

Abstract
Lung cancer causes the highest mortality in cancer-related deaths. As these cancers often become resistant to existing therapies, definition of novel molecular targets is needed. Epigenetic modifiers may provide such targets. Recent reports suggest that the histone acetyltransferase (HAT) module within the transcriptional coactivator SAGA complex plays a role in cancer, creating a new link between epigenetic regulators and this disease. GCN5 serves as a coactivator for MYC target genes, and here we investigate links between GCN5 and c-MYC in non-small cell lung cancer (NSCLC). Our data indicate that both GCN5 and c-MYC proteins are upregulated in mouse and human NSCLC cells compared to normal lung epithelial cells. This trend is observable only at the protein level, indicating that this upregulation occurs post-transcriptionally. Human NSCLC tissue data provided by The Cancer Genome Atlas (TCGA) indicates that GCN5 and c-MYC expression are positively associated with one another and with the expression of c-MYC target genes. Depletion of GCN5 in NSCLC cells reduces c-MYC expression, cell proliferation, and increases the population of necrotic cells. Similarly, inhibition of the GCN5 catalytic site using a commercially available probe reduces c-MYC expression, cell proliferation, and increases the percentage of cells undergoing apoptosis. Our findings suggest that GCN5 might provide a novel target for inhibition of NSCLC growth and progression.
Keywords: Histone acetyltransferases, GCN5, oncoprotein, c-MYC, non-small cell lung cancer, SAGA complex
Introduction
Approximately 85% of all lung cancer cases are non-small cell lung cancers (NSCLCs), including squamous cell carcinomas, adenocarcinomas and less commonly large cell carcinomas [1]. Treatment for lung cancer, including surgery, radiation therapy, chemotherapy and targeted therapies, has improved survival, but novel targets for lung cancer are still needed since these cancers often become resistant to therapies [2]. Elucidation of epigenetic mechanisms that play a role in lung cancer holds promise to improve targeted therapy for this cancer as well as other cancer types [3].
Spt-Ada-Gcn5-Acetyl transferase (SAGA) is a multi-functional, multi-protein complex that post-translationally modifies histones [4-6]. The SAGA complex is composed of multiple functional units including the histone acetyltransferase (HAT), deubiquitinase (DUB), SPT and TAF modules [4-6]. These modules regulate histone acetylation and deubiquitination, maintain SAGA architecture, and facilitate interactions with transcriptional machinery and transcription factors [4-6]. In mammals, SAGA plays a role in regulating transcriptional initiation and elongation and also targets non-histone substrates that serve in diverse biological roles [4-7]. SAGA is important for normal embryonic development in mice and plays important roles in cell growth, signaling pathways, genome integrity, metabolic control and stress responses [4-6]. Many of these processes are hallmarks of cancer, and dysregulation of these activities may contribute to oncogenesis [8].
The SAGA histone acetylation module contains GCN5 (also known as KAT2A) as the catalytic subunit and ADA proteins that regulate HAT activity [4-6]. PCAF (also known as KAT2B), a homologous transcriptional co-activator to GCN5, is also incorporated in a SAGA-like complex [9]. In addition, GCN5 and PCAF are incorporated into ATAC complexes [10], as well as smaller ADA complexes [11]. In all cases, complexes contain either GCN5 or PCAF, never both. Prior work indicates that GCN5 and PCAF can compensate for one another in some biological settings, but these HATs also have distinct functions [12]. GCN5, the first identified transcription-related HAT, acetylates lysine residues in histone proteins to facilitate transcription initiation and elongation [5]. GCN5-mediated acetylation occurs on multiple resides in histone H3 including K9, K14, K18, K23, K27, K36, and additional lysines in histones H2B and H4 [13]. A variety of studies have shown that both GCN5 expression and proper function are essential for normal development [14-17]. Embryonic stem cells (ESCs) null for Gcn5 show cell-cycle arrest in the G2/M phase and premature loss of transcription factors essential for ESC identity upon differentiation, indicating that GCN5 is required for maintaining both ES cell self-renewal and differentiation [18]. Conditional knock out (KO) of Gcn5 in neural progenitor cell populations reduces brain mass, similar to phenotypes observed upon deletion of c-Myc or N-myc, suggesting that GCN5 positively regulates the expression or the function of MYC proteins [19]. Indeed, MYC mutant mice exhibit global changes in gene expression linked to altered expression of GCN5 [20].
Our previous work revealed that loci bound by GCN5 in mouse ESCs are highly enriched for c-MYC and/or E2F1 consensus binding sites. In addition, a large percentage of genes affected by GCN5 loss are known to be regulated by c-MYC and E2F1 [21]. Interestingly, c-MYC regulates expression of GCN5 and several other SAGA components [21]. GCN5 is one of the earliest genes induced by c-MYC during reprogramming of fibroblasts to induced pluripotent (iPS) cells, and together c-MYC and GCN5 activate genes required for alternative splicing events necessary for reprogramming [21]. These findings indicate that c-MYC and GCN5 work together in a feed forward loop where c-MYC increases the expression of GCN5, which functions as a coactivator of c-MYC target genes [21]. Since GCN5 is required for the functions of c-MYC in ESCs and as a Yamanaka factor during somatic cell reprogramming [21], it may also contribute to c-MYC functions in oncogenesis. c-MYC is frequently amplified and aberrantly expressed in human lung cancers, raising the question of whether GCN5 may contribute to the formation or progression of these cancers [22].
Previous studies have interrogated the effect of GCN5 knockdown in various cancer cells [23-26]. These studies reveal that GCN5 serves as an oncoprotein in various cancer types and that its inhibition represses certain cancer phenotypes [23-26]. Thus far, only a small number of studies have probed GCN5 functions in lung cancer. Inhibition of GCN5 was found to reduce viability of lung cancer stem-like cells [27]. A separate study revealed that repression of GCN5 reduced NSCLC growth by suppressing key oncoproteins such as E2F1, cyclin D1 and cyclin E1 [23]. However, neither study analyzed the relationship between GCN5 and c-MYC.
Here we examine links between GCN5 and c-MYC in NSCLC cell lines and tissues, in order to determine whether the GCN5 catalytic center might provide a novel target for therapy development.
Materials and methods
Cell culture
The murine (ED1, 344P, 344SQ, 393P, KC2 and LKR13) lung cancer cell lines, the human (H1355, A549, H520, H1299 and LUDLU1) lung cancer cell lines and the immortalized murine pulmonary epithelial cell line C10 were cultured in RPMI-1640 medium (HyClone) supplemented with 10% fetal bovine serum (FBS) (HyClone) and 1% penicillin-streptomycin (HyClone). The immortalized human pulmonary epithelial cell line BEAS2B was cultured in LHC-9 medium (Gibco) supplemented with 1% penicillin-streptomycin. The immortalized human pulmonary epithelial cell lines HBEC was cultured in Keratinocyte Serum-Free Media (KSFM, Gibco) supplemented with 1% penicillin-streptomycin, Estrogen Growth Factor (EGF, 5 ng/μL, Invitrogen) and Bovine Pituitary Extract (BPE, 50 ng/μL, Invitrogen). Immortalized murine fibroblast cell lines (MCAFs and MLFs) were cultured in Alpha-MEM medium (Corning) supplemented with 20% FBS, 1% Sodium Pyruvate (Gibco), 1% L-glutamine (HyClone) and 1% penicillin-streptomycin. The immortalized human fibroblast cell line (IMR90) was cultured in DMEM (Corning) supplemented with 10% FBS, 1% L-glutamine and 1% penicillin-streptomycin. 293T human embryonic kidney cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. Cells were cultured at 37°C in a humidified incubator with 5% CO2. Cell lines were purchased and authenticated by the American Type Culture Collection (ATCC). The murine lung cancer cell line ED1 was derived from lung cancers arising from wild-type cyclin E engineered mice and was previously authenticated [28]. The murine lung cancer cell lines 344P, 344SQ, 393P, KC2 and LKR13 were derived from lung cancers arising from mutations in Kras or both Kras and p53 [29]. The immortalized murine fibroblast cell lines were derived as previously described [30,31].
3-Methylcyclopentylidene-[4-(4’-chlorophenyl)thiazol-2-yl] hydrazone (CPTH6) [32] was purchased from Cayman Chemical, dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich) for a final concentration of 10 mM (10 mmol/L) and diluted to final concentrations in complete medium similar to previous studies [27,33]. Equal volumes of DMSO were used as vehicle control (1% DMSO maximum). After 24 hours from seeding, logarithmically growing cells were treated with CPTH6 at concentrations ranging from 40-100 μM for 24-96 hours.
Immunoblot analyses
Cells were washed with phosphate buffered saline (PBS, Corning) and lysed with ice-cold Pierce RIPA Lysis and Extraction Buffer (Thermo Scientific) supplemented with Protease Inhibitor Cocktail (Sigma-Aldrich) and PhosSTOPTM Phosphatase Inhibitor (Sigma-Aldrich). Total histone extracts from cells were isolated using the Histone Purification Kit (Active Motif) according to manufacturer’s protocol. Proteins were resolved on SDS-PAGE before transfer to nitrocellulose membranes (Bio-Rad). Membranes were blocked in 5% nonfat milk (Equate Dry Milk, Wal-Mart) dissolved in Tris-buffered saline with 0.1% Tween 20 (TBS-T) for at least 1 hour before overnight incubation at 4°C with primary antibody diluted in 5% nonfat milk or 5% bovine serum albumin (New England Biolabs). This was followed by a series of washes with TBS-T and an incubation (1 hour) with secondary antibody diluted in 5% nonfat milk at room temperature. After additional washes with TBS-T, antibody binding was visualized by ECLTM Prime Western Blotting System (GE Healthcare) or by an Odyssey Blot Imager (LI-COR Biosciences) and quantified by ImageJ software (version 1.51m9, imagej.nih.gov/ij). Antibodies used for immunoblot analyses were: anti-ADA2B (Abcam, #57953), anti-β-Actin (Santa Cruz, #sc47778), anti-c-Myc (Cell Signaling Technology, #5605 and #9402), anti-GCN5 (Cell Signaling Technology, #3305), anti-H3 (Abcam, #1791), anti-H3-K9Ac (Abcam, #32129), and anti-PCAF (Cell Signaling Technology, #3378). Secondary HRP anti-mouse and anti-rabbit antibodies and secondary Alexa Fluor Plus anti-mouse (#A32729) and anti-rabbit (#A32735) antibodies were purchased from Invitrogen (Thermo Scientific). Immunoblots were stripped using Restore PLUS Western Blot Stripping Buffer (Thermo Scientific). Protein expression levels were quantified by ImageJ software (version 1.52k, imagej.nih.gov/ij).
Proliferation and apoptosis assays
Cell proliferation was measured using the CellTiter-Glo Assay kit (Promega) according to manufacturer’s protocol. Apoptotic and necrotic cells were measured using FITC Annexin V Apoptosis Detection Kit with Propidium Iodide (PI, BioLegend) according to manufacturer’s protocol.
Quantitative real-time PCR assays
Quantitative real-time PCR (qRT-PCR) assay primers are listed in Table 1. Total RNA was isolated using the RNeasy minikit (Qiagen). RNA was converted to cDNA prior to qPCR using SYBR green PCR master mix (Roche).
Table 1.
Quantitative RT-PCR Primers. A list of target genes along with respective forward and reverse primer sequences
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Mouse Gcn5 | 5’-CTTCTGTGCCGTCACCTCAA-3’ | 5’-TGGTACTCCTTTAGGTGGTTCATCA-3’ |
| Mouse c-Myc | 5’-AATCCTGTACCTCGTCCGATTCCA-3’ | 5’-TTTGCCTCTTCTCCACAGACACCA-3’ |
| Mouse Pcaf | 5’-GGAGAAACTCGGCGTGTACT-3’ | 5’-CTGGAGGTCTCCTCTTGGTG-3’ |
| Mouse Gapdh | 5’-TCACCACCATGGAGAAGGC-3’ | 5’-GCTAAGCAGTTGGTGGTGCA-3’ |
| Human GCN5 | 5’-GTGCTGTCACCTCGAATGAG-3’ | 5’-CGGCGTAGGTGAGGAAGTAG-3’ |
| Human c-MYC | 5’-TTTCGGGTAGTGGAAAACCA-3’ | 5’-CACCGAGTCGTAGTCGAGGT-3’ |
| Human PCAF | 5’-GGCCAAGAAACTGGAGAAACT-3’ | 5’-GGTGAGGGGTTAGGGTTTTT-3’ |
| Human GAPDH | 5’-AGGTGAAGGTCGGAGTCAACG-3’ | 5’-CGTTCTCAGCCTTGACGGTG-3’ |
Stable cell lines with repressed GCN5 expression
Five candidate TRC pLKO.1 lentiviral shRNAs for murine and five TRC pLKO.1 lentiviral shRNAs for human repressing GCN5 or c-MYC were purchased (Dharmacon) and two shRNAs with the greatest knockdown efficiency as determined by immunoblots (> 50%) were chosen. Lentiviral psPAX2 and pMD2-G plasmids were used to generate lentiviral particles, as described previously [14]. Lentiviral particles used for individual GCN5 knockdown experiments in murine and human cell lines were generated in 293T cells, as described previously [14].
DepMap
Genetic screens and gene dependencies for human lung cancer cell lines were obtained from public repositories of DepMap (https://depmap.org/portal/). Dependency scores were directly recorded from genome-wide-loss-of-function RNAi (DEMETER2) [34] and CRISPR (CERES) [35] screen datasets. Lung cancer cell lines containing dependency scores of ~ -0.3 or lower were considered as dependent on GCN5 or ADA2B, respectively.
The cancer genome atlas (TCGA) and oncomine
Expression data (mRNA) for human NSCLC (both lung adenocarcinomas and squamous cell carcinomas) and adjacent normal lung tissues were obtained from public repositories of TCGA. High and low two-gene combination predictions were analyzed as previously reported [36]. To compare expression levels, transcripts per million (TPM) units were used, which was found optimal for comparing expression data from RNA sequencing [37].
To confirm these analyses, expression data (mRNA) for human normal and non-small cell lung cancer tissues (both lung adenocarcinomas and squamous cell carcinomas) were analyzed using the Oncomine database (www.oncomine.org). Both normal lung and NSCLC tissue mRNA expression data (average mRNA Log2 median-centered intensity) was analyzed from a previously published data set [38].
Statistics
Two-tailed Student’s t test was used to compare differences between groups. Spearman rank correlation measured the strength of association between two variables. One-way ANOVA with Bonferroni correction was used for multiple comparisons. The logrank test was used to compare the survival distributions shown in Kaplain-Meier plots. The IC50 values for CPTH6 treatment were calculated as previously described [39]. Data are presented as mean with SD bars, unless otherwise stated. Results of independent experiments were pooled to assess statistical significance. Statistical significance was defined as P < 0.05.
Results
GCN5 and c-MYC are positively associated in human NSCLC tissues
First, we conducted a comprehensive analysis of GCN5 mRNA expression in human NSCLC tissues, comparing GCN5 mRNA levels in adjacent normal and NSCLC tissues using publicly available databases including The Cancer Genome Atlas (TCGA) and Oncomine. These analyses revealed that GCN5 mRNA expression is upregulated in lung cancers (both adenocarcinomas and squamous cell carcinomas) versus normal tissues (Figures 1A, S1). These findings are consistent with previous work that indicated GCN5 protein levels are elevated in NSCLC tissues [23]. Interestingly, PCAF mRNA levels were significantly (P < 0.001) downregulated in NSCLC compared to adjacent normal tissue (Figure S2). In contrast to GCN5 mRNA levels, both ADA2B, another SAGA HAT subunit component important for GCN5/PCAF HAT activity [40], and c-MYC mRNA levels showed either no significant differences or inconsistent trends when comparing expression in adenocarcinomas or squamous cell carcinomas to adjacent normal lung tissues (Figures S3, S4).
Figure 1.

GCN5 mRNA levels are upregulated and associated with c-MYC and c-MYC target genes in human NSCLC tissues. Public repositories of TCGA were analyzed for (A) GCN5 mRNA levels in human adjacent normal lung (n = 59) and lung adenocarcinoma (n = 517) as well as adjacent normal lung (n = 51) and lung squamous cell carcinoma (n = 501) tissues, (B) GCN5 mRNA levels in different stages (I-IV) of lung adenocarcinoma or lung squamous cell carcinoma, (C) associations between GCN5 and c-MYC mRNA levels in human lung adenocarcinoma and lung squamous cell carcinoma tissues, (D) significant associations between GCN5 and c-MYC to known c-MYC target genes, or (E) GCN5 and c-MYC mRNA levels in relation to NSCLC (both lung adenocarcinoma and lung squamous cell carcinoma) patient survival. Data are represented by (A) box-and-whisker plots, (B) scatter plots, (C) a heat map representing negative (green) versus positive (red) associations, (D) a Kaplan-Meier survival curve where: purple = GCN5 high, c-MYC low (P = ns), green = GCN5 low, c-MYC low (P = ns), red = GCN5 high, c-MYC high (P = ns), and yellow = GCN5 low, c-MYC high (*P < 0.05). High versus low levels are relative to median expression. Symbols refer to ***P < 0.001.
Late stage lung cancers are associated with reduced positive response to therapy and higher mortality rates [41]. Since GCN5 mRNA levels were upregulated in NSCLC tissues, we next sought to determine whether GCN5 mRNA levels were associated with stage in NSCLC, using TCGA to compare expression of GCN5 in both lung adenocarcinomas and squamous cell carcinomas categorized by stages I-IV. A slight, but significant (P < 0.001), decrease in GCN5 mRNA levels was only observed in later stage lung squamous cell carcinomas only, relative to earlier stage tumors (Figure 1B). Variability in GCN5 expression levels between different NSCLC types and stages suggest that GCN5 expression is not associated specifically with early or advance stages of NSCLC.
Next, GCN5 and c-MYC mRNA levels were examined in individual adenocarcinomas and squamous cell carcinomas data in TCGA to determine if expression of these factors is associated in these NSCLCs. GCN5 and c-MYC mRNA levels were found to be significantly (P < 0.001, r = 0.150 for lung adenocarcinomas and r = 0.295 for lung squamous cell carcinomas), positively associated in human NSCLC tissues (Figure 1C). In contrast, PCAF and c-MYC mRNA levels were not found to be associated in the same human NSCLC tissues (Figure S5). The association between GCN5 and c-MYC mRNA levels in human NSCLC tissues suggests that there may also be an association between GCN5 and c-MYC target gene expression levels.
Prior work has shown that direct target genes of GCN5 overlap with those of c-MYC in ES cells [21]. We next asked whether GCN5 and c-MYC show similar associations with known c-MYC target genes [42] in human NSCLC tissues. TCGA was again interrogated, and expression of some known c-MYC target genes was found to be similarly associated to both GCN5 and c-MYC expression (Figures 1D, S6). These target genes, with their respective Spearman Rho values and statistically significant p-values, are also listed in Table S1. Most of these genes have functions in cell growth, apoptosis, and cell metabolism. The associations between expression of GCN5 and c-MYC, and between expression of GCN5, c-MYC, and known c-MYC target genes, suggest that GCN5 may serve as an important cofactor for c-MYC in NSCLC, consistent our prior work in ES cells and in iPS cells [21].
Lastly, since amplification of c-MYC correlates with poor prognosis in NSCLC [43], we sought to determine the relationship between GCN5 and c-MYC mRNA levels and NSCLC patient survival over a two-hundred-month period. However, no significant differences in survival were found among patients with high vs low expression of GCN5 and c-MYC (Figure 1E).
GCN5 and c-MYC protein expression levels are up regulated in NSCLC cell lines
To complement our in-silico analyses, we next determined whether GCN5 protein levels are up regulated in lung cancer cell lines, and whether GCN5 levels are elevated along with c-MYC. Both GCN5 and c-MYC protein levels were examined in murine (ED1, 344P, 344SQ, 393P, KC2, LKR13) and human (A549, H520, H1299, H1355, LUDLU1) NSCLC lines compared to murine (C10, MCAFs, MLFs) and human (HBEC, BEAS2B, IMR90) normal immortalized controls. In murine and most human NSCLC cell lines, both GCN5 and c-MYC protein levels were up regulated compared to normal controls (Figure 2A, 2B). Such trends were more robust in the murine cell lines. Protein expression of ADA2B was also upregulated in murine NSCLC cell lines compared to normal lines, but this trend was not observed in human cell lines. PCAF protein levels were found to be highly variable in both murine and human NSCLC cells (Figure S7A, S7B). Next, we sought to determine whether c-MYC affects GCN5 expression in NSCLC lines. Knockdown of c-MYC levels in ED1 and H1355 decreased GCN5 protein levels, consistent with previous findings that c-MYC regulates GCN5 expression other cells [21].
Figure 2.
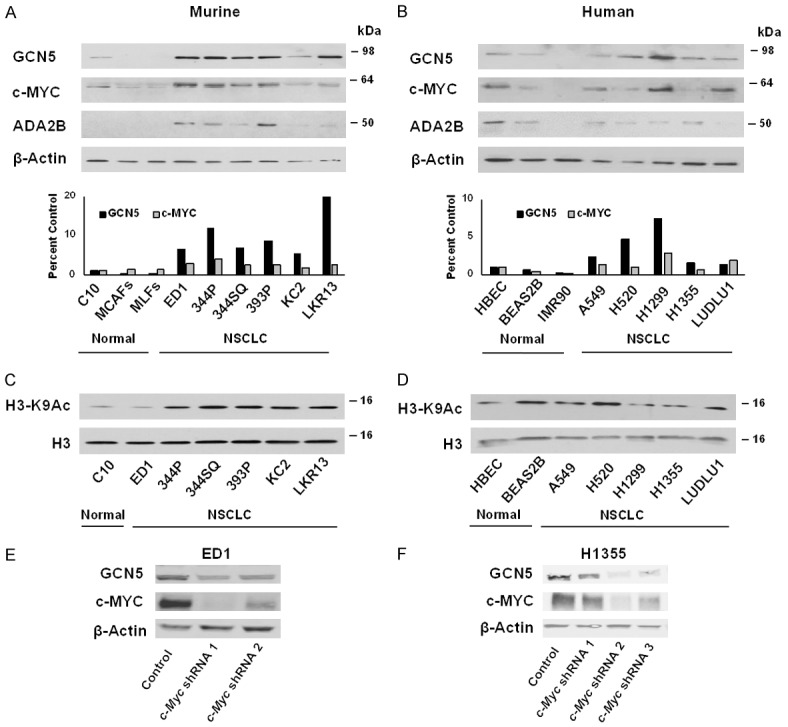
GCN5 and c-MYC proteins are upregulated and positively associated in NSCLC cell lines. Steady state levels of GCN5, c-MYC, and ADA2B proteins were analyzed in (A) murine and (B) human immortalized epithelial and fibroblast lung controls and NSCLC cell lines. Immunoblotting was performed using anti-GCN5, anti-c-MYC, and anti-ADA2B antibodies. β-Actin served as a loading control. GCN5 and c-MYC expression levels were normalized to β-Actin expression and compared between immortalized lung controls and NSCLC cell lines. Steady state levels of histone H3 K9 acetylation were detected by immunoblotting using anti-H3K9 acetylation and anti-H3 antibodies in (C) murine and (D) human cell lines. Total H3 protein served as a loading control. Stable reduction of endogenous c-MYC expression by independent shRNAs introduced into murine ED1 (E) and human H1355 (F) lung cancer cells decreased endogenous GCN5 protein levels. Immunoblotting was performed using anti-GCN5 and anti-c-MYC antibodies. β-Actin served as a loading control.
Histone H3K9 acetylation is largely dependent on GCN5 and PCAF expression in other cell types [13], so we compared levels of this modification in normal immortalized cells and NSCLC cells. Since GCN5 protein levels were undetectable in normal fibroblast immortalized lung controls, we focused our acetylation analyses on epithelial cells. Histone H3K9 acetylation was upregulated in murine NSCLC lines compared to normal control cells (Figure 2C), consistent with increased GCN5 expression and HAT activity in these cell lines. Again, this trend was not apparent in human cell lines (Figure 2D). Such differences between mouse and human cell lines is not uncommon, since human cell lines harbor many different mutations that may affect protein steady state levels [44].
To determine whether GCN5 and c-MYC mRNA levels were also upregulated in murine and human NSCLC cell lines, we performed qRT-PCR analyses on RNA extracted from murine (ED1, 393P, KC2, LKR13) and human (H520, H1299, H1355, A549, LUDLU1) NSCLC lines and compared the results to those using RNA extracted from murine (C10) and human (HBEC and BEAS2B) normal immortalized lung cells as controls. Again, we focused attention on normal epithelial immortalized lung cells since expression levels of GCN5 and c-MYC were almost undetectable in normal fibroblast immortalized cells. No consistent differences in either GCN5 or c-MYC mRNA expression in the NSCLC lines relative to the normal controls were observed (Figure S8A, S8B), although decreased levels of Gcn5 mRNA (P < 0.05) were observed in two murine NSCLC lines (393P and LKR13). We also analyzed expression of PCAF in both murine and human NSCLC lines, and again no clear, consistent trends were observed amongst the cell lines examined (Figure S9A, S9B).
After observing a positive association between GCN5 and c-MYC expression in human NSCLC tissues in TCGA, we next asked whether such association between GCN5 and c-MYC RNA levels also occurred in murine and human NSCLC cell lines. In murine cells, Gcn5 and c-Myc mRNA levels appear similar to each other in most lines, except for LKR13 (Figure S8C), and Spearman rank correlation tests indicate that Gcn5 and c-Myc mRNA levels have a weak, positive association (r = 0.2, P = 0.80) in these murine NSCLC and normal control cells. Similar GCN5 and c-MYC mRNA levels were observed in some of the human cell lines but were notably different in others (Figure S8D). Spearman rank correlation tests indicate a moderate, negative association between the mRNA levels of these genes (r = -0.67, P = 0.17) in human cells.
Taken together, our analyses in murine and human NSCLC cell lines indicate that expression of GCN5 and c-MYC were coordinately upregulated in NSCLC cells relative to normal lung cells mainly at the protein level. The strong associations between GCN5 and c-MYC in normal lung and NSCLC cell lines indicates that the upregulation of both proteins occurs post-transcriptionally.
NSCLC cell lines are dependent on GCN5 function
To explore whether viability of human lung cancer cell lines is dependent on GCN5 expression, two independent genome-wide loss-of-function screens using either RNAi [34] or CRISPR [35] were interrogated using the DepMap database. We also examined dependency data for ADA2B, another component of the HAT module in SAGA, and for PCAF, a GCN5 homolog. Out of ~127 human lung cancer cell lines analyzed in the RNAi screen, 10 exhibited a dependency of GCN5 (KAT2A) expression, as indicated by a dependency score of ~ -0.3 or lower [45], and the majority of these are NSCLC cell lines (Figure 3A, 3B). Only 3 lung cancer cell lines in the RNAi screen exhibited a dependency of ADA2B (TADA2B) expression, as also indicated by a dependency score of ~ -0.3 or lower (Figure 3C). The small cell lung cancer cell line DMS53 was the only line in the RNAi screen that showed a dependency on PCAF (KAT2B), with a dependency score of -0.35 (data not shown).
Figure 3.
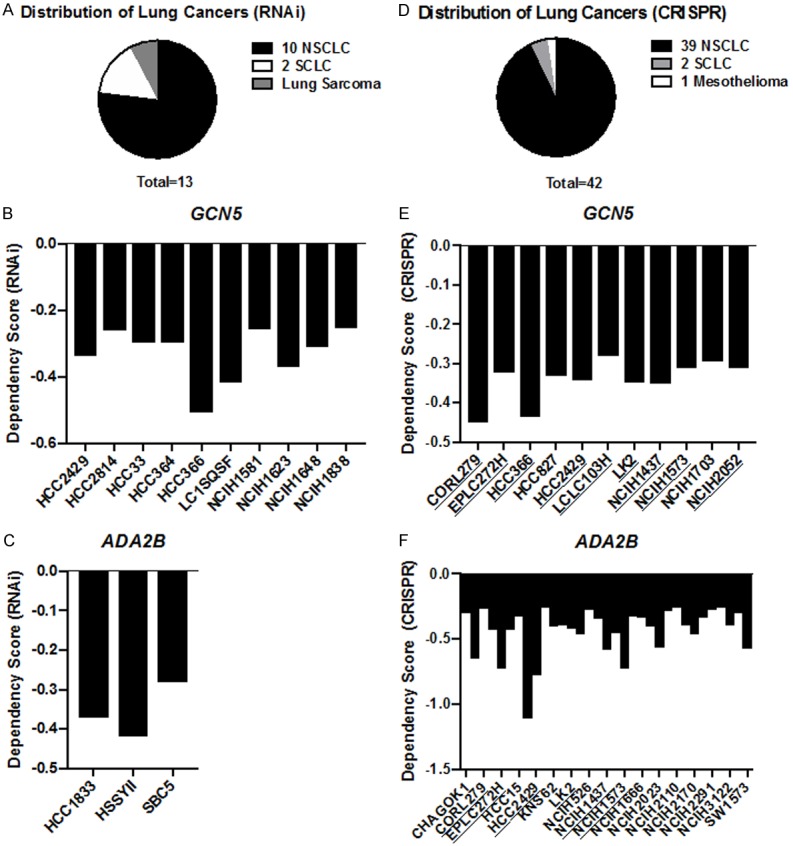
Human NSCLC cell lines are dependent on GCN5 and ADA2B functions. Genome-loss-of-function dependency data of human lung cancer cell lines were analyzed using RNAi and CRISPR screen datasets. (A) The breakdown of human lung cancer cell types dependent on GCN5 and ADA2B in the RNAi screen and (B) GCN5 or (C) ADA2B dependent cell lines and dependency scores were analyzed. Cell lines with dependency scores of ~ -0.3 or lower are represented. (D) The breakdown of human lung cancer cell types dependent on GCN5 and ADA2B in the CRISPR screen and (E) GCN5 as well as (F) ADA2B dependent cell lines and dependency scores were analyzed. Cell lines with dependency scores of ~ -0.3 or lower are represented. Lung cancer cell lines exhibiting a dependency for both GCN5 and ADA2B are underlined.
To gain further insights to SAGA HAT module dependencies in human lung cancer cell lines, we examined data from CRISPR genetic screens for both GCN5 and ADA2B and found 42 lung cancer cell lines out of the 70 lines used in the screen exhibited dependency on one or both of these factors (Figure 3D). The majority of these lines were again NSCLC cells lines; 11 lines had a dependency score for GCN5 of -0.3 or lower (Figure 3E) and 33 had a dependency score for ADA2B of -0.3 or lower (Figure 3F). Only 9 of these cell lines exhibited dependency on both GCN5 and ADA2B (Figure 3E, 3F, underlined cell lines). Moreover, 12 lung cancer lines exhibited a very high dependency on ADA2B, with a score -0.5 or lower, which may reflect the fact that ADA2B is required for both GCN5 and PCAF containing SAGA complexes. However, no lung cancer cell lines in the CRISPR screen showed dependency on PCAF, suggesting that GCN5 is most crucial in these cells. Altogether, these data suggest that SAGA HAT module functions are required for proliferation or survival of multiple NSCLC cell lines.
Repression of GCN5 expression or activity affects proliferation and reduces c-MYC protein levels in NSCLC cell lines
We next determined how depletion of Gcn5 affects NSCLC cell proliferation and survival using two independent shRNAs to repress GCN5 expression. Representative results from Gcn5 depletion in murine ED1 cells and human H1355 cells are shown (Figure 4A), but similar results were also seen in other cell lines including 393P, LKR13, H520, and A549 (data not shown). Proliferation assays were performed in ED1 and H1355 over a four-day period. Similar to prior reports [23], we found that depletion of GCN5 significantly (P < 0.01) reduced proliferation of both murine and human NSCLC cell lines (Figure 4A) but does not affect the percentage of cells undergoing apoptosis (Figure 4B). Interestingly, our data indicate that repression of GCN5 significantly (P < 0.05) increased the population of necrotic cells. Repression of GCN5 also inhibited cell proliferation and increased the percentage of necrotic cells in normal control cells (Figure S10A, S10C). These data suggest that that GCN5 is critical for the growth and survival of both normal lung and NSCLC cell lines.
Figure 4.
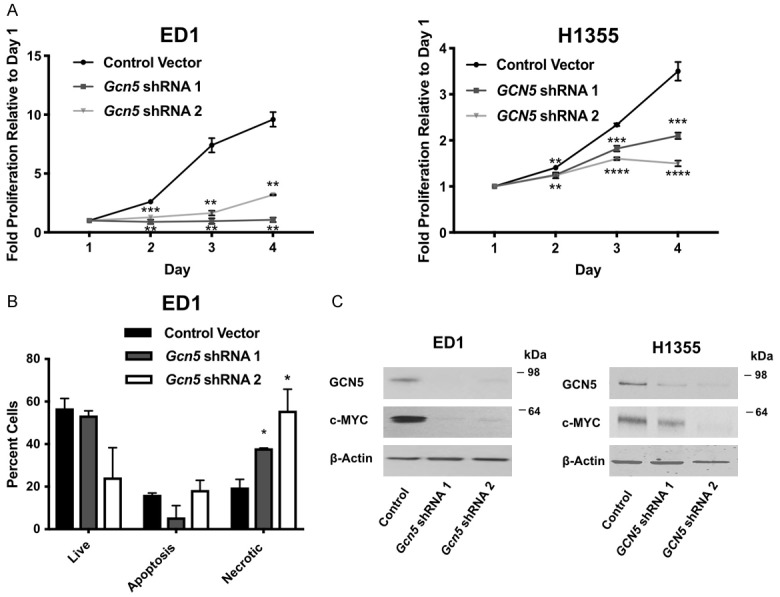
Reduction of GCN5 protein represses cell proliferation, increases the percentage of necrotic cells, and decreases c-MYC protein expression. A. Proliferation of murine ED1 and human H1355 NSCLC cell lines with stable knockdown of GCN5 by two independent shRNAs was monitored over 4 days. Proliferation rate was normalized to day 1. Representative immunoblots confirming GCN5 knockdown are shown in panel 3C. B. Apoptosis assays were performed in the murine ED1 NSCLC cell line with stable knockdown of GCN5 by shRNAs 3 days after seeding cells. Percentage of live, apoptotic and necrotic cells are shown. C. Stable reduction of endogenous GCN5 expression by two independent shRNAs introduced into murine and human lung cancer cells decreased endogenous c-MYC protein. Immunoblotting was performed using anti-GCN5 and anti-c-MYC antibodies. β-Actin served as a loading control. Symbols refer to *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Regulatory relationships between GCN5 and c-MYC have been observed in multiple different model systems [21,46-50]. In 293T cells, MYC directly interacts with GCN5 and the SAGA complex [47]. GCN5 also acetylates c-MYC in lung cancer cell lines, thereby increasing c-MYC protein stability [48,51,52]. Lastly, loss of GCN5 reduces levels of c-MYC in mouse embryonic stem cells [49]. Consistent with these reports, we found that loss of GCN5 decreased c-MYC protein levels in NSCLC cell lines (Figure 4C).
To determine if the observed effects of Gcn5 depletion on cell proliferation is linked to loss of HAT activity, we treated NSCLC cells with the GCN5/PCAF HAT domain inhibitor CPTH6 [27,33]. The murine ED1 cells and human H1355 cells were each treated with varying doses of CPTH6 (40-100 μM) found to be effective in prior studies [27], or with vehicle control (DMSO). Cell proliferation assays were performed after four days of treatment. Repression of GCN5 by CPTH6 treatment significantly (P < 0.01) reduced cell proliferation of both ED1 (IC50 = 38.54) and H1355 (IC50 = 29.67) NSCLC cell lines (Figure 5A). Reduction in cell proliferation was also observed in normal control (C10, IC50 = 13.33) cells treated with CPTH6 (Figure S11A). We tested effects of multiple concentrations of CPTH6 at multiple time points, and found that the effect of CPTH6 on cell proliferation was most robust after four-days of treatment (Figure S11B). Inhibition of GCN5 HAT activity by CPTH6 treatment significantly (P < 0.001) increased the percentage of NSCLC cells (Figure 5B) and normal control cells (Figure S11C) undergoing apoptosis. To confirm inhibition of GCN5 HAT activity by CPTH6 in these cells, we isolated histones from NSCLC cells treated with vehicle control (DMSO) or CPTH6 and analyzed H3K9 acetylation levels via immunoblot. As expected, CPTH6 treatment led to decreased H3K9 acetylation levels (Figure 5C). We also found that CPTH6 treatment caused reduction in c-MYC protein levels as early as 1-day after treatment and becoming most pronounced 4-days after treatment (Figure 5D).
Figure 5.
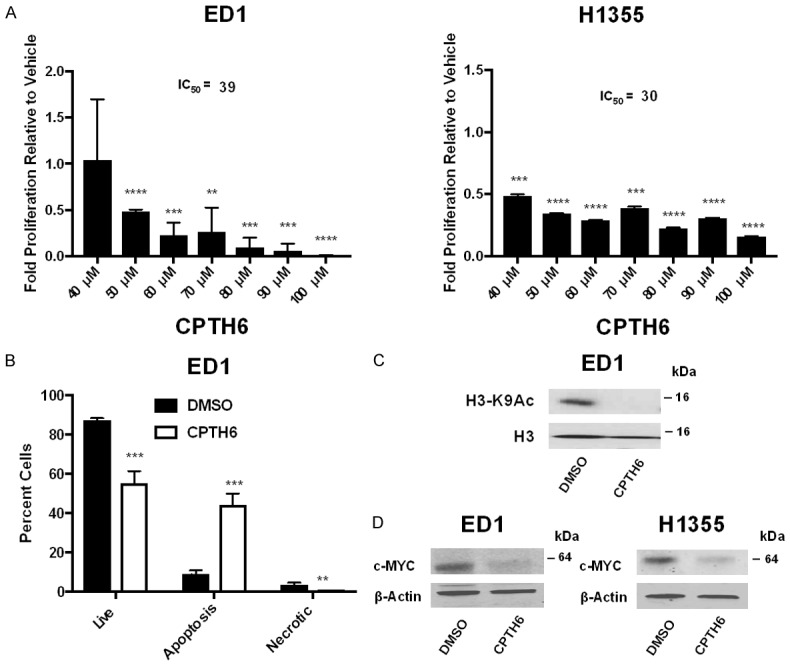
Inhibition of GCN5 activity by CPTH6 represses cell proliferation, increases apoptosis, and decreases c-MYC protein expression. A. Proliferation of murine ED1 and human H1355 NSCLC cell lines at multiple doses (40-100 mM) of CPTH6 treatment was normalized to proliferation of vehicle (DMSO) treated cells after 4 days. B. Apoptosis assays were performed using 100 mM of CPTH6 or vehicle control and analyzed after 3 days of treatment. Percentage of live, apoptotic and necrotic cells are shown. C. Immunoblotting was performed to analyze H3K9 acetylation levels in murine ED1 NSCLC cell line treated with 100 mM of CPTH6 for 1 day. H3 protein served as a loading control. D. Immunoblotting was performed to analyze c-MYC protein levels in murine ED1 and human H1355 NSCLC cell lines treated with 100 mM of CPTH6 for 4 days. Total H3 protein served as a loading control. Symbols refer to **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Overall, our data are consistent with prior work [27] with the addition that total c-MYC protein levels are reduced in NSCLC cell lines when GCN5 protein levels are repressed or GCN5 activity is inhibited. This further supports literature indicating GCN5 stabilizes c-MYC [48] and that GCN5 and c-MYC work together in a feed-forward system [21]. Our study is the first to form a link between c-MYC and cancer phenotypes in non-small cell lung cancers with reduced GCN5 levels.
Discussion
c-MYC over expression promotes tumor growth, proliferation, and altered metabolism and protein synthesis in many different cancers including breast, lymphoma and lung [53]. However, c-MYC is challenging to directly target since, as a transcription factor, it lacks druggable enzymatic domains [54]. New targeted approaches focusing on c-MYC cofactors show potential for successful strategies [53,54]. Our prior work and the work of others have revealed a positive relationship between GCN5 and c-MYC [21,47-49], suggesting that GCN5 might be a targetable cofactor for c-MYC in cancer therapy. Our current study further contributes to this body of literature by linking GCN5, the HAT enzyme of the SAGA complex, to c-MYC in NSCLC cell lines and tissues.
In human NSCLC tissues, GCN5 mRNA levels are upregulated, however, in murine and human NSCLC cell lines, the association between GCN5 and c-MYC are mainly observed at the protein level. Upregulation of GCN5 mRNA in tissues and not NSCLC cell lines indicates that the heterogeneous nature (both cell type and mutation status) of NSCLC tissues plays a role. Additional work is needed to determine whether the c-MYC target genes associated with GCN5 in NSCLC tissues are also direct targets of GCN5 in these tissues and NSCLC cell lines. Our work indicates that NSCLC cell lines are dependent on GCN5 and ADA2B functions and that depletion of GCN5 in both murine and human NSCLC lines decreases cell proliferation with no effect on the percentage of cells undergoing apoptosis. However, we did find that there was a much higher proportion of necrotic cells in the GCN5 repressed cell lines, which is most likely a long-term consequence of the G1/S phase arrest [23]. Functional studies also conducted using normal immortalized lung epithelial controls revealed a similar effect with GCN5 knockdown. Since GCN5 is a cofactor of c-MYC with functions in normal cells, it is not surprising that knockdown of GCN5 alters phenotypes in normal lung cells as well. Lastly, recent work has revealed that GCN5 promotes E2F1, cyclin D1, and cyclin E1 expression in NSCLC cell lines [23]. Our work extends this by showing that loss of GCN5 expression levels or activity decreases the total levels of c-MYC protein.
Other co-factors besides GCN5 interact with c-MYC and serve as co-activators for c-MYC target genes. The TIP60 HAT complex associates with c-MYC, recruits c-MYC to chromatin and is recruited to MYC-target genes where it contributes to histone acetylation [55]. Interestingly, TIP60 exhibits tumor suppressive functions in NSCLCs by inhibiting cell viability and invasion and down-regulating AKT signaling [56]. An association between TIP60 and c-MYC in NSCLC has not yet been investigated. The co-activators CBP and p300 interact directly with c-MYC and stimulate its function [57]. Acetylation by CBP/p300 alters c-MYC stability and modulates transcription by influencing interactions with other factors [57]. Knockdown of CBP in NSCLC cells inhibits cell growth, increases apoptosis, and inactivates MAPK signaling [58]. However, the effect of CBP knockdown on c-MYC in these NSCLC cell lines has not been explored.
There are currently several HAT inhibitors such as natural product derivatives, small molecules, protein-protein interaction inhibitors, and bi-substrate inhibitors [59]. These HAT inhibitors hold therapeutic potential for diseases including inflammatory and neurological disorders [59]. Inhibitors against HATs implicated in promoting tumorigenesis hold therapeutic value for cancer therapy. However, the biological properties of HAT inhibitors and their therapeutic effects are still unclear. Current HAT inhibitors show low potency, instability, and a lack of specificity [59]. Other proteins associated with and regulating the function of HATs may also affect the potency of HAT inhibitors [59]. In addition to a HAT domain, GCN5 also contains a bromodomain that facilitates cooperative and cross-tail acetylation of nucleosomes [60]. Additional studies are needed to determine whether the HAT domain, bromodomain [61], or both are critical for GCN5 functions in oncogenesis. By understanding which domains are most critical for GCN5 oncogenic functions, inhibitors can be generated against these GCN5 domains for therapeutic purposes. The interplay between GCN5 and PCAF in these cancers is also critical to understand whether PCAF compensates for GCN5 when inhibited.
The work presented in this study provides insight into how the GCN5 and c-MYC relationship previously identified in mouse ESCs extends to human cancers. This study also provides evidence for generating additional, selective GCN5 inhibitors that may be used for lung cancer therapy in the clinic. The KRAS oncogene is frequently mutated and serves as a driver of NSCLCs [1]. Multiple studies suggest that KRAS and c-MYC cooperate in oncogenicity and that inhibition of c-MYC eradicates KRAS-driven lung cancers [62,63]. The relationship between GCN5 and c-MYC may have greater relevance in cancers more dependent on c-MYC amplifications and overexpression. MYC family amplifications are a frequent oncogenic event in small cell lung cancer (SCLCs) and B-cell lymphoma [64,65]. Future, more comprehensive studies should be extended to cancers dependent on c-MYC to identify whether GCN5 inhibition reverses oncogenic phenotypes observed in these cancers.
Acknowledgements
We thank all members of the Dent (MD Anderson Cancer Center), Shi (Van Andel Research Institute) and Barton (MD Anderson Cancer Center) Laboratories for their helpful consultation. We thank Drs. Ethan Dmitrovsky (Frederick National Laboratory), Jonathan M. Kurie (MD Anderson Cancer Center) and John V. Heymach (MD Anderson Cancer Center) for generously providing cell lines used in this study. This work was supported by the Postdoctoral Fellowship 131779-PF-18-034-01-DMC from the American Cancer Society, the Postdoctoral Fellowship Award from the Center for Cancer Epigenetics at MD Anderson Cancer Center and the Tobacco Pilot Grant from MD Anderson Cancer Center. Flow cytometry experiments were performed using the Flow Cytometry & Cellular Imaging Facility in Smithville, which is supported by the CPRIT Core Facility Support Grant (RP170628).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–80. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reck M, Heigener DF, Mok T, Soria JC, Rabe KF. Management of non-small-cell lung cancer: recent developments. Lancet. 2013;382:709–19. doi: 10.1016/S0140-6736(13)61502-0. [DOI] [PubMed] [Google Scholar]
- 3.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 4.Wang L, Dent SY. Functions of SAGA in development and disease. Epigenomics. 2014;6:329–39. doi: 10.2217/epi.14.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koutelou E, Hirsch CL, Dent SY. Multiple faces of the SAGA complex. Curr Opin Cell Biol. 2010;22:374–82. doi: 10.1016/j.ceb.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker SP, Grant PA. The SAGA continues: expanding the cellular role of a transcriptional co-activator complex. Oncogene. 2007;26:5329–40. doi: 10.1038/sj.onc.1210603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Downey M, Johnson JR, Davey NE, Newton BW, Johnson TL, Galaang S, Seller CA, Krogan N, Toczyski DP. Acetylome profiling reveals overlap in the regulation of diverse processes by sirtuins, gcn5, and esa1. Mol Cell Proteomics. 2015;14:162–76. doi: 10.1074/mcp.M114.043141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 9.Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY, Ge K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30:249–62. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang YL, Faiola F, Xu M, Pan S, Martinez E. Human ATAC is a GCN5/PCAF-containing acetylase complex with a novel NC2-like histone fold module that interacts with the TATA-binding protein. J Biol Chem. 2008;283:33808–15. doi: 10.1074/jbc.M806936200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soffers JHM, Li X, Saraf A, Seidel CW, Florens L, Washburn MP, Abmayr SM, Workman JL. Characterization of a metazoan ADA acetyltransferase complex. Nucleic Acids Res. 2019;47:3383–94. doi: 10.1093/nar/gkz042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeitany M, Bakhos-Douaihy D, Silvestre DC, Pineda JR, Ugolin N, Moussa A, Gauthier LR, Busso D, Junier MP, Chneiweiss H, Chevillard S, Desmaze C, Boussin FD. Opposite effects of GCN5 and PCAF knockdowns on the alternative mechanism of telomere maintenance. Oncotarget. 2017;8:26269–80. doi: 10.18632/oncotarget.15447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cieniewicz AM, Moreland L, Ringel AE, Mackintosh SG, Raman A, Gilbert TM, Wolberger C, Tackett AJ, Taverna SD. The bromodomain of Gcn5 regulates site specificity of lysine acetylation on histone H3. Mol Cell Proteomics. 2014;13:2896–910. doi: 10.1074/mcp.M114.038174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atanassov BS, Evrard YA, Multani AS, Zhang Z, Tora L, Devys D, Chang S, Dent SY. Gcn5 and SAGA regulate shelterin protein turnover and telomere maintenance. Mol Cell. 2009;35:352–64. doi: 10.1016/j.molcel.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu W, Edmondson DG, Evrard YA, Wakamiya M, Behringer RR, Roth SY. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat Genet. 2000;26:229–32. doi: 10.1038/79973. [DOI] [PubMed] [Google Scholar]
- 16.Lin W, Zhang Z, Chen CH, Behringer RR, Dent SY. Proper Gcn5 histone acetyltransferase expression is required for normal anteroposterior patterning of the mouse skeleton. Dev Growth Differ. 2008;50:321–30. doi: 10.1111/j.1440-169X.2008.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin W, Zhang Z, Srajer G, Chen YC, Huang M, Phan HM, Dent SY. Proper expression of the Gcn5 histone acetyltransferase is required for neural tube closure in mouse embryos. Dev Dyn. 2008;237:928–40. doi: 10.1002/dvdy.21479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez-Cerdeno V, Lemen JM, Chan V, Wey A, Lin W, Dent SR, Knoepfler PS. N-Myc and GCN5 regulate significantly overlapping transcriptional programs in neural stem cells. PLoS One. 2012;7:e39456. doi: 10.1371/journal.pone.0039456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin W, Srajer G, Evrard YA, Phan HM, Furuta Y, Dent SY. Developmental potential of Gcn5 (-/-) embryonic stem cells in vivo and in vitro. Dev Dyn. 2007;236:1547–57. doi: 10.1002/dvdy.21160. [DOI] [PubMed] [Google Scholar]
- 20.Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. EMBO J. 2006;25:2723–34. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirsch CL, Coban Akdemir Z, Wang L, Jayakumaran G, Trcka D, Weiss A, Hernandez JJ, Pan Q, Han H, Xu X, Xia Z, Salinger AP, Wilson M, Vizeacoumar F, Datti A, Li W, Cooney AJ, Barton MC, Blencowe BJ, Wrana JL, Dent SY. Myc and SAGA rewire an alternative splicing network during early somatic cell reprogramming. Genes Dev. 2015;29:803–16. doi: 10.1101/gad.255109.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Little CD, Nau MM, Carney DN, Gazdar AF, Minna JD. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature. 1983;306:194–6. doi: 10.1038/306194a0. [DOI] [PubMed] [Google Scholar]
- 23.Chen L, Wei T, Si X, Wang Q, Li Y, Leng Y, Deng A, Chen J, Wang G, Zhu S, Kang J. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J Biol Chem. 2013;288:14510–21. doi: 10.1074/jbc.M113.458737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Majaz S, Tong Z, Peng K, Wang W, Ren W, Li M, Liu K, Mo P, Li W, Yu C. Histone acetyl transferase GCN5 promotes human hepatocellular carcinoma progression by enhancing AIB1 expression. Cell Biosci. 2016;6:47. doi: 10.1186/s13578-016-0114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu K, Zhang Q, Lan H, Wang L, Mou P, Shao W, Liu D, Yang W, Lin Z, Lin Q, Ji T. GCN5 potentiates glioma proliferation and invasion via STAT3 and AKT signaling pathways. Int J Mol Sci. 2015;16:21897–910. doi: 10.3390/ijms160921897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, Mupo A, Grinkevich V, Li M, Mazan M, Gozdecka M, Ohnishi S, Cooper J, Patel M, McKerrell T, Chen B, Domingues AF, Gallipoli P, Teichmann S, Ponstingl H, McDermott U, Saez-Rodriguez J, Huntly BJP, Iorio F, Pina C, Vassiliou GS, Yusa K. A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep. 2016;17:1193–205. doi: 10.1016/j.celrep.2016.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Martile M, Desideri M, De Luca T, Gabellini C, Buglioni S, Eramo A, Sette G, Milella M, Rotili D, Mai A, Carradori S, Secci D, De Maria R, Del Bufalo D, Trisciuoglio D. Histone acetyltransferase inhibitor CPTH6 preferentially targets lung cancer stem-like cells. Oncotarget. 2016;7:11332–48. doi: 10.18632/oncotarget.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Sempere LF, Ouyang H, Memoli VA, Andrew AS, Luo Y, Demidenko E, Korc M, Shi W, Preis M, Dragnev KH, Li H, Direnzo J, Bak M, Freemantle SJ, Kauppinen S, Dmitrovsky E. MicroRNA-31 functions as an oncogenic microRNA in mouse and human lung cancer cells by repressing specific tumor suppressors. J Clin Invest. 2010;120:1298–309. doi: 10.1172/JCI39566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wislez M, Fujimoto N, Izzo JG, Hanna AE, Cody DD, Langley RR, Tang H, Burdick MD, Sato M, Minna JD, Mao L, Wistuba I, Strieter RM, Kurie JM. High expression of ligands for chemokine receptor CXCR2 in alveolar epithelial neoplasia induced by oncogenic kras. Cancer Res. 2006;66:4198–207. doi: 10.1158/0008-5472.CAN-05-3842. [DOI] [PubMed] [Google Scholar]
- 30.Pankova D, Chen Y, Terajima M, Schliekelman MJ, Baird BN, Fahrenholtz M, Sun L, Gill BJ, Vadakkan TJ, Kim MP, Ahn YH, Roybal JD, Liu X, Parra Cuentas ER, Rodriguez J, Wistuba II, Creighton CJ, Gibbons DL, Hicks JM, Dickinson ME, West JL, Grande-Allen KJ, Hanash SM, Yamauchi M, Kurie JM. Cancer-associated fibroblasts induce a collagen cross-link switch in tumor stroma. Mol Cancer Res. 2016;14:287–95. doi: 10.1158/1541-7786.MCR-15-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roybal JD, Zang Y, Ahn YH, Yang Y, Gibbons DL, Baird BN, Alvarez C, Thilaganathan N, Liu DD, Saintigny P, Heymach JV, Creighton CJ, Kurie JM. miR-200 inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol Cancer Res. 2011;9:25–35. doi: 10.1158/1541-7786.MCR-10-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chimenti F, Bizzarri B, Maccioni E, Secci D, Bolasco A, Chimenti P, Fioravanti R, Granese A, Carradori S, Tosi F, Ballario P, Vernarecci S, Filetici P. A novel histone acetyltransferase inhibitor modulating Gcn5 network: cyclopentylidene-[4-(4’-chlorophenyl) thiazol-2-yl) hydrazone. J Med Chem. 2009;52:530–6. doi: 10.1021/jm800885d. [DOI] [PubMed] [Google Scholar]
- 33.Trisciuoglio D, Ragazzoni Y, Pelosi A, Desideri M, Carradori S, Gabellini C, Maresca G, Nescatelli R, Secci D, Bolasco A, Bizzarri B, Cavaliere C, D’Agnano I, Filetici P, Ricci-Vitiani L, Rizzo MG, Del Bufalo D. CPTH6, a thiazole derivative, induces histone hypoacetylation and apoptosis in human leukemia cells. Clin Cancer Res. 2012;18:475–86. doi: 10.1158/1078-0432.CCR-11-0579. [DOI] [PubMed] [Google Scholar]
- 34.McFarland JM, Ho ZV, Kugener G, Dempster JM, Montgomery PG, Bryan JG, Krill-Burger JM, Green TM, Vazquez F, Boehm JS, Golub TR, Hahn WC, Root DE, Tsherniak A. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat Commun. 2018;9:4610. doi: 10.1038/s41467-018-06916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, Goodale A, Lee Y, Ali LD, Jiang G, Lubonja R, Harrington WF, Strickland M, Wu T, Hawes DC, Zhivich VA, Wyatt MR, Kalani Z, Chang JJ, Okamoto M, Stegmaier K, Golub TR, Boehm JS, Vazquez F, Root DE, Hahn WC, Tsherniak A. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49:1779–84. doi: 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gianni M, Qin Y, Wenes G, Bandstra B, Conley AP, Subbiah V, Leibowitz-Amit R, Ekmekcioglu S, Grimm EA, Roszik J. High-throughput architecture for discovering combination cancer therapeutics. JCO Clin Cancer Inform. 2018;2:1–12. doi: 10.1200/CCI.17.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131:281–5. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 38.Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, Grosveld F, Philipsen S. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawakami M, Mustachio LM, Rodriguez-Canales J, Mino B, Roszik J, Tong P, Wang J, Lee JJ, Myung JH, Heymach JV, Johnson FM, Hong S, Zheng L, Hu S, Villalobos PA, Behrens C, Wistuba I, Freemantle S, Liu X, Dmitrovsky E. Next-generation CDK2/9 inhibitors and anaphase catastrophe in lung cancer. J Natl Cancer Inst. 2017;109 doi: 10.1093/jnci/djw297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gamper AM, Kim J, Roeder RG. The STAGA subunit ADA2b is an important regulator of human GCN5 catalysis. Mol Cell Biol. 2009;29:266–80. doi: 10.1128/MCB.00315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chakraborty S, Rahman T. The difficulties in cancer treatment. Ecancermedicalscience. 2012;6:ed16. doi: 10.3332/ecancer.2012.ed16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwakawa R, Kohno T, Kato M, Shiraishi K, Tsuta K, Noguchi M, Ogawa S, Yokota J. MYC amplification as a prognostic marker of early-stage lung adenocarcinoma identified by whole genome copy number analysis. Clin Cancer Res. 2011;17:1481–9. doi: 10.1158/1078-0432.CCR-10-2484. [DOI] [PubMed] [Google Scholar]
- 44.Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci U S A. 2003;100:776–81. doi: 10.1073/pnas.0334858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, Meyers RM, Ali L, Goodale A, Lee Y, Jiang G, Hsiao J, Gerath WFJ, Howell S, Merkel E, Ghandi M, Garraway LA, Root DE, Golub TR, Boehm JS, Hahn WC. Defining a cancer dependency map. Cell. 2017;170:564–76. e16. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin YW, Jin HJ, Zhao W, Gao B, Fang J, Wei J, Zhang DD, Zhang J, Fang D. The histone acetyltransferase GCN5 expression is elevated and regulated by c-Myc and E2F1 transcription factors in human colon cancer. Gene Expr. 2015;16:187–96. doi: 10.3727/105221615X14399878166230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang N, Ichikawa W, Faiola F, Lo SY, Liu X, Martinez E. MYC interacts with the human STAGA coactivator complex via multivalent contacts with the GCN5 and TRRAP subunits. Biochim Biophys Acta. 2014;1839:395–405. doi: 10.1016/j.bbagrm.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen CJ, Rakowski C, Chatterjee C, Lieberman PM, Lane WS, Blobel GA, McMahon SB. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol Cell Biol. 2004;24:10826–34. doi: 10.1128/MCB.24.24.10826-10834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, Koutelou E, Hirsch C, McCarthy R, Schibler A, Lin K, Lu Y, Jeter C, Shen J, Barton MC, Dent SYR. GCN5 regulates FGF signaling and activates selective MYC target genes during early embryoid body differentiation. Stem Cell Rep. 2018;10:287–99. doi: 10.1016/j.stemcr.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kenneth NS, Ramsbottom BA, Gomez-Roman N, Marshall L, Cole PA, White RJ. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc Natl Acad Sci U S A. 2007;104:14917–22. doi: 10.1073/pnas.0702909104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qiao L, Zhang Q, Zhang W, Chen JJ. The lysine acetyltransferase GCN5 contributes to human papillomavirus oncoprotein E7-induced cell proliferation via up-regulating E2F1. J Cell Mol Med. 2018;22:5333–45. doi: 10.1111/jcmm.13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flinn EM, Wallberg AE, Hermann S, Grant PA, Workman JL, Wright AP. Recruitment of Gcn5-containing complexes during c-Myc-dependent gene activation. Structure and function aspects. J Biol Chem. 2002;277:23399–406. doi: 10.1074/jbc.M201704200. [DOI] [PubMed] [Google Scholar]
- 53.Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014;4 doi: 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun L, Gao P. Small molecules remain on target for c-Myc. Elife. 2017:6. doi: 10.7554/eLife.22915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, Livingston DM, Amati B. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003;4:575–80. doi: 10.1038/sj.embor.embor861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Y, Sun J, Chen T, Tao Z, Zhang X, Tian F, Zhou X, Lu D. Tat-interactive protein-60KDA (TIP60) regulates the tumorigenesis of lung cancer in vitro. J Cancer. 2017;8:2277–81. doi: 10.7150/jca.19677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, Austen M, Luscher B. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 2003;4:484–90. doi: 10.1038/sj.embor.embor821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang Z, Yu W, Zhang C, Zhao S, Yu Z, Xiao X, Tang R, Xuan Y, Yang W, Hao J, Xu T, Zhang Q, Huang W, Deng W, Guo W. CREB-binding protein regulates lung cancer growth by targeting MAPK and CPSF4 signaling pathway. Mol Oncol. 2016;10:317–29. doi: 10.1016/j.molonc.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wapenaar H, Dekker FJ. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin Epigenetics. 2016;8:59. doi: 10.1186/s13148-016-0225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li S, Shogren-Knaak MA. The Gcn5 bromodomain of the SAGA complex facilitates cooperative and cross-tail acetylation of nucleosomes. J Biol Chem. 2009;284:9411–7. doi: 10.1074/jbc.M809617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Humphreys PG, Bamborough P, Chung CW, Craggs PD, Gordon L, Grandi P, Hayhow TG, Hussain J, Jones KL, Lindon M, Michon AM, Renaux JF, Suckling CJ, Tough DF, Prinjha RK. Discovery of a potent, cell penetrant, and selective p300/CBP-associated factor (PCAF)/general control nonderepressible 5 (GCN5) bromodomain chemical probe. J Med Chem. 2017;60:695–709. doi: 10.1021/acs.jmedchem.6b01566. [DOI] [PubMed] [Google Scholar]
- 62.Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10:57–67. doi: 10.4161/cc.10.1.14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, Swigart LB, Evan GI. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504–13. doi: 10.1101/gad.205542.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alves Rde C, Meurer RT, Roehe AV. MYC amplification is associated with poor survival in small cell lung cancer: a chromogenic in situ hybridization study. J Cancer Res Clin Oncol. 2014;140:2021–5. doi: 10.1007/s00432-014-1769-1. [DOI] [PubMed] [Google Scholar]
- 65.Nguyen L, Papenhausen P, Shao H. The role of c-MYC in B-cell lymphomas: diagnostic and molecular aspects. Genes (Basel) 2017;8 doi: 10.3390/genes8040116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.