

Abstract
RUNX1 is frequently mutated as chromosomal translocations in a variety of hematological malignancies. Recent studies show that RUNX1 is also mutated somatically in many solid tumors. We have recently identified a 260 kb un-spliced intragenic overlapping long noncoding RNA RUNXOR in the RUNX1 locus, yet its role as an epigenetic regulator in tumors remains to be characterized. To delineate this RUNXOR-RUNX1 regulatory interplay in breast cancer cells, we devised a novel “gene in situ cis-activation” approach to activate the endogenous RUNXOR gene. We found that the in situ activation of RUNXOR lncRNA upregulated RUNX1 in cis from the P1 promoter. The preferred activation of the P1 promoter caused a shift to the RUNX1c isoform expression. Using a chromatin conformation capture (3C) approach, we showed that RUNXOR lncRNA epigenetically activated the RUNX1 P1 promoter in cis by altering the local chromatin structure. The binding of RUNXOR lncRNA triggered DNA demethylation and induced active histone modification markers in the P1 CpG island. Changes in RUNX1 isoform composition correlated with a trend to cell cycle arrest at G0/G1, although cell proliferation rate, apoptosis, and migration ability were not significantly changed. Our results reveal an underlying epigenetic mechanism by which the lncRNA regulates in cis the RUNX1 promoter usage in breast cancer cells, thereby shedding light on potential genetic therapies in malignancies in which RUNX1 loss-of-function mutations frequently occur.
Keywords: Epigenetics, long noncoding RNA, RUNXOR, breast cancer, RUNX1
Introduction
The RUNX1 gene encodes a high-affinity DNA binding protein that heterodimerizes with CBFβ to form a core binding factor complex (CBF). This gene is commonly disrupted by chromosome translocations in acute myeloid leukemia (AML) [1-11]. The classic t(8;21) translocation juxtaposes a fragment of the RUNX1 gene on chromosomal 21 with a fragment of the RUNX1T1 gene on chromosome 8, resulting in the production of a RUNX1-RUNX1T1 chimeric protein in 12% of AML patients [5,12]. Moreover, RUNX1 is also commonly mutated in the absence of chromosome translocation [13,14]. The role of RUNX1 mutations in epithelial tumors [15-17] has recently been recognized. In breast cancers, however, RUNX1 seems to work in a context-dependent manner, with evidence supporting both tumor suppressor and oncogenic roles [18-26].
The RUNX1 gene is regulated by two promoters, the distal promoter (P1) and the proximal promoter (P2), which are separated by 150 kb [16,27,28]. Transcription from each promoter generates transcripts with different amino termini. Together with alterative splicing and posttranslational modifications [29,30], activation of different promoters can finally lead to RUNX1 proteins with varied structure and interaction profiles. There are three common RUNX1 isoforms (Figure S1) [4]. RUNX1c, transcribed from promoter P1, is the largest RUNX1 protein and is primarily expressed in hematopoietic tissues. RUNX1a and RUNX1b, which are transcribed from promoter P2, are the predominant isoforms in non-hematopoietic tissues. All three isoforms contain a conserved DNA binding domain (Runt domain). Several elements have been identified within the RUNX1 locus that presumably regulate the expression of RUNX1 in a cis manner [31-34]. Varying promoter usage and alternative splicing give rise to a mixed pool of RUNX1 proteins with different amino or carboxyl termini; however, the corresponding mechanisms of which are currently unknown [31,35,36].
To delineate the transcriptional regulation of RUNX1, we recently identified an intragenic long noncoding RNA RUNXOR that overlaps with the entire RUNX1 RNA sequence [37]. RUNXOR is a 260 kb un-spliced noncoding RNA that is widely expressed in numerous cell lines. Using a reverse transcription-associated trap (RAT) assay [38], we showed that the 3’-fragment of RUNXOR interacted with multiple sites within the RUNX1 locus [37]. However, the specific role of this RUNXOR-RUNX1 regulatory axis in breast cancer remains elusive. As a 260 kb un-spliced lncRNA, RUNXOR is very hard to be studied using conventional vectors. In this study, we devised a novel “gene in situ cis-activation” approach to activate the huge RUNXOR lncRNA gene in breast cancer cells. Using this approach, we examined the role of RUNXOR lncRNA in the regulation of RUNX1 and tumor phenotypes in cultured breast cancer cells. Because of the flexibility of gene-specific gRNAs, this in situ activation approach may be used broadly to activate other endogenous genes in tumor studies and basic research.
Materials and methods
Overexpression of RUNXOR by endogenous gene in situ activation
It was assumed that RUNXOR and RUNX1 were co-regulated based on their spatial proximity and sequence overlapping. Due to the difficulty of overexpressing the 260 kb RUNXOR in a lentiviral vector, we devised a novel “endogenous gene in situ activation” (EGIA) approach to overexpress this lncRNA in breast cancer cells. Specifically, we utilized the CRISPR Cas9 gene editing system to insert a potent CMV promoter in front of RUNXOR. Using this approach, the endogenous RUNXOR lncRNA was overexpressed under the control of the inserted CMV promoter.
To precisely target the RUNXOR promoter region for homologous recombination, we constructed a Cas9-gRNA-RUNXOR promoter targeting vector and a RUNXOR Arm-pCMV-puromycin donor vector. The primers used for vector construction was listed in Table S1. The RUNXOR gRNAs were selected using online resources (http://crispr.mit.edu). To increase the efficiency of the CRISPR system, we jointed the pU6-gRNA1-pH1-gRNA2 cassette that contained two target gRNAs in a Cas9 expression vector [39]. RUNXOR gRNA1 was 5’-GGAACAGAAGTTACCAGGAG-3’, and gRNA2 was 5’-GAGCCAGTGATGGCTTTATG-3’.
The donor vector was constructed by PCR amplification of two arm fragments from the RUNXOR locus, and cloned into a targeting vector containing both the pCMV-loxP-puromycin-GFP-loxP positive selection cassette and the pPGK-thymidine kinase negative selection marker [39]. The two arm fragments were amplified from the genome and the primers used are listed in the supplementary materials Table S1. They were cloned into the donor vector using ClaI/NheI and BsrG1/SwaI, respectively. The targeting plasmids were confirmed by DNA sequencing.
RUNXOR targeting
Human breast cancer MCF7 cells, purchased from ATCC, were co-transfected with cas9-gRNA vector and donor vector simultaneously using Lipofectamine 3000 Reagent. Two days after transfection, we collected the cells and reseeded them at low density in puromycin-containing medium (4 µg/ml). Ganciclovir were added to the medium seven days after reseeding when individual clones had formed. The addition of ganciclovir eliminated those clones in which the vectors were randomly inserted into the genome [39]. After selection, stable clones were collected, and the genomic DNA was extracted for screening using primers listed in Table S1.
Quantitative analysis of RUNXOR/RUNX1 expression
Total RNA was extracted from cell pellets by TRI-REAGENT (Sigma, CA), and was reverse transcribed following the protocol as previously described [40]. The resulting cDNA was used for quantitative PCR to determine the relative expression of RUNXOR, RUNX1 and other genes using the primers listed in Table S2. For comparative analysis, the mRNA levels of the tested genes were quantified by normalizing the Ct value of the target gene over that of β-Actin as previously described [32].
Western blotting analysis of RUNX1
Western blot was performed to validate the upregulated RUNX1 level in the RUNXOR targeted MCF7 cells. Total cell protein was extracted with RIPA buffer (P0013B, Beyotime) supplemented with 1×Protease Inhibitor Tablets (Sigma), and protein concentration was determined using Pierce BCA protein kit. Immunoblot was incubated with anti-RUNX1 rabbit polyclonal IgG antibody (Abcam, ab23980), anti-β-Actin mouse monoclonal IgG antibody (Abcam, ab8226) diluted in 5% BSA overnight at 4°C. For secondary antibodies we used IRDye 680 goat anti-rabbit IgG and IRDye 800 goat anti-mouse IgG (LI-COR). The compatible Odyssey Infrared Imaging System was used for proper imaging.
Chromosome conformation capture (3C)
3C analysis was performed as previously described [41]. Briefly, chromatin was digested with MboI, ligated with T4 DNA Ligase, and the ligated 3C products were quantitated by Q-PCR. The amplicon ERCC3 was chosen to serve as an internal standard accounting for the differences in the efficiency of crosslinking/restriction digestion/ligation as well as quantities of DNA samples obtained from different cells. The primers used was listed in Table S3.
DNA methylation analysis
To assess the DNA methylation status of the promoters and enhancers of the RUNX1 gene, sodium bisulfite sequencing was performed using reagents provided in the EZ DNA Methylation-GoldTM Kit (ZYMO Research Corporation, D5005), as described elsewhere [39,42]. PCR products were cloned into a pJET vector (Thermo ScientificTM K1231) for sequencing. After DNA sequencing, comparison between different samples was achieved by calculating the T/C+T ratios at selected sites. The primers used for PCR was listed in Table S4.
ChIP-qPCR
As previously described [43], the chromatin complex was incubated with anti-H3K4me3, anti-H3K9me2 and anti-H3K27me3 antibodies (9727S, 13969S, and 9733S Cell Signaling Technology) and immunoprecipitated by 20 µl prewashed protein A/G magnetic beads (88802, Thermo ScientificTM). The immunoprecipitated chromatin DNA was quantitated by Q-PCR, and calculated as fold enrichment after normalization over the signals of the IgG group. The primers were listed in Table S5.
Cell cycle and apoptosis analysis
Cells cycle was analyzed using the method as previously described [44]. To measure apoptosis, cells were stained with FITC Annexin V and 7-AAD consecutively, and were analyzed using the FACScan flow cytometer (BD Biosciences, CA). Data on cell cycle distribution and apoptosis were analyzed using ModFit LT 3.0 software [44] and FlowJo software, respectively.
Cell migration and proliferation assays
Cell migration was examined by transwell migration assay [32,40]. Cell proliferation was measured using the CCK-8 kit [44]. Viable cells were incubated with 10 µl CCK-8 reagent and the absorbance was measured at 450 nm. The proliferation rate was calculated based on the ratio of absorbance tested between two time points of the same well.
Statistical analysis
All experiments were performed in triplicates, and all experimental data were presented as mean ± S.E.M. Student’s t test was chosen to compare statistical differences for variables between different groups using PRISM software. Results were considered significant when P<0.05.
Results
Positive correlation between RUNX1 and its intragenic lncRNA RUNXOR
RUNXOR is an intragenic lncRNA that is transcribed from a promoter that is 3.8 kb upstream of the RUNX1 promoter (Figure 1A). RUNXOR spans the entire RUNX1 locus, including its P1 and P2 promoters, and shares the majority of its sequence with RUNX1. Despite the spatial proximity of RUNXOR with the RUNX1 coding gene, it is unclear if and how they are co-regulated in breast cancer cells.
Figure 1.

The RUNX1-RUNXOR epigenetic interplay in breast cancer cells. A. Structure of the RUNX1/RUNXOR locus. RUNX1 is controlled by two promoters: distal promoter P1 and proximal promoter P2, which are separated by 150 kb. RUNXOR is transcribed from a promoter 3.8 kb upstream of the RUNX1 distal promoter P1. These two genes are transcribed in the same direction and share most of the sequence (colored red and green, respectively). Black block: RUNX1 exon 1-9; blue ovals: RUNX1 enhancer; purple oval: RUNX1 silencer, RE1, RE2: enhancers 1-2; pRUNX1: RUNX1 promoters; pRUNXOR: RUNXOR lncRNA promoter. B. Positive correlation between RUNX1 mRNA and RUNXOR lncRNA in cancer cells. Dot plot showed the relative levels of RUNX1 and RUNXOR in cell lines of different tissue origins calculated as (1/ave Ct) ×100). Pearson correlation analysis was performed using PRISM 6. C. RUNXOR lncRNA-mediated epigenetic regulation of oncogenic RUNX1 mRNA gene in breast cancer cells.
To investigate the relation between these two genes, we compared the expression of RUNXOR and RUNX1 in a large group of tumor cell lines of different tissue origins. To distinguish the RUNXOR lncRNA from the RUNX1 coding RNA, we performed PCR using primers specific to each transcript (Table S2). We found that there was a significant positive correlation between RUNXOR and RUNX1 expression levels, with the Pearson correlation coefficient of 0.8790, P<0.0001 (Figure 1B). We attempted to test the hypothesis that as a long noncoding RNA, RUNXOR may participate in the regulation of its overlapping RUNX1 mRNA gene in cis, which is involved in chromosome translocations in hematological malignancies and somatic mutations in solid tumors (Figure 1C).
CRISPR Cas9 mediated RUNXOR endogenous in situ activation
RUNXOR is a 260 kb un-spliced noncoding RNA. It was difficult to decipher its functions through overexpression using traditional methods. We thus devised a novel CRISPR cas9-mediated gene in situ cis-activation system to activate the endogenous RUNXOR (Figure 2A). With the aid of the CRISPR-Cas9 gene editing system, we inserted a strong cytomegalovirus promoter (pCMV) in front of RUNXOR. Using this approach, we hoped to achieve the endogenous overexpression of RUNXOR in situ.
Figure 2.

The cis-overexpression of RUNXOR by CRISPR Cas9 knock-in system. Activation of the endogenous RUNXOR lncRNA gene by the “gene in situ cis-activation” system. A. Schematic diagram illustrating the gene in situ cis-activation strategy. Cas9: CRISPR Cas9; gRNA: Cas9 guiding RNA; pCMV: CMV promoter; pEF1: EF1 promoter; pRUNXOR: RUNXOR promoter; pH1: RNA polymerase III H1 promoter; Cre: Cre recombinase; loxP: the locus of X-over P1 recombination site recognized by Cre; Arm 1, Arm2: the genomic sequences used for Cas9-mediated recombination. Under the guidance of gRNA, Cas9 mediated genomic recombination at the RUNXOR promoter loci, resulting in the insertion of the strong CMV promoter in front of RUNXOR. After transfection, cell clones positive for puromycin and negative for ganciclovir were selected and validated by sequencing. The chosen clones were then transiently transfected with a vector expressing Cre recombinase to eliminate the puromycin/GFP selection cassette. B. Initial screening of targeted clones by PCR. Positive clones were selected by targeting primers located in the targeting vector and the RUNXOR region beyond Arm 1 and Arm 2, respectively. Primers were designed from the RUNXOR arm, selection marker, and genomic regions. Arm 1: JH5859/JH5860; Arm 2: JH745/JH5826. Using these pairs of primers, only the targeted clones can be amplified. Different lanes represent different clones. C. Cis-overexpression of RUNXOR lncRNA in the selected knock-in clones. Total mRNAs extracted from the aforementioned clones were subjected to RT-qPCR. The control was set as 1 for standardization. The data were shown as mean ± S.E.M of three independent experiments. Results were considered significant if P<0.05. *P<0.05, **P<0.01; ***P<0.001.
To achieve this goal, we constructed a targeting vector by inserting two gRNAs in a Cas9 vector to target the region in front of RUNXOR and a donor vector that contains a strong pCMV-puromycin/GFP selection markers cassette flanked by two arm sequences from the targeting region of RUNXOR [39]. Both vectors were co-transfected into breast cancer MCF7 cells. With the guidance of gRNAs, Cas9 nuclease introduced a genomic break at the targeting site in front of RUNXOR. Through the donor vector dependent homologous recombination repair (HDR), the pCMV-puromycin/GFP cassette was inserted in front of the RUNXOR promoter. Puromycin was used to select the positive recombination clones. Ganciclovir selection was used to reduce the possibility of random integration of the donor vector into the genome (Figure 2A).
After puromycin and ganciclovir double selection, stable cells clones were collected, and genomic DNA was isolated to confirm the proper knock-in using PCR sequencing. The primer pairs were designed to cover the 5’-upstream genomic arm or the 3’-downstream genomic arm and the inserted CMV-puro/GFP cassette (JH5860/JH5859 for the 5’-arm; JH5826/JH745 for the 3’-arm). In total, seven positive clones were selected (Figure 2B). DNA sequencing was used to confirm the homologous insertion of the pCMV-puro/GFP cassette (Figure S2).
We then used qRT-PCR to examine the expression of RUNXOR in selected knock-in clones. Two groups of controls were used for comparison, including an untreated control (Ctrl) and a random insertion control (C2), in which the pCMV-puro/GFP cassette was randomly inserted in the genome of MCF7 cells. Compared with these two controls, all 7 clones expressed high levels of RUNXOR, with clone A3 showing the highest RUNXOR level (Figure 2C). Thus, clone A3 was chosen for the subsequent studies.
RUNXOR lncRNA preferentially upregulates RUNX1 P1 transcription
We then evaluated if the overexpressed RUNXOR could affect the expression of its overlapping coding gene RUNX1. Notably, we found that compared with the Ctrl and the randomly-inserted C2 controls, RUNXOR lncRNA in situ overexpression significantly upregulated RUNX1 coding mRNA abundance (Figure 3A). Using western blotting, we confirmed the upregulation of RUNX1 at the protein level (Figure 3B). The multiple bands in western blot represents distinct RUNX1 isoforms which are generated through transcription from different promoters and alternative splicing.
Figure 3.
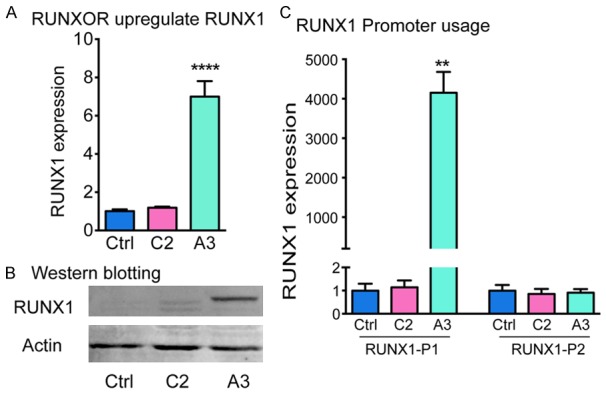
Cis-overexpression of RUNXOR specifically activates the RUNX1 P1 transcripts. A. Expression of RUNX1 mRNAs in the RUNXOR cis-overexpressing cell clone. Total mRNA was extracted for RT-qPCR. Ctrl: MCF7 control cells; C2: MCF7 clone cells with CMV/puromycin/GFP random insertion; A3: The RUNXOR knock-in MCF7 clone cells. Note the activation of RUNX1 in the RUNXOR-overexpressing A3 cells. B. Western blot analysis of RUNX1. The protein level of RUNX1 was measured by Western blot. Beta-ACTIN was used as the loading control. C. Distinct expression of RUNX1 isoforms. The expression of RUNX1 isoforms was quantitated by qPCR using primers specific for each promoter. RUNX1 P1 represents the transcripts from the P1 promoter. RUNX1 P2 represents the transcript from the P2 promoter. All results were performed in triplicates and were shown as mean ± S.E.M of three independent experiments. The values of the control (Ctrl) were set as 1 for standardization. Results were considered significant if P<0.05. *P<0.05, **P<0.01.
We then sought to determine which promoter accounted for the elevated RUNX1 mRNA. By using forward primers specific to P1- and P2-derived transcripts and a common reverse primer located in the second exon of the Runt domain (exon 5), we were able to distinguish between different RUNX1 transcripts. We found that the transcripts from the RUNX1 P1 promoter were significantly increased upon RUNXOR elevation, while the transcripts derived from RUNX1 P2 promoter remained unchanged (Figure 3C).
We also tested the correlation between the abundance of different RUNX1 transcripts with RUNXOR in cancer cell lines (Figure S3). Consistent with the aforementioned experiments, these results showed that RUNXOR was positively correlated with RUNX1 transcripts from promoter P1. No correlation was found between RUNXOR and RUNX1 transcribed from promoter P2. Together, these data suggested that RUNXOR lncRNA upregulated RUNX1 primarily through the activation of the RUNX1 P1 promoter.
RUNXOR alters the spatial chromatin structure of the RUNX1 gene
Previous studies have demonstrated the existence of a common transcription hub at the RUNX1 locus composed of P1, P2, two enhancers RE1 and RE2. This functional three-dimensional organization was believed to be important for the regulation of RUNX1 transcription and alternative promoter usage. To explore the possible roles of RUNXOR in orchestrating the RUNX1 chromosome structure, we used chromatin conformation capture (3C) to assess the potential DNA interactions (Figure 4A).
Figure 4.

Changes of RUNX1 local chromatin structures upon RUNXOR overexpression. RUNXOR triggers the alteration of RUNX1 local chromatin structures. A. Location of the 3C PCR primer sets used to quantitate the intrachromosomal loop between the cis-regulating elements within the RUNX1 locus. B. Quantitation of the 3C products by qPCR. ERCC3, a housekeeping gene, was used as the 3C internal control for normalization of the discrepancies of digestion/ligation efficiency and DNA quantities between the control and A3 groups. Q-PCR was performed in triplicate and shown as mean ± S.E.M. C. Confirmation of the 3C products by DNA sequencing. Red arrows: the site of ligated Mbo1 restriction enzyme site flanked by genomic DNAs from two remote areas, respectively.
The cells were fixed with formaldehyde, digested with restriction enzyme MboI, and re-ligated with T4 DNA ligase. The ligated 3C products were quantitated with 3C PCR primers that are located at two remote regions. The 3C data were quantitated by normalizing over that of the house-keeping control gene ERCC3 as previously reported [45]. We observed decreased interaction between the P1/P2 promoters in the RUNXOR-overexpressing cells. On the other hand, more interactions were observed between P1 and RE2. The interaction between P1 and RE1 was also decreased (Figure 4B). The 3C interactions were confirmed by DNA sequencing (Figure 4C). These data suggested that RUNXOR altered the local 3D chromatin structure of RUNX1.
RUNXOR induces DNA demethylation in the RUNX1 P1 promoter
We further examined whether the altered RUNX1 promoter preference was related to DNA methylation in the locus. Sodium bisulfite sequencing was used to determine the status of DNA methylation in the RUNX1 promoters and enhancers (Figure 5A). CpG islands near the promoter P1 were heavily methylated in control groups Ctrl and C2. The RUNXOR in situ overexpression group A3, however, shifted to a relatively hypomethylated status in the P1 promoter (Figure 5B). Similar patterns of DNA demethylation were also observed at the CpG sites in enhancer RE1, in parallel with the increased transcriptional activity of promoter P1 (Figure 5B). In contrast, the CpG islands in promoter P2 were hypomethylated in control cells and were not affected by RUNXOR overexpression (Figure 5B).
Figure 5.
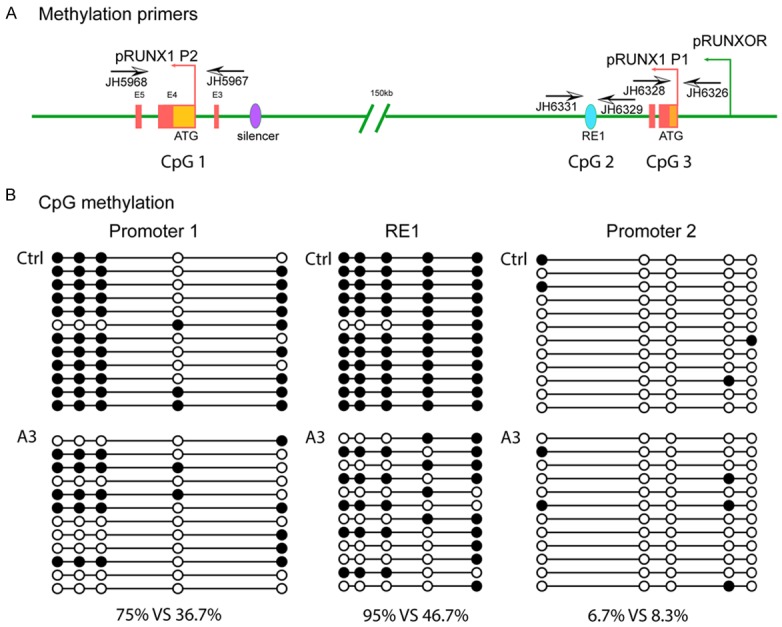
RUNXOR overexpression changes the methylation status of RUNX1. RUNXOR changes the methylation status of RUNX1. A. Schematic diagram of the RUNX1/RUNXOR locus and the primers used for methylation analysis. B. DNA CpG methylation status of the RUNX1 P1 promoter, RE1 and P2 promoter. P1 promoter: DNA methylation was decreased from 75% to 36.7% in the RUNX1 P1 promoter after RUNXOR overexpression. RE1: There was a significant demethylation in the A3 group (46.7%) as compared with the control group (95%). P2 promoter: Genomic DNA was hypomethylated in both groups. Open circles: unmethylated CpGs; solid circles: methylated CpGs.
RUNXOR increases histone H3K4me3 epigenetic marks in promoter P1
To examine if histone modification is involved in the RUNX1 promoter usage regulation, we performed chromatin immunoprecipitation qPCR (ChIP-qPCR) to quantify the changes of histone methylation marker in promoter P1, including H3K4me3, H3K9me2, and H3K27me3. We found that the H3K4me3 mark was increased more than twentyfold in RUNXOR overexpressing cells (Figure 6A). However, no significant changes were seen in H3K9me2 and H3K27me3 (Figure 6B, 6C).
Figure 6.

Alteration of histone methylation in the RUNX1 distal promoter P1. RUNXOR alters histone methylation in the RUNX1 distal promoter P1. The chromatin complex was fixed with formaldehyde and was immunoprecipitated with antibodies targeting H3K4me3 (A), H3K9me2 (B) and H3K27me3 (C), respectively. DNA from the pulled down chromatin was purified and subjected to qPCR using primers that targeted the RUNX1 P1. Results are shown as relative to control for standardization. P<0.05 was regarded as significant. **P<0.01.
Targeted overexpression of RUNX1 isoform failed to alter the phenotype of MCF7 breast tumor cells
We then asked whether the elevated RUNX1, specifically the RUNX1c transcript, could affect the tumor phenotype of MCF7 cells. First, we compared apoptosis by Annexin V labeling, but failed to detect any differences in the RUNXOR-overexpressed cells (Figure 7A). However, cell cycle analysis revealed that there was a trend, towards the arrest of cells at G0/G1 phase in the treated group where RUNXOR was overexpressed (Figure 7B).
Figure 7.

Phenotypic characterization of MCF7 cells upon RUNXOR cis-overexpression. A. Apoptotic analysis. Cells stained by Annexin V and 7-AAD were subjected for analysis using flow cytometer. Apoptotic cells were characterized as Annexin V positive/7-AAD negative (marked as Q3 at the fourth quadrant), and comparisons made were in triplicates. B. Cell cycle analysis. Cells stained with propidium iodide were analyzed using flow cytometry and data processing was performed using ModFit LT.
RUNXOR overexpression-induced upregulation of RUNX1, specifically RUNX1c, did not affect cell proliferation rate in MCF7 breast cancer cells (Figure S4A). In addition, a transwell migration assay did not show any significant differences in the RUNXOR over-expressing cells compared with control groups (Figure S4B).
Discussion
RUNX1 is a master regulator of hematopoiesis and its spatiotemporal expression plays a crucial role in normal hematopoietic development. Mutations of RUNX1 are frequently involved in malignant diseases. In this study, using a novel CRISPR cas9 mediated endogenous in situ activation approach we demonstrate that intragenic lncRNA RUNXOR plays a key role in the epigenetic regulation of RUNX1 expression in breast cancer MCF7 cells. LncRNA RUNXOR overexpression increases the transcription of RUNX1 from the distal promoter P1, while the activity of the proximal promoter P2 is not affected. We further show that RUNXOR functions through a cis epigenetic mechanism, primarily by altering the local chromatin structure and epigenotypes in the promoter. This study suggests a close epigenetic interplay between the lncRNA RUNXOR and the oncogenic RUNX1 in breast cancer cells.
The RUNX1 gene is transcriptionally controlled by two promoters [27]. The two promoters and their corresponding transcripts are thought to be non-redundant, and transcription is triggered at varying developmental stages [2]. However, the mechanisms controlling the alternative promoter usages are unknown. Although several cis-regulating elements have been located in the RUNX1 locus, for example, a highly conserved enhancer within the first intron has been found to be responsible for enhancing transcription activities of the RUNX1 gene during hematopoiesis [31,34]. Besides, Cheng et al recently reported a P2-specific silencer near the P2 within the intron 1 [33]. Currently, few studies related to the P1 promoter have been published.
The long intragenic lncRNA RUNXOR we recently discovered, is transcribed 3.8 kb upstream of the RUNX1 promoter and overlaps the whole RUNX1 transcripts [37]. Reverse transcription-associated trap (RAT) experiment demonstrated multiple interactions between the 3’ segment of RUNXOR and the RUNX1 locus, indicating its possible role in regional transcription regulation. Thus, we made the assumption that RUNXOR might interfere with RUNX1 transcription in cis through mechanisms, such as “transcription interference” [41,46-49], judging by their spatial propinquity [49]. Using Pearson correlation analysis, we found that there was indeed a positive correlation between RUNX1 and RUNXOR transcript levels (r=0.879, P<0.0001). This positive relationship led us to wonder whether RUNXOR was responsible for the transition between RUNX1 promoter changes. We performed qRT-PCR using the aforementioned isoform specific primers in a series of cell lines of different origins. We found that RUNXOR was positively correlated with RUNX1 transcribed from promoter P1 but not from promoter P2. These results suggest that RUNXOR could be a crucial element in charge of P1/P2 switch during hematopoiesis and potentially tumorigenesis.
To test our hypothesis, we decided to further explore the roles of RUNXOR in cell models. However, given its enormous length, it is hard to investigate RUNXOR using traditional loss/gain of function strategies, such as RNA interference or overexpression. Particularly, it would be impossible to construct a vector containing the 260 kb long sequence. In addition, we recently demonstrate that the trans-expressed lncRNA may function differently from the cis-expressed lncRNA [39]. Thus, we used a Cas9-gRNA mediated homology recombination method to insert a strong promoter in front of RUNXOR, so that RUNXOR overexpression can be achieved by transcription from a potent promoter. Most importantly, this overexpression happens in situ, which can better mimic the endogenous environment [50].
In accord with the aforementioned correlation analysis, both RUNX1 RNA and protein were significantly elevated after RUNXOR overexpression in situ. To further clarify where the “extra” RUNX1 was originated, we used isoform specific primers to distinguish between transcripts from different promoters. Results indicated that transcripts from P1 were elevated while transcription from P2 remained uninfluenced. This was in accordance with the western blot results in which a band of a larger size were detected in the RUNXOR overexpressing cells. Together, these results indicate that lncRNA RUNXOR could elevate the expression of RUNX1 specifically through increasing transcription from the promoter P1, suggesting its possible roles in the regulation of the alternative RUNX1 promoter usages.
Mechanisms through which RUNXOR favors P1 over P2 were further explored. Chromosome conformation capture assay showed a change of RUNX1 chromatin spatial organization after RUNXOR overexpression. The crosslinking frequency was decreased between P1/P2 and P1/RE1, and increased between P1 and RE2. No crosslinking ligation product could be detected between the newly reported silencer with any promoters [33]. The RUNX1 gene locus is quite interesting since reports have shown that both promoters and enhancers were assembled into an active chromatin hub [34], which was supposed to be important for its regulation. The enhancer RE1 has hematopoietic enhancer activities yet has no promoter specificity. On the contrary, RE2 lacks any enhancer activity and appears to be essential for the assembly of these hubs [34]. The local changes of chromatin interactions we observed here indicate a possible role of RUNXOR in the maintenance and regulation of functional chromosome structures, which might be responsible for the observed promoter switch.
DNA methylation is a major epigenetic means of transcriptional regulation of the RUNX1 locus [28,51]. Promoter P2 is nested in an evolutionally conserved CpG enriched region, which was shown to remain unmethylated among all cells tested. This is consistent with our results here, thus provided little contributions in the observed promoter switch. Promoter P1, however, is comparatively CpG poor, and a higher degree of methylation was observed in the cells tested. Overexpression of RUNXOR led to a significantly lower levels of methylation, consistent with the higher level of transcription upon RUNXOR elevation. Besides, there was also a clear trend of demethylation at the enhancer RE1. Together, these data suggested that RUNXOR activated RUNX1 distal promoter P1 by DNA demethylation of the P1 promoter and enhancer RE1.
A group of researchers recently developed a new class of RNA named DNMT1-interacting RNA. The DNA methylation levels of CEBPA promoter were inversely correlated with a lncRNA ecCEBPA that encompassed the entire CEBPA mRNA sequence in the same sense orientation, and further studies demonstrated that the lncRNA ecCEBPA had higher affinity with DNA methyltransferase 1 (DNMT1) than the DNA sequence of its overlapping coding gene CEBPA, and could function in cis as a decoy by sequestering DNMT1 from the CEBPA promoter [41]. There is no reason to doubt that the lncRNA RUNXOR we described here could work in a similar way as ecCEBPA, and our results is a critical support to the hypothesis that RNA can participate in the establishment of genomic methylation patterns in a site-specific manner, although further experiments such as anti-DNMT1 RNA Immunoprecipitation (RIP) and electrophoretic mobility shift assay (EMSA) are essentially needed.
Methylation modifications of histones are another kind of epigenetic regulation critical to development. We also show that more H3K4me3 appeared at the P1 promoter, leading to a more open chromatin structure that favors active transcription. Together, we showed that lncRNA RUNXOR selectively increased RUNX1 transcription from the distal promoter P1 through demethylation of the P1 CpG island and increasing H3K4me3 modifications. This is the first article describing a possible mechanism for the involvement of noncoding RNA in the regulation of alternative RUNX1 promoter usage. The LncRNA RUNXOR we discussed here is enormous long, and is supposed to overlap the two RUNXOR promoters. The fact that only the distal promoter was activated makes us wonder if the regulatory role of RUNXOR in RUNX1 promoter switch was more of a local function than an actual sequence dependent event. Further experiments are needed to dissect the details.
RUNX1 plays its part in a context dependent manner [15], and it can switch from a tumor suppressor to an oncogene depending upon its environment. Estrogen receptor status [23,52,53], intact p53 [54,55] or Rb1 [20], homeostasis of other related signaling factors [22,56], and even proper expression of other RUNX family members are important in determining its activity [25]. We are the first ones to study the effect of different RUNX1 isoforms on ER+ breast cancer malignancies which is an important supplement to the current studies. Using our in situ overexpression approach, we showed that RUNXOR lncRNA mediated alteration in RUNX1 isoform composition, which subsequently induced cell cycle arrest at G0/G1. However, other tumor phenotypes seemed unaltered in this breast cancer cell model, including cell proliferation rate, apoptosis, and migration ability. This also reminds us of the somehow contradictory results concerning the roles of RUNX1 in solid tumors that have been published so far.
In conclusion, our study demonstrates a novel lncRNA RUNXOR-oncogenic RUNX1 epigenetic interplay mechanism in breast cancers. Notably, the lncRNA RUNXOR epigenetically activates the oncogenic RUNX1 mRNA selectively from the promoter P1. Mechanistically, RUNXOR lncRNA regulates the RUNX1 mRNA gene by altering the local chromatin structure, triggering DNA demethylation, and inducing active histone modification markers in the P1 CpG island. The activation of this RUNXOR-RUNX1 epigenetic axis induced a block in G0/G1 phase in MCF7 breast cancer cells. Our studies may shed light on potential genetic therapies in malignancies in which RUNX1 loss-of-function mutations frequently occur.
Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFA0106902), the National Natural Science Foundation of China (31430021, 81372835, 81670143, 81672275, 81500116, 81601449), the National Basic Research Program of China (973 Program) (2015CB943303), Natural Science Foundation of Jilin Science and Technique (20180101117JC), and California Institute of Regenerative Medicine (CIRM) grant (RT2-01942), Jilin Science and Technique International Collaboration grant (20130413010GH), and the Department of Veterans Affairs (BX002905).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature. 2009;457:887–891. doi: 10.1038/nature07619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bee T, Swiers G, Muroi S, Pozner A, Nottingham W, Santos AC, Li PS, Taniuchi I, de Bruijn MF. Nonredundant roles for Runx1 alternative promoters reflect their activity at discrete stages of developmental hematopoiesis. Blood. 2010;115:3042–3050. doi: 10.1182/blood-2009-08-238626. [DOI] [PubMed] [Google Scholar]
- 3.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 4.Lam K, Zhang DE. RUNX1 and RUNX1-ETO: roles in hematopoiesis and leukemogenesis. Front Biosci (Landmark Ed) 2012;17:1120–1139. doi: 10.2741/3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peterson LF, Zhang DE. The 8;21 translocation in leukemogenesis. Oncogene. 2004;23:4255–4262. doi: 10.1038/sj.onc.1207727. [DOI] [PubMed] [Google Scholar]
- 6.Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. 2002;2:502–513. doi: 10.1038/nrc840. [DOI] [PubMed] [Google Scholar]
- 7.Lichtinger M, Hoogenkamp M, Krysinska H, Ingram R, Bonifer C. Chromatin regulation by RUNX1. Blood Cells Mol Dis. 2010;44:287–290. doi: 10.1016/j.bcmd.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Link KA, Chou FS, Mulloy JC. Core binding factor at the crossroads: determining the fate of the HSC. J Cell Physiol. 2010;222:50–56. doi: 10.1002/jcp.21950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedman AD. Leukemogenesis by CBF oncoproteins. Leukemia. 1999;13:1932–1942. doi: 10.1038/sj.leu.2401590. [DOI] [PubMed] [Google Scholar]
- 10.Yamagata T, Maki K, Mitani K. Runx1/AML1 in normal and abnormal hematopoiesis. Int J Hematol. 2005;82:1–8. doi: 10.1532/IJH97.05075. [DOI] [PubMed] [Google Scholar]
- 11.Rossetti S, Sacchi N. RUNX1: A MicroRNA Hub in Normal and Malignant Hematopoiesis. Int J Mol Sci. 2013;14:1566–1588. doi: 10.3390/ijms14011566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niebuhr B, Fischer M, Tager M, Cammenga J, Stocking C. Gatekeeper function of the RUNX1 transcription factor in acute leukemia. Blood Cells Mol Dis. 2008;40:211–218. doi: 10.1016/j.bcmd.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 13.Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, Ardlie KG, Auclair D, Bautista-Pina V, Duke F, Francis J, Jung J, Maffuz-Aziz A, Onofrio RC, Parkin M, Pho NH, Quintanar-Jurado V, Ramos AH, Rebollar-Vega R, Rodriguez-Cuevas S, Romero-Cordoba SL, Schumacher SE, Stransky N, Thompson KM, Uribe-Figueroa L, Baselga J, Beroukhim R, Polyak K, Sgroi DC, Richardson AL, Jimenez-Sanchez G, Lander ES, Gabriel SB, Garraway LA, Golub TR, Melendez-Zajgla J, Toker A, Getz G, Hidalgo-Miranda A, Meyerson M. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, Snider J, Ballman K, Weber J, Chen K, Koboldt DC, Kandoth C, Schierding WS, McMichael JF, Miller CA, Lu C, Harris CC, McLellan MD, Wendl MC, DeSchryver K, Allred DC, Esserman L, Unzeitig G, Margenthaler J, Babiera GV, Marcom PK, Guenther JM, Leitch M, Hunt K, Olson J, Tao Y, Maher CA, Fulton LL, Fulton RS, Harrison M, Oberkfell B, Du F, Demeter R, Vickery TL, Elhammali A, Piwnica-Worms H, McDonald S, Watson M, Dooling DJ, Ota D, Chang LW, Bose R, Ley TJ, Piwnica-Worms D, Stuart JM, Wilson RK, Mardis ER. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheitz CJ, Tumbar T. New insights into the role of Runx1 in epithelial stem cell biology and pathology. J Cell Biochem. 2013;114:985–993. doi: 10.1002/jcb.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blyth K, Cameron ER, Neil JC. The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer. 2005;5:376–387. doi: 10.1038/nrc1607. [DOI] [PubMed] [Google Scholar]
- 17.Ito Y, Bae SC, Chuang LS. The RUNX family: developmental regulators in cancer. Nat Rev Cancer. 2015;15:81–95. doi: 10.1038/nrc3877. [DOI] [PubMed] [Google Scholar]
- 18.Chimge NO, Frenkel B. The RUNX family in breast cancer: relationships with estrogen signaling. Oncogene. 2013;32:2121–2130. doi: 10.1038/onc.2012.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kadota M, Yang HH, Gomez B, Sato M, Clifford RJ, Meerzaman D, Dunn BK, Wakefield LM, Lee MP. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS One. 2010;5:e9201. doi: 10.1371/journal.pone.0009201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Brugge JS, Janes KA. Intersection of FOXO- and RUNX1-mediated gene expression programs in single breast epithelial cells during morphogenesis and tumor progression. Proc Natl Acad Sci U S A. 2011;108:E803–812. doi: 10.1073/pnas.1103423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janes KA. RUNX1 and its understudied role in breast cancer. Cell Cycle. 2011;10:3461–3465. doi: 10.4161/cc.10.20.18029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong D, Fritz AJ, Finstad KH, Fitzgerald MP, Weinheimer A, Viens AL, Ramsey J, Stein JL, Lian JB, Stein GS. Suppression of breast cancer stem cells and tumor growth by the RUNX1 transcription factor. Mol Cancer Res. 2018;16:1952–1964. doi: 10.1158/1541-7786.MCR-18-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Bragt MP, Hu X, Xie Y, Li Z. RUNX1, a transcription factor mutated in breast cancer, controls the fate of ER-positive mammary luminal cells. Elife. 2014;3:e03881. doi: 10.7554/eLife.03881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chimge NO, Ahmed-Alnassar S, Frenkel B. Relationship between RUNX1 and AXIN1 in ER-negative versus ER-positive breast cancer. Cell Cycle. 2017;16:312–318. doi: 10.1080/15384101.2016.1237325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulkarni M, Tan TZ, Syed Sulaiman NB, Lamar JM, Bansal P, Cui J, Qiao Y, Ito Y. RUNX1 and RUNX3 protect against YAP-mediated EMT, stem-ness and shorter survival outcomes in breast cancer. Oncotarget. 2018;9:14175–14192. doi: 10.18632/oncotarget.24419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrari N, Mohammed ZM, Nixon C, Mason SM, Mallon E, McMillan DC, Morris JS, Cameron ER, Edwards J, Blyth K. Expression of RUNX1 correlates with poor patient prognosis in triple negative breast cancer. PLoS One. 2014;9:e100759. doi: 10.1371/journal.pone.0100759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levanon D, Groner Y. Structure and regulated expression of mammalian RUNX genes. Oncogene. 2004;23:4211–4219. doi: 10.1038/sj.onc.1207670. [DOI] [PubMed] [Google Scholar]
- 28.Levanon D, Glusman G, Bangsow T, Ben-Asher E, Male DA, Avidan N, Bangsow C, Hattori M, Taylor TD, Taudien S, Blechschmidt K, Shimizu N, Rosenthal A, Sakaki Y, Lancet D, Groner Y. Architecture and anatomy of the genomic locus encoding the human leukemia-associated transcription factor RUNX1/AML1. Gene. 2001;262:23–33. doi: 10.1016/s0378-1119(00)00532-1. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Zhang Q, Zhang DE, Zhou C, Xing H, Tian Z, Rao Q, Wang M, Wang J. Overexpression of an isoform of AML1 in acute leukemia and its potential role in leukemogenesis. Leukemia. 2009;23:739–745. doi: 10.1038/leu.2008.350. [DOI] [PubMed] [Google Scholar]
- 30.Wu H, Zheng J, Deng J, Zhang L, Li N, Li W, Li F, Lu J, Zhou Y. LincRNA-uc002yug.2 involves in alternative splicing of RUNX1 and serves as a predictor for esophageal cancer and prognosis. Oncogene. 2015;34:4723–4734. doi: 10.1038/onc.2014.400. [DOI] [PubMed] [Google Scholar]
- 31.Ng CE, Yokomizo T, Yamashita N, Cirovic B, Jin H, Wen Z, Ito Y, Osato M. A Runx1 intronic enhancer marks hemogenic endothelial cells and hematopoietic stem cells. Stem Cells. 2010;28:1869–1881. doi: 10.1002/stem.507. [DOI] [PubMed] [Google Scholar]
- 32.Cavalcante de Andrade Silva M, Krepischi ACV, Kulikowski LD, Zanardo EA, Nardinelli L, Leal AM, Costa SS, Muto NH, Rocha V, Velloso EDRP. Deletion of RUNX1 exons 1 and 2 associated with familial platelet disorder with propensity to acute myeloid leukemia. Cancer Genet. 2018;222-223:32–37. doi: 10.1016/j.cancergen.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 33.Cheng CK, Wong THY, Wan TSK, Wang AZ, Chan NPH, Chan NCN, Li CK, Ng MHL. RUNX1 upregulation via disruption of long-range transcriptional control by a novel t(5;21)(q13;q22) translocation in acute myeloid leukemia. Mol Cancer. 2018;17:133. doi: 10.1186/s12943-018-0881-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Markova EN, Kantidze OL, Razin SV. Transcriptional regulation and spatial organisation of the human AML1/RUNX1 gene. J Cell Biochem. 2011;112:1997–2005. doi: 10.1002/jcb.23117. [DOI] [PubMed] [Google Scholar]
- 35.Martinez M, Hinojosa M, Trombly D, Morin V, Stein J, Stein G, Javed A, Gutierrez SE. Transcriptional Auto-Regulation of RUNX1 P1 Promoter. PLoS One. 2016;11:e0149119. doi: 10.1371/journal.pone.0149119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sroczynska P, Lancrin C, Kouskoff V, Lacaud G. The differential activities of Runx1 promoters define milestones during embryonic hematopoiesis. Blood. 2009;114:5279–5289. doi: 10.1182/blood-2009-05-222307. [DOI] [PubMed] [Google Scholar]
- 37.Wang H, Li W, Guo R, Sun J, Cui J, Wang G, Hoffman AR, Hu JF. An intragenic long noncoding RNA interacts epigenetically with the RUNX1 promoter and enhancer chromatin DNA in hematopoietic malignancies. Int J Cancer. 2014;135:2783–2794. doi: 10.1002/ijc.28922. [DOI] [PubMed] [Google Scholar]
- 38.Sun J, Li W, Sun Y, Yu D, Wen X, Wang H, Cui J, Wang G, Hoffman AR, Hu JF. A novel antisense long noncoding RNA within the IGF1R gene locus is imprinted in hematopoietic malignancies. Nucleic Acids Res. 2014;42:9588–9601. doi: 10.1093/nar/gku549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pian L, Wen X, Kang L, Li Z, Nie Y, Du Z, Yu D, Zhou L, Jia L, Chen N, Li D, Zhang S, Li W, Hoffman AR, Sun J, Cui J, Hu JF. Targeting the IGF1R pathway in breast cancer using antisense lncRNA-mediated promoter cis competition. Mol Ther Nucleic Acids. 2018;12:105–117. doi: 10.1016/j.omtn.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen N, Zhao G, Yan X, Lv Z, Yin H, Zhang S, Song W, Li X, Li L, Du Z, Jia L, Zhou L, Li W, Hoffman AR, Hu JF, Cui J. A novel FLI1 exonic circular RNA promotes metastasis in breast cancer by coordinately regulating TET1 and DNMT1. Genome Biol. 2018;19:218. doi: 10.1186/s13059-018-1594-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J, Figueroa ME, De Figueiredo Pontes LL, Alberich-Jorda M, Zhang P, Wu M, D’Alo F, Melnick A, Leone G, Ebralidze KK, Pradhan S, Rinn JL, Tenen DG. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature. 2013;503:371–376. doi: 10.1038/nature12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, Zhang L, Liu L, Sun S, Zhao X, Wang Y, Zhang Y, Du J, Gu L. Rasip1 is a RUNX1 target gene and promotes migration of NSCLC cells. Cancer Manag Res. 2018;10:4537–4552. doi: 10.2147/CMAR.S168438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H, Guo R, Du Z, Bai L, Li L, Cui J, Li W, Hoffman AR, Hu JF. Epigenetic targeting of granulin in hepatoma cells by synthetic CRISPR dCas9 epi-suppressors. Mol Ther Nucleic Acids. 2018;11:23–33. doi: 10.1016/j.omtn.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, Li W, Chen N, Zhao H, Xu G, Zhao Y, Pan X, Zhang X, Zhou L, Yu D, Li A, Hu JF, Cui J. FLI1 exonic circular RNAs as a novel oncogenic driver to promote tumor metastasis in small cell lung cancer. Clin Cancer Res. 2019;25:1302–1317. doi: 10.1158/1078-0432.CCR-18-1447. [DOI] [PubMed] [Google Scholar]
- 45.Hagege H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W, Forne T. Quantitative analysis of chromosome conformation capture assays (3C-qPCR) Nat Protoc. 2007;2:1722–1733. doi: 10.1038/nprot.2007.243. [DOI] [PubMed] [Google Scholar]
- 46.Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonen H, Kol N, Shomron N, Leibowitz-Amit R, Quagliata L, Lorber T, Sidi Y, Avni D. Promoter-associated RNAs regulate HSPC152 gene expression in malignant melanoma. Noncoding RNA. 2016;2 doi: 10.3390/ncrna2030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han J, Kim D, Morris KV. Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proc Natl Acad Sci U S A. 2007;104:12422–12427. doi: 10.1073/pnas.0701635104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016;539:452–455. doi: 10.1038/nature20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407. doi: 10.1016/j.cell.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Webber BR, Iacovino M, Choi SH, Tolar J, Kyba M, Blazar BR. DNA methylation of Runx1 regulatory regions correlates with transition from primitive to definitive hematopoietic potential in vitro and in vivo. Blood. 2013;122:2978–2986. doi: 10.1182/blood-2013-03-489369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chimge NO, Little GH, Baniwal SK, Adisetiyo H, Xie Y, Zhang T, O’Laughlin A, Liu ZY, Ulrich P, Martin A, Mhawech-Fauceglia P, Ellis MJ, Tripathy D, Groshen S, Liang C, Li Z, Schones DE, Frenkel B. RUNX1 prevents oestrogen-mediated AXIN1 suppression and beta-catenin activation in ER-positive breast cancer. Nat Commun. 2016;7:10751. doi: 10.1038/ncomms10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stender JD, Kim K, Charn TH, Komm B, Chang KC, Kraus WL, Benner C, Glass CK, Katzenellenbogen BS. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol Cell Biol. 2010;30:3943–3955. doi: 10.1128/MCB.00118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wotton SF, Blyth K, Kilbey A, Jenkins A, Terry A, Bernardin-Fried F, Friedman AD, Baxter EW, Neil JC, Cameron ER. RUNX1 transformation of primary embryonic fibroblasts is revealed in the absence of p53. Oncogene. 2004;23:5476–5486. doi: 10.1038/sj.onc.1207729. [DOI] [PubMed] [Google Scholar]
- 55.Krishnan V, Ito Y. A Regulatory Role for RUNX1, RUNX3 in the maintenance of genomic integrity. Adv Exp Med Biol. 2017;962:491–510. doi: 10.1007/978-981-10-3233-2_29. [DOI] [PubMed] [Google Scholar]
- 56.Canel M, Byron A, Sims AH, Cartier J, Patel H, Frame MC, Brunton VG, Serrels B, Serrels A. Nuclear FAK and Runx1 cooperate to regulate IGFBP3, cell-cycle progression, and tumor growth. Cancer Res. 2017;77:5301–5312. doi: 10.1158/0008-5472.CAN-17-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.