

Abstract
Epidermal growth factor receptor pathway substrate 8 (EPS8), which acts as an oncoprotein in various carcinomas, is associated with tumor progression. However, its impact on multiple myeloma (MM) has not been determined. Here, we investigate the role of EPS8 in MM and consider the potential of EPS8 as an anti-MM target. We confirmed overexpression of EPS8 in MM cells compared with plasma cells derived from healthy volunteers. Knockdown of EPS8 significantly abrogated MM cell survival, migration and invasion. Moreover, depletion of EPS8 overcomes drug resistance. TNFα or bone marrow stromal cell culture supernatants induce EPS8, which is blocked by the IKKβ inhibitor MLN120B, suggesting that EPS8 is regulated by NF-κB signaling in MM cells. Mithramycin (MTM), a selective EPS8 inhibitor, suppressed MM cell proliferation and exerted potent anti-MM activity in xenograft tumor models. A synergistic effect of MTM and bortezomib (BTZ) was also observed in vitro and in vivo. Mechanistically, treatment of MM cells with MTM reduced the expression of EPS8 and related pathways. Additionally, the EPS8-knockdown phenotype can be rescued by shRNA-resistant EPS8. Taken together, we describe overexpression of EPS8 in MM by highlighting its role as a potential target and reveal therapeutic targeting of EPS8 by MTM as a novel therapy for MM.
Keywords: Multiple myeloma, EPS8, mithramycin, bortezomib
Introduction
Multiple myeloma (MM) arises from the clonal growth of malignant plasma cells in the bone marrow associated with immunoglobulin in the serum and urine [1,2]. The overall outcome of MM patients has markedly improved due to the application of novel agents including proteasome inhibitors and immunomodulatory drugs [3]. Despite this progress, myeloma remains incurable with most patients eventually relapsing. Moreover, nearly all patients will ultimately develop resistance to currently available agents [4,5]. Therefore, there is a need to decipher the pathogenesis of MM to identify novel therapeutic targets for better prevention and treatment.
EGFR signal transduction plays a critical role in normal cell physiology [6]. Epidermal growth factor receptor pathway substrate 8 (EPS8) was initially identified as a novel substrate for EGFR kinase [7]. Recently, an increasing number of studies show that EPS8 is involved in many signaling pathways that promote proliferation, tumorigenesis, and the development of metastases [8,9]. EPS8 extensively functions as an oncogene in various types of human carcinomas, including lung cancer, cervical cancer, squamous cell carcinoma and leukemia [10-14]. High levels of EPS8 in cancer patients serve as a biomarker of poor prognosis or decreased overall survival [15,16].
EPS8 is implicated in the pathogenesis of certain carcinomas in a context-dependent fashion; however, to date the biological function of EPS8 in MM remains to be determined. In the present, we explore the biological impact of EPS8 in MM. We demonstrate that EPS8 is highly expressed in myeloma patients compared with healthy donors. Depletion of EPS8 leads to inhibition of MM cell survival, migration and invasion. EPS8 is activated by NF-κB signaling in MM cells. Furthermore, we demonstrated that inhibition of EPS8 by mithramycin (MTM) significantly increased the efficacy of BTZ in vitro and in vivo. Taken together, our data delineate the biological sequelae of EPS8, and validate it as a novel therapeutic target in MM.
Materials and methods
Chemicals
Bortezomib and mithramycin were obtained from SelleckChem (Houston, TX, USA) and Sigma-Aldrich (St Louis, MO, USA), respectively. Both chemicals were dissolved in DMSO and stored at -80°C. MLN120B was purchased from ApexBio (Houston, TX, USA). TNFα was obtained from PEPROTECH (Rocky Hill, NJ, USA).
Cell lines and culture conditions
Human MM cell lines MM.1S, RPMI-8226, U266 and NCI-H929 were cryopreserved in the Hematological Laboratory of Zhujiang Hospital (Guangzhou, China). The MM.1S cell line was purchased from ATCC. RPMI-8226 and NCI-H929 were purchased from the Guangzhou Jennio Biotech CO., LTD. U266 was purchased from COBIOERBIOSCIENCES CO., LTD. The bortezomib-resistant 8226/BR cell line was developed by the incremental addition of bortezomib. The identity of these cell lines was confirmed before use by STR profiling. The sequence of the annealed oligonucleotide fragment encoding short hairpin transcript corresponding to EPS8 was AACTTCTAATCGCCATATA. The nontargeting empty plasmid was used as the control shRNA plasmid. We purchased open reading frame (ORF) of wild-type EPS8 (EPS8W) and an shRNA-resistant form of EPS8 (EPS8R) that harbors nine silent mutations within the sequence targeted by shEPS8. The shEPS8 targeting sequence in EPS8 was mutated from AACTTCTAATCGCCATATA to CACGAGCAACCGTCACATC by site-directed mutagenesis. MM cells were transfected with lentivirus according to the manufacturer’s protocol. The cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum at 37°C with 5% CO2.
Clinical samples
Bone marrow was obtained from MM patients or healthy volunteers with informed consent; this protocol was approved by the Institutional Ethics Committee. After separating mononuclear cells from bone marrow by Ficoll density gradient centrifugation, cells were further purified by CD138-positive selection using anti-CD138 magnetic activated cell separation microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). MM patient BMSCs were established and maintained by culturing CD138-negative bone marrow mononuclear cells in DMEM supplemented with 20% fetal bovine serum.
Quantitative RT-PCR
Total RNA was extracted from fresh-frozen cells in Trizol (Invitrogen). Reverse transcription of total RNA was performed using the PrimeScript TM RT reagent Kit with gDNA Eraser (Takara) according to the manufacturer’s instructions. Polymerase chain reaction amplifications of the respective genes were performed with 100 ng cDNA, 10 μM forward and reverse primers, and 2 × SYBR®Select Master Mix (Takara) in a final volume of 20 μl.
Cell viability assays
MM cells were incubated in 96-well plates with or without treatments in culture medium for different time points, and then 10 μl of the CCK-8 solution (Dojindo Laboratories) was added to each well. After a 4-h incubation at 37°C and 5% CO2, the optical density (OD) was measured at 450 nm. All experiments were performed in quintuplicate.
Apoptotic and cell cycle analysis
MM cells were collected after treatment with DMSO, MTM (250 nM), BTZ (25 nM) or MTM combined with BTZ for 48 h. Apoptosis was assessed by flow cytometry (FCM) according to the manufacturer’s protocol (BD, Annexin-V-APC & PI Apoptosis Detection Kit). The cell cycle was analyzed by propidium iodide (PI) staining and quantified using FCM.
Western blotting
Cells were collected and lysed by RAPI lysis buffer supplementation with proteinase and phosphatase inhibitors at 4°C for 10 min. The supernatants were collected after centrifuged at 4°C for 10 min at 12,000 rpm. The protein concentrations were determined using a Bradford protein assay kit (Beyotime, China). After the addition of loading buffer, protein samples were separated by 10% SDS-PAGE and transferred onto the PVDF membranes (Millipore, Bedford, MA). Membranes were blocked in 5% nonfat milk/TBST and incubated with indicated antibodies overnight at 4°C. After three washes with TBST, the blots were incubated with HRP-conjugated secondary antibody for 1 h at room temperature. The HRP signal was detected using the ECL kit (Fdbio science).
Colony formation assay
Cells were seeded on 24-well plate at a density of 1000 cells. Methylcellulose (Sigma) in final concentration of 0.9% was added and mixed. After 14 days, the number of colonies was calculated by microscopy.
Transwell migration assay and invasion studies
For migration studies, 20% FBS media was added to the lower chambers of transwell plates (pore size 0.8 um; Costar-Corning). MM cells (1 × 105 cells) suspended in serum-free media were placed in the upper chambers. After 24 h at 37°C, the number of cells that migrated to the lower chambers was calculated.
MM-cell invasion was evaluated by determining their ability to migrate through transwell chambers separated by an 8-um pore-size polycarbonate membrane, over which a thin layer of Matrigel (BD Biosciences) was coated. Five hundred microliters of RPMI 1640 supplemented with 20% FBS was added to the lower chamber. MM cells (1 × 105 cells) resuspended in 300 μl serum-free media, were added to the upper chamber of the transwell plates and incubated for 24 h at 37°C. Cells that invaded the Matrigel-coated filters were fixed in methanol, stained with 0.2% crystal violet, and counted using an inverted microscope [17,18].
In vivo study
Five-week-old NSG (NOD-PrkdcscidIL2rgtm1/Bcgen) female mice were purchased from Jiangsu Biocytogen Co., Ltd. (Jiangsu, China). The in vivo experiments were performed strictly in accordance with Southern Medical University’s Policy on the Care and Use of Laboratory Animals. Briefly, 8226/NC and 8226/sh cells were harvested and injected subcutaneously (5 × 106 cells in 100 μl of PBS) into mice. Tumor volume was measured twice a week. Mice were sacrificed on day 28, and subcutaneous tumors were excised. In addition, 8226 cells were harvested and injected subcutaneously (5 × 106 cells in 100 μl of PBS) into mice. Then, 8226-injected mice were treated with BTZ (0.5 mg/kg) and/or MTM (0.5 mg/kg) twice a week. PBS was injected as a control. All mice were sacrificed at the 28th day, and the tumors were removed. Tumor volumes were determined by measuring tumor length (L) and width (W) and calculating the volume (V = 0.5 × L × W2) [19]. Tumors were cut into small pieces and stored in liquid nitrogen immediately, and a portion of each tumor was used for protein extraction.
Statistical analysis
Data are presented as the means ± standard deviations. Comparison between two groups assuming normal distribution was made with two-tailed Student’s t-test, and Welch’s correction was applied when the variances were unequal, which was determined by an F-test. All statistical analyses were performed using SPSS software. P < 0.05 was considered statistically significant.
Results
EPS8 is overexpressed in multiple myeloma
We first evaluated EPS8 expression in MM and normal plasma cells separated from bone marrow. EPS8 mRNA levels were significantly elevated in MM patients compared with healthy donors (Figure 1A). EPS8 mRNA was constitutively expressed at higher levels in most MM cell lines compared with normal plasma cells (Figure 1B), suggesting a role for EPS8 in the pathogenesis of MM. EPS8 protein expression was also verified by western blotting in 4 MM cell lines and 3 primary MM samples; EPS8 protein was not detected in 3 healthy volunteer derived-BMMNCs (Figure 1C). These results demonstrate that EPS8 is overexpressed in MM cells from MM patients compared with normal plasma cells.
Figure 1.
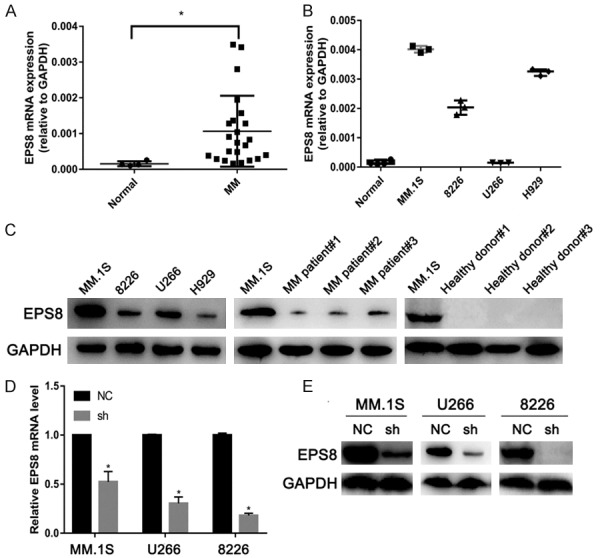
EPS8 is overexpressed in multiple myeloma. (A) EPS8 mRNA expression in normal plasma cells from 4 volunteers (Normal) and MM cells from 23 MM patients or (B) 4 MM cell lines. CD138+ cells enriched from bone marrow by immunomagnetic cell sorting method (MACS). EPS8 mRNA in the cells was measured by quantitative real-time PCR. (C) Whole cell lysates were extracted from MM cell lines, MM patient samples and healthy donor samples and subjected to immunoblot analysis with the indicated antibodies. (D and E) EPS8 expression was analyzed by real-time PCR and immunoblotting in EPS8 knockdown (sh) and control cells (NC).
EPS8 confers MM cell growth in vitro and in vivo
To explore the function of EPS8 in MM, we transduced short hairpin (sh) RNA against EPS8 or control shRNA into MM cell lines. Efficient silencing of EPS8 was confirmed by qRT-PCR and western blotting (Figure 1D and 1E). In stably transfected cells, the CCK-8 assay was performed to determine viable cell numbers. Loss of EPS8 resulted in significant inhibition of MM cell growth (Figure 2A). Next, to investigate whether EPS8 also affects the progenitor activity of MM cells, myeloma cells were evaluated for colony formation in methylcellulose. Compared to control, EPS8 knockdown reduced colony formation (Figure 2B). Western blotting results showed that phosphorylated Akt and phosphorylated Erk were downregulated after EPS8 knockdown in MM cells (Figure 2C). FCM analysis demonstrated that depletion of EPS8 induced blockade of cell cycle progression from G1 to S phase (Figure 2D). Moreover, CyclinD1 expression was decreased and P21 expression was increased in EPS8-silenced cells (Figure 2C). The effect of EPS8 on the tumorigenic potential of MM cells was evaluated in vivo. Briefly, 8226/NC and 8226/sh cells were subcutaneously injected into the right flank of NSG mice, and tumor formation was monitored. Twenty-eight days after injection, the average volume and weight of the 8226/sh group were markedly reduced compared with the values in the 8226/NC group (Figure 2E and 2F). Using IHC, 8226/NC-derived tumors displayed elevated Eps8, CD31 (PECAM1) staining and microvessel density (MVD) compared with 8226/sh-derived tumors, indicating that upregulation of Eps8 enhanced angiogenic processes (Figures S1 and S2). Taken together, these findings indicate a potent effect of EPS8 on the survival and progenitors of MM cells, a relevant population of disease-initiating cells.
Figure 2.
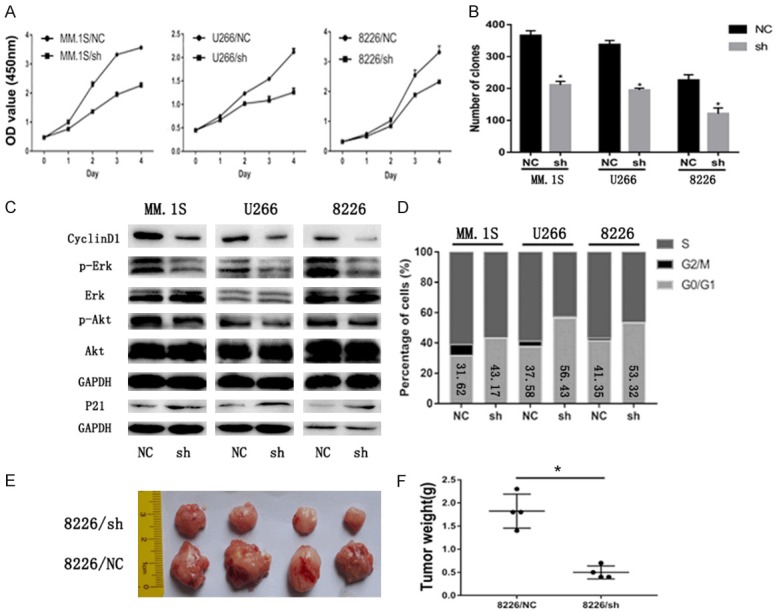
EPS8 confers MM cell growth in vitro and in vivo. A. Cell viability was determined by CCK-8 assay, and viability of shRNA-transfected myeloma cells decreased in a time-dependent manner. B. Colony formation of myeloma cells in methylcellulose was performed according to the manufacturer’s instructions. Number of colonies was counted by microscopy on the 14th day. C. CyclinD1, P21, p-Akt, Akt, p-Erk and Erk expression were analyzed by immunoblotting. GAPDH served as the loading control for each membrane. D. Cell cycle was analyzed by flow cytometry. E and F. The tumors excised from NSG mice after being inoculated subcutaneously with 8226/NC and 8226/sh cells for 28 days. Tumor weight was measured on the 28th day.
EPS8 enhances cell survival and drug resistance
To investigate the potential mechanism of cell growth inhibition in EPS8-knockdown cells, we quantified apoptotic cells by FCM using Annexin V staining. As shown in Figure 3A, percentages of apoptotic cells were markedly increased after EPS8 knockdown in MM cells. Moreover, cleaved forms of caspase-3, caspase-7 and PARP were elevated after EPS8 knockdown (Figure 3B). We further explored whether the extrinsic or intrinsic pathway is involved in the apoptosis induced by EPS8 knockdown. Immunoblotting showed increased cleavage of caspase-8 and caspase-9 in EPS8-knockdown cells (Figure 3B), indicating involvement of both extrinsic and intrinsic apoptosis pathways. Next, we evaluated the influence of EPS8 on the chemotherapeutic sensitivity of MM treatment. Bortezomib, a proteasome inhibitor, has significant anti-myeloma activity in MM. Blocking the expression of EPS8 significantly enhanced the therapeutic effects of bortezomib in MM cells (Figure 3C). We further asked whether EPS8 contributes to bortezomib resistance in MM cells. Bortezomib-resistant cells were developed by exposing parental cells to serially increased drug concentrations. As shown in Figure 3D, BR cells were confirmed by increased IC50 (approximately 10-fold) values compared with parental cells. EPS8 mRNA and protein levels were upregulated in BR cells. Further knocking down EPS8 resensitized BR cells to bortezomib (Figure 3E and 3F). Taken together, these results indicate that knockdown of EPS8 induces apoptosis and enhances MM cells sensitivity to bortezomib.
Figure 3.
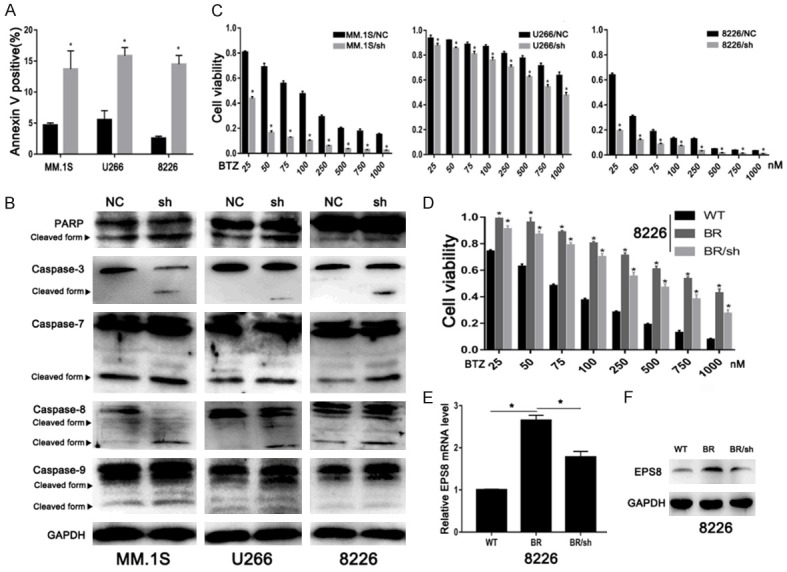
EPS8 enhances cell survival and drug resistance. A. The percentage of apoptotic cells was examined by FCM. B. Whole cell lysates were extracted and subjected to immunoblot analysis with the indicated antibodies. GAPDH served as the loading control for each membrane. C and D. Cell viability was determined by CCK-8 assay at various bortezomib concentrations for 48 h. E and F. EPS8 expression was analyzed by real-time PCR and immunoblotting.
EPS8 regulates MM cell migration and invasion
Previous studies showed that EPS8 is a master regulator of cell migration and invasion, by participating in the formation of EPS8-Abi1-Sos1 and EPS8-IRSp53 complexes, which converge multiple signaling pathways on the Rho-GTPase family [20-22]. Therefore, we further investigate the role of EPS8 on the capacity of MM cells to migrate and invade. In a transwell migration assay, EPS8-positive cells showed markedly increased migration rates compared with those transfected by lentivirus-delivered shRNA (Figure 4A). As MM cells need to actively penetrate through the subendothelial basement membrane of the BM sinus to migrate in and out of the BM, we examined whether EPS8 is required for MM-cell invasion. Experiments using Matrigel-coated invasion chambers revealed that depletion of EPS8 was considerably more effective in inhibiting MM cell invasion (Figure 4B and 4C). Next, western blotting was used to explore the mechanism involved in EPS8-mediated MM migration and invasion. As shown in Figure 4D, EPS8 silencing reduced the expression of proteins related to cell migration and invasion. Taken together, these data indicate that EPS8 is a major regulator of MM tumor migration and invasion.
Figure 4.
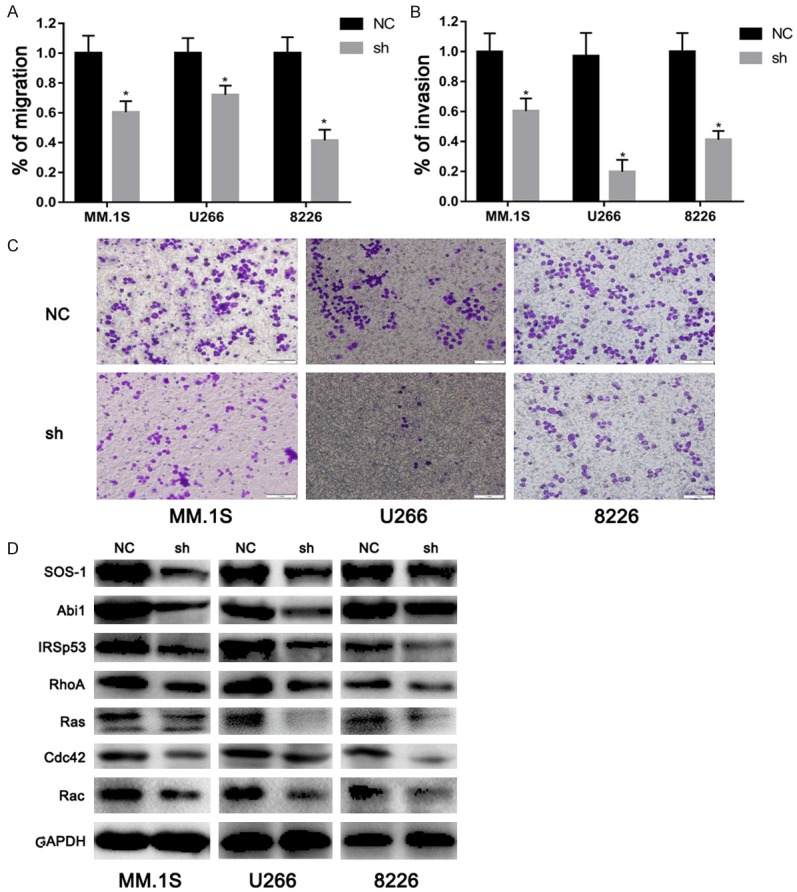
EPS8 regulates MM cell migration and invasion. A. Transwell migration of myeloma cells was calculated after 24 h. B and C. Shown is a representative transwell Matrigel invasion of myeloma cells under the conditions described in “Transwell migration assay and invasion studies.” Cells that invaded the Matrigel-coated filters were stained with crystal violet and counted using an inverted microscope. Images were acquired with a bright light Olympus CKX41 microscope. D. Whole cell lysates were extracted and subjected to immunoblot analysis with the indicated antibodies. GAPDH served as the loading control for each membrane.
EPS8 is upregulated by the NF-κB pathway in MM
The NF-κB pathway plays an important role in the survival of MM [23]. We next explored whether NF-κB mediates MM cell growth through EPS8. TNFα activates the NF-κB pathway by increasing phospho-IκBa and phospho-p65 as well as IkBa protein degradation, thus inducing EPS8 in 8226 cells (Figure 5A and 5B). This EPS8 induction was blocked by the IκB kinase (IKK) β inhibitor MLN120B (Figure 5B), suggesting that EPS8 expression is induced by NF-κB in MM cells. Previous studies showed that BMSCs activate the NF-κB pathway through cytokines, such as BAFF, TNFα and APRIL. Therefore, we further tested whether BMSCs induce EPS8 expression in MM cells. As shown in Figure 5C, MM patient-derived BMSC CM upregulated EPS8 expression in 8226 cells. Similar to TNFα stimulation, BMSC CM-mediated EPS8 induction was markedly inhibited in the presence of MLN120B, indicating that EPS8 is induced by BMSCs, at least in part, via the NF-κB signaling pathway in MM cells.
Figure 5.
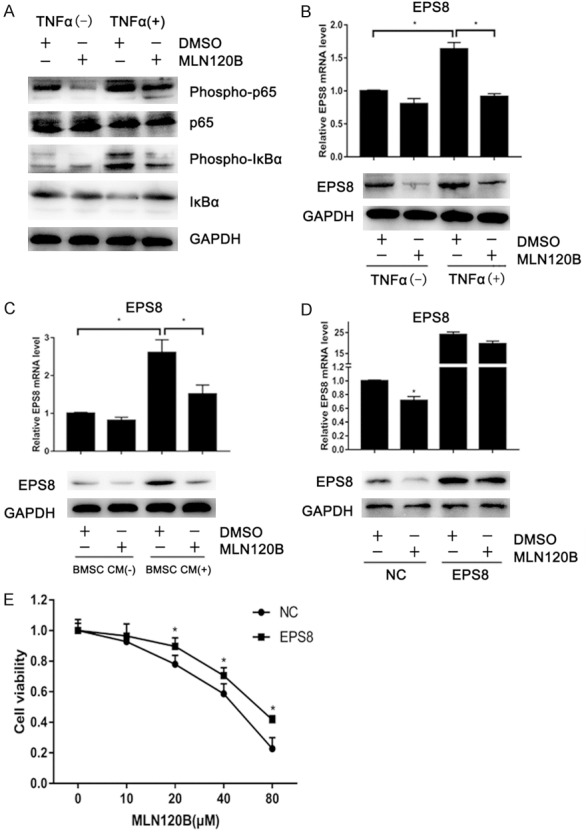
EPS8 is upregulated by the NF-κB pathway in MM. A and B. 8226 cells were incubated with or without 30 ng/ml TNFα and 20 μM MLN120B or DMSO. After 4 h of treatment, total RNA and whole cell lysates were extracted and subjected to qRT-PCR and immunoblotting. C. 8226 cells were treated with conditioned medium from MM patient-derived bone marrow stromal cells or control culture medium for 4 h. D. 8226 cells stably expressing EPS8 or empty vector-transduced control cells were incubated with 20 μM of MLN120B or DMSO for 72 h. Cells were then harvested for qRT-PCR and immunoblotting. E. 8226 cells stably expressing EPS8 or control cells were incubated with the indicated dose of MLN120B for 96 h.
We next examined whether EPS8 mediates MM cell growth under the NF-κB pathway. Endogenous EPS8 expression was obviously decreased after 72-h MLN120B treatment (Figure 5D) and associated with growth inhibition in 8226 cells (Figure 5E). The ectopic expression of EPS8 partially rescued 8226 cells from this growth inhibition (Figure 5D), indicating that EPS8 is at least one of the downstream effectors of the NF-κB pathway in MM.
EPS8 inhibitor enhances the effect of BTZ on myeloma cells
The selective EPS8 inhibitor mithramycin (MTM), has been demonstrated to sensitize cells to cytotoxics [10,24]. To investigate the function of MTM in multiple myeloma, MM cells were treated for 48 h with increasing concentrations of MTM. A dose-dependent anti-proliferative effect of MTM was determined using CCK-8 assay (Figure 6A). EPS8 expression levels were markedly decreased following MTM exposure (Figure 6B and 6C). To explore whether the inhibition of EPS8 by MTM can enhance the effect of BTZ in MM, cells were treated with BTZ, MTM or BTZ combined with MTM. Compared with the control group, cells in the BTZ and MTM groups showed a lower growth rate. The combination of BTZ and MTM dramatically inhibited cell growth (Figure 6D). MTM remarkably facilitated BTZ-induced apoptosis in MM cells (Figure 6E). Cell cycle analysis indicated that MTM promoted cell cycle arrest at the G0/G1 phase induced by MTM (Figure 6F). Next, to reveal the molecular mechanisms involved in EPS8 inhibition-mediated apoptosis and cell cycle arrest, immunoblotting was conducted, and the expression of associated proteins were detected. As shown in (Figure 6G), the combination of MTM and BTZ induced downregulation of Eps8 and CyclinD1 and upregulation of p21, p53, PARP and Caspase-3 cleavage products compared to treatment with MTM or BTZ alone.
Figure 6.

EPS8 inhibitor enhances the effect of BTZ on myeloma cells. A. MM cells were incubated with the indicated dose of MTM for 48 h. Cell viability was determined by CCK-8 assay. B and C. MM cells were incubated with 250 nM MTM or DMSO for 48 h. Cells were then harvested for qRT-PCR and immunoblotting. D. MM cells were treated with MTM (250 nM) with or without 25 nM BTZ for 48 h, and cell viability was determined by CCK-8 assay. E. The apoptotic rate was evaluated by FCM. F. Cell cycle distribution was examined by FCM. G. Eps8, CyclinD1, P53, P21 and Cleaved caspase-3 and PARP expression were detected by immunoblotting.
EPS8 inhibitor synergizes with bortezomib in vivo
To further validate the synergistic effect of MTM with proteasome inhibitor in vivo, we employed an NSG mouse xenograft assay by subcutaneously implanting 8226 cells into NSG mice. Mice were treated with MTM (0.5 mg/kg), BTZ (0.5 mg/kg) or MTM combined with BTZ when we detected palpable tumors. PBS treatment served as the control group (Figure 7A). The tumor volumes (Figure 7B and 7D) and weights (Figure 7C) at the terminal point were considerably more decreased in the combination group than in both MTM and BTZ groups. Moreover, the combination of MTM and BTZ dramatically induced downregulation of Eps8 protein levels compared to MTM or BTZ treatment alone (Figure 7E).
Figure 7.
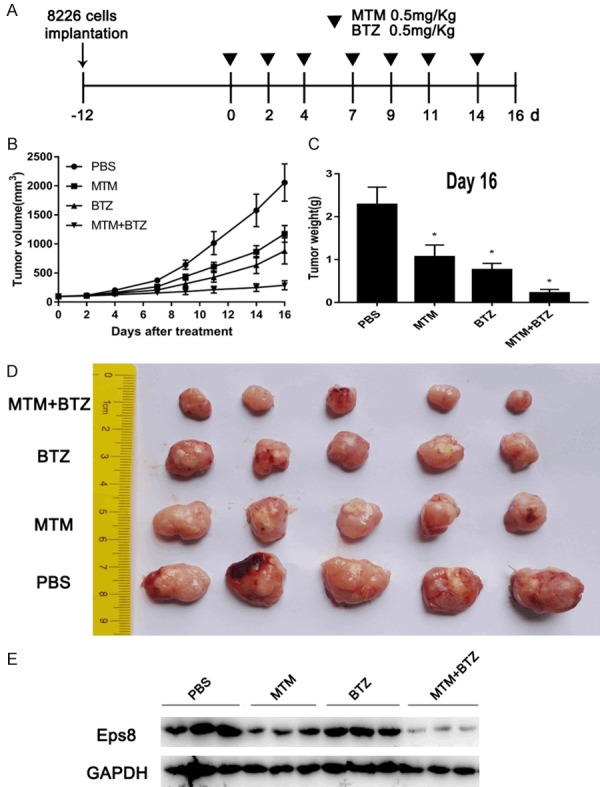
EPS8 inhibitor synergizes with bortezomib in vivo. A. 8226 cells (5 × 106 per flank) were injected into the right flank of NSG mice. When the tumor volumes reached 100 mm3, the mice were randomly divided into four groups (5 mice in each group). Then, MTM, BTZ or MTM combined with BTZ were intraperitoneally injected into the mice thrice a week. Mice injected with PBS were used as a control. B. The tumor volumes were measured at the indicated time for 16 days. C. The weight of each tumor was measured after extraction from mice at the experimental endpoint. D. Image of tumors excised from mice after 16 days of treatment. E. Immunoblotting for tumor sections with similar volume were used for Eps8 assessment.
Rescue of EPS8 knockdown phenotype by expression of shRNA-resistant EPS8
To further demonstrate the specificity of shRNA manipulations, rescue experiments were performed by constructing a silent mutant EPS8 that is resistant to EPS8 shRNA. MM cells with shEPS8 lenti-viruses stably transfected were infected by EPS8W lentiviruses and EPS8R lentiviruses. EPS8 RNA and protein levels were higher in the shEPS8+EPS8W and shEPS8+EPS8R groups than the shEPS8 groups, with particularly more obvious effects in shEPS8+EPS8R groups (Figure 8A and 8B). The OD450 values on day 4 were higher in the shEPS8+EPS8W and shEPS8+EPS8R groups than in the shEPS8 group (Figure 8C). The clone numbers (159 ± 12 vs. 200 ± 11 and 269 ± 10 in MM.1S and 181 ± 10 vs. 222 ± 11 and 279 ± 13 in U266 and 96 ± 9 vs. 143 ± 11 and 168 ± 12 in 8226, P < 0.05, respectively) indicated a similar trend (Figure 8D). Furthermore, EPS8R expression significantly reduced the therapeutic effects of bortezomib in MM cells (Figure 8E). EPS8 overexpression markedly restored cell migratory and invasive functions that were suppressed by shRNA-mediated EPS8 silencing (Figure 8F and 8G). Additionally, these results showed that EPS8R had a stronger ability to rescue the EPS8-knockdown phenotype than EPS8W.
Figure 8.
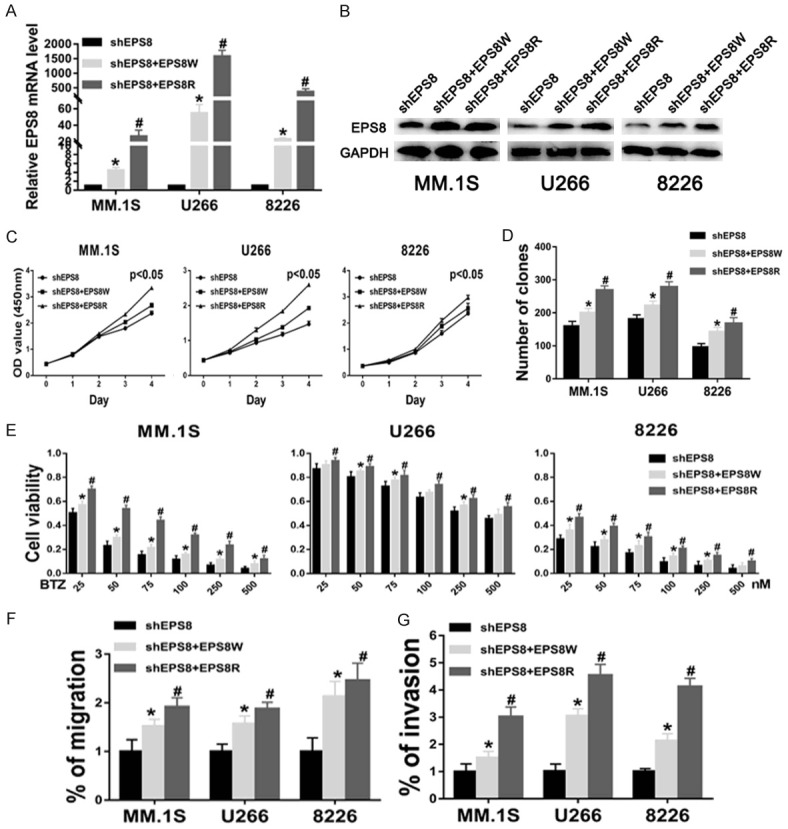
Rescue of EPS8 knockdown phenotype by expression of shRNA-resistant EPS8. A and B. EPS8 expression was analyzed by real-time PCR and immunoblotting in shEPS8, shEPS8+EPS8W and shEPS8+EPS8R cells. C. Cell viability was determined by CCK-8 assay. D. Colony formation by myeloma cells in methylcellulose was performed according to the manufacturer’s instructions. The number of colonies was counted by microscopy on the 14th day. E. Cell viability was determined by CCK-8 assay at various bortezomib concentrations for 48 h. F and G. Transwell migration and invasion assays in shEPS8, shEPS8+EPS8W and shEPS8+EPS8R cells.
Discussion
EPS8 is widely overexpressed and involved in regulating the development and progression of human solid tumors and hematological malignancies. Previous studies show that EPS8 activates MEK and stimulates Erk kinase via the Raf/MEK/Erk cascade, leading to cellular transformation and further influencing tumorigenesis and cell survival [25]. EGF activates EGFR through the recruitment of PI3K, which stimulates EPS8 and subsequently activates Akt [12,26]. Moreover, the PI3K/Akt pathway is involved in EPS8-associated tumor growth and metastasis by activating downstream targets, such as FOXM1, CXCL5 and caspase-9 [12,27]. EPS8 also participates in the EPS8/mTOR/STAT3 pathway, which regulates tumor progression [6,28]. All these data indicated that EPS8 is a significant therapeutic target in tumors. Nevertheless, prior to this study, the role of EPS8 in MM was unclear.
In this study, we showed that EPS8 expression was upregulated in MM cell lines and BMMNCs from MM patients, which makes it an attractive target in MM. We then used a lentivirus-based RNAi system to knock down EPS8 expression in MM cells (Figure 1). Genetic inhibition of EPS8 efficaciously suppressed cell proliferation, reduced colony formation and induced cell cycle arrest at G1 phase in MM cells (Figure 2). Both extrinsic and intrinsic apoptosis pathways were involved in promoting apoptosis after depletion of EPS8. EPS8 attenuation increases the sensitivity of MM cells to proteasome inhibitor. Moreover, knockdown of EPS8 reverses drug resistance in MM cells (Figure 3). Our data indicated that EPS8 expression is crucial to MM cell growth and survival in vitro and in vivo.
Previous studies indicated that EPS8 plays significant roles in the Rho-GTPase family and tumor migration. Activated Ras binds to PI3K and mediates signaling via subsequent activation of Rac through the tricomplex EPS8-Abi-1-Sos-1 [29,30]. Cdc42 regulates the synergic bundling activity of the EPS8-IRSp53 complex, contributes to the generation of actin bundles and stimulates filopodia formation through Mena [22,31]. We further investigated the role of EPS8 on the capacity of MM cells to migrate and invade. Inhibition of EPS8 reduced migration, invasion and downregulated relative Rho-GTPase expression (Figure 4). Recent studies using whole-genome and whole-exome sequencing have identified driver mutations in MM, and revealed relevant pathways in myelomagenesis [32]. The NF-κB pathway is mutated in approximately 20% of patients. Our study indicated that TNFα or bone marrow stromal cell culture supernatants induce EPS8, which is blocked by the IKKβ inhibitor MLN120B, suggesting that EPS8 is regulated by NF-κB signaling in MM cells. Inhibition of the NF-κB pathway could be a potential strategy since EPS8 is partially regulated by NF-κB (Figure 5).
It was reported that mithramycin inhibited tumor proliferation and synergized with other chemotherapeutic drugs via the downregulation of EPS8 expression [10,24]. In our study, we observed that both EPS8 mRNA and protein levels were downregulated by mithramycin, which suggests that EPS8 expression was regulated by mithramycin at the transcription level. Meanwhile, pharmacological inhibition of EPS8 efficaciously suppressed cell proliferation, induced cell cycle arrest in G1 phase and promoted apoptosis in MM cells. Moreover, a significant synergistic effect of mithramycin and bortezomib was also observed both in vitro and in vivo (Figures 6 and 7). The overexpression of EPS8 showed an opposite effect of shEPS8 on MM cells. Furthermore, the specificity of shEPS8 was verified by rescue experiments (Figure 8).
Acknowledgements
The authors are grateful to Liang Wang (Department of hematology, Zhujiang Hospital, Guangzhou, China) and Sanfang Tu (Department of hematology, Zhujiang Hospital, Guangzhou, China) for providing clinical samples. This work was supported by the National Natural Science Foundation of China [grant number 81372249], the Science and Technology Planning Project of Guangdong Province, China [grant number 2016A020213005], the Science and Technology Program of Guangzhou, China [grant number 201704020216], Guangdong Provincial Department of Education High-level University Construction Funding Southern Medical University Clinical Research Startup Program [grant number LC2016ZD027], the Natural Science Foundation of Guangdong Province, China [grant number 2018B030311042], and the Frontier Research Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory [grant number 2018GZR110105014]. The study was approved by the ethical review board of Zhujiang Hospital (Guangzhou, China).
Disclosure of conflict of interest
None.
Abbreviations
- MM
Multiple myeloma
- BTZ
Bortezomib
- MTM
Mithramycin
- EGF
Epidermal growth factor
- EPS8
Epidermal growth factor receptor pathway substrate 8
- IHC
Immunohistochemistry
- MVD
Microvessel density
- BM
Bone marrow
- BMSCs
Bone marrow stromal cells
Supporting Information
References
- 1.Fairfield H, Falank C, Avery L, Reagan MR. Multiple myeloma in the marrow: pathogenesis and treatments. Ann N Y Acad Sci. 2016;1364:32–51. doi: 10.1111/nyas.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364:1046–60. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 3.Larocca A, Palumbo A. How I treat fragile myeloma patients. Blood. 2015;126:2179–85. doi: 10.1182/blood-2015-05-612960. [DOI] [PubMed] [Google Scholar]
- 4.Goldberg AL. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans. 2007;35:12–7. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- 5.Orlowski RZ. Novel agents for multiple myeloma to overcome resistance in phase III clinical trials. Semin Oncol. 2013;40:634–51. doi: 10.1053/j.seminoncol.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maa MC, Lee JC, Chen YJ, Chen YJ, Lee YC, Wang ST, Huang CC, Chow NH, Leu TH. EPS8 facilitates cellular growth and motility of colon cancer cells by increasing the expression and activity of focal adhesion kinase. J Biol Chem. 2007;282:19399–409. doi: 10.1074/jbc.M610280200. [DOI] [PubMed] [Google Scholar]
- 7.Fazioli F, Minichiello L, Matoska V, Castagnino P, Miki T, Wong WT, Di Fiore PP. EPS8, a substrate for the epidermal growth factor receptor kinase, enhances EGF-dependent mitogenic signals. EMBO J. 1993;12:3799–808. doi: 10.1002/j.1460-2075.1993.tb06058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Logue JS, Cartagena-Rivera AX, Baird MA, Davidson MW, Chadwick RS, Waterman CM. Erk regulation of actin capping and bundling by Eps8 promotes cortex tension and leader bleb-based migration. Elife. 2015;4:e08314. doi: 10.7554/eLife.08314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cattaneo MG, Cappellini E, Vicentini LM. Silencing of Eps8 blocks migration and invasion in human glioblastoma cell lines. Exp Cell Res. 2012;318:1901–12. doi: 10.1016/j.yexcr.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 10.Gorsic LK, Stark AL, Wheeler HE, Wong SS, Im HK, Dolan ME. EPS8 inhibition increases cisplatin sensitivity in lung cancer cells. PLoS One. 2013;8:e82220. doi: 10.1371/journal.pone.0082220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen YJ, Shen MR, Chen YJ, Maa MC, Leu TH. EPS8 decreases chemosensitivity and affects survival of cervical cancer patients. Mol Cancer Ther. 2008;7:1376–85. doi: 10.1158/1535-7163.MCT-07-2388. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Patel V, Miyazaki H, Gutkind JS, Yeudall WA. Role for EPS8 in squamous carcinogenesis. Carcinogenesis. 2009;30:165–74. doi: 10.1093/carcin/bgn252. [DOI] [PubMed] [Google Scholar]
- 13.Kang H, Wilson CS, Harvey RC, Chen IM, Murphy MH, Atlas SR, Bedrick EJ, Devidas M, Carrol AJ, Robinson BW, Stam RW, Valsecchi MG, Pieters R, Heerema NA, Hilden JM, Felix CA, Reaman GH, Camitta B, Winic N, Carroll WL, Dreyer ZE, Hunger SP, Willman CL. Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a Children’s Oncology Group study. Blood. 2012;119:1872–81. doi: 10.1182/blood-2011-10-382861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Xie X, Wu A, Wang L, Hu Y, Zhang H, Li Y. A synthetic cell-penetrating peptide derived from nuclear localization signal of EPS8 exerts anticancer activity against acute myeloid leukemia. J Exp Clin Cancer Res. 2018;37:12. doi: 10.1186/s13046-018-0682-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu PY, Liou JH, Lin YM, Chen CJ, Chen MK, Lin SH, Yeh CM, Wang HK, Maa MC, Leu TH, Chang NW, Hsu NC, Yeh KT. Expression of EPS8 correlates with poor survival in oral squamous cell carcinoma. Asia Pac J Clin Oncol. 2012;8:e77–81. doi: 10.1111/j.1743-7563.2011.01459.x. [DOI] [PubMed] [Google Scholar]
- 16.He YZ, Liang Z, Wu MR, Wen Q, Deng L, Song CY, Wu BY, Tu SF, Huang R, Li YH. Overexpression of EPS8 is associated with poor prognosis in patients with acute lymphoblastic leukemia. Leuk Res. 2015;39:575–81. doi: 10.1016/j.leukres.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, Lin C, Duggan P, Tassone P, Mansoor A, Stewart DA, Boise LH, Ghobrial IM, Bahlis NJ. Integrin β7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood. 2011;117:6202–13. doi: 10.1182/blood-2010-06-292243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamorte S, Ferrero S, Aschero S, Monitillo L, Bussolati B, Omedè P, Ladetto M, Camussi G. Syndecan-1 promotes the angiogenic phenotype of multiple myeloma endothelial cells. Leukemia. 2012;26:1081–90. doi: 10.1038/leu.2011.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie X, Zhou W, Hu Y, Chen Y, Zhang H, Li Y. A dual-function epidermal growth factor receptor pathway substrate 8 (Eps8)-derived peptide exhibits a potent cytotoxic T lymphocyte-activating effect and a specific inhibitory activity. Cell Death Dis. 2018;9:379. doi: 10.1038/s41419-018-0420-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yap LF, Jenei V, Robinson CM, Moutasim K, Benn TM, Threadgold SP, Lopes V, Wei W, Thomas GJ, Paterson IC. Upregulation of Eps8 in oral squamous cell carcinoma promotes cell migration and invasion through integrin-dependent Rac1 activation. Oncogene. 2009;28:2524–34. doi: 10.1038/onc.2009.105. [DOI] [PubMed] [Google Scholar]
- 21.Chen H, Wu X, Pan ZK, Huang S. Integrity of SOS1/EPS8/ABI1 tri-complex determines ovarian cancer metastasis. Cancer Res. 2010;70:9979–90. doi: 10.1158/0008-5472.CAN-10-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Funato Y, Terabayashi T, Suenaga N, Seiki M, Takenawa T, Miki H. IRSp53/Eps8 complex is important for positive regulation of Rac and cancer cell motility/invasiveness. Cancer Res. 2004;64:5237–44. doi: 10.1158/0008-5472.CAN-04-0327. [DOI] [PubMed] [Google Scholar]
- 23.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, Dave S, Hurt EM, Tan B, Zhao H, Stephens O, Santra M, Williams DR, Dang L, Barlogie B, Shaughnessy JD Jr, Kuehl WM, Staudt LM. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–30. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang TP, Chiou HL, Maa MC, Wang CJ. Mithramycin inhibits human epithelial carcinoma cell proliferation and migration involving downregulation of Eps8 expression. Chem Biol Interact. 2010;183:181–6. doi: 10.1016/j.cbi.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 25.Li YH, Xue TY, He YZ, Du JW. Novel oncoprotein EPS8: a new target for anticancer therapy. Future Oncol. 2013;9:1587–94. doi: 10.2217/fon.13.104. [DOI] [PubMed] [Google Scholar]
- 26.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–8. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 27.Laoukili J, Kooistra MR, Brás A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–36. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- 28.Hanks SK, Calalb MB, Harper MC, Patel SK. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc Natl Acad Sci U S A. 1992;89:8487–91. doi: 10.1073/pnas.89.18.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scita G, Nordstrom J, Carbone R, Tenca P, Giardina G, Gutkind S, Bjarnegård M, Betsholtz C, Di Fiore PP. EPS8 and E3B1 transduce signals from Ras to Rac. Nature. 1999;401:290–3. doi: 10.1038/45822. [DOI] [PubMed] [Google Scholar]
- 30.Lanzetti L, Rybin V, Malabarba MG, Christoforidis S, Scita G, Zerial M, Di Fiore PP. The Eps8 protein coordinates EGF receptor signalling through Rac and trafficking through Rab5. Nature. 2000;408:374–7. doi: 10.1038/35042605. [DOI] [PubMed] [Google Scholar]
- 31.Disanza A, Mantoani S, Hertzog M, Gerboth S, Frittoli E, Steffen A, Berhoerster K, Kreienkamp HJ, Milanesi F, Di Fiore PP, Ciliberto A, Stradal TE, Scita G. Regulation of cell shape by Cdc42 is mediated by the synergic actin-bundling activity of the Eps8-IRSp53 complex. Nat Cell Biol. 2006;8:1337–47. doi: 10.1038/ncb1502. [DOI] [PubMed] [Google Scholar]
- 32.Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14:100–113. doi: 10.1038/nrclinonc.2016.122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.