

Abstract
Heat shock factors (HSFs) are essential for all organisms to survive exposures to acute stress. Recent years have witnessed the progress in uncovering the importance of HSFs in cancer cell oncogenesis, progression and metastasis. However, their roles in hepatocellular carcinoma (HCC) proliferation and the underlying mechanism have seldom been discussed. The present study aims to uncover the two important HSFs members HSF1 and HSF2 in hepatocellular carcinoma (HCC). By using the Cancer Genome Atlas (TCGA) dataset analysis, we investigated the prognosis value of HSF1 and HSF2 in HCC and identified HSF2 as a prediction factor of overall survival of HCC. In vitro cell line studies demonstrated that silencing HSF2 expression could decrease the proliferation in HCC cells. In depth mechanism analysis demonstrated that HSF2 promoted cell proliferation via positive regulation of aerobic glycolysis, and HSF2 interacted with euchromatic histone lysine methyltransferase 2 (EHMT2) to epigenetically silence fructose-bisphosphatase 1 (FBP1), which is a tumor suppressor and negative regulator of aerobic glycolysis in HCC. HSF2 expression indicated unfavorable prognosis of HCC patients and it could regulate aerobic glycolysis by suppression of FBP1 to support uncontrolled proliferation of HCC cells.
Keywords: Hepatocellular carcinoma, heat shock factor 2, aerobic glycolysis, fructose-bisphosphatase 1, euchromatic histone lysine methyltransferase 2
Introduction
HCC is among the most common and lethal cancer types worldwide, and it is ranked the third leading cause of cancer death and the fifth prevalent human cancer [1]. Although significant progress has been made in the therapeutic options of HCC, including early diagnosis, surgical operations and chemotherapy, the 5-year overall survival rates remains poor and stay at approximately at 30%. Therefore, there is an urgent need for a better understanding the molecular mechanism of HCC tumorigenesis, progression and identify novel predictive and treatment targets for HCC [2].
Heat-shock factors (HSFs) are multifaceted transcription factors that regulate the proteotoxic stress, namely the heat-shock response [3]. HSF1 is the master regulator of stress responses and its targets include molecular chaperones, which contains cis-acting sequences call heat shock elements (HSEs) and these chaperons played vitals roles in maintaining protein homeostasis. HSF1 has been reported to regulate cell cycle progression, protein synthesis, ribosome biogenesis and glucose metabolism [4]. Recent years have witnessed the importance of HSF1 in cancer, including oncogenesis, progression, metastasis and metabolism reprogramming. The clinical significance of HSF1 was highlighted in a cohort of cancer patients, such as breast cancer, prostate cancer, etc [5,6]. In contrast to HSF1, another HSF family member HSF2, which participated in the regulation of HSF1-meidated stress response, has seldom been studied in cancer.
It is well acknowledged that proliferating human cells require more energy and material supply to sustain the proliferative process. However, rapidly proliferative solid tumor cells lack supplies including oxygen and nutrient due to limited oxygen supply caused by severely hypoxic conditions. To survive under such hypoxic conditions, tumor cells must shift their metabolic pattern to adapt to the hostile microenvironment, the phenomenon is known as metabolic reprogramming [7]. The best characterized metabolic reprogramming is aerobic glycolysis, which has been discovered by Otto Warburg nighty years ago, also known as “Warburg effect” [8]. Aerobic glycolysis is thought to enable the generation of building blocks for macromolecule synthesis, such as nucleotide, proteins, lipids, that are necessary to maintain the malignant proliferative state [9]. HCC is a solid tumor with severe hypoxic microenvironment, thus aerobic glycolysis might be a prominent characteristic of HCC and could be utilized to generate novel predictive and treatment targets [10,11].
In the present study, by using the cancer genome atlas (TCGA) dataset analysis, we demonstrated that HSF2 could be used to reflect overall survival of HCC. In vitro studies demonstrated that HSF2 could regulate proliferation of HCC cells. Mechanism explorations demonstrated that HSF2 could regulate aerobic glycolysis by suppression of FBP1 through interaction with EHMT2. In conclusion, the present study uncovered novel predictive markers and provided that possible mechanism, which might help improve the dilemma for the prediction and treatment of HCC.
Material and methods
Cell culture
HEK293T cells were purchased from the American Type Culture Collection (ATCC, Manassas, Virginia, USA). PLC, HepG2, Hep3B, SMMC-7721 and Li-7 cells were obtained from the China Center of Type Culture Collection (Shanghai, China). HEK293T and SMMC-7721 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Thermo Fisher Scientific, California, USA) with 10% fetal bovine serum (FBS) and antibiotics. Huh-7 cells was purchased from JCRB cell bank (Tokoyo, Japan) and cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium (Thermo Fisher Scientific) with 10% new born calf serum.
RNA isolation and quantitative real-time PCR
Total RNA was prepared using TRIzol reagent (Invitrogen, USA). To obtain cDNA, TaKaRa PrimeScript RT reagent Kit was used for reverse transcription. Expression status of candidate genes and β-actin were determined by quantitative real-time PCR using an ABI 7900HT Real-Time PCR system (Applied Biosystems, USA). All reactions were run in triplicate. Primers sequences are listed in below (Table 1):
Table 1.
Primers sequences
| GLUT1F | TCCTATCTGAGCATCGTGGCCAT |
| GLUT1R | CCAGGAGCACAGTGAAGATGATG |
| HK2F | GATCAGTGGAATGTACCTGGGTG |
| HK2R | CAGGGCCAGGCAGTCACTCTCAAT |
| LDHAF | TGGTGTCTCTCTGAAGACTCTGCA |
| LDHAR | GCAAGGAACACTAAGGAAGACATC |
| FBP1F | TCTACAGCCTTAACGAGGGCTAC |
| FBP1R | AGCCTTCTCCATGACGTAGGCCA |
| HSF2F | GACCCAGATCTCCTGGTTGATCTT |
| HSF2R | CGAGGAATGCAAGAAGTGGAAAGG |
| β-actinF | TTCCAGCTCCTCCCTGGAGAAGAG |
| β-actinR | GACAGCACTGTGTTGGCGTACAGG |
Protein extraction and western blot analysis
Cells were washed twice with ice-cold PBS and lysed in RIPA buffer for 10 min followed by sonication to obtain complete lysates. Cell debris was removed by centrifugation at 12000 rpm for 20 min at 4°C. 20 μg total protein lysate were subjected to electrophoresis in denaturing 10% SDS-polyacrylamide gel, and then transferred to a membrane for subsequent blotting with specific antibodies. Antibodies against HSF2, EHMT2, FBP1 and β-actin were purchased from Proteintech.
Lentivirus production and stable cell line selection
For the generation of shRNA expression constructs against HSF2 and EHMT2, pLKO.1 TRC cloning vector (Addgene plamid: 10878) was used. 21 bp targets against HSF2 were 5’-CGCAGATGTAATGCACATTAT-3’ and 5’-TAACCAACTTGTGAGTTTAAA-3’ respectively. 21 bp targets against EHMT2 were 5’-CTCCAGGAATTTAACAAGATT-3’ and 5’-CGAGAGAGTTCATGGCTCTTT-3’ respectively. Scramble shRNA or Scr was obtained from Addgene (Addgene plasmid:1864). Lentiviral particles were produced by co-transfection of pLKO.1-shHSF2 or pLKO.1-shEHMT2 constructs with psPAX2 and pMD2.G into HEK-293T cells in a ratio of 4:3:1. Stable cell lines were obtained by infection of SMMC-7721 and Huh7 cells with lentiviral particles followed by puromycin selection.
CCK-8 proliferation assay
Cell proliferation was determined by CCK-8 assays by using CCK-8 reagents (Dojindo, Japan) and performed according to the manufacturer’s protocol.
Colony-formation assay
SMMC-7721 and Huh7 cells (5 × 102) stably expressing shRNA targets against HSF2 and their relative control cells were seeded into 6-well plates. After cultivating for 14 days, 4% paraformaldehyde was used to fix the cells followed by staining with 1% crystal violet. The colonies were counted subsequently.
Measurement of glycolysis
In order to assess the impact of HSF2 and EHMT2 on HCC cell glycolysis and mitochondrial respiration, we measured ECAR and OCR by using the Seahorse Bioscience XF96 Extracellular Flux Analyzer, all according to the manufacturer’s instructions of seahorse XF Glycolysis Stress Test Kit and Cell Mito stress test kit.
Dual-luciferase assay
The impact of HSF2 on FBP1 promoter activity regulation was assessed by dual-luciferase assay. The core promoter region of FBP1 was amplified from Huh7 genomic DNA, according to a previous report. The core promoter region of FBP1 was ligated into pGL3-basic vector (Promega). The promoter luciferase construct that harbors the mutated HSF binding element were generated by using the KOD-Plus-Mutagenesis Kit (Toyobo, Japan). The impact of HSF2 and EHMT2 on pGL3-FBP1 activity was measured by using Promoga’s dual luciferase assay kit.
Chromatin immunoprecipitation assay
To explore the occupancy status of HSF2 and EHMT2 on FPB1 promoter region, we performed ChIP assay by using Millipore’s EZ-ChIP kit, all according to the manufacture’s protocol. The PCR primers to amplify FBP1 promoter were F: 5’-GGCCAGGCAGAGTGGACGGGGCTT-3’ and R: 5’-CGCCTGCTTGGATCTTCAGACAAGGT-3’ [12].
TCGA dataset analysis
TCGA-LIHC on RNA expression (Level 3) of liver hepatocellular carcinoma patients in terms of RNA-seq by Expectation-Maximization was downloaded from the Cancer Genomics Brower of the University of California, Santa Cruz (UCSC) (https://genome-cancer.ucsc.edu/). In total, 360 liver hepatocellular carcinoma samples from patients with detailed expression data were chosen from the updated TCGA database according to parameters mentioned. Detailed demographics of these patients, characterized by the TCGA consortium.
Statistical analyses
Statistical analyses were performed by SPSS software (version 17.0, IBM Corp., Armonk, NY, USA) using independent Student’s t-test (two-tailed) or one-way analysis of variance (ANOVA). Statistical significance was based on two-sided P values of <0.05.
Results
HSF2 predicts prognosis and regulates proliferation of HCC cancer cells
The expression of HSF2 on HCC proliferation has seldom been discussed previously, thus we first assessed the expression of HSF2 on HCC prognosis. By using the TCGA dataset analysis, we demonstrated that patients with higher levels of HSF2 display worse survival, indicating that HSF2 might be a prognostic marker for HCC (Figure 1A). Next, we measure the influence of HSF2 on HCC cancer cell proliferation. We assessed the expression status of HSF2 in liver cancer cells by quantitative PCR, including L02, PLC, HepG2, Hep3B, Huh7, SMMC7721 and Li-7. Our Real-time PCR results demonstrated that HSF2 mRNA levels exhibited a lower expression in normal liver cells of L02, and displayed a highest expression status in Huh7 and SMMC7721 cells (Figure S1A). Subsequent western blot results demonstrated that HSF2 expression was higher in Huh7 and SMMC7721 cells (Figure 1B). Then, we silenced HSF2 expression by lentiviral-mediated transfection method, and the efficacy of knockdown was confirmed by quantitative real-time PCR and western blot (Figure 1C and 1D). Subsequently, we performed CCK-8 assay, and our results demonstrated that silencing HSF2 expression inhibited cells viability in Huh7 and SMMC7721 cells (Figure 1E and 1F). Moreover, in HSF2-low HepG2 cells, overexpression of HSF2 could increase cell viability (Figure S1B-D). In the end, we carried out colony formation assay, and results suggested that silencing HSF2 expression in Huh7 and SMMC7721 cells inhibited colony formation capacity (Figure 1G and 1H). Collectively, these results suggested that HSF2 reflected prognosis and could regulate proliferation of HCC cancer cells.
Figure 1.

HSF2 predicts prognosis and regulates proliferation of HCC cancer cells. A. Patients with higher levels of HSF2 displayed worse prognosis as revealed by TCGA-LIHC cohort analysis. B. HSF2 expression in liver cancer cell lines was assayed by western blot analysis. C. Real-time PCR results confirmed that HSF2 was silenced in Huh7 and SMMC7721 cells. D. Western blot analysis further confirmed that HSF2 was effectively silenced in Huh7 and SMMC7721 cells. E and F. CCK8 assay results indicated that knockdown of HSF2 inhibited cell variability of Huh7 and SMMC7721 cells. G and H. Silencing HSF2 expression inhibited colony formation capacity of liver cancer cells.
HSF2 regulates aerobic glycolysis in HCC cancer cells
Recently years have witnessed the significance of aerobic glycolysis for its decisive roles in maintaining proliferation of solid tumors under hypoxia conditions. Thus, we assessed the impact of HSF2 on HCC cells. ECAR measurement could reflect glycolysis rate by measuring extracellular acidification rates that caused by lactate accumulation. As shown, silencing HSF2 expression in Huh7 and SMMC7721 cells could inhibit ECAR rates, reflecting that HSF2 was a positive regulator of glycolysis and glycolytic capacity (Figure 2A-D). In HSF2-low HepG2 cells, introduction of HSF2 into HepG2 cells promoted glycolysis, as reflected by ECAR measurement (Figure S2A, S2B). Solid tumor reprograms glucose metabolism by shifting from mitochondrial respiration to glycolysis, and in the process of glycolysis, the mitochondrial respiration was inhibited that could be detected by examining oxygen consumption rates. In HSF2 silenced Huh7 and SMMC7721 cells, we observed an increase in OCR, indicating that HSF2 was a negative regulator of mitochondrial respiration (Figure 2E and 2H). In consistent with these observation, instruction of HSF2 into HepG2 cells inhibited mitochondrial respiration, as measured by OCR measurement (Figure S2C, S2D).
Figure 2.
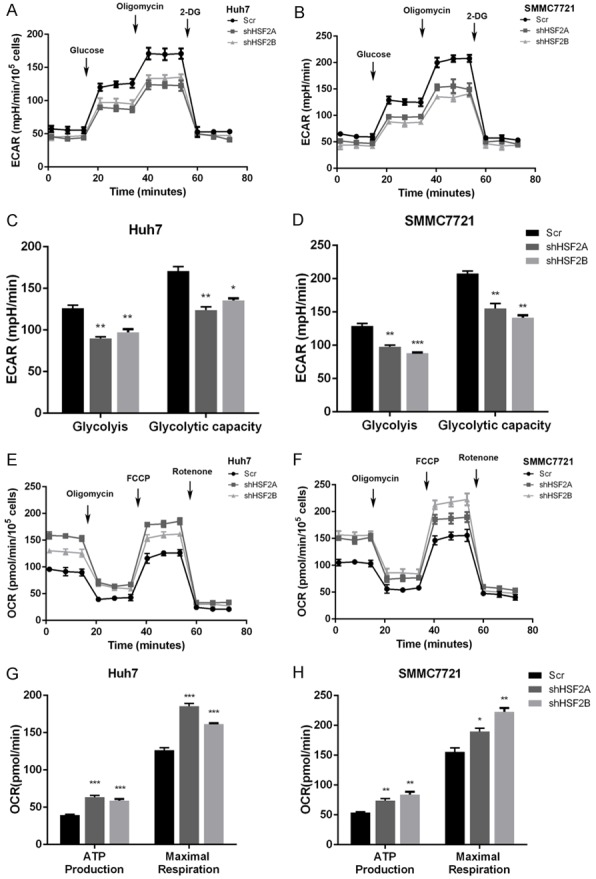
HSF2 regulates aerobic glycolysis in HCC cancer cells. A and B. Silencing HSF2 expression inhibited ECAR that reflected by Seahorse extracellular flux analyzer in Huh7 and SMMC7721 cells. C and D. Knockdown of HSF2 decreased glycolysis and glycolytic capacity in Huh7 and SMMC7721 cells. E and F. Decreasing HSF2 expression increased OCR values in Huh7 and SMMC7721 cells. G and H. Silencing HSF2 increase mitochondrial respiration as reflected by ATP production and maximal respiration.
HSF2 regulated GLUT1, HK2 and LDHA expression, which could also reflect prognosis of HCC patients
Glycolysis was a multi-step enzymatic reaction that catalyzed by glycolysis enzymes. Among them, glucose transporter 1 (GLUT1) is responsible for glucose intake, hexokinase 2 (HK2) phosphorylates glucose to produce glucose-6-phosphate, lactate dehydrogenase A (LDHA) converts L-lactate to pyruvate in the final step of glycolysis. In HSF2 silenced Huh7 and SMMC7721 cells, we observed a decrease in GLUT1, HK2 and LDHA in mRNA levels (Figure 3A and 3B). Subsequently, we asked whether GLUT1, HK2 and LDHA had prognostic values in HCC overall survival. In the TCGA-LIHC cohort, we demonstrated that patients with higher levels of GLUT1, HK2 and LDHA displayed worse prognosis respectively (Figure 3C-E). In the end, we analyzed the correlation between HSF2 with GLUT1, HK2 and LDHA in LIHC patients. As shown, HSF2 positively and significantly correlated with SLC2A1 or GLUT1 and HK2 respectively in liver cancer patients (Figure 3F and 3G). However, we observed no significant correlation between HSF2 with LDHA in TCGA cohort liver cancer patients (Figure 3H).
Figure 3.

HSF2 regulated GLUT1, HK2 and LDHA expression, which could also reflect prognosis of HCC patients. A and B. Silencing HSF2 expression decreased expression of glycolytic genes including GLUT1, HK2 and LDHA in Hun7 and SMMC7721 cells. C-E. Expression of SLC2A1, HK2 and LDHA predicted prognosis and patients with higher levels of these genes displayed worse overall survival respectively. F. HSF2 positively correlated with SLC2A1 expression in TCGA-LIHC cohort. G. HSF2 positively correlated with HK2 expression in TCGA-LIHC cohort. H. No significant correlation between HSF2 with LDHA was observed.
FBP1 was a downstream target gene of HSF2
FBP1 was reported to be a negative regulator of aerobic glycolysis and could act as a negative factor in hypoxia inducible factor 1α (HIF1α) that govern the expression of GLUT1, HK2 and LDHA, which all contain hypoxia response elements (HRE) in their promoter region. Therefore, we asked whether HSF2 could regulate FBP1 to enhance aerobic glycolysis in liver cancer. In HSF2 silenced Huh7 and SMMC7721 cells, we observed an increase in FBP1 mRNA levels (Figure 4A). Immunoblotting with FBP1 antibody further confirmed the real-time PCR results (Figure 4B). Next, we cloned FBP1 promoter region covering from -2000 to +200 and performed dual-luciferase assay. Luciferase assay results demonstrated that HSF2 could suppress pGL3-FBP1 promoter activity in a dose-dependent manner in Huh7 and SMMC7721 cells (Figure 4C). In the promoter region of FBP1, there exists a non-canonical heat-shock response element (HSE) TTCTGAACTTC from -205 to -195. We constructed the deletion mutant (del) and mutated GAA into GTT (HSEMut), subsequent luciferase assay results demonstrated that HSF2 lost its suppressive effect on these mutated luciferase constructs (Figure 4D). Subsequently, we performed ChIP assay to confirm whether HSF2 could bind HSE in FBP1 promoter region. ChIP results demonstrated that HSF2 could occupy the HSE in FBP1 promoter region in Huh7 and SMMC7721 cells (Figure 4E). Subsequently, we assessed the roles of FBP1 in liver cancer overall survival and TCGA-LIHC dataset analysis results demonstrated that patients with lower levels of FBP1 displayed worse prognosis (Figure 4F). Moreover, HSF2 negatively and significantly correlated with FBP1 expression in liver cancer patients (Figure 4G).
Figure 4.
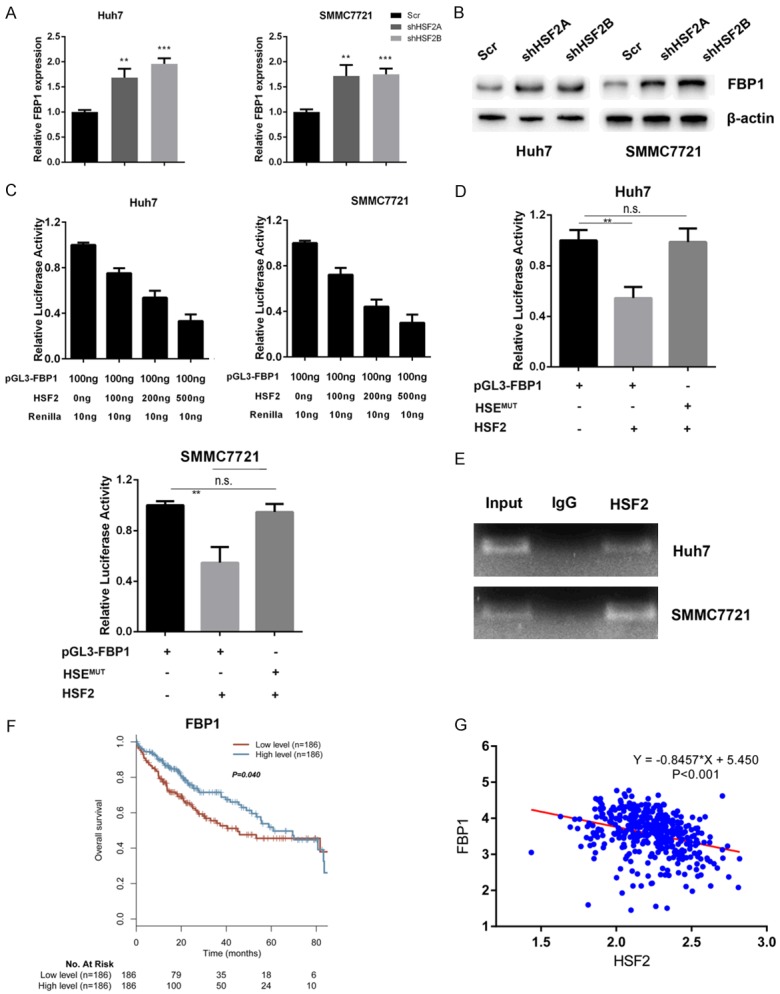
FBP1 was a downstream target gene of HSF2. A. Silencing HSF2 expression increased FBP1 expression in mRNA levels. B. Knockdown of HSF2 increased FBP1 protein levels in Huh7 and SMMC7721 cells. C. HSF2 suppressed FBP1 promoter activity in a dose-dependent manner. D. We mutated HSE on FBP1 promoter from TTCTGAACTTC to TTCTGTTCTTC to generate the HSEMut construct and luciferase assay results demonstrated that HSF2 lost its suppressive impact of HSEMut. E. HSF2 occupied the HSE in FBP1 promoter region. F. Patients with low levels of FBP1 expression have worse prognosis. G. HSF2 negatively correlated with FBP1 expression in TCGA-LIHC cohort.
EHMT2 interacts with HSF2 and regulates proliferation of liver cancer cells
FBP1 could be epigenetically silenced by EHMT2 or G9a in cancer cells, therefore we asked whether EMHT2 could interact with HSF2 to silence FBP1 expression. By co-immunoprecipitation assay, we demonstrated that HF2 interacted with EHMT2 in liver cancer cells (Figure 5A). Next, we assessed the roles of EMHT2 in liver cancer cell proliferation. Firstly, EHMT2 was silenced in Huh7 and SMMC7721 cells and the knockdown effect was confirmed by real-time PCR and western blot assay (Figure 5B and 5C). CCK-8 cell variability assay results demonstrated that knockdown of EHMT2 inhibited cell variability of Huh7 and SMMC7721 cells (Figure 5D). Colony formation assay results further confirmed that knockdown of EHMT2 in Huh7 and SMMC7721 cells attenuated colony formation capacity of these cells (Figure 5E and 5F). In the end, we measured the impact of EMHT2 in liver cancer overall survival and higher levels of EHMT2 was an unfavorable marker for overall survival in liver cancer patients (Figure 5G). In conclusion, EMHT2 was an interacting partner for HSF2 and regulated proliferation of liver cancer cells.
Figure 5.

EHMT2 interacts with HSF2 and regulates proliferation of liver cancer cells. A. HSF2 interacted with EHMT2 in Huh7 and SMMC7721 cells as reflected by co-immunoprecipitation assay. B and C. EHMT2 was effectively silenced in Huh7 and SMMC7721 cells as validated by real-time PCR and western blot. D. Knockdown of EHMT2 expression suppressed cell variability that measured by CCK-8 assay. E and F. Knockdown of EHMT2 decreased colony formation capacity of Huh7 and SMMC7721 cells. G. Patients with higher levels of EMHT2 had shorter overall survival.
EHMT2 regulates aerobic glycolysis in liver cancer cells
By using Seahorse extracellular flux analyzer, we demonstrated that silencing of EHMT2 expression decreased ECAR in Huh7 and SMMC7721 cells (Figure 6A and 6B). In depth analysis of the ECAR results reflected that silencing EHMT2 inhibited glycolysis and glycolytic capacity in Huh7 and SMMC7721 cells (Figure 6C and 6D). Subsequently, we examined the roles of EHMT2 in mitochondrial respiration by measuring OCR with Seahorse MitoStress kit. Our results demonstrated that silencing EHMT2 expression increased OCR vales in Huh7 and SMMC7721 cells (Figure 6E and 6F). Moreover, with the OCR date we demonstrated that silencing EHMT2 increased ATP production and maximal respiration in liver cancer cells, which further confirmed the negative roles of EHMT2 in mitochondrial respiration in liver cancer (Figure 6G and 6H). Therefore, we conclude that EHTM2 was a positive regulator of aerobic glycolysis in liver cancer cells.
Figure 6.
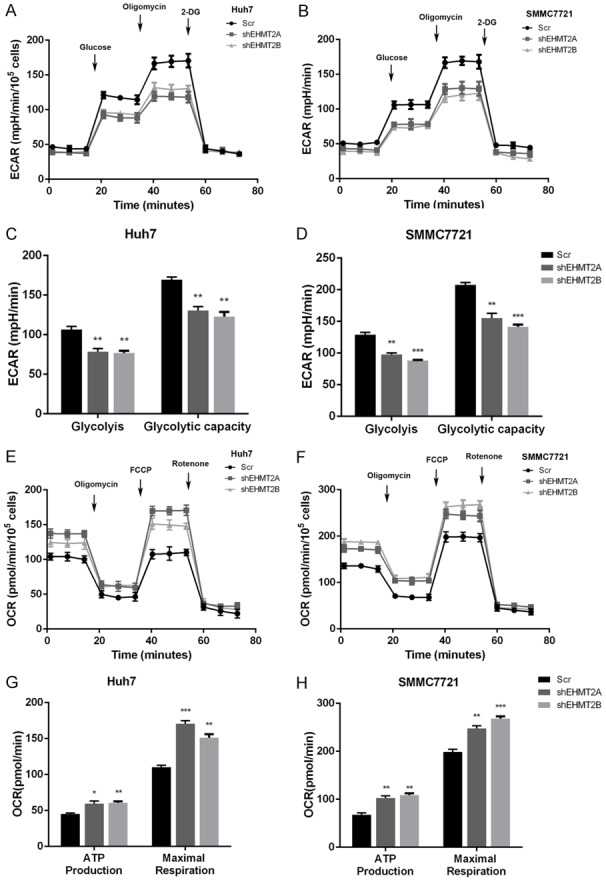
EHMT2 regulates aerobic glycolysis in liver cancer cells. A and B. Silencing EHMT2 expression decreased ECAR values. C and D. Knockdown of EHMT2 decreased glycolysis and glycolytic capacity of Huh7 and SMMC7721 cells. E and F. Decreasing EHMT2 expression increased OCR that measured by Seahorse extracellular flux analyzer. G and H. Knockdown of EHMT2 increased ATP production and maximal respiration in Huh7 and SMMC7721 cells.
EHMT2 negatively regulated FBP1 expression in liver cancer cells
Based on the positive roles of EHMT2 in aerobic glycolysis, we asked whether EHMT2 could suppress FBP1 expression, a negative regulator of aerobic glycolysis. In EHMT2 silenced Huh7 and SMMC7721 cells, we observed an up-regulation of FBP1 in mRNA and protein levels (Figure 7A and 7B). Dual luciferase assay results demonstrated that EHMT2 could suppress FBP1 promoter activity in a dose-dependent manner (Figure 7C and 7D). By performing ChIP assay, we demonstrated that EHMT2 could occupy the HSE region in FBP1 promoter, confirming it negative roles in FBP1 expression (Figure 7E). In the end, we performed TCGA dataset analysis, and uncovered that EHTM2 negatively correlated with FBP1 expression in liver cancer patients (Figure 7F). Therefore, we reported EHMT2 as a negative regulator of FBP1 expression in liver cancer.
Figure 7.
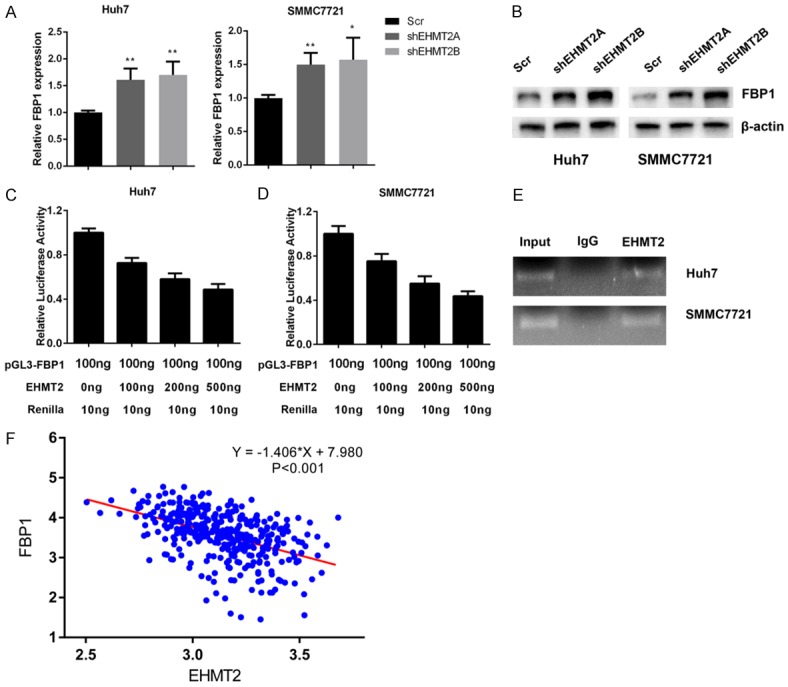
EHMT2 negatively regulated FBP1 expression in liver cancer cells. A. Silencing of EHMT2 expression increased FBP1 expression in mRNA levels. B. The protein levels of FBP1 increased in EHMT2-silenced Huh7 and SMMC7721 cells. C and D. EHMT2 suppressed promoter activity of FBP1 in a dose-dependent manner. E. EHMT2 occupied the HSE in FBP1 promoter region that demonstrated by ChIP assay. F. EHMT2 negatively correlated with FBP1 expression in TCGA-LIHC cohort patients.
HSF2 interacted with EHMT2 to epigenetically silence FBP1 expression
To further explore the influence of HSF2 and EMTH2 on FBP1 expression, we performed dual-luciferase assay, and results demonstrated that co-transfection of HSF2 and EMHT2 suppressed FBP1 promoter activity to the most extent compared to transfection of HSF2 or EMHT2 alone in Huh7 and SMMC7721 cells (Figure 8A). Then, we performed ChIP and reChIPassay, and results suggested that EMHT2 and HSF2 co-occupy the same HSE region on FBP1 promoter (Figure 8B). In the end, by performing quantitative ChIP assay, we demonstrated that silencing of HSF2 expression decreased occupancy of EHMT2 on FBP1 promoter. Furthermore, heterochromatin markers of tir-methylation H3K9 decreased and euchromatin marker AcH3 that stands for active transcription increased in HSE region when HSF2 was silenced (Figure 8C and 8D). Therefore, HSF2 was responsible for recruiting EHMT2 to HSE in FBP1 promoter to epigenetically silence its expression.
Figure 8.
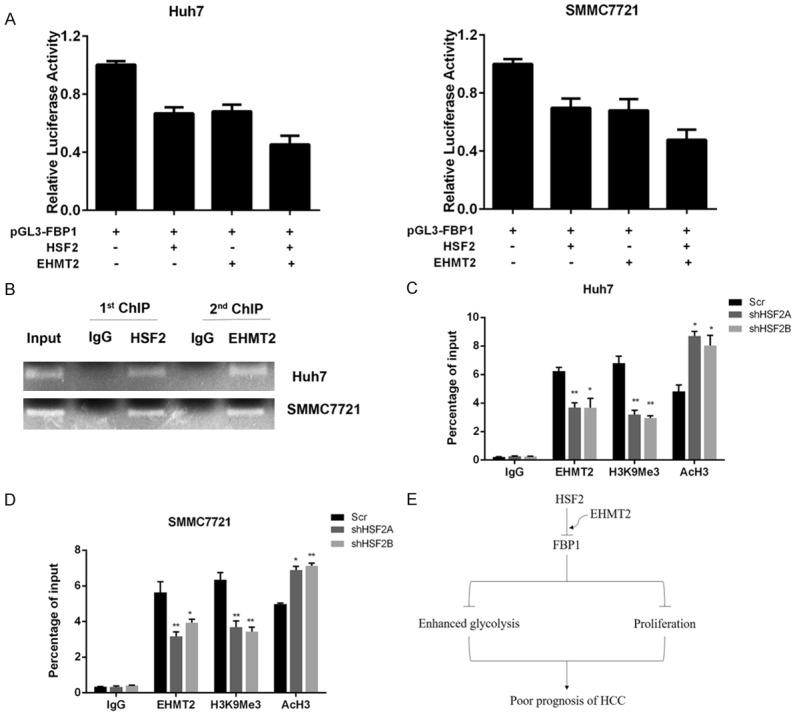
HSF2 interacted with EHMT2 to epigenetically silence FBP1 expression. A. Co-transfection of HSF2 and EHMT2 could suppress FBP1 promoter activity to a more extent. B. ReChIP assay results demonstrated that EHT2 and HSF2 co-occupied the same HSE region in FBP1 promoter. C and D. Silencing HSF2 expression decreased occupancy of EHMT2 and H3K9Me3 in FBP1 promoter and increased euchromatin marker AcH3. E. Schematic representation of the working model.
In conclusion of the whole study, we uncovered HSF2 as a novel prognostic marker for HCC. Mechanism studies demonstrated that HSF2 could interact and recruit EMHT2 to the promoter region of FBP1, leading to decreased expression of FBP1 and enhanced aerobic glycolysis, which supported the proliferation of HCC cancer cells (Figure 8E).
Discussion
Hepatocellular carcinoma ranks as the second leading cause of cancer-related deaths worldwide and is responsible for approximately 700000 deaths worldwide [13]. Although significant progress by using surgically hepatic resection, local ablation, liver transplantation and targeted therapies has been made for the treatment of HCC, the patients’ prognosis remains poor [14]. Thus, there is an urgent need for a better understanding of the molecular mechanisms that governing malignancies of HCC. One prominent feature of HCC is the alterations in glucose metabolism that received more and more attention recently. Therefore, uncovering targets that regulate aberrant HCC glucose metabolism might offer promising therapeutic strategies for HCC in the future [15]. In the present study, we demonstrated that HSF2 could be used to reflect prognosis of HCC, mechanism explorations suggested that HSF2 could regulate aerobic glycolysis via suppression of FBP, which was a negative regulator of aerobic glycolysis. Moreover, HSF2 recruited epigenetic factor EHME2 to FBP1 promoter region to epigenetically silence FBP1 expression.
HSFs are multifaceted transcription factors that regulate the response upon proteotoxic stress. Moreover, HSFs have been implicated in cell cycle, protein synthesis, ribosome biogenesis [16]. One new function of HSFs is its impact on glucose metabolism. For example, HSF1 could modulate tumor energy metabolism via suppression of thioredoxin interacting protein (TXNIP), which is a powerful negative regulator of glucose uptake and is a well-established regulator of cellular energy status [17]. What is more, HSF1 has been reported to play central roles in controlling cellular bioenergetic and mediate adaptation to nutrient availability. Hypoxia inducible factor 1α (HIF1α) and cMyc are master regulator of metabolism reprogramming for cancer cells, and HSF1 has been reported to regulate HIF1α and cMyc, reinforcing the role of HSFs on cancer cells metabolism [18-20]. But there were little reports linking HSFs to glucose metabolism. In the present study, we demonstrated a novel role of HSF2 in regulating glycolysis of HCC cells. Aerobic glycolysis has been regarded as one decisive factors that governing malignancies of cancer cells. Besides providing cancer cells with constitutive requirement of nutrient supply, aerobic glycolysis also participate to other malignancies of HCC cells. Phosphoglycerate kinase 1 (PGK1) is an important enzyme in the pathway of metabolic glycolysis, could also regulate metastasis of HCC [21,22]. Monocarboxylate transporter 1 (MCT1) plays an important role in lactic acid transport and Wnt/β-catenin signaling, indicating its roles in glycolysis, proliferation and metastasis [23]. The acidic microenvironment that caused by accumulation of lactate is the hallmark of invasive tumors, which also renders the ability to escape adaptive immune surveillance and contribute to local inflammation [24]. Therefore, targeting HCC cancer cell metabolism might provide novel strategies for the treatment of HCC. In this study, we pointed out that HSF2 was overexpressed in HCC and could regulate aerobic glycolysis, which adds the potential for targeting HSF2 as novel therapeutic strategies.
As a negative regulator of aerobic glycolysis, FBP1 gained more and more attention for its important roles in many hallmarks of cancer. FBP1 opposes renal carcinoma progression by antagonizing glycolytic flux and inhibiting “Warburg effect” [25]. In gastric, ovarian cancer and breast cancer, FBP1 expression indicated a better prognosis and can negatively regulate the EMT process [26-29]. In cervical cacner, decreased FBP1 expression promoted carcinogenesis and chemotherapy resistance [30]. In HCC, FBP1 could reduce 18F-FDG uptake, suggesting its negative roles in glycolysis [31,32]. Overall survival analysis demonstrated that decreased FBP1 expression predicted worse prognosis of HCC. In vitro and in vivo studies indicated that FBP1 expression was responsible for proliferation and metastasis of HCC cells [33]. Therefore, strategies to restore the levels and activity of FBP1 might help to develop novel treatment targets for HCC. Hepatocyte nuclear factor 4α (HNF4α) is a transcription factor and could bind to promoter region of FBP1 to suppress expression in HepG2 cells [34]. In colorectal cancer, transcription factor forkhead box C1 (FOXC1) could specifically bind to FBP1 promoter to silence its expression [35]. Here, we identified a novel transcription factor HSF2 to regulate FBP1 expression and targeting HSF2 might restore FBP1 expression to reverse malignancies of HCC. Transcription factors inhibit or activate target gene expression via interaction with epigenetic factors like DNA methyltransferase, histone lysine methyltransferase or demethylase, histone acetyltransferase of deacetylase and chromatin modifiers. As to FBP1, histone deacetylase 1 and 2 (HDAC1/2) could regulated histone H3 lysine 27 acetylation, and inhibiting HDAC1/2 activity could restore FBP1 expression [36]. Histone H3 lysine 27 methyltransferase enhancer of zeste homolog 2 (EZH2) could trimethylated chromatin of FBP1 promoter, resulting in decreased FBP1 expression in osteosarcoma cells [37]. Based on negative roles of HSF2 on FBP1 expression, it is necessary to carry out protein interaction screening to identify HSF2 interacting partners in the hope of elucidate the mechanism that govern HSF2 in regulation of HSF2.
Epigenetic changes such as DNA hypermethylation or hypomethylation, dysregulation of histone modification patterns, chromatin remodeling, and aberrant expression of microRNAs and long noncoding RNAs are associated with HCC [38]. Due to versatility of histone modification, there comes up with the notion of “histone code” which is of paramount importance in gene regulation and tumorigenesis [39]. EHMT2 is a specific histone lysine methyltransferase that could both mono- and dimethylate H3K9. Loss-of-function analysis by silencing EHMT2 and HCC tissue sample analysis demonstrated that EHMT2 expression could regulate carcinogenesis and reflect prognosis of HCC [40]. Moreover, pharmacological inhibition of EHMT2 with BIX-01294 could inhibit proliferation and tumorigenesis of HCC in culture and in vivo xenograft models [41]. Here we identified that in HCC cancer cells, EHMT2 could suppress FBP1 expression and was a positive regulator of aerobic glycolysis via interaction with HSF2. Further investigations are needed to verify whether EHMT2 could directly interact with HSF2 to suppress FPB1 expression. Moreover, developing compounds like BIX-01294 might be used to restore FBP1 expression and cutting fuel supply for HCC. In conclusion, we have demonstrated that HSF2 could interact with EHMT2 to silence FBP1 expression, rendering proliferative and metabolic advantages to HCC cells. Further analysis are necessary to elucidate the in-depth regulator mechanism of HSF2/EHMT2 complex on FPB1 silence and maintenance of malignant hallmarks of HCC.
Acknowledgements
This study was supported by National Natural Science Foundation of China (81803895) and by 2017 hospital fund of Fudan University Shanghai Cancer Center (YJ201702).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Lamarca A, Mendiola M, Barriuso J. Hepatocellular carcinoma: exploring the impact of ethnicity on molecular biology. Crit Rev Oncol Hematol. 2016;105:65–72. doi: 10.1016/j.critrevonc.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Niu ZS, Niu XJ, Wang WH. Genetic alterations in hepatocellular carcinoma: an update. World J Gastroenterol. 2016;22:9069–9095. doi: 10.3748/wjg.v22.i41.9069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gomez-Pastor R, Burchfiel ET, Thiele DJ. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat Rev Mol Cell Biol. 2018;19:4–19. doi: 10.1038/nrm.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barna J, Csermely P, Vellai T. Roles of heat shock factor 1 beyond the heat shock response. Cell Mol Life Sci. 2018;75:2897–2916. doi: 10.1007/s00018-018-2836-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai C, Sampson SB. HSF1: guardian of proteostasis in cancer. Trends Cell Biol. 2016;26:17–28. doi: 10.1016/j.tcb.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang S, Tu K, Fu Q, Schmitt DC, Zhou L, Lu N, Zhao Y. Multifaceted roles of HSF1 in cancer. Tumour Biol. 2015;36:4923–31. doi: 10.1007/s13277-015-3674-x. [DOI] [PubMed] [Google Scholar]
- 7.Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen X, Qian Y, Wu S. The Warburg effect: evolving interpretations of an established concept. Free Radic Biol Med. 2015;79:253–63. doi: 10.1016/j.freeradbiomed.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones W, Bianchi K. Aerobic glycolysis: beyond proliferation. Front Immunol. 2015;6:227. doi: 10.3389/fimmu.2015.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin D, Wu J. Hypoxia inducible factor in hepatocellular carcinoma: a therapeutic target. World J Gastroenterol. 2015;21:12171–8. doi: 10.3748/wjg.v21.i42.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shang RZ, Qu SB, Wang DS. Reprogramming of glucose metabolism in hepatocellular carcinoma: progress and prospects. World J Gastroenterol. 2016;22:9933–9943. doi: 10.3748/wjg.v22.i45.9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T, Lorkiewicz P, St Clair D, Hung MC, Evers BM, Zhou BP. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23:316–31. doi: 10.1016/j.ccr.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 14.Bruix J, Sherman M Practice Guidelines Committee, American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–36. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 15.Alves AP, Mamede AC, Alves MG, Oliveira PF, Rocha SM, Botelho MF, Maia CJ. Glycolysis inhibition as a strategy for hepatocellular carcinoma treatment? Curr Cancer Drug Targets. 2019;19:26–40. doi: 10.2174/1568009618666180430144441. [DOI] [PubMed] [Google Scholar]
- 16.Fujimoto M, Nakai A. The heat shock factor family and adaptation to proteotoxic stress. FEBS J. 2010;277:4112–25. doi: 10.1111/j.1742-4658.2010.07827.x. [DOI] [PubMed] [Google Scholar]
- 17.Santagata S, Mendillo ML, Tang YC, Subramanian A, Perley CC, Roche SP, Wong B, Narayan R, Kwon H, Koeva M, Amon A, Golub TR, Porco JA Jr, Whitesell L, Lindquist S. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science. 2013;341:1238303. doi: 10.1126/science.1238303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–13. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabai VL, Meng L, Kim G, Mills TA, Benjamin IJ, Sherman MY. Heat shock transcription factor Hsf1 is involved in tumor progression via regulation of hypoxia-inducible factor 1 and RNA-binding protein HuR. Mol Cell Biol. 2012;32:929–40. doi: 10.1128/MCB.05921-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cigliano A, Pilo MG, Li L, Latte G, Szydlowska M, Simile MM, Paliogiannis P, Che L, Pes GM, Palmieri G, Sini MC, Cossu A, Porcu A, Vidili G, Seddaiu MA, Pascale RM, Ribback S, Dombrowski F, Chen X, Calvisi DF. Deregulated c-Myc requires a functional HSF1 for experimental and human hepatocarcinogenesis. Oncotarget. 2017;8:90638–90650. doi: 10.18632/oncotarget.21469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie H, Tong G, Zhang Y, Liang S, Tang K, Yang Q. PGK1 drives hepatocellular carcinoma metastasis by enhancing metabolic process. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18081630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu H, Zhu W, Qin J, Chen M, Gong L, Li L, Liu X, Tao Y, Yin H, Zhou H, Zhou L, Ye D, Ye Q, Gao D. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology. 2017;65:515–528. doi: 10.1002/hep.28887. [DOI] [PubMed] [Google Scholar]
- 23.Fan Q, Yang L, Zhang X, Ma Y, Li Y, Dong L, Zong Z, Hua X, Su D, Li H, Liu J. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/beta-catenin signaling pathway activation in hepatocellular carcinoma cells. J Exp Clin Cancer Res. 2018;37:9. doi: 10.1186/s13046-018-0673-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lacroix R, Rozeman EA, Kreutz M, Renner K, Blank CU. Targeting tumor-associated acidity in cancer immunotherapy. Cancer Immunol Immunother. 2018;67:1331–1348. doi: 10.1007/s00262-018-2195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, Mathew LK, Mancuso A, Gade TP, Keith B, Nissim I, Simon MC. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 2014;513:251–5. doi: 10.1038/nature13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Wang Y, Li QG, Xue JJ, Wang Z, Yuan X, Tong JD, Xu LC. Downregulation of FBP1 promotes tumor metastasis and indicates poor prognosis in gastric cancer via regulating epithelial-mesenchymal transition. PLoS One. 2016;11:e0167857. doi: 10.1371/journal.pone.0167857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiong X, Zhang J, Hua X, Cao W, Qin S, Dai L, Liu W, Zhang Z, Li X, Liu Z. FBP1 promotes ovarian cancer development through the acceleration of cell cycle transition and metastasis. Oncol Lett. 2018;16:1682–1688. doi: 10.3892/ol.2018.8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi L, He C, Li Z, Wang Z, Zhang Q. FBP1 modulates cell metabolism of breast cancer cells by inhibiting the expression of HIF-1alpha. Neoplasma. 2017;64:535–542. doi: 10.4149/neo_2017_407. [DOI] [PubMed] [Google Scholar]
- 29.Li K, Ying M, Feng D, Du J, Chen S, Dan B, Wang C, Wang Y. Fructose-1,6-bisphosphatase is a novel regulator of Wnt/beta-Catenin pathway in breast cancer. Biomed Pharmacother. 2016;84:1144–1149. doi: 10.1016/j.biopha.2016.10.050. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Li M, Pang Y, Liu F, Sheng D, Cheng X. Fructose-1,6-bisphosphatase-1 decrease may promote carcinogenesis and chemoresistance in cervical cancer. Mol Med Rep. 2017;16:8563–8571. doi: 10.3892/mmr.2017.7665. [DOI] [PubMed] [Google Scholar]
- 31.Chen R, Li J, Zhou X, Liu J, Huang G. Fructose-1,6-bisphosphatase 1 reduces (18)F FDG uptake in hepatocellular carcinoma. Radiology. 2017;284:844–853. doi: 10.1148/radiol.2017161607. [DOI] [PubMed] [Google Scholar]
- 32.Yang J, Wang C, Zhao F, Luo X, Qin M, Arunachalam E, Ge Z, Wang N, Deng X, Jin G, Cong W, Qin W. Loss of FBP1 facilitates aggressive features of hepatocellular carcinoma cells through the Warburg effect. Carcinogenesis. 2017;38:134–143. doi: 10.1093/carcin/bgw109. [DOI] [PubMed] [Google Scholar]
- 33.Hirata H, Sugimachi K, Komatsu H, Ueda M, Masuda T, Uchi R, Sakimura S, Nambara S, Saito T, Shinden Y, Iguchi T, Eguchi H, Ito S, Terashima K, Sakamoto K, Hirakawa M, Honda H, Mimori K. Decreased expression of fructose-1,6-bisphosphatase associates with glucose metabolism and tumor progression in hepatocellular carcinoma. Cancer Res. 2016;76:3265–76. doi: 10.1158/0008-5472.CAN-15-2601. [DOI] [PubMed] [Google Scholar]
- 34.Wattanavanitchakorn S, Rojvirat P, Chavalit T, MacDonald MJ, Jitrapakdee S. CCAAT-enhancer binding protein-alpha (C/EBPalpha) and hepatocyte nuclear factor 4alpha (HNF4alpha) regulate expression of the human fructose-1,6-bisphosphatase 1 (FBP1) gene in human hepatocellular carcinoma HepG2 cells. PLoS One. 2018;13:e0194252. doi: 10.1371/journal.pone.0194252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Q, Wei P, Wu J, Zhang M, Li G, Li Y, Xu Y, Li X, Xie D, Cai S, Xie K, Li D. The FOXC1/FBP1 signaling axis promotes colorectal cancer proliferation by enhancing the Warburg effect. Oncogene. 2019;38:483–496. doi: 10.1038/s41388-018-0469-8. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Jin X, Yan Y, Shao Y, Pan Y, Roberts LR, Zhang J, Huang H, Jiang J. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci Rep. 2017;7:43864. doi: 10.1038/srep43864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiong X, Zhang J, Liang W, Cao W, Qin S, Dai L, Ye D, Liu Z. Fuse-binding protein 1 is a target of the EZH2 inhibitor GSK343, in osteosarcoma cells. Int J Oncol. 2016;49:623–8. doi: 10.3892/ijo.2016.3541. [DOI] [PubMed] [Google Scholar]
- 38.Wahid B, Ali A, Rafique S, Idrees M. New insights into the epigenetics of hepatocellular carcinoma. Biomed Res Int. 2017;2017:1609575. doi: 10.1155/2017/1609575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol. 2016;8:a019521. doi: 10.1101/cshperspect.a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei L, Chiu DK, Tsang FH, Law CT, Cheng CL, Au SL, Lee JM, Wong CC, Ng IO, Wong CM. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J Hepatol. 2017;67:758–769. doi: 10.1016/j.jhep.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 41.Yokoyama M, Chiba T, Zen Y, Oshima M, Kusakabe Y, Noguchi Y, Yuki K, Koide S, Tara S, Saraya A, Aoyama K, Mimura N, Miyagi S, Inoue M, Wakamatsu T, Saito T, Ogasawara S, Suzuki E, Ooka Y, Tawada A, Otsuka M, Miyazaki M, Yokosuka O, Iwama A. Histone lysine methyltransferase G9a is a novel epigenetic target for the treatment of hepatocellular carcinoma. Oncotarget. 2017;8:21315–21326. doi: 10.18632/oncotarget.15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.