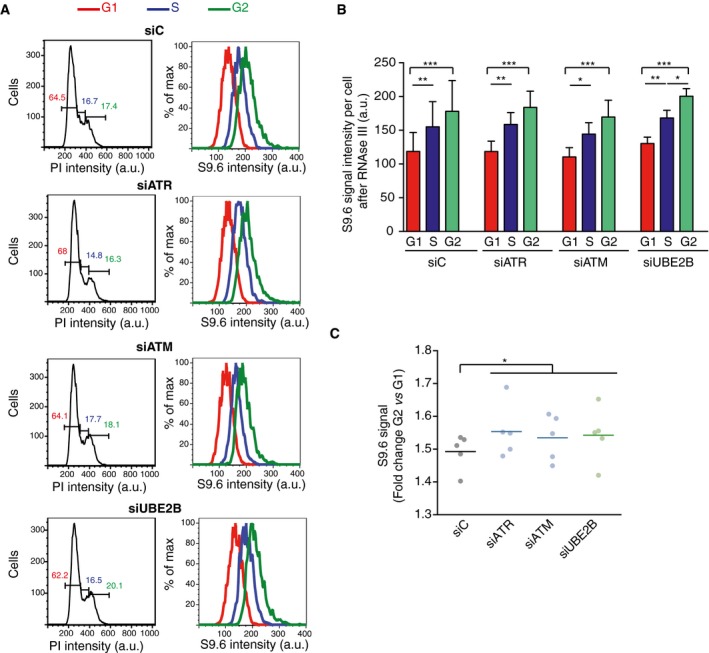
- Left column: flow cytometry profiles showing DNA content of the indicated RNase III‐treated cells. G1 (red), S (blue), and G2 (green) phases were calculated from the profile. Right column: flow cytometry histograms depicting intensity of S9.6 signals in each phase of the cell cycle for the indicated cells.
- Quantification of panel (A). The mean ± SD of the S9.6 mean intensity of five experiments is shown. ***P < 0.001, **P < 0.01, *P < 0.05 (repeated measures ANOVA test with Bonferroni's post‐test).
- Graph represents fold change of S9.6 mean signal in G2 with respect to G1 cells. Values for five independent experiments together with the mean are shown. *P < 0.05 (one‐tailed paired t‐test).
Discussion
By screening an siRNA library targeting DDR genes, we have uncovered three DDR pathways that are important for the prevention of DNA–RNA hybrid accumulation in human cells. These are, the 9‐1‐1/ATR/CHK1 pathway, known to respond to ssDNA accumulation; the ATM/CHK2 pathway, which responds to DSBs; and the PRR pathway, which triggers the filling of ssDNA gaps left in the daughter strands after replication bypass of damaged DNA. Despite its mixed specificity for DNA–RNA hybrids 48, the S9.6 antibody has enabled us to detect new functions in the DDR whose depletion leads to DNA–RNA hybrid accumulation and genetic instability by focusing on the top candidates of our screening (Fig 1). Importantly, we have reproducibly seen that the depletion of FA genes leads to an increase in S9.6 signal, in agreement with previous reports 25, 28.
The fact that 9‐1‐1/ATR/CHK1 depletion led to DNA–RNA hybrid accumulation and that RNase H partially reduced the damage observed (Fig 7A) indicates that DNA–RNA hybrids are a frequent source of replication‐associated DNA damage, likely through transcription–replication conflicts 57. Indeed, DNA–RNA hybrids have been shown to accumulate at human common fragile sites (CFSs) 34, 58, 59, which suggest that they can also promote fragility at these sites. Our results suggest that persistent DNA–RNA hybrids themselves or some derivative intermediates could be sensed during replication. In agreement, R loop‐accumulating yeast cells have been shown to activate the S‐phase checkpoint and to require it for survival 60 and head‐on transcription–replication collisions enhanced by R loops have been recently shown to activate ATR in human cells 21. Although R loop‐driven replication fork stalling is likely the reason behind ATR activation, the displaced ssDNA of R loops in principle could also promote local ATR activation as it has been reported at centromeres in mitotic cells 61. Together with our recent data 11, our results support that DNA–RNA hybrids not necessarily impact replication fork velocity. Indeed, so far, both faster and slower forks have been detected in conditions of transcription–replication conflicts. Thus, slower fork speed was detected in cells depleted for FACT 26, histone H1 62, 63, ASF/SF2, and TOP1 23, whereas faster fork speed was reported in SIN3A‐ and THOC1‐depleted cells 7, 11. Instead, obstacles in the DNA should cause replication fork stalling. Indeed, fork asymmetry has been reported in all cases of cells accumulating R loops and analyzed, regardless of whether forks move faster (such as THOC1‐ or SIN3‐depleted cells) or slower (such as in histone H1‐, ASF/SF2‐, or TOP1‐depleted cells). In support of the conclusion that R loops are a frequent source of spontaneous fork stalling, we observed RNase H‐sensitive fork stalling after 9‐1‐1/ATR/CHK1 depletion (Fig 7A).
Figure 7. Integrative view and model showing how the lack of DNA Damage Checkpoint and Post‐replicative Repair factors can affect DNA–RNA hybrid homeostasis.
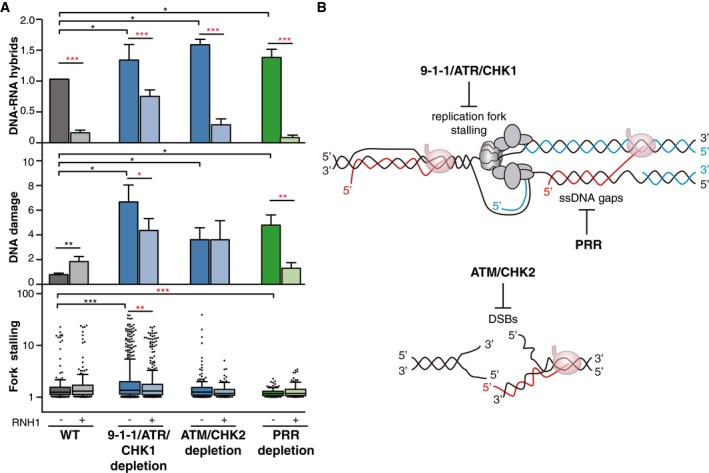
- DNA–RNA hybrids, DNA damage, and fork stalling in 9‐1‐1/ATR/CHK1, ATM/CHK2, and PRR‐deficient cells. DNA–RNA hybrids represent the mean ± SEM of all the DRIP data from Fig 2A. ***P < 0.001, *P < 0.05 (paired t‐test). DNA damage represents the mean of medians ± SEM of all the comet tail moment data from Fig 3B. **P < 0.01, *P < 0.05 (paired t‐test). Fork stalling represents the median, 25–75 (boxes) and 10–90 percentiles (whiskers) of all the fork asymmetry data from Fig 5B. ***P < 0.001, **P < 0.01 (Mann–Whitney U‐test). Black stars denote significant increases, whereas red stars denote significant decreases.
- A model to show that spontaneous DNA–RNA hybrids impairing replication fork progression would require the 9‐1‐1/ATR/CHK1 for dissolution. Additionally, unrepaired DSBs (accumulated as a consequence of ATM/CHK2 depletion) and post‐replicative ssDNA gaps (present in PRR‐defective cells) could favor DNA–RNA hybrid accumulation without stalling replication forks.
ATM/CHK2 might also have a role in R loop resolution. However, the accumulation of DNA–RNA hybrids observed in ATM/CHK2‐depleted cells does not seem to be a major problem for replication fork progression, as we were not able to detect any increase in fork asymmetry (Fig 7A). Also, the fact that RNase H overexpression had no effect on the number of breaks induced by ATM/CHK2 depletion (Fig 7A) suggests that most breaks occurring in the absence of ATM/CHK2 are independent on DNA–RNA hybrids. Given the views and recent observations supporting that DNA breakage, whether single‐ or double‐stranded, is a driving force for DNA–RNA hybrid formation 15, 56, 64, 65, 66, the accumulation of DNA–RNA hybrids in ATM/CHK2‐depleted cells might rather be a consequence of such unrepaired DSBs, which would not imply any additional consequences in fork progression.
More strikingly than in ATM‐ and ATR‐depleted cells, we observed that all the DNA breaks occurring after depletion of PRR were reduced by RNase H overexpression (Fig 7A). This result indicates that most of the DNA damage observed in the absence of these factors is due to DNA–RNA hybrids. However, they did not cause any increase in fork stalling, suggesting that hybrids might also form after replication fork passage. This study certainly opens a new view on R loop formation (Fig 7B). Whereas DNA–RNA hybrids are formed in any cell cycle phase regardless of replication, but causing instability as a consequence of their posterior impact on replication fork progression 8, 11, 19, 20, 21, 22, 23, 24, 52, they may also form after the passage of the replication fork. Supporting the de novo formation of DNA–RNA hybrids, we detected an increased S9.6 signal in cells in S‐G2 (Fig 6). Although it could be argued that gene duplication could double the amount of transcripts, this is known not to be the case due to the gene dosage balance 67. Furthermore, given that most transcription takes place in G1 and that DNA–RNA hybrid formation is likely a very infrequent stochastic event, genome duplication by itself is not expected to lead to any increase in DNA–RNA hybrids. Consequently, we interpret that the S9.6 enrichment detected is caused by the formation of DNA–RNA hybrids during or after replication. Importantly, these DNA–RNA hybrids also lead to genetic instability, but this instability would be replication‐independent.
In this context, we believe that the PRR pathway would have a key role in preventing post‐replicative R loop formation and their harmful impact on genome integrity. Bacterial cells are known to re‐prime DNA synthesis after persistent blocks, leaving daughter‐strand gaps behind the fork 68. Although the mechanism of PRR in human cells is largely unknown, some evidence supports that forks can also restart in eukaryotes 69, 70 and mutants in yeast RAD6 and RAD18 (orthologs of human UBE2B and RAD18) are defective in gap filling 71, 72, 73. Similarly, the absence of the human PRR machinery could lead to the accumulation of unrepaired post‐replicative ssDNA gaps that would be prone to RNA hybridization. We thus propose that PRR is required to prevent the formation of DNA–RNA hybrids at this ssDNA gaps generated after the replication fork (Fig 7B). These DNA–RNA hybrids would not cause fork stalling, since they are not encountered by new forks, but they are probably stabilized in the absence of the PRR machinery.
Finally, we have recently observed that RNA–DNA hybrids that cause genome instability are accompanied or require additional events to become harmful, a key one being the acquisition of histone modifications, such as histone H3S10‐P and H3K9me2 associated with chromatin condensation and heterochromatin 31, 32. To test the validity of this hypothesis, we have determined the overall levels of these chromatin marks in cell depleted of the three DDR pathways and we found that, regardless of having or not an effect in fork asymmetry, ATR/CHK1, ATM/CHK2, and PRR depletion tend to accumulate such chromatin compaction marks (Fig 4). This supports our model by which DNA–RNA hybrids trigger chromatin alterations that would potentially be responsible for the genome instability observed in those cases.
In summary, our study revealing the important role of DDR in preventing DNA–RNA hybrid accumulation and genome instability suggests that hybrids can be formed spontaneously throughout the cell cycle and are a source of spontaneous DNA damage. Our results suggest that DNA–RNA hybrids have different consequences depending on whether they form before replication thus impairing replication fork progression, at putative sites of spontaneous DNA breaks thus putatively interfering with the repair of such breaks, or post‐replicatively, likely facilitated by transient formation of ssDNA gaps. In contrast to cells depleted of mRNP biogenesis factors such as THO or of histone deacetylases 11, DDR depletion would not necessarily induce formation of hybrids. We interpret that the accumulation of hybrids in DDC‐depleted cells, as in cells depleted of FACT, BRCA2, or Fanconi anemia proteins 8, 25, 26, 27, 28, would respond to inefficient repair of the intermediate containing the hybrid, such as a replication fork block or a DSB, which leads to the accumulation of such intermediate together with the hybrid, as we have previously discussed 56. The association with chromatin compaction marks of such hybrids reinforces the idea that the hybrid, regardless of its different spontaneous origin, leads to genetic instability associated with chromatin alterations, and opens new perspectives on the mechanisms and biological meaning of such association that would need to be explored in the future.
Materials and Methods
Cell cultures and transfection
HeLa cells used in this study were obtained from ATCC and maintained in DMEM (Gibco) supplemented with 10% heat‐inactivated fetal bovine serum at 37°C (5% CO2). Every 6 months, they were tested for mycoplasma contamination. Transient transfection of plasmid (2 μg) or siRNA (100 nM) was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) or Dharmafect, respectively, according to the manufacturer's instructions. ON‐TARGET SMARTpool of siRNA from Thermo Scientific was used for all depletions.
Immunofluorescence staining
S9.6 (hybridoma cell line HB‐8730) and nucleolin (ab50279; Abcam) immunofluorescence (IF) was performed as previously described 42 72 h after siRNA transfection. The S9.6 signal in the nucleoli was subtracted from the integrated nuclear S9.6 signal to perform the analysis. S9.6 IF in cells treated with the transcription inhibitors 5,6‐dichloro‐1‐β‐D‐ribofuranosylbenzimidazole (DRB) and cordycepin (Cord) was performed after a 4‐h treatment with 100 μM DRB or 50 μM Cord and cells were treated with a pre‐extraction solution (0.5% Triton X‐100, 20 mM HEPES‐KOH, 50 mM NaCl, 3 mM MgCl2, and 300 mM sucrose) before fixation. Treatment with RNase III and RNase H1 was performed before blocking in pre‐extracted cells, incubating each coverslip with 1.2 U of RNase III and/or 9 U of RNase H1, for 30 min at 37°C. If both treatments are needed, they are performed consecutively.
For fluorescence quantification analysis of the H3S10‐P and H3K9me2 signal, 72 h after siRNA transfection cells were fixed and permeabilized with 2% formaldehyde in PBS, 70% ethanol −20°C, and 70% ethanol 4°C. Cells were washed 2 × 3% BSA in PBS and incubated in 0.5% Triton X‐100 in PBS for 20 min at room temperature. After washing and blocking with 3% BSA in PBS, cells were incubated with anti‐H3S10P (06‐570; Merck) or anti‐H3K9me2 (07‐212; Millipore) for 1 h at room temperature.
Secondary antibodies conjugated with Alexa Fluor 594, Alexa Fluor 488, and Alexa Fluor 568 were used. Nuclei were counterstained with DAPI. Images were acquired with a Leica DM6000 microscope equipped with a DFC390 camera (Leica) at x63 magnification. Metamorph v7.5.1.0 software (Molecular Probes) was used to quantify foci or signal intensity. When taking the images at the microscope, all the fields were randomly chosen in DAPI staining and the quantitation was automatized, so that the investigator could not be biased.
Replication analysis by DNA combing
DNA combing was performed as described 74, except that both iododeoxyuridine (IdU) and chlorodeoxyuridine (CldU) labels were added for 20 min each. Anti‐ssDNA from Developmental Studies Hybridoma Bank (DSHB) was used instead of the one described. Track length was calculated measuring all green tracks. Fork asymmetry was calculated by dividing the longest green track by the shortest in divergent CldU tracks. Fork velocity was calculated as reported 75.
DNA–RNA immunoprecipitation (DRIP)
DRIP assay was performed as described in HeLa cells 72 h after siRNA transfection 42. Briefly, DNA–RNA hybrids were immunoprecipitated using the S9.6 antibody from gently extracted and enzymatically digested DNA, treated or not with RNase H. Quantitative PCR was performed at the indicated regions of RPL13A, APOE, MIB2, RHOT2, and SNRPN genes with the corresponding primers listed in Table EV1. Means and SEM from at least three independent experiments were calculated.
Quantitative PCR analysis
For real‐time (RT)–qPCR analysis, cDNA was synthesized using QuantiTect Reverse Transcription Kit (Qiagen). mRNA expression values of the indicated genes were normalized with mRNA expression of the HPRT housekeeping gene. RT–qPCR was performed with iTaq Universal SYBR Green Supermix (Bio‐Rad) and analyzed on 7500 FAST Real‐Time PCR system (Applied Biosystems, Carlsbad, CA). Primers are listed in Table EV1.
Single‐cell electrophoresis
Single‐cell alkaline gel electrophoresis was performed with CometAssay Kit (Trevigen) following manufacturer's instructions. Images were acquired with a Leica DM6000 microscope equipped with a DFC390 camera (Leica). Means and SEM of the medians from at least three independent experiments were obtained and are shown in each case. Comet tail moments were analyzed using Comet‐score software (version 1.5). More than 100 cells from each experiment were scored. Experiments in which the median of the tail moment was higher than 30 units were discarded as outliers.
EdU incorporation
Cells were pulse‐labeled 20 min with EdU 10 μM added directly to the growing medium and stained with a Click‐iT EdU Alexa Fluor 488 Imaging Kit (Invitrogen) according to manufacturer's instructions. After three washes with 1% BSA and 0.1% Triton X‐100 in PBS, DNA was counterstained with 7AAD (51‐68981, BD), diluted 1:50, and treated with 0.5 μg/μl of RNase A in PBS for 30 min. Cells were examined by flow cytometry (FACSCalibur, BD).
Flow cytometry
Cells were harvested and fixed with methanol 100% at −20°C for 20 min, washed with PBS, and incubated with RNAse III (40 U/ml) for 30 min at 37°C. Samples were then washed with PBS and immunostained with S9.6 antibody as previously described 42 in cells in suspension. Cells were then stained with 50 μg/ml PI (Invitrogen) overnight at 4°C and acquired in BD FACSCalibur cell analyzer (BD). Data were analyzed in FlowJo 9.3.2 (Tree Star). Data from S9.6 and PI signals were compensated to avoid bleed‐through.
Cell sorting
Cells were harvested and incubated in pre‐extraction buffer (0.5% Triton X‐100, 20 mM HEPES‐KOH pH 7.9, 50 mM NaCl, 300 mM sucrose, and PBS) for 5 min at 4°C and then fixed with 4% formaldehyde for 10 min at room temperature. S9.6 and anti‐nucleolin (Abcam) staining was performed as previously described 42 in cells in suspension. Samples were then stained with 1 μg/ml DAPI at 4°C overnight. G1 and S/G2 cells were sorted in a BD influx sorter, recovered in PBS, cytocentrifuged in a Cytospin 4 (Thermo Scientific), and mounted with ProLong Gold antifade reagent.
Statistical methods
To estimate sample size when means or medians were calculated, the following formula was used: n = [(Z*S)/E]2, where Z is z‐score for 95% of confidence; E is the margin error (5%); and S is standard deviation. To estimate sample size when proportions were calculated, the following formula was used: n = [Z2*p*(1‐p)]/E2, where p is the expected proportion. Variations among biological replicas are expected to have normal distributions and equal variances. One‐tailed Student's t‐tests were applied for comparisons of two independent groups when the results were hypothesized “a priori”. Paired tests were used to minimize the effect of variation among replicas when indicated. In comet assay and H3S10‐P or FANCD2 foci analysis, the means of medians were compared and t‐test was applied. For S9.6 and H3K9me2 IF analysis, differences between samples variances were calculated with F‐test and distribution of intensities was revealed as not Gaussian in siC‐treated cells, with KS normality test. Statistical significant differences between samples were assessed with non‐parametric Mann–Whitney U‐tests.
In DRIP assays, the percentage of inputs were normalized to the siC value in each replica. Means of normalized values were represented. One‐tailed paired Student's t‐tests were used to ensure statistical significant differences.
For DNA combing assays, the sample size was determined following the recommendations of 76. KS normality test was used to ensure that the data distribution of velocity, track length, and asymmetry was not Gaussian. Statistical significant differences were assessed with non‐parametric Mann–Whitney U‐tests.
In flow cytometry assays, at least 10,000 cells were acquired as usually done for cell cycle analysis 77. For comparisons between multiple groups, repeated measures ANOVA test and Bonferroni's post‐test were applied. Repeated measurement test were used to minimize differences between replicas.
Statistical analyses were performed in Prism v4.0 (GraphPad Software). The specific analysis used in each experiment is indicated in the corresponding Figure legend.
Author contributions
SB, EH‐M, SM, and MG‐R conducted the experiments; SB, BG‐G, and AA designed the experiments; and BG‐G and AA wrote the manuscript. All authors read, discussed, and agreed with the final version of this manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
We would like to thank R. Luna for her help selecting the library of siRNAs to screen. Research was funded by the European Research Council (ERC2014 AdG669898 TARLOOP) and the Worldwide Cancer Research (WCR15‐00098). S.B. was supported by the WCR and B.G‐G by a post‐doctoral grant from the Scientific Foundation of the Spanish Association Against Cancer (AECC).
EMBO Reports (2019) 20: e47250
References
- 1. Aguilera A, Garcia‐Muse T (2012) R loops: from transcription byproducts to threats to genome stability. Mol Cell 46: 115–124 [DOI] [PubMed] [Google Scholar]
- 2. Santos‐Pereira JM, Aguilera A (2015) R loops: new modulators of genome dynamics and function. Nat Rev Genet 16: 583–597 [DOI] [PubMed] [Google Scholar]
- 3. Skourti‐Stathaki K, Proudfoot NJ (2014) A double‐edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev 28: 1384–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chedin F (2016) Nascent connections: R‐loops and chromatin patterning. Trends Genet 32: 828–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huertas P, Aguilera A (2003) Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription‐associated recombination. Mol Cell 12: 711–721 [DOI] [PubMed] [Google Scholar]
- 6. Li X, Manley JL (2005) Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 122: 365–378 [DOI] [PubMed] [Google Scholar]
- 7. Dominguez‐Sanchez MS, Barroso S, Gomez‐Gonzalez B, Luna R, Aguilera A (2011) Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet 7: e1002386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bhatia V, Barroso SI, Garcia‐Rubio ML, Tumini E, Herrera‐Moyano E, Aguilera A (2014) BRCA2 prevents R‐loop accumulation and associates with TREX‐2 mRNA export factor PCID2. Nature 511: 362–365 [DOI] [PubMed] [Google Scholar]
- 9. Stirling PC, Chan YA, Minaker SW, Aristizabal MJ, Barrett I, Sipahimalani P, Kobor MS, Hieter P (2012) R‐loop‐mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev 26: 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morales JC, Richard P, Patidar PL, Motea EA, Dang TT, Manley JL, Boothman DA (2016) XRN2 links transcription termination to DNA damage and replication stress. PLoS Genet 12: e1006107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salas‐Armenteros I, Perez‐Calero C, Bayona‐Feliu A, Tumini E, Luna R, Aguilera A (2017) Human THO‐Sin3A interaction reveals new mechanisms to prevent R‐loops that cause genome instability. EMBO J 36: 3532–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Skourti‐Stathaki K, Proudfoot NJ, Gromak N (2011) Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2‐dependent termination. Mol Cell 42: 794–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sollier J, Stork CT, Garcia‐Rubio ML, Paulsen RD, Aguilera A, Cimprich KA (2014) Transcription‐coupled nucleotide excision repair factors promote R‐loop‐induced genome instability. Mol Cell 56: 777–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chakraborty P, Grosse F (2011) Human DHX9 helicase preferentially unwinds RNA‐containing displacement loops (R‐loops) and G‐quadruplexes. DNA Repair 10: 654–665 [DOI] [PubMed] [Google Scholar]
- 15. Li L, Germain DR, Poon HY, Hildebrandt MR, Monckton EA, McDonald D, Hendzel MJ, Godbout R (2016) DEAD Box 1 facilitates removal of RNA and homologous recombination at DNA double‐strand breaks. Mol Cell Biol 36: 2794–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hodroj D, Recolin B, Serhal K, Martinez S, Tsanov N, Abou Merhi R, Maiorano D (2017) An ATR‐dependent function for the Ddx19 RNA helicase in nuclear R‐loop metabolism. EMBO J 36: 1182–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sridhara SC, Carvalho S, Grosso AR, Gallego‐Paez LM, Carmo‐Fonseca M, de Almeida SF (2017) Transcription dynamics prevent RNA‐mediated genomic instability through SRPK2‐dependent DDX23 phosphorylation. Cell Rep 18: 334–343 [DOI] [PubMed] [Google Scholar]
- 18. Song C, Hotz‐Wagenblatt A, Voit R, Grummt I (2017) SIRT7 and the DEAD‐box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev 31: 1370–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Castellano‐Pozo M, Garcia‐Muse T, Aguilera A (2012) R‐loops cause replication impairment and genome instability during meiosis. EMBO Rep 13: 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, Li X (2011) R‐loop‐mediated genomic instability is caused by impairment of replication fork progression. Genes Dev 25: 2041–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA (2017) Transcription‐replication conflict orientation modulates R‐loop levels and activates distinct DNA damage responses. Cell 170: 774–786 e719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lang KS, Hall AN, Merrikh CN, Ragheb M, Tabakh H, Pollock AJ, Woodward JJ, Dreifus JE, Merrikh H (2017) Replication‐transcription conflicts generate R‐loops that orchestrate bacterial stress survival and pathogenesis. Cell 170: 787–799 e718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tuduri S, Crabbe L, Conti C, Tourriere H, Holtgreve‐Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C et al (2009) Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol 11: 1315–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wellinger RE, Prado F, Aguilera A (2006) Replication fork progression is impaired by transcription in hyperrecombinant yeast cells lacking a functional THO complex. Mol Cell Biol 26: 3327–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia‐Rubio ML, Perez‐Calero C, Barroso SI, Tumini E, Herrera‐Moyano E, Rosado IV, Aguilera A (2015) The fanconi anemia pathway protects genome integrity from R‐loops. PLoS Genet 11: e1005674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herrera‐Moyano E, Mergui X, Garcia‐Rubio ML, Barroso S, Aguilera A (2014) The yeast and human FACT chromatin‐reorganizing complexes solve R‐loop‐mediated transcription‐replication conflicts. Genes Dev 28: 735–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Madireddy A, Kosiyatrakul ST, Boisvert RA, Herrera‐Moyano E, Garcia‐Rubio ML, Gerhardt J, Vuono EA, Owen N, Yan Z, Olson S et al (2016) FANCD2 facilitates replication through common fragile sites. Mol Cell 64: 388–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, Niedzwiedz W (2015) The fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol Cell 60: 351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hatchi E, Skourti‐Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz‐Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML et al (2015) BRCA1 recruitment to transcriptional pause sites is required for R‐loop‐driven DNA damage repair. Mol Cell 57: 636–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bhatia V, Herrera‐Moyano E, Aguilera A, Gomez‐Gonzalez B (2017) The role of replication‐associated repair factors on R‐loops. Genes 8: E171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garcia‐Pichardo D, Canas JC, Garcia‐Rubio ML, Gomez‐Gonzalez B, Rondon AG, Aguilera A (2017) Histone mutants separate R loop formation from genome instability induction. Mol Cell 66: 597–609 e595 [DOI] [PubMed] [Google Scholar]
- 32. Castellano‐Pozo M, Santos‐Pereira JM, Rondon AG, Barroso S, Andujar E, Perez‐Alegre M, Garcia‐Muse T, Aguilera A (2013) R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol Cell 52: 583–590 [DOI] [PubMed] [Google Scholar]
- 33. Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, Jaffrey SR (2014) Promoter‐bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343: 1002–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Groh M, Lufino MM, Wade‐Martins R, Gromak N (2014) R‐loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet 10: e1004318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ginno PA, Lim YW, Lott PL, Korf I, Chedin F (2013) GC skew at the 5′ and 3′ ends of human genes links R‐loop formation to epigenetic regulation and transcription termination. Genome Res 23: 1590–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, Chedin F (2016) Prevalent, dynamic, and conserved R‐loop structures associate with specific epigenomic signatures in mammals. Mol Cell 63: 167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C et al (2005) DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature 434: 864–870 [DOI] [PubMed] [Google Scholar]
- 38. Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B et al (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434: 907–913 [DOI] [PubMed] [Google Scholar]
- 39. Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cimprich KA, Cortez D (2008) ATR: an essential regulator of genome integrity. Nat Rev 9: 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liang Z, Liang F, Teng Y, Chen X, Liu J, Longerich S, Rao T, Green AM, Collins NB, Xiong Y et al (2019) Binding of FANCI‐FANCD2 complex to RNA and R‐loops stimulates robust FANCD2 monoubiquitination. Cell Rep 26: 564–572 e565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garcia‐Rubio M, Barroso SI, Aguilera A (2018) Detection of DNA‐RNA hybrids in vivo . Methods Mol Biol 1672: 347–361 [DOI] [PubMed] [Google Scholar]
- 43. Song IY, Palle K, Gurkar A, Tateishi S, Kupfer GM, Vaziri C (2010) Rad18‐mediated translesion synthesis of bulky DNA adducts is coupled to activation of the Fanconi anemia DNA repair pathway. J Biol Chem 285: 31525–31536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Y, Zhou X, Zhao L, Li C, Zhu H, Xu L, Shan L, Liao X, Guo Z, Huang P (2011) UBE2W interacts with FANCL and regulates the monoubiquitination of Fanconi anemia protein FANCD2. Mol Cells 31: 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koken M, Reynolds P, Bootsma D, Hoeijmakers J, Prakash S, Prakash L (1991) Dhr6, a Drosophila homolog of the yeast DNA‐repair gene RAD6. Proc Natl Acad Sci USA 88: 3832–3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Somasagara RR, Spencer SM, Tripathi K, Clark DW, Mani C, Madeira da Silva L, Scalici J, Kothayer H, Westwell AD, Rocconi RP et al (2017) RAD6 promotes DNA repair and stem cell signaling in ovarian cancer and is a promising therapeutic target to prevent and treat acquired chemoresistance. Oncogene 36: 6680–6690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lyakhovich A, Shekhar MP (2004) RAD6B overexpression confers chemoresistance: RAD6 expression during cell cycle and its redistribution to chromatin during DNA damage‐induced response. Oncogene 23: 3097–3106 [DOI] [PubMed] [Google Scholar]
- 48. Hartono SR, Malapert A, Legros P, Bernard P, Chedin F, Vanoosthuyse V (2018) The affinity of the S9.6 antibody for double‐stranded RNAs impacts the accurate mapping of R‐loops in fission yeast. J Mol Biol 430: 272–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kinney JS, Viscidi RP, Vonderfecht SL, Eiden JJ, Yolken RH (1989) Monoclonal antibody assay for detection of double‐stranded RNA and application for detection of group A and non‐group A rotaviruses. J Clin Microbiol 27: 6–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Silva S, Camino LP, Aguilera A (2018) Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc Natl Acad Sci USA 115: 11024–11029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Garcia‐Muse T, Aguilera A (2016) Transcription‐replication conflicts: how they occur and how they are resolved. Nat Rev Mol Cell Biol 17: 553–563 [DOI] [PubMed] [Google Scholar]
- 52. Garcia‐Rubio M, Aguilera P, Lafuente‐Barquero J, Ruiz JF, Simon MN, Geli V, Rondon AG, Aguilera A (2018) Yra1‐bound RNA‐DNA hybrids cause orientation‐independent transcription‐replication collisions and telomere instability. Genes Dev 32: 965–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hamperl S, Cimprich KA (2016) Conflict resolution in the genome: how transcription and replication make it work. Cell 167: 1455–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Petermann E, Maya‐Mendoza A, Zachos G, Gillespie DA, Jackson DA, Caldecott KW (2006) Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol 26: 3319–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gaillard H, Aguilera A (2016) Transcription as a threat to genome integrity. Annu Rev Biochem 85: 291–317 [DOI] [PubMed] [Google Scholar]
- 56. Aguilera A, Gomez‐Gonzalez B (2017) DNA‐RNA hybrids: the risks of DNA breakage during transcription. Nat Struct Mol Biol 24: 439–443 [DOI] [PubMed] [Google Scholar]
- 57. Gomez‐Gonzalez B, Aguilera A (2019) Transcription‐mediated replication hindrance: a major driver of genome instability. Genes Dev 33: 1008–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Helmrich A, Ballarino M, Tora L (2011) Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell 44: 966–977 [DOI] [PubMed] [Google Scholar]
- 59. Loomis EW, Sanz LA, Chedin F, Hagerman PJ (2014) Transcription‐associated R‐loop formation across the human FMR1 CGG‐repeat region. PLoS Genet 10: e1004294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gomez‐Gonzalez B, Felipe‐Abrio I, Aguilera A (2009) The S‐phase checkpoint is required to respond to R‐loops accumulated in THO mutants. Mol Cell Biol 29: 5203–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kabeche L, Nguyen HD, Buisson R, Zou L (2018) A mitosis‐specific and R loop‐driven ATR pathway promotes faithful chromosome segregation. Science 359: 108–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bayona‐Feliu A, Casas‐Lamesa A, Reina O, Bernues J, Azorin F (2017) Linker histone H1 prevents R‐loop accumulation and genome instability in heterochromatin. Nat Commun 8: 283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Almeida R, Fernandez‐Justel JM, Santa‐Maria C, Cadoret JC, Cano‐Aroca L, Lombrana R, Herranz G, Agresti A, Gomez M (2018) Chromatin conformation regulates the coordination between DNA replication and transcription. Nat Commun 9: 1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Britton S, Dernoncourt E, Delteil C, Froment C, Schiltz O, Salles B, Frit P, Calsou P (2014) DNA damage triggers SAF‐A and RNA biogenesis factors exclusion from chromatin coupled to R‐loops removal. Nucleic Acids Res 42: 9047–9062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ohle C, Tesorero R, Schermann G, Dobrev N, Sinning I, Fischer T (2016) Transient RNA‐DNA hybrids are required for efficient double‐strand break repair. Cell 167: 1001–1013 e1007 [DOI] [PubMed] [Google Scholar]
- 66. Roy D, Zhang Z, Lu Z, Hsieh CL, Lieber MR (2010) Competition between the RNA transcript and the nontemplate DNA strand during R‐loop formation in vitro: a nick can serve as a strong R‐loop initiation site. Mol Cell Biol 30: 146–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Voichek Y, Bar‐Ziv R, Barkai N (2016) Expression homeostasis during DNA replication. Science 351: 1087–1090 [DOI] [PubMed] [Google Scholar]
- 68. Heller RC, Marians KJ (2006) Replisome assembly and the direct restart of stalled replication forks. Nat Rev 7: 932–943 [DOI] [PubMed] [Google Scholar]
- 69. Elvers I, Johansson F, Groth P, Erixon K, Helleday T (2011) UV stalled replication forks restart by re‐priming in human fibroblasts. Nucleic Acids Res 39: 7049–7057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lopes M, Foiani M, Sogo JM (2006) Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21: 15–27 [DOI] [PubMed] [Google Scholar]
- 71. Gangavarapu V, Prakash S, Prakash L (2007) Requirement of RAD52 group genes for postreplication repair of UV‐damaged DNA in Saccharomyces cerevisiae . Mol Cell Biol 27: 7758–7764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hishida T, Kubota Y, Carr AM, Iwasaki H (2009) RAD6‐RAD18‐RAD5‐pathway‐dependent tolerance to chronic low‐dose ultraviolet light. Nature 457: 612–615 [DOI] [PubMed] [Google Scholar]
- 73. Huang D, Piening BD, Paulovich AG (2013) The preference for error‐free or error‐prone postreplication repair in Saccharomyces cerevisiae exposed to low‐dose methyl methanesulfonate is cell cycle dependent. Mol Cell Biol 33: 1515–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bianco JN, Poli J, Saksouk J, Bacal J, Silva MJ, Yoshida K, Lin YL, Tourriere H, Lengronne A, Pasero P (2012) Analysis of DNA replication profiles in budding yeast and mammalian cells using DNA combing. Methods 57: 149–157 [DOI] [PubMed] [Google Scholar]
- 75. Bialic M, Coulon V, Drac M, Gostan T, Schwob E (2015) Analyzing the dynamics of DNA replication in Mammalian cells using DNA combing. Methods Mol Biol 1300: 67–78 [DOI] [PubMed] [Google Scholar]
- 76. Técher H, Koundrioukoff S, Azar D, Wilhelm T, Carignon S, Brison O, Debatisse M, Le Tallec B (2013) Replication dynamics: biases and robustness of DNA fiber analysis. J Mol Biol 425: 4845–4855 [DOI] [PubMed] [Google Scholar]
- 77. Shankey TV, Rabinovitch PS, Bagwell B, Bauer KD, Duque RE, Hedley DW, Mayall BH, Wheeless L, Cox C (1993) Guidelines for implementation of clinical DNA cytometry. International Society for Analytical Cytology. Cytometry 14: 472–477 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File