

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is considered to be the deadliest cancer type in the world. Chemotherapy resistance, including gemcitabine, is the main reason for poor prognosis in PDAC patients. Increased aerobic glycolysis is involved in chemotherapy resistance in PDAC. Fructose-1,6-bisphosphatase (FBP1) is one of the key enzymes in the process of gluconeogenesis and negatively regulates aerobic glycolysis. FBP1 loss is common in PDAC patient specimens and is associated with gemcitabine resistance by activating the MAPK pathway. While the regulatory mechanism of FBP1 in pancreatic cancer remains un-elucidated. Here, we found that ubiquitin-specific protease 44 (USP44) was down-regulated in PDAC patients, and USP44 might be a prognostic marker for PDAC patients. USP44 inhibit tumor cells progression and regulated gemcitabine resistance in PDAC. Importantly, we revealed USP44 promoted FBP1 deubiquitination to increase FBP1 protein expression in pancreatic cancer, which might be one of the underlying mechanisms of USP44 impeding the progression of pancreatic cancer. Collectively, the recognition of USP44 in the stabilization of FBP1 indicates USP44 might be considered as a new prognostic marker for pancreatic cancer therapy.
Keywords: Fructose-1,6-bisphosphatase; ubiquitin-specific protease 44; deubiquitination; gemcitabine; chemotherapy resistance; pancreatic cancer
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is considered to be the deadliest cancer type in the world [1]. Surgical resection combined with systemic chemo-radiotherapy is applied for PDAC patients [2]. Despite the standard treatment is surgical intervention followed by adjuvant chemotherapy with or without radiotherapy, a considerable portion of the patient cannot benefit from this approach due to inoperable disease and therefore, their treatment is considered palliative rather than curative intent [3,4]. Gemcitabine, a DNA synthesis inhibitor, based chemotherapy has been used for the treatment of advanced pancreatic cancer patients and demonstrated a positive effect on prolonging the survival time of those patients from 1990s [5]. Moreover, it has been reported that nab-Paclitaxel (a potent anticancer medication which can promote microtubule (MT) assembly, inhibit MT depolymerization, and change MT dynamics required for mitosis and cell proliferation) plus gemcitabine regimens show better outcomes than gemcitabine alone [6]. Besides, FOLFIRINOX [(5-fluorouracil, leucovorin, irinotecan (a topoisomerase I inhibitor), oxaliplatin (a DNA synthesis inhibitor))] regimen significantly improves the survival rate compared with gemcitabine alone [7]. While inherent resistance to chemo-therapy leads to a poor prognosis in PDAC patients [8]. Although genetic mutation, tumor microenvironment and cancer stem cell were responsible for chemotherapy resistance in pancreatic cancer, the underlying mechanism of chemotherapy resistance is not fully understood [9]. Thus, understanding the pathogenesis of PDAC and developing new therapeutics would improve the survival of pancreatic cancer patients.
Increased aerobic glycolysis is involved in promoting tumor progression and chemotherapy resistance in pancreatic cancer [10]. Gluconeogenesis antagonizes the process of aerobic glycolysis [11]. Fructose-1,6-bisphosphatase (FBP1), one of the key enzymes of gluconeogenesis, help fructose-1,6-bisphosphate transforming into fructose-6-phosphate [11]. Previous researches revealed FBP1 loss is commonly in PDAC patients [12,13]. Recently, we reported FBP1 down-regulation activates the IQGAP1-ERK-Myc axis, which is responsible for gemcitabine and BET inhibitors resistance [12,14]. As a result, understanding the regulatory mechanism of FBP1 would help develop attractive therapeutic strategies for PDAC treatment. Studies have documented TRIM28 promoted degradation of FBP1 in liver cancer cells [15], but the underlying mechanism of FBP1 loss in PDAC has not been well understood.
Deubiquitinases could remove ubiquitin modifications to antagonize E3 ligase induced degradation of target proteins [16]. In the present study, we show that ubiquitin-specific protease 44 (USP44) was downregulated in pancreatic cancer patient specimens. USP44 could inhibit cancer cell progression and regulate the sensitivity of gemcitabine by suppressing glycolysis. Moreover, we determined that USP44 directly deubiquitinated FBP1 and blocked the activation of ERK. To sum up, our results highlight that the USP44 modulates the sensitivity of gemcitabine by stabilizing FBP1 in pancreatic cancer.
Materials and methods
Cell culture
Pancreatic cancer cell lines (PANC-1 and BxPC-3) were purchased from the Chinese Academy of Science Cell Bank. All cell lines were cultured in Dulbecco’s modified Eagle’smedium (DMEM) medium (Invitrogen, USA) containing 10% fetal bovine serum (FBS) (HyClone, USA) at 37°C in a 5% CO2 incubator.
Plasmids, antibodies, and chemicals
Mammalian expression vectors for Myc-USP44 recombinant proteins were generated using the pcDNA3.1 backbone vector. USP44 mutants were generated by using the KOD-Plus-Mutagenesis Kit (Toyobo, Japan). The USP44 antibody (ab205032) was purchased from Abcam (working dilution 1:1000); FBP1 antibody (ab109732) was purchased from Abcam (working dilution 1:1000); GAPDH antibody (ab8245) was from Abcam (working dilution 1:5000); Phospho-p44/42 MAPK (ERK1/2) antibody (9101) was from Cell signaling Technology and p44/42 MAPK (ERK1/2) antibody (9102) was from Cell signaling Technology. Gemcitabine was obtained from Eli Lilly and Company (Indianapolis, USA).
Western blot of cells and tissue specimens
The ethics of using human tissue (11 pairs of matched pancreatic cancer/adjacent noncancerous tissues) was approved by the local ethics committee (Tongji Medical College, China), and written informed consent was obtained from patients prior to surgery exactly as described previously [14]. The cells or the tissue specimens were subjected to lysis buffer (Beyotime, China) containing 1% protease and phosphatase inhibitors. The protein concentration was quantified via a protein assay kit (Pierce Biotechnology, USA). Equal amounts of protein for each sample were loaded and separated using SDS-PAGE gels, followed by transferring to PVDF membranes (Pierce Biotechnology, USA). The membranes were subsequently blocked in 5% not-fat milk for 1 h at room temperature, followed by incubation with primary antibody overnight at 4°C. The membranes were then washed with 1 × TBST and incubated with a secondary antibody for 1 h. Finally, the membranes were treated with ECL detection reagents and exposed to X-ray films.
Real-time RT-PCR
Total RNA was isolated from the cells using Trizol reagent (Thermo Fisher Scientific, USA). cDNA was synthesized from 1 μg RNA using a cDNA Reverse Transcription kit (PrimeScript™ RT reagent Kit, Code No. RR037A), and real-time PCR analysis was carried out with a PCR kit (TB Green™ Fast qPCR Mix, Code No. RR430A) according to the manufacturer’s protocols [17]. The two kits were purchased from Takara Bio Inc. (Shigo, Japan). All the values were normalized to β-actin, and the 2-ΔΔCt method was used to quantify the fold change. The primers used for RT-qPCR are provided as follows: FBP1, forward (5’-3’): ACATCGATTGCCTTGTGTCC, reverse (5’-3’): CCACCAAAATGAACTCCCCG; USP44, forward (5’-3’): CAGGACTAAGTGGAGCA, reverse (5’-3’): CCACGAAAGGCAGGAATGAG; β-actin, forward (5’-3’): CCCTGGCTCCTAGCACCAT, reverse (5’-3’): AGAGCCACCAATCCACACAGA.
Tissue microarray and immunohistochemistry (IHC)
The tissue microarray slides were purchased from Outdo Biobank (Shanghai, China) (HPan-Ade060CD-01) [17]. The tissue microarray specimens were immunostained with USP44 (Abcam, ab205032, dilution 1:1000) and FBP1 antibodies (Abcam, ab109732, dilution 1:4000) as described previously [17]. Staining intensity was scored in a blinded fashion: 1 = weak staining at 100 × magnification but little or no staining at 40 × magnification; 2 = medium staining at 40 × magnification; 3 = strong staining at 40 × magnification [15]. The degree of immunostaining was scored by two independent pathologists who were blinded to the clinical details. The IHC scores were calculated by the percentage of positive cells x the staining intensity.
RNA interference
The lentivirus-based control and gene-specific shRNAs were purchased from Sigma-Aldrich. 293T cells were transfected with shRNA plasmids and viral packaging plasmids (pVSV-G and pEXQV) by Lipofectamine 2000. 24 h after transfection, the medium was replaced with fresh DMEM, containing 10% FBS and 1 mM of sodium pyruvate. Next, 48 h -post-transfection, the virus culture medium was collected and added to the PANC-1 and BxPC-3 cells supplemented with 12 µg/ml of polybrene. 24 h after infection, the infected cells were selected with 10 µg /ml of puromycin. The shRNA sequence information is provided as follows: shUSP44#1: 5’-CCGGCGGATGATGAACTTGTGCAATCTCGAGATT GCACAAGTTCATCATCCGTTTTTG-3’; shUSP44#2: 5’-CCGGTACACTGCCTA CTGCTATAATCTCGAGATTATAGCAGTAGGCAGTGTATTTTTG-3’; shFBP1#1: 5’-CCGGCCTTGATGGATCTTCCAACATCTCGAGATGTTGGAAGATCCATCAAGGTTTTTG-3’; shFBP1#2: 5’-CCGGCGACCTGGTTATGAACATGTTCTCGA GAACATGTTCATAACCAGGTCGTTTTTG-3’.
Cell proliferation assay
MTS[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] assay was performed to detect the cell viability according to the manufacturer’s instructions (Abcam, USA). In brief, cells (1 × 103 cells) were plated in 96-well plates with 100 μl of culture medium. After 72 h, 20 μl of MTS reagent (Abcam, USA) was added to each well of the cells and incubated for 1 hour at 37°C in standard culture conditions. The absorbance was measured in a microplate reader at 490 nm.
Generation of PDAC xenografts in nude mice
The BALB/c-nu mice (4-5 weeks of age, 18-20 g) were purchased from Vitalriver (Beijing, China) and randomly divided into four groups (n = 4/group) for the subcutaneous inoculation with 5 × 106 of PANC-1 cells infected with shControl or shFBP1, shUSP44 or both shFBP1 plus shUSP44 lentivirus in the left dorsal flank of the mice. The tumors were examined every other day for 21 days; the length and width measurements were obtained with calipers to calculate the tumor volumes by using the equation (L × W2)/2. On day 21, the animals were euthanized, and the tumors were excised and weighed. All the animal experimental procedures were approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology.
Correlation analysis using the GEPIA web tool
The online database Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancerpku.cn/index.html) [18] was used to analyze the RNA sequencing expression data described previously [17]. GEPIA performs a pairwise gene correlation analysis for any given set expression data using Pearson correlation statistics.
In vitro deubiquitin assay
PANC-1 cells were transfected with Flag-FBP1 and HA-UB for 24 h. The whole-cell lysate was undergoing immunoprecipitaion by using flag antibody to get the purified FBP1. Myc-USP44 protein was translated in vitro following the manufacture protocol of TNT ® Quick coupled Translation System Technical (Promega, #TM045). The in vitro translated USP44 and the purified FBP1 were mixed in a buffer (50 mMTris-HCl, pH 7.5 and 4 mM DTT) and incubated for 2 h at 37°C. Then, the reaction and samples were sent for western blotting analysis.
Statistical analysis
Statistical analyses were performed with one-sided or two-sided paired Student’s t-test for single comparison and one-way ANOVA with a post hoc test for multiple comparisons. A P value < 0.05 was considered statistically significant. All the values are expressed as the mean ± SD.
Results
USP44 inhibits pancreatic cancer progression
The biologic role and underlying mechanism of the USP44 in pancreatic cancer have not been studied. Firstly, we used the tissue microarray (TMA) (n = 41 PDAC specimens, n = 25 normal pancreatic specimens) to examine the expression level of USP44 in PDAC tissues. The immunohistochemistry (IHC) data indicated that USP44 expression was lower in PDAC than in non-tumor pancreatic tissues (Figure 1A and 1B). Meanwhile, the Western blot analysis showed USP44 was decreased in cancer tissues compared with adjacent non-tumor pancreatic tissues (Figure 1C and 1D). Furthermore, we analyzed whether USP44 was associated with the prognosis of PDAC patients through The Human Protein Atlas dataset, and found that USP44 down-regulation was negatively correlated with prognosis in pancreatic cancer patients (Figure 1E).
Figure 1.
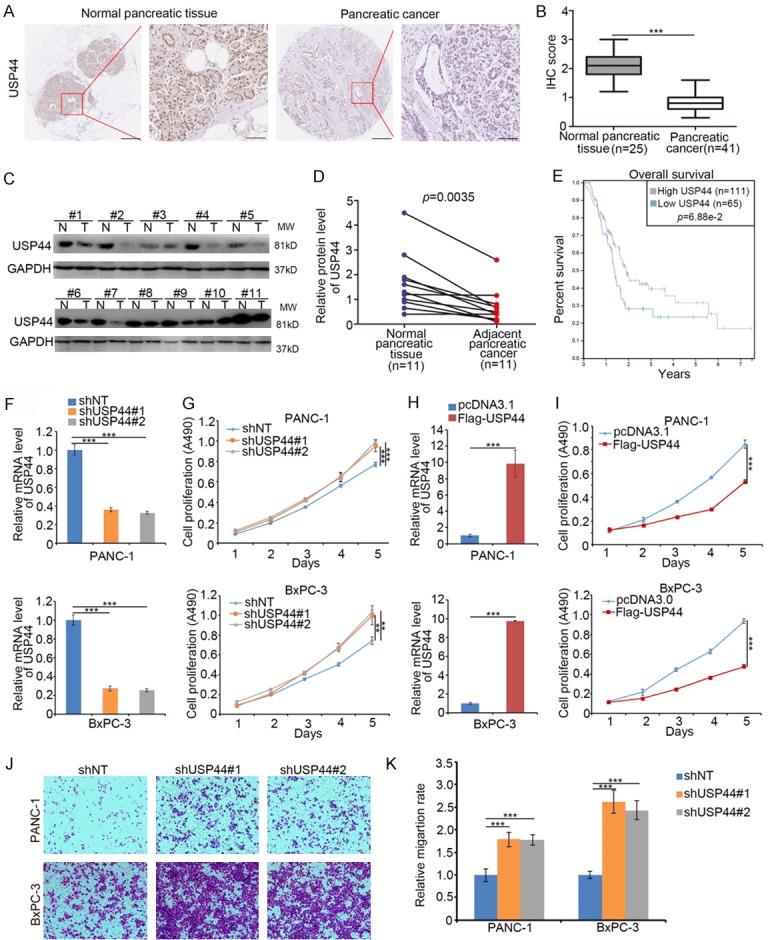
USP44 inhibits pancreatic cancer progression. (A and B) Tissue microarray specimens were stained with USP44, the IHC image (A) and the IHC score of USP44 (B) are shown. ***P < 0.001. (C and D) 11 paired pancreatic adenocarcinoma tissues (T) and the adjacent non-tumor tissues (N) was subjected to Western blot analysis (C). USP44 expression levels were quantified by using Image J software. P = 0.0035. (E) The overall survival rate of PDAC patients was determined by the Human Protein Atlas. The P valued was shown. (F and G) BxPC-3 and PANC-1 cells were infected with indicated constructs for 48 h. The cells underwent RT-qPCR analysis (F) and MTS assay (G). Data are presented as the mean values ± SD (n = 3). ***P < 0.001. (H and I) BxPC-3 and PANC-1 cells were transfected with indicated plasmids for 48 h. The cells were sent for RT-qPCR analysis (H) and MTS assay (I). Data are presented as the mean values ± SD from 3 replicates. ***P < 0.001. (J and K) BxPC-3 and PANC-1 cells were infected with indicated shRNA. 48 h post-infection, the cells were collected for the transwell migration assay. The image of transwell is shown in (J) and the relative qualification data are present in (K) Data are present as the mean values ± SD (n = 3). ***P < 0.001.
Given that USP44 functions as a positive biomarker for the prognosis of PDAC, the biological role of USP44 need to be further elucidatedin pancreatic cancer cells. We first performed knocking down USP44 in both BxPC-3 and PANC-1 cells (Figure 1F). We demonstrated USP44 knockdown significantly promoted pancreatic cancer cell proliferation (Figure 1G). Conversely, the ectopic expressed USP44 inhibited the growth of pancreatic cancer cells (Figure 1H and 1I). Moreover, we demonstrated the knockdown of USP44 increased the invasive activities of pancreatic cancer cells (Figure 1J and 1K). Therefore, our results suggest USP44 inhibits cancer cell progression in PDAC.
USP44 overcomes gemcitabine resistance by suppressing glycolysis in pancreatic cancer cells
Gemcitabine based chemotherapy is the first choice for pancreatic cancer in the last decades [8]. While, due to the acquisition of chemoresistance, gemcitabine could not improve the outcome of PDAC patients [5]. To determine whether USP44 regulated the sensitivity of gemcitabine, the IC50 values of gemcitabine after USP44 knocked down or overexpressed in pancreatic cancer cells (PANC-1 and BxPC-3, respectively) was evaluated (Figure 2A and 2B). We found that the IC50 values of gemcitabine in the USP44-knockdown group were higher than which in the control group (Figure 2A). In contrast, the overexpression of USP44 decreased the IC50 values of gemcitabine (Figure 2B). The above data indicated USP44 was one of the key mediators for modulating gemcitabine sensitivity, but the mechanism was unclear. It has been well documented that pancreatic cancer cells present increased glucose consumption and shift cellular metabolism from oxidative phosphorylation to glycolysis, which leads to gemcitabine resistance [19]. Intriguingly, we found down-regulation of USP44 enhanced the ability of pancreatic cancer cells to utilize glucose and produce lactate (Figure 2C). Conversely, USP44 overexpression decreased glucose utilization and lactate production in pancreatic cancer cells (Figure 2D). To further explore the mechanism of USP44 regulating glucose metabolism, we examined many key enzymes (such as FBP1, PKM2, and HK2) in glycolysis or gluconeogenesis and revealed that the knockdown of USP44 down-regulated FBP1 but not PKM2 or HK2 in PANC-1 cells (Figure 2E). Moreover, USP44 inhibition only decreased the protein level FBP1 in PANC-1 cells (Figure 2E and 2F). On the contrary, overexpressed USP44 up-regulated the protein level of FBP1 in PANC-1 cells (Figure 2G and 2H). Previous studies have suggested that FBP1 acts as a key enzyme in the process of gluconeogenesis and undermined FBP1 results in gemcitabine resistance. In our study, FBP1 was regulated by USP44, and our data also indicated the knockdown of FBP1 attenuated the decrease in the IC50 values of gemcitabine-induced by USP44 overexpression (Figure 2I). Thus, our findings indicate that FBP1 play a significant role in the process of USP44-mediated gemcitabine sensitivity and glucose metabolism changes in PDAC.
Figure 2.
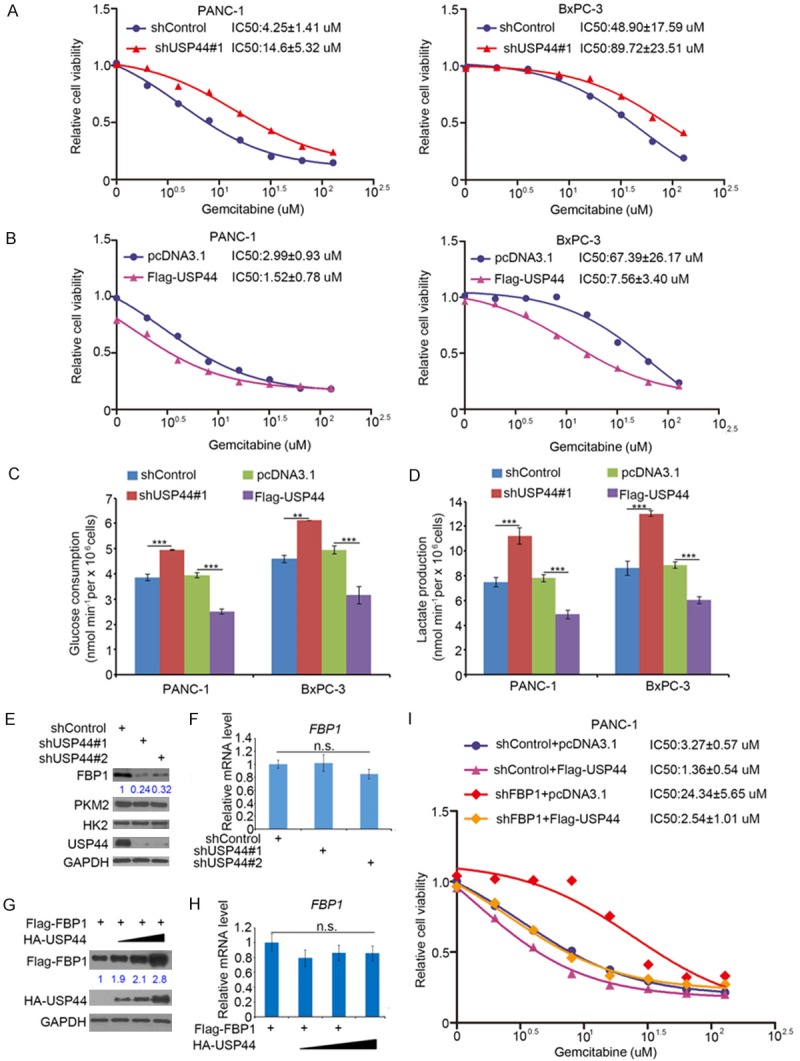
USP44 overcomes gemcitabine resistance by suppressing the glycolysis in pancreatic cancer cells. (A) Both PANC-1 and BxPC-3 cells were infected with indicated shRNAs. After 48 h, the cells were treated with a serial concentration of gemcitabine for another 24 h. The cell growth rate was evaluated using the MTS assay. The IC50 values of gemcitabine in each group are also shown. (B) BxPC-3 and PANC-1 cells were transfected with indicated plasmids for 24 h. The cells were treated with various concentration of gemcitabine for another 24 h. The cell proliferation rate was evaluated by MTS assay. The IC50 values of gemcitabine in each group are also shown. (C and D) PANC-1 and BxPC-3 cells were infected with indicated shRNA. 48 h post-infection, the spent medium was sent for analysis of glucose utilization (C) and lactate secretion (D). Data are present as the mean values ± SD (n=3). ***P < 0.001. (E and F) PANC-1 cells were infected with indicated shRNAs. After 72 h, the cells were collected for Western blot analysis (E) and RT-qPCR analysis (F). The data are shown as the mean values ± SD (n = 3). n.s., not significant. (G and H) PANC-1 cells were transfected with Flag-FBP1 and a serial dose of HA-USP44. After 24 h, the cells were collected for Western blot analysis (G) and RT-qPCR analysis (H). The data are presented as the mean values ± SD (n = 3). n.s., not significant. (I) PANC-1 cells were infected with indicated shRNAs. 48 h post-infection, the cells were transfected with indicated plasmids for another 24 h. The cells growth rate was evaluated using the MTS assay for calculating the IC50 values of gemcitabine in each group.
USP44 stabilizes FBP1 in pancreatic cancer
USP44 up-regulated FBP1 protein expression without affecting its mRNA levels (Figure 2E-H). Since USP44 is a well-known deubiquitinase to stabilize its target protein [20], we want to verify whether FBP1 is the direct target of USP44. Firstly, reciprocal co-IP assays between FBP1 and USP44 was performed and we found that both ectopically expressed and endogenous USP44 interacted with FBP1 in 293T and PANC-1 cells respectively (Figure 3A and 3B). To further confirm the interaction between FBP1 and USP44, the FBP1 recombinant constructs were synthesized as reported previously [14]. GST pull-down assay was performed to determine which region of FBP1 mediate its interaction with USP44. We found that FBP1 E1 (aa 1-57) bind with USP44 in vitro (Figure 3C). Inconsistent with the results in PANC-1 cells (Figure 2E), USP44 knockdown decreased FBP1 protein expression in BxPC-3 cells (Figure 3D). Then, overexpressed wild-type (WT) USP44 but not the deubiquitinase-activity-deficient mutant Cl [21] increased the level FBP1 protein in pancreatic cancer cells (Figure 3F). We also found neither the overexpression nor knockdown of USP44 had any overt effect on the mRNA level of FBP1 (Figure 3E and 3G). In agreement with these results, the USP44 knockdown shortened the half-life of FBP1 (Figure 3H) and diminished the ubiquitination level of FBP1 (Figure 3I). Furthermore, we also observed that USP44 wide type but not deubiquitinase-activity-deficient mutant Cl decreased the ubiquitination level of FBP1 in PANC-1 cells (Figure 3J). Besides, the direct deubiquitination of FBP1 by USP44 was determined in Figure 3K. Taken together, we identified that USP44 as a deubiquitinase of FBP1 in pancreatic cancer cells.
Figure 3.

USP44 stabilizes FBP1 in pancreatic cancer. (A) 293T cells were transfected with Flag-FBP1 and Myc-USP44 for 24 h. Whole-cell lysate (WCL) were collected for Western blot analysis of reciprocal co-immunoprecipitation (co-IP). (B) The WCL of PANC-1 were used for Western blot analysis of reciprocal co-immunoprecipitation of endogenous FBP1 and USP44 proteins. (C) Schematic diagram depicting a set of GST-FBP1 recombinant protein constructs. Western blot analysis of USP44 proteins in PANC-1 pulled down by GST or GST-FBP1 recombinant proteins. (D and E) BxPC-3 cells were infected with indicated shRNAs for 72 h. The cells were collected for Western blot analysis (C) and RT-qPCR analysis (D). The data are presented as the mean values ± SD (n = 3). n.s., not significant. (F and G) PANC-1 cells were transfected with indicated plasmids for 48 h. The cells were collected for Western blot analysis (E) and RT-qPCR analysis (F). The data are presented as the mean values ± SD (n = 3). n.s., not significant. (H) PANC-1 cells were infected with indicated shRNAs for 72 h. 50 μg/μl of cycloheximide (CHX) was added to the culture-medium of PANC-1 cells. The WCL of cells were collected for Western blot analysis at different time points as indicated. The protein expression level of FBP1 was normalized to the protein level of GAPDH and then to the value at the 0 h time point. (I) PANC-1 cells transfected with the indicated plasmids for 24 h. The cells were treated with 20 μM of MG132 for 8 h before harvest for Western blotting analysis. (J) PANC-1 cells transfected with the indicated plasmids for 24 h. The cells were treated with 20 μM of MG132 for 8 h before harvest for Western blotting analysis. (K) Purified FBP1, and USP44 were incubated for 2 h and the reaction was harvested for Western blotting analysis. (L) Western blot analysis of WCL after PANC-1 cells were transfected with indicated constructs for 24 h. (M) PANC-1 cells transfected with the indicated constructs for 48 h. The cells were treated with 20 μM of MG132 for 8 h before harvest for Western blotting analysis.
We recently reported that TRIM28 is a bona fide E3 ubiquitin ligase of FBP1 [15]. We were interested in determining whether USP44 influences the TRIM28-mediated proteasomal degradation of FBP1. Our data showed that TRIM28 destabilized FBP1 by decreasing the protein expression and increasing the polyubiquitination of FBP1, but this effect was abolished by the overexpression of USP44 (Figure 3L and 3M). Therefore, our findings demonstrate that USP44 stabilizes FBP1 and that the TRIM28-mediated ubiquitination of FBP1 can be diminished by USP44.
FBP1 protein levels rather than mRNA levels are positively correlated with USP44 in PDAC specimens
To determine whether FBP1 protein expression is correlated with USP44 protein levels in PDAC specimens, the TMA of PDAC patients (n = 39) was employed to test these two proteins’ expression through IHC assay. The IHC score was calculated through multiplying the staining intensity and the percentage of positively stained cells [14]. The IHC images of FBP1 and USP44 are shown in Figure 4A. USP44 level was correlated with the FBP1 expression positively in the PDAC specimens (Figure 4B). Next, the correlation between the mRNA levels of FBP1 and USP44 was determined based on the GEPIA web tool. This result showed there was no correlation between FBP1 mRNA and USP44 mRNA in pancreatic cancer patient specimens (Figure 4C), which was consistent with our findings in both BxPC-3 and PANC-1 cells. Together, our findings suggest that FBP1 protein levels rather than FBP1 mRNA levels are positively correlated with USP44 in PDAC patient specimens.
Figure 4.
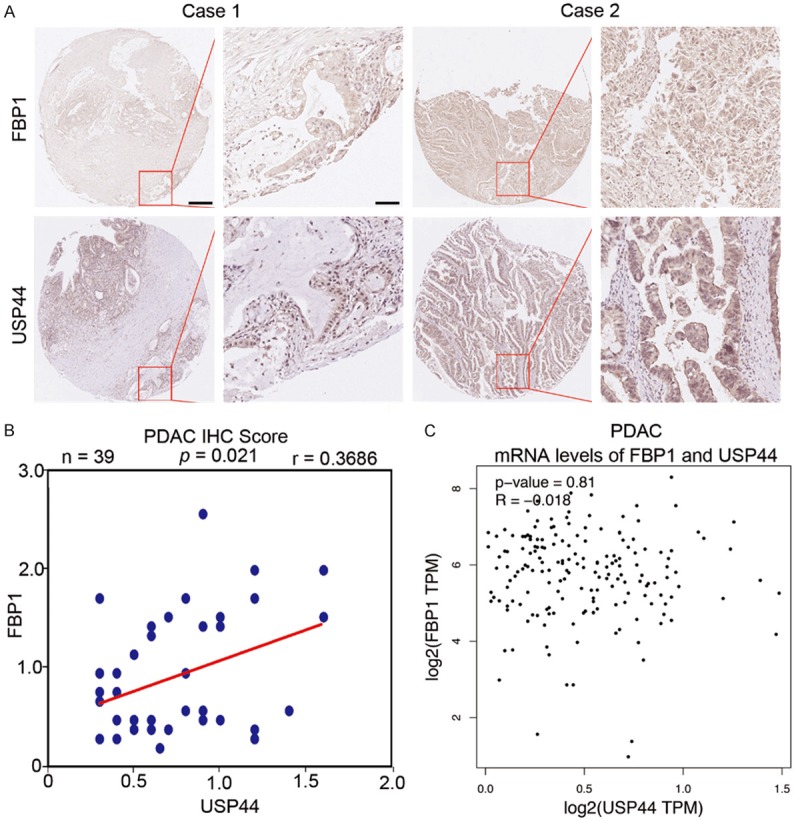
FBP1 protein levels rather than FBP1 mRNA levels are positively correlated with USP44 in PDAC specimens. A. The TMA sections (n = 39 for PDAC) were stained with FBP1 or USP44 respectively. B. Correlation analysis of the IHC scores of FBP1 and USP44 in TMA sections. Pearson’s product-moment correlation co-efficiency and the p-values are also shown. C. Correlation between the mRNA expression levels of USP44 and FBP1 in human pancreatic cancer samples was determined by The GEPIA web tool.
USP44 impedes pancreatic cancer cell growth through the FBP1-MAPK pathway
Previous studies have shown that FBP1 loss up-regulates the phosphorylation of ERK, and the restoration of FBP1 inhibits tumor cell proliferation in pancreatic cancer cells [12]. Since USP44 promoted FBP1 deubiquitination and stabilized FBP1 protein expression, we wanted to determine whether USP44 regulates tumor growth via the FBP1-MAPK pathway. FBP1, USP44 or both were overexpressed in PANC-1 cells with the indicated transfection plasmids. We found that the co-over expression of USP44 and FBP1 decreased the phosphorylation of ERK to a greater extent than the overexpression of USP44 or FBP1 alone in PANC-1 cells (Figure 5A). The MTS assay also demonstrated the co-overexpression of USP44 and FBP1 had the lowest cell proliferation rate (Figure 5B). In contrast, the knockdown of FBP1 and USP44 alone increased the phosphorylation of ERK in PANC-1 cells (Figure 5C). Importantly, the increased phosphorylation of ERK caused by the co-knockdown of FBP1 and USP44 was less than that of the sum of the knockdown of either FBP1 or USP44 alone (Figure 5D). Furthermore, the MTS assay and animal studies showed that the co-knockdown of FBP1 and USP44 induced less tumor cell growth than that of the sum of the knockdown of either FBP1 or USP44 alone in vivo and in vitro (Figure 5D-G). These data suggest that USP44 impedes tumor cell growth through the FBP1-MAPK pathway in pancreatic cancer.
Figure 5.

USP44 impedes pancreatic cancer cells growth through FBP1-MAPK pathway.( A and B) PANC-1 cells were transfected with indicated constructs for 48 h. The cells were collected for Western blot analysis (A) and MTS assay (B). Data are presented as the mean values ± SD (n = 3). **P < 0.01; ***P < 0.001. (C and D) PANC-1 cells were infected with indicated shRNAs for 72 h. The cells were collected for Western blot analysis (C) and MTS assay (D). Data are presented as the mean values ± SD (n = 3). **P < 0.01; ***P < 0.001. (E-G) PANC-1 cells were infected with indicated shRNAs for 72 h. The cells were harvested and injected subcutaneously into the nude mice. After 21 days, the tumors were collected, photographed and measured. Data are presented as the mean values ± SD (n = 4). n.s., not significant; *P < 0.05; ***P < 0.001.
Discussion
The specific role of USP44 in cancer is still controversial. It has been documented that USP44 inhibits tumor progression through regulating centrosome positioning and suppresses cancer cell progression in human lung cancer [22]. USP44 was also acted as a key regulator of APC/C by repressing Cdc20-APC/C activity [23]. Moreover, the USP44 down-regulation by its CpG island promoter methylation was found as an early event in colorectal neoplasia [24]. In contrast, some researchers have demonstrated that USP44 stabilizes histone H2Bto enhance the function of N-CoR, which is required for breast cancer cells invasion [20]. Moreover, up-regulated USP44 is found in cancer stem cell (CSC) subpopulations and contributes to aggressive behaviors in breast cancer [25]. Furthermore, USP44 promotes cancer cell progression by stabilizing securin in glioma [26]. The specific role of USP44 in PDAC is still elucidated. Here, our findings suggest USP44 is down-regulated in pancreatic cancer, and the depletion of USP44 contributes to a poor prognosis of PDAC patients. Our results further demonstrate USP44 impedes cancer cell proliferation and glycolysis through stabilizing FBP1 in pancreatic cancer.
Gemcitabine based chemotherapy could improve the survival time of patients with resected PDAC [8]. While resistance to gemcitabine is a major issue of pancreatic cancer chemotherapy [5]. The mechanism of gemcitabine resistance is complicated [27]. Kras mutation is found in 90% of pancreatic cancer patients [28]. Kras mutation activates the MAPK pathway, which is one of the key reasons for gemcitabine resistance in pancreatic cancer [29]. We have previously reported that FBP1 could bind with IQGAP1 and block the activation of the MAPK pathway by down-regulation of the phosphorylation level of ERK in pancreatic cancer cells [12]. In our study, we demonstrated USP44 modulated glycolysis and was closely related to gemcitabine resistance. We screen some of the key enzymes in regulating the glucose metabolism and occasionally found that FBP1 was decreased after knockdown of USP44 in PANC-1 cells. Then, we have proved that FBP1 is a bona fide substrate of USP44.
Collectively, we systematically investigate the role of USP44 in PDAC. Our results showed USP44 is decreased in PDAC specimens and is associated with poor prognosis of pancreatic cancer patients. USP44 impedes tumor cell progression and modulates the sensitivity of gemcitabine in PDAC. Especially, we showed USP44 acts as a negative regulator in glycolysis by stabilizing FBP1. The recognition of USP44 as the deubiquitinase of FBP1 made USP44 as a new candidate for pancreatic cancer therapy.
Acknowledgements
This work was supported by grants from the Chinese National Natural Science Foundation Grant No. 81702374.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Cheng D, Fan J, Ma Y, Zhou Y, Qin K, Shi M, Yang J. LncRNA SNHG7 promotes pancreatic cancer proliferation through ID4 by sponging miR-342-3p. Cell Biosci. 2019;9:28. doi: 10.1186/s13578-019-0290-2. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Adamska A, Domenichini A, Falasca M. Pancreatic ductal adenocarcinoma: current and evolving therapies. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18071338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu B, Wang Y, Liu S, Zhao T, Zhao B, Jiang X, Ye L, Zhao L, Lv W, Zhang Y, Zheng T, Xue Y, Chen L, Chen L, Wu Y, Li Z, Yan J, Wang S, Sun X, Gao G, Qu Y, He S. A randomized controlled study of preoperative oral carbohydrate loading versus fasting in patients undergoing elective craniotomy. Clin Nutr. 2018 doi: 10.1016/j.clnu.2018.11.008. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 5.Binenbaum Y, Na’ara S, Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist Update. 2015;23:55–68. doi: 10.1016/j.drup.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohammed S, Van Buren G 2nd, Fisher WE. Pancreatic cancer: advances in treatment. World J Gastroenterol. 2014;20:9354–9360. doi: 10.3748/wjg.v20.i28.9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reni M, Zanon S, Peretti U, Chiaravalli M, Barone D, Pircher C, Balzano G, Macchini M, Romi S, Gritti E, Mazza E, Nicoletti R, Doglioni C, Falconi M, Gianni L. Nab-paclitaxel plus gemcitabine with or without capecitabine and cisplatin in metastatic pancreatic adenocarcinoma (PACT-19): a randomised phase 2 trial. Lancet Gastroenterol Hepatol. 2018;3:691–697. doi: 10.1016/S2468-1253(18)30196-1. [DOI] [PubMed] [Google Scholar]
- 9.Chiorean EG, Coveler AL. Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther. 2015;9:3529–3545. doi: 10.2147/DDDT.S60328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo J, Hao J, Jiang H, Jin J, Wu H, Jin Z, Li Z. Proteasome activator subunit 3 promotes pancreatic cancer growth via c-Myc-glycolysis signaling axis. Cancer Lett. 2017;386:161–167. doi: 10.1016/j.canlet.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 11.Timson DJ. Fructose 1,6-bisphosphatase: getting the message across. Biosci Rep. 2019;39 doi: 10.1042/BSR20190124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin X, Pan Y, Wang L, Ma T, Zhang L, Tang AH, Billadeau DD, Wu H, Huang H. Fructose-1,6-bisphosphatase Inhibits ERK activation and bypasses gemcitabine resistance in pancreatic cancer by blocking IQGAP1-MAPK Interaction. Cancer Res. 2017;77:4328–4341. doi: 10.1158/0008-5472.CAN-16-3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen LY, Cheng CS, Qu C, Wang P, Chen H, Meng ZQ, Chen Z. CBX3 promotes proliferation and regulates glycolysis via suppressing FBP1 in pancreatic cancer. Biochem Biophys Res Commun. 2018;500:691–697. doi: 10.1016/j.bbrc.2018.04.137. [DOI] [PubMed] [Google Scholar]
- 14.Wang B, Fan P, Zhao J, Wu H, Jin X, Wu H. FBP1 loss contributes to BET inhibitors resistance by undermining c-Myc expression in pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res. 2018;37:224. doi: 10.1186/s13046-018-0888-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin X, Pan Y, Wang L, Zhang L, Ravichandran R, Potts PR, Jiang J, Wu H, Huang H. MAGE-TRIM28 complex promotes the Warburg effect and hepatocellular carcinoma progression by targeting FBP1 for degradation. Oncogenesis. 2017;6:e312. doi: 10.1038/oncsis.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clague MJ, Urbe S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol. 2019;20:338–352. doi: 10.1038/s41580-019-0099-1. [DOI] [PubMed] [Google Scholar]
- 17.Fan P, Zhao J, Meng Z, Wu H, Wang B, Wu H, Jin X. Overexpressed histone acetyltransferase 1 regulates cancer immunity by increasing programmed death-ligand 1 expression in pancreatic cancer. J Exp Clin Cancer Res. 2019;38:47. doi: 10.1186/s13046-019-1044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai IL, Chou CC, Lai PT, Fang CS, Shirley LA, Yan R, Mo X, Bloomston M, Kulp SK, Bekaii-Saab T, Chen CS. Targeting the Warburg effect with a novel glucose transporter inhibitor to overcome gemcitabine resistance in pancreatic cancer cells. Carcinogenesis. 2014;35:2203–2213. doi: 10.1093/carcin/bgu124. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Lan X, Atanassov BS, Li W, Zhang Y, Florens L, Mohan RD, Galardy PJ, Washburn MP, Workman JL, Dent SYR. USP44 is an integral component of N-CoR that contributes to gene repression by deubiquitinating histone H2B. Cell Rep. 2016;17:2382–2393. doi: 10.1016/j.celrep.2016.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosbech A, Lukas C, Bekker-Jensen S, Mailand N. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J Biol Chem. 2013;288:16579–16587. doi: 10.1074/jbc.M113.459917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Foreman O, Wigle DA, Kosari F, Vasmatzis G, Salisbury JL, van Deursen J, Galardy PJ. USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J Clin Invest. 2012;122:4362–4374. doi: 10.1172/JCI63084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, van Deursen J, Galardy PJ. Overexpression of ubiquitin-specific protease 44 (USP44) induces chromosomal instability and is frequently observed in human T-cell leukemia. PLoS One. 2011;6:e23389. doi: 10.1371/journal.pone.0023389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sloane MA, Wong JW, Perera D, Nunez AC, Pimanda JE, Hawkins NJ, Sieber OM, Bourke MJ, Hesson LB, Ward RL. Epigenetic inactivation of the candidate tumor suppressor USP44 is a frequent and early event in colorectal neoplasia. Epigenetics. 2014;9:1092–1100. doi: 10.4161/epi.29222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu T, Sun B, Zhao X, Li Y, Zhao X, Liu Y, Yao Z, Gu Q, Dong X, Shao B, Lin X, Liu F, An J. USP44+ cancer stem cell subclones contribute to breast cancer aggressiveness by promoting vasculogenic mimicry. Mol Cancer Ther. 2015;14:2121–2131. doi: 10.1158/1535-7163.MCT-15-0114-T. [DOI] [PubMed] [Google Scholar]
- 26.Zou Y, Qiu G, Jiang L, Cai Z, Sun W, Hu H, Lu C, Jin W, Hu G. Overexpression of ubiquitin-specific proteases 44 promotes the malignancy of glioma by stabilizing tumor-promoter securin. Oncotarget. 2017;8:58231–58246. doi: 10.18632/oncotarget.16447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsesmetzis N, Paulin CBJ, Rudd SG, Herold N. Nucleobase and nucleoside analogues: resistance and Re-Sensitisation at the level of pharmacokinetics, pharmacodynamics, and metabolism. Cancers (Basel) 2018;10 doi: 10.3390/cancers10070240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ, Kalluri R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503. doi: 10.1038/nature22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Xi Z, Chen X, Cai S, Liang C, Wang Z, Li Y, Tan H, Lao Y, Xu H. Natural compound Oblongifolin C confers gemcitabine resistance in pancreatic cancer by downregulating Src/MAPK/ERK pathways. Cell Death Dis. 2018;9:538. doi: 10.1038/s41419-018-0574-1. [DOI] [PMC free article] [PubMed] [Google Scholar]