

Abstract
Cancer metastasis involves the adhesion of cancer cells to the endothelium. This process can be mediated by integrins which are surface receptors responsible for interactions with ECM proteins. Integrins β1 and αVβ3 represent factors are involved in cancer progression and metastasis. Activation of integrins can be promoted by thiol-disulfide exchanges initiated by Protein Disulfide Isomerase (PDI). The purpose of this study was to prove the involvement of disulfide rearrangements in the molecules of integrins in the course of cancer cell adhesion and migration through the endothelium. We present the evidence which proves that highly metastatic MDA-MB-231 breast cancer cell lines adhere to endothelial cells are more effective than non-invasive MCF-10A and MCF-7 cell lines and that the attachment of MDA-MB-231 to the endothelium can be attenuated either by the agents blocking free thiol groups (DTNB, cystamine or PCMBS) or by PDI inhibitors (Q3Rut, 16F16 or PACMA-31). Furthermore, we prove that the transendothelial migration of MDA-MB-231 cells and contraction of collagen can be blocked by thiol blockers or PDI inhibitors and that these factors affect exposition of free thiols on integrin molecules.
Keywords: Disulfide bonds, oxidation-reduction, integrins, PDI, migration
Introduction
Metastasis is the leading cause of mortality among cancer patients. Invasiveness of circulating tumor cells depends on their ability to attach to the vascular endothelium of different distant organs. Such interactions are supported by cell membrane embedded adhesion molecules. Two major cell adhesion molecule families, i.e. selectins and integrins, have been identified as crucial factors in cancer metastasis [1,2]. Integrins are glycoprotein receptors responsible for cells attachment to extracellular matrix (ECM) proteins: fibronectin, fibrinogen, vitronectin, laminins, collagens, plasminogen, osteopontin and von Willebrand factor [3]. Integrins dependent cell attachment to ECM proteins initiates activation of signalling pathways that regulate behaviour and motility of cells [4]. Among different integrin receptors, it is mainly the expressions of αVβ3, αVβ5, α5β1, α2β1 and α6β4 that are associated with metastatic progression in melanoma, breast carcinoma, prostate, pancreatic and lung cancer [2]. In despite of the roles they play in contact with ECM proteins, integrins are also involved in the interactions between cancer cells and platelets, which is another important stage of the metastatic process [5].
The role of β1 integrin in cancer metastasis is not clear. There is evidence for α2β1-dependent suppression of metastasis in a mouse model of breast cancer [6]. We are also aware of the findings suggesting that α2β1 may be crucial during cancer progression [7-9]. MDA-MB-23, i.e. highly metastatic breast cancer cells, express higher levels of α2β1 as compared to non-invasive breast cancer cells MCF-7 [10,11]. Moreover, α2β1 simulates the attachment of MDA-MB-231 to endothelial cells by promoting phosphorylation of VE-Cadherin and dissociation of β-catenin from the VE-Cadherin complex. This process is involved in cell-cell interaction and can be promoted by the H-Ras/Raf/MEK/ERK signalling cascade. It has been suggested that the difference in integrin expression of α2β1 is one of numerous features responsible for the highly-metastatic character of MDA-MB-231 [10].
The invasive nature of MDA-MB-231 may also be related to the expression of αVβ3 integrin, which is very poorly expressed on MCF-7 [11]. αVβ3 integrin has been shown to be involved in the growth and invasiveness of pulmonary metastatic melanoma [12], breast cancer [13] and prostate cancer [14]. In MDA-MB-231 cells, αVβ3 plays an important role in cell adhesion and invasion to the HUVEC cells under low shear stress. Shear stress-dependent activation of αVβ3 integrin leads to imitation of PI3K/AKT signal, consequently resulting in the overexpression of MMP-2, MMP-9 and VEGF. A similar effect may be observed in the presence of platelets activated by thrombin [15] or after LPS-induced TLR4 activation [16].
A potential role of thiol-disulfide exchange in integrin activation was demonstrated on αVβ3 [17], αIIbβ3 [18] α11 [19] and β1 [20] receptors. Activation of these integrins can be triggered by modifications of extracellular disulfide bonds. This process is promoted by Protein Disulfide Isomerase (PDI) or by integrins themselves since some integrin receptors has an endogenous thiol isomerase activity [54,55]. PDI is a thiol oxidoreductase that catalyses the formation, breakage or rearrangements of disulfide bonds and thus it is responsible for regulation of posttranscriptional protein folding in ER [21,22]. Apart from its intracellular localization, PDI can be found on the surface of several cell types, including endothelial cells [23], platelets [24,25], leukocytes [26,27], fibroblasts [19] and cancer cells [28]. Inhibition of PDI activity triggers the accumulation of misfolded proteins which results in ER stress and finally leads to cell death. These observations encourage researchers to use PDI-dependent ER stress in enforcing apoptosis of cancer cells [29]. Increased PDI levels have been observed in different cancers including ovarian [30], prostate [31] and lung cancer [32] as well as in lymphoma [33], glioma [34] and melanoma [35]. PDI knockdown can cause apoptosis in human breast cancer cells MCF-7 [36] and PDI inhibitors, like PACMAs, are also known to interrupt the cell cycle progression in breast cancer cells. In vivo, PACMAs express a therapeutic potential in a mouse xenograft model of human breast cancer [37]. Another PDI inhibitor, i.e. bacitracin, has been used in chemotherapy as a factor increasing efficiency of pro-apoptotic drugs [29]. Moreover, administration of bacitracin in conjunction with chemotherapy can overcome the temozolomide resistance in malignant glioma [38].
In this study, three cancer cell lines: MCF-10A, MCF-7 and MDA-MB-231 were analysed. The MCF10A human breast epithelial cell line is the most commonly used normal breast cell model. MCF-10A cells were derived from benign proliferative breast tissue and spontaneously immortalized without defined factors. They are not tumorigenic and do not express the oestrogen receptor [39]. MCF-10A exhibit a basal-like phenotype and display a non-invasive character, but share some features of mesenchymal cancer cell lines [40,41]. MCF-7 is a hormone-dependent cell line, non-tumorigenic and non-invasive on Matrigel [41]. MDA-MB-231 cells, initially isolated from a pleural effusion of breast adenocarcinoma are categorised as a very invasive and aggressive phenotype capable of metastasizing in the lung [39,42]. In this study, we explored the role of thiol-disulfide exchanges in breast cancer adhesion and migration to the endothelium. The blocking effect of PDI inhibitors and free-thiol blockers on breast cancer adhesion and migration suggest that PDI or perhaps other thioredoxin-like disulphide isomerases are involved in breast cancer metastasis mediated by integrins.
Experimental procedures
Materials
Horseradish conjugated secondary antibodies were purchased from Santa Cruz biotechnology (Santa Cruz, CA, USA), Alexa Fluor conjugated antibodies, antibodies anti-β1 clone 12G10 were purchased from Abcam (Cambridge, UK), chemiluminescence detection system was purchased from Cyanagen (Bologna, BO, Italy), antibodies anti-PDI RL77, Pierce BCA Protein Assay Kit, Pierce IP Lysis Buffer, Pierce Control Agarose, protein A/G-agarose, CellTracker Green CMFDA Dye, protease inhibitor cocktail and neutravidin-agarose were purchased from Thermo Scientific Pierce (Waltham, MA, USA), GSH, iodoacetamide, DTNB, MPB, cystamine, rutin hydrate (Q3Rut), 16F16, PACMA-31, NEM, TCEP were purchased from Sigma-Aldrich LTD (St. Louis, MO), vitronectin, antibodies anti-β1 AB1952 and anti-β1 MAB1987, anti-αVβ3 clone LM609 and anti-αVβ3 clone CBL544 were purchased from Merck Milipore (Billerica, MA, USA); Bovine Collagen Solution PureCol was purchased from Advanced BioMatrix Inc. (San Diego, CA, USA); all standard tissue culture reagents, including DMEM, fetal bovine serum, trypsin/EDTA and others were purchased from GIBCO (Waltham, MA, USA). DMEM 2 × concentrated was purchased from Biological Industries USA (Cromwell, CT, USA), PCMBS was purchased from Toronto Research Chemicals Inc. (Ontario, Canada).
Cell cultures
MCF-10A cells were cultured using Dulbecco’s modified eagle’s medium/ham’s nutrient mixture F-12 with the addition of 10% heat inactivated fetal bovine serum (FBS), 1 ng/ml cholera toxin, 10 μg/ml human insulin, 10 ng/ml epidermal growth factor and 0.5 μg/ml hydrocortisone. MCF-7 and MDA-MB-231 cells were cultured using Dulbecco’s modified eagle’s medium with 10% FBS in a humidified 5% CO2 atmosphere at 37°C. To make the complete growth medium for MCF-7 10 ng/ml human recombinant, insulin was added. EA.hy926 cells were cultured using Dulbecco’s modified eagle’s medium with 10% FBS and HAT (100 μM hypoxanthine, 0.4 μM aminopterin, 16 μM thymidine). HMEC-1 cells were cultured using MCDB131 with the addition of 10% FBS, 10 ng/ml epidermal growth factor and 1 μg/ml hydrocortisone. After reaching an appropriate level of confluency, cells were harvested using PBS as a washing buffer and 0.05% trypsin-EDTA solution as a detaching factor. Cells viability was determined microscopically by trypan blue exclusion.
Cell viability and proliferation assay using PrestoBlue
Proliferation assay with PrestoBlue was performed according to the manufacturer’s protocol. Briefly, the cells were seeded at 1 × 104/well in a 96-well plate and allowed to attach for 3 hours. Subsequently, the medium was replaced with fresh DMEM/10% FBS containing tested reagents. After 24 hours of incubation at 37°C and 5% CO2 in a humidified atmosphere, 10 μl of the PrestoBlue reagent dissolved in a fresh medium was added directly to each well. An alternative version includes incubation with tested reagents dissolved in serum-free medium for 2 hours at 37°C to examine cytotoxicity of the used compounds. The medium-PrestoBlue mixture was incubated for 2 hours at 37°C and absorbance was measured at 540-570/580-610 nm on a multifunctional plate reader Perkin Elmer Victor 3 microplate reader.
Adhesion assay
48-well plates were coated with collagen type I (10 μg/ml) or vitronectin (10 μg/ml) overnight at 4°C. After washing with TBS, plates were incubated with 1% BSA/TBS for 2 hours at 37°C to block nonspecific binding sites. Cells were detached using trypsin/EDTA and trypsin was removed by washing with DMEM/10% FBS and serum-free DMEM. The cells were incubated in serum-free DMEM with different concentrations of thiol group blockers: p-chloromercuribenzoic acid (PCMBS), cystamine dihydrochloride (cystamine), dithionitrobenzoic acid (DTNB) and PDI inhibitors: quercetin-3-rutinoside (Q3Rut), 2-(2-Chloroacetyl)-2,3,4,9-tetrahydro-1-methyl-1H-pyrido[3,4-b]indole-1-carboxylic acid methyl ester (16F16) and N-(2,4-Dimethoxyphenyl)-N-(1-oxo-2-propyn-1-yl)-2-(2-thienyl)glycyl-glycine ethyl ester (PACMA-31) for 30 minutes at 37°C with 5% CO2. After incubation, the cells were transferred into plates at a count of 0.1 × 106 cells/well and incubated at 37°C and 5% CO2 for 2 hours. Not-adherent cells were washed out using TBS and TBS/0.05% Tween 20. Number of adherent cells was measured using Pierce BCA Protein Assay Kit (Thermo Scientific Pierce) according to the manufacturer’s instructions. The absorbance at 562 nm was measured using a Perkin Elmer Wallac 1420 Victor 2 microplate reader.
Scratch migration assay
12-well plates were coated with collagen type I (10 μg/ml) or vitronectin (10 μg/ml) overnight at 4°C. Cells were seeded on plates at the count of 1 × 105 cells/well and when the monolayer reaches 80-90% of confluency, cells were washed with PBS and starved in serum-free medium for 4 hours. After starving, cells were incubated with tested factors in serum-free DMEM for 30 minutes at 37°C and 5% CO2. After incubation, two separate scratches were made in every well using a sterile 200 ml pipette tip. The wells were gently washed with PBS in order to remove detached or damaged cells. After washing, the first photograph was taken, which corresponded to the control time 0 hour. Subsequent photographs were taken after 24 hours of migration in serum-free DMEM containing tested factors, which was the time corresponding to near 100% of scratch area overgrown. The pictures were taken using an inverted Olympus CKX41 microscope combined with an Olympus C3040 camera. Decreasing scratch area was measured using the National Institutes of Health (NIH) ImageJ program with MiToBo plugin.
Cell-cell adhesion assay
HMEC-1 and EAhy.926 cells were seeded on a 24-well plate at a count of 5 × 105 cells/ml and incubated until reaching a confluent monolayer. MCF-10A, MCF-7 and MDA-MB-231 cells after overnight starving in serum-free medium were washed using PBS and detached using trypsin/EDTA. Trypsin was removed by multiple washing with DMEM/10% FBS and serum-free DMEM. Cells were stained in serum-free DMEM using CellTracker Green CMFDA Dye for 30 minutes at 37°C and 5% CO2. CellTracker was removed by washing with DMEM/10% FBS and centrifugation. The stained cells were incubated in serum-free DMEM supplemented with the tested inhibitors for 30 minutes at 37°C. After incubation cells were transferred into plates at a count of 2 × 105 cells/well and allowed to adhere to the endothelium for 2 hours. Not-adherent cells were washed out using warm PBS. Adherent cells were dissolved in 2% SDS and the absorbance at 492/517 nm was measured using a Perkin Elmer Wallac 1420 Victor 2 microplate reader. Alternative version includes cells adhesion in the presence of blocking anti-integrins antibodies: anti-β1 (clone HUTS-4) or anti-αVβ3 (clone LM609) at the final concentration 1:50.
Transwell migration assay
The upper chamber of a transwell system containing the polycarbonate membrane (diameter 6.5 mm, thickness 10 mm, pore diameter 8 mm) was coated with gelatin (0.1% w/v) overnight at 4°C. The chamber was transferred into the 24-well plate filled with DMEM/10% FBS. MCF-10A, MCF-7 and MDA-MB-231 cells were starved for 24 hours, detached using trypsin/EDTA and resuspended in serum-free DMEM. The cells were incubated with the appropriate inhibitors for 30 minutes and with the addition of 5 mg/ml LPS were transferred into the upper chamber of the transwell at the final count of 1 × 105 cells/well. The cells were allowed to migrate through the polycarbonate membrane for 24 hours at 37°C and 5% CO2. After the removal of not-migrated cells with a sterile cotton swab, the cells were washed with PBS, fixed with ice-cold methanol (95% methanol in PBS) for 10 minutes at 4°C and stained using crystal violet. The membrane with the stained cells was viewed under the inverted Olympus CKX41 microscope combined with an Olympus C3040 camera and the cells from ten randomly selected pictures were counted.
Transendothelial migration assay
A transwell system, similar to the one previously described, was prepared. HMEC-1 cells were seeded on gelatine-coated chamber at the final count of 5 × 105 cells/ml and incubated for 24 hours until reaching a confluent monolayer. EAhy.926 were seeded at a count of 5 × 105 cells/ml and incubated for 8 hours until reaching a confluent monolayer. The chamber was transferred into the 24-well plate filled with 1:1 mixture of DMEM/10% FBS and serum-free DMEM from human skin fibroblasts hTERT BJ stimulated with 50 mg/L ascorbate for 24 hours, as a source of chemoattractants. MDA-MB-231 cells were starved for 24 hours, detached using trypsin/EDTA and resuspended in serum-free DMEM. After staining with CellTracker Green CMFDA Dye MDA-MB-231 cells with the addition of 5 mg/ml LPS and the appropriate inhibitors, were transferred into the upper chamber of the transwell at the final count of 1 × 105 cells/well. The cells were allowed to migrate through the polycarbonate membrane for 24 hr at 37°C and 5% CO2. After the removal of non-migrated cells with a sterile cotton swab, the cells were washed with PBS and fixed with ice-cold methanol. The cells stained with CellTracker Green CMFDA Dye were viewed under the microscope and the cells from ten randomly selected pictures were counted. Alternative version includes cell migration in the presence of blocking anti-integrins antibodies: anti-β1 (clone HUTS-4) or anti-αVβ3 (clone LM609) at the final concentration 1:50.
Collagen gel contraction
24-well plates were coated with 2% BSA overnight at 37°C. Collagen gels composed of 2 × concentrated DMEM, 3.2 mg/ml collagen type I, glutamine, antibiotics and 0.1 M NaOH were mixed on ice. The cells, after detaching with trypsin/EDTA and washing with DMEM/10% FBS, were added to the collagen gel and allowed to polymerize for 2 hours at 37°C and 5% CO2. The collagen-cell mix was detached from the wells with serum-free DMEM and transferred to a culture dish with a diameter of 8.8 cm2 filled with DMEM containing 2% FBS. Collagen gels were incubated with tested factors and pictures were taken respectively after 2, 6, 12, 24, 48 and 72 hours using a Samsung ES25 digital camera. The decreasing gel area was measured using the National Institutes of Health (NIH) ImageJ program. Since collagen gel contraction depends on the presence of fibronectin in the environment, gel contraction stops after 24 hours without changing the medium. Therefore, the time point at 24 hours was taken as an optimal time point of measurement.
Immunostaining
MCF-10A, MCF-7 and MDA-MB-231 cells adhere to EA.hy926 cells like described in the cell-cell adhesion assay. After 2 hours of adhesion, cells were fixed with 1% methanol-free paraformaldehyde (PFA) for 10 minutes at RT. After fixing, the cells were washed with a PHEM buffer (60 mM PIPES, pH 6.9, 25 mM HEPES, 10 mM EGTA, 4 mM MgCl2) and blocked using 3% BSA/PHEM for 1 hour at RT. Subsequently, the cells were incubated overnight at 4°C with primary antibodies diluted in 3% BSA/PHEM. Finally, the cells were washed three times with PHEM buffer, stained for 1 hr at RT with respective secondary antibodies prepared in 3% BSA/PHEM and again washed three times with PHEM buffer. The tested antibodies included antibodies for the active form of integrin β1 (clone 12G10 and HUTS-4) and for the inactive form of β1 (clone P4C10), as well the active form of integrin αVβ3 (clone LM609) and the inactive form of αVβ3 (clone CBL544). All photographs were taken using Fluorescent Cell Imager ZOE (Bio-Rad).
Sulfhydryl group labelling
Labelling of sulfhydryl groups was performed using the membrane impermeable maleimide reagent N-(3-Maleimidopropionyl) biocytin MPB. The cells were washed with PBS and labelled with 100 mM MPB for 30 minutes at 37°C with gentle mixing. The labelling reaction was quenched by adding of 200 mM GSH for 15 minutes and after replacing to a fresh medium by incubation with 200 mM iodoacetamide for 10 minutes. All the labelling steps were performed in a serum-free medium to strictly maintain a neutral pH, being aware that a different pH can influence free thiols labelling. After washing with PBS to remove MPB and iodoacetamide, the cells were lysed using a lysis/wash buffer (Thermo Scientific Pierce) supplemented with protease inhibitors cocktail (Thermo Scientific Pierce) for 10 minutes on ice. The cell debris was removed after centrifugation at 14000 × g for 15 minutes at 4°C. Proteins labelled with MPB were precipitated using a high capacity Neutretavidin-Agarose resin overnight at 4°C with gentle mixing/shaking. After washing the resin with ice-cold PBS and lysis buffer, the labelled proteins were detached from the resin by reducing agent β-mercaptoethanol and boiling the samples at 95°C for 5-10 minutes. The detached proteins were separated using 8% or 10% SDS-PAGE and transferred to a nitrocellulose membrane. The biotinylated proteins were visualised using avidin-horseradish peroxidase with a chemiluminescent substrate or using specific antibodies. Alternative version of labelling include pre-incubation with free thiol blockers and PDI inhibitors to examine the role of tested subjects on free thiols exposition on surface proteins.
Statistical analysis
Data is presented as a mean + SD or median and IQR (interquartile range: from lower [25%] to upper quartile [75%]), depending on data distribution (the Shapiro-Wilk test). Heteroscedasticity was verified based on the Brown-Forsythe test. For some variables departing from normal distribution, we subjected the data to Box-Cox transformation for further analyses. The analysis of outliers was performed with the use Grubb’s and Tukey’s tests. The conventional or bootstrap-boosted student’s t-test, Mann-Whitney U-test or one- or two-way ANOVA were used as the inference tests, according to assumptions of data normality and homoscedasticity. Due to the relatively small sample sizes and the low statistical power of the estimated inferences in some calculations, the resampling bootstrap technique (1000-10000 iterations) was used to make sure that the revealed differences were not observed due to pure chance. In such circumstances, we refer to the bootstrap-boosted test statistics instead of the classical approach. The following software was used for the statistical analyses: Statistica v.13.1 (Statsoft), StatsDirect v.3.0.182, Resampling Stats Add-in for Excel v.4, GraphPad Prism v.5.
Results
Cell viability and proliferation assay using PrestoBlue
Blocking effect of PDI inhibitors and thiol blockers on cells proliferation was measured in the course of 24-hour incubation (Figure 1A). A potential cytotoxic effect was estimated after 2 hours (Figure 1B), which corresponds to short-time assays like adhesion to ECM matrix or cell-cell adhesion. None of the analysed thiol blockers or PDI inhibitors displays any significant cytotoxic effect on MCF-10A, MCF-7 and MDA-MB-231 cells after short-time incubation. Of all the tested factors, only PCMBS at the concentration of 100 μM inhibits the proliferation of MCF-10A cells. Proliferation of the MCF-7 cell line can be inhibited by PCMBS or PDI inhibitor PACMA-31 (Figure 1A). PACMA-31 at the concentration of 5 μM displays a significant anti-proliferating effect also on MDA-MB-231 (Figure 1B). The anti-proliferating effect of PACMA-31 on MDA-MB-231 seems to be independent of the used concentrations, since the doses within the range of 1 μM and 10 μM display similar effects (Figure 1C). However, the cytotoxic effect of PACMA-31 on MDA-MB-231 increases dramatically when the concentration exceeds 10 μM (IC50=10 μM). Complete analysis of anti-proliferating and cytotoxic effect of tested subjects in different concentrations is included in supplementary data (Supplementary Figure 1).
Figure 1.
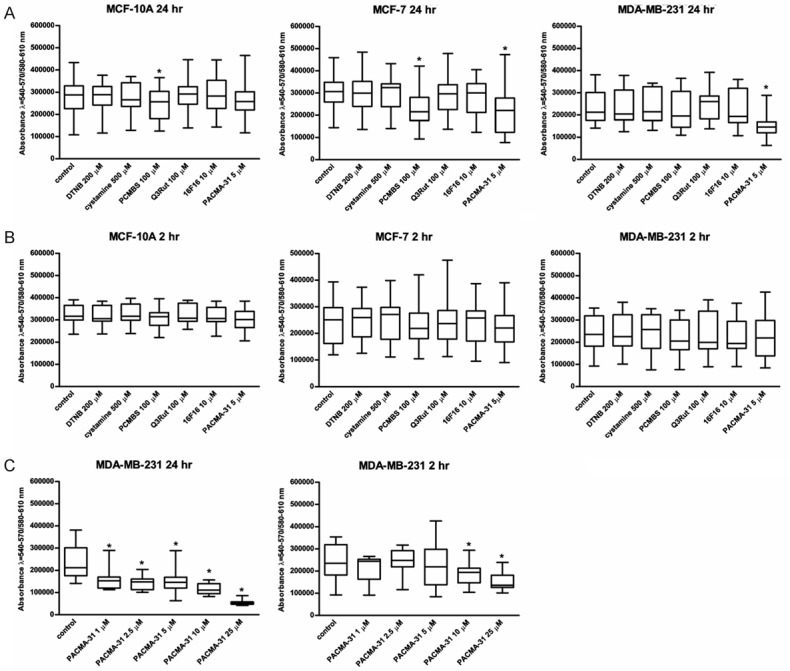
Anti-proliferative and cytotoxic effects of various PDI inhibitors and free thiol blockers on three cell lines, MCF-7, MCF-10A and MDA-MB-231. Data presented as mean + SD; mini-max ranges marked as whiskers; n=6. Long-time cytotoxicity (A) was tested following 24-hour incubation. Short-time cytotoxicity (B) was tested following 2-hour incubation. Anti-proliferative and cytotoxic effects of the increasing concentrations of PACMA31 (C) were tested following 2-hour or 24-hour incubation. Total number of living cells was estimated using PrestoBlue assay. Significance of differences were calculated by using one-way ANOVA and the post-hoc multiple comparisons Tukey’ test. Planned comparisons were verified the bootstrap-boosted unpaired student’s t test (10000 iterations) with the Bonferroni’s correction for multiple comparisons.
Adhesion assay
Adhesion of MCF-10A, MCF-7 and MDA-MB-231 cells to ECM proteins was used to examine the role of thiol-disulfide exchanges in cell attachment mediated by integrin receptors. Figure 2A shows that incubation with free thiol blockers and PDI inhibitors diminishes integrin β1-mediated adhesion to collagen type I as well as αVβ3-mediated adhesion to vitronectin of MCF-10A cells. Only DTNB displays no significant inhibitory effect on MCF-10A adhesion to vitronectin. Figure 2B shows that the MCF-7 adhesion, similarly to MCF-10A adhesion, can be attenuated by PDI inhibitors and thiol blockers, but the attachment of MCF-7 to ECM is less effective as compared to MCF-10A. Figure 2C presents blocking effect of cystamine and PCMBS, as well as Q3Rut and PACMA-31 on MDA-MB-231 adhesion to collagen. 16F16 despite attenuating the adhesion process displays no significant effect on MDA-MB-231 adhesion to collagen and DTNB exhibits no blocking effect on both: adhesion to collagen and adhesion to vitronectin. Solvents, like DMSO and ethanol demonstrate no significant effect on cell adhesion at tested concentrations (Supplementary Figure 2).
Figure 2.

Adhesion of MCF-10A (A), MCF-7 (B) and MDA-MB-231 (C) Cells to collagen type I and vitronectin in the presence of PDI inhibitors and free thiol blockers. Data presented as mean + SD; mini-max ranges marked as whiskers; n=6. Total numbers of adherent cells were estimated with BCA Protein Assay. Significance of differences was analysed with one-way ANOVA and the post-hoc multiple comparisons Tukey’s test and planned comparisons were verified the bootstrap-boosted unpaired student’s t test (10000 iterations) with the Bonferroni’s correction for multiple comparisons.
Scratch migration assay
In the scratch migration assay, also referred to as the “wound healing” assay, cells were allowed to migrate on plates coated with ECM proteins: Collagen type I and vitronectin. Figure 3A shows that all the tested agents display an inhibitory effect on MCF-10A migration on ECM proteins but only PCMBS and PACMA-31 exhibit a statistically significant effect. Likewise, the migration of MCF-7 can be attenuated by the tested factors. However, only PCMBS and PACMA-31 display a significant blocking effect (Figure 3B). Figure 3C presents the results on blocking the migration of MDA-MB-231 cells. As in the case of adhesion, the migration of MDA-MB-231 is much more effective than the migration of non-invasive cells MCF-10A and MCF-7. The migration of MDA-MB-231 on collagen type I can be significantly blocked by cystamine, PCMBS and PACMA-31, whereas the migration on vitronectin can be additionally blocked by Q3Rut. Solvents, like DMSO and ethanol at tested concentration display no significant effect on cells adhesion (Supplementary Figure 3).
Figure 3.

Migration of MCF-10A (A), MCF-7 (B) and MDA-MB-231 (C) Cells to collagen type I and vitronectin in the presence of PDI inhibitors and free thiol blockers. Data presented as mean + SD; mini-max ranges marked as whiskers; n=6. Pictures of overgrowing scratches were taken following 24-hour incubation with the tested inhibitors/blockers using an inverted light microscopy. Significance of differences was analysed with one-way ANOVA and the post-hoc multiple comparisons Tukey’s test and planned comparisons were verified the bootstrap-boosted unpaired student’s t test (10000 iterations) with the Bonferroni’s correction for multiple comparisons.
Cell-cell adhesion assay
MCF-10A, MCF-7 and MDA-MB-231 after staining with CellTracker Green CMFDA Dye were allowed to adhere to two different endothelial cell lines: HMEC-1 and EA.hy926. It is important to mention that all the tested cell lines adhere to HMEC-1 endothelial cells with higher efficiency than to EA.hy926. Only DTNB exhibits no significant blocking effect on MCF-10A adhesion to endothelial cell lines (Figure 4A). MCF-7 more effectively adheres to the endothelium than MCF-10A (Figure 4B). The interaction between MCF-7 and HMEC-1 can be blocked by cystamine, PCMBS and PACMA-31, whereas the adhesion of MCF-7 to EA.hy926 is affected additionally by Q3Rut. MDA-MB-231 cells adhere to the endothelium more effectively than non-invasive cells and, as the only cell line, MDA-MB-231 cells adapts a typical long shape after adhesion to the endothelium layer (Figure 4C). All the tested factors block the adhesion of MDA-MB-231 to the endothelium with the strongest inhibitory effect observed after incubation with cystamine or PCMBS. Solvents, like DMSO and ethanol at tested concentration display no significant effect on cells migration (Supplementary Figure 4).
Figure 4.

Adhesion of MCF-10A (A), MCF-7 (B) and MDA-MB-231 (C) Cells to endothelial cells HMEC-1 and EA.hy926 in the presence of PDI inhibitors and free thiol blockers. Data presented as mean + SD; mini-max ranges marked as whiskers; n=10. Total numbers of adherent cells were estimated with a multifunctional plate reader. Significance of differences was analysed with one-way ANOVA and the post-hoc multiple comparisons Tukey’s test and planned comparisons were verified the bootstrap-boosted unpaired student’s t test (10000 iterations) with the Bonferroni’s correction for multiple comparisons.
Transwell migration assay
A transwell system is often employed to examine cell invasiveness. Figure 5A shows that non-invasive MCF-10A and MCF-7 cells, even after LPS stimulation, very poorly migrate through a transwell chamber coated with gelatine, which significantly limits the possibilities of investigation of the role of thiol-disulfide modifications on MCF-10A and MCF-7 migration. LPS-stimulated migration of invasive cells MDA-MB-231 can be attenuated by all the tested free thiol blockers as well as PDI inhibitors (Figure 5B). Solvents, like DMSO and ethanol demonstrate no significant effect on cell migration (Supplementary Figure 5).
Figure 5.
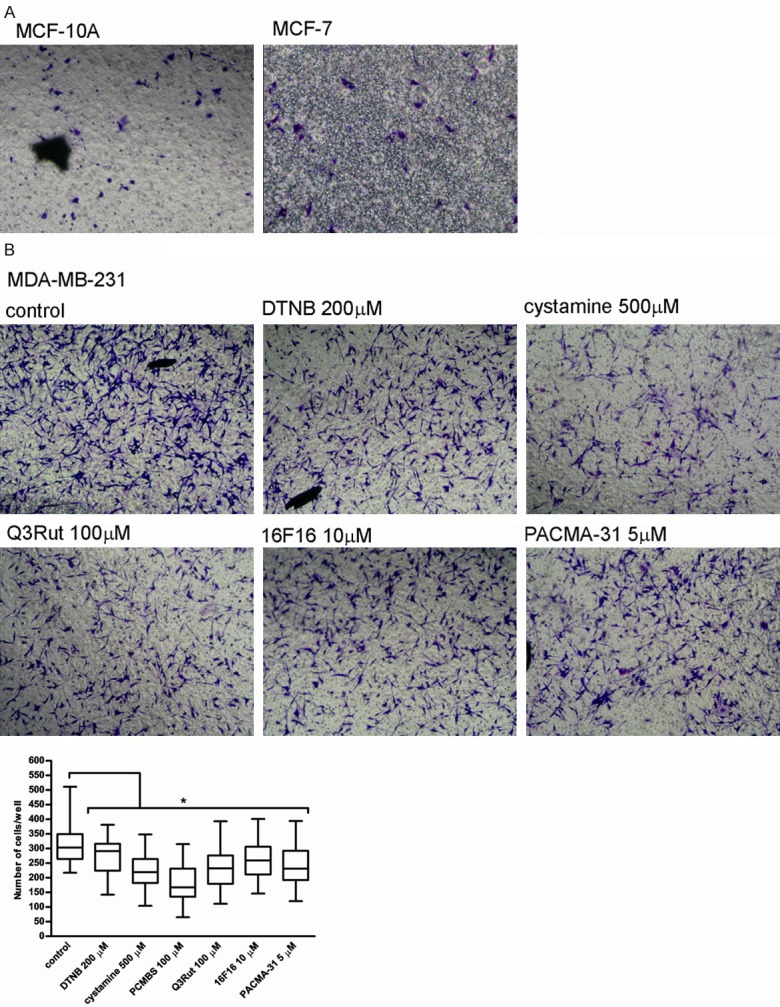
Migration of MCF-10A, MCF-7 and MDA-MB-231 cells through the gelatin-coated transwell chamber. Data presented as mean + SD; mini-max ranges marked as whiskers; n=5. MCF-10A, MCF-7 (A) and MDA-MB-231 cells (B) Were incubated with PDI inhibitors and free thiol blockers following their prior stimulation with 5 mg/ml LPS and allowed to migrate through the gelatine-coated chamber for 24 hr. Significance of differences was analysed with one-way ANOVA and the post-hoc multiple comparisons Tukey’s test and planned comparisons were verified the bootstrap-boosted unpaired student’s t test (10000 iterations) with the Bonferroni’s correction for multiple comparisons.
Transendothelial migration assay
Similarly to the transwell migration, non-invasive MCF-7 and MCF-10A are unable to migrate through the endothelium monolayer. Transendothelial migration of invasive MDA-MB-231 through HMEC-1 (Figure 6A) or through EA.hy926 (Figure 6B) can be attenuated by the incubation with thiol blockers or with PDI inhibitors. Despite a lower efficiency of migration through the endothelium as compared to the migration through gelatine itself, the tested factors blocking effects are comparable. The highest blocking efficiency, regardless of the type of endothelial cell line used for the experiment, is displayed by using PCMBS and the lowest after incubation with DTNB or 16F16. Solvents, like DMSO and ethanol demonstrate no significant effect on cell migration (Supplementary Figure 5).
Figure 6.

Transendothelial migration of MDA-MB-231 cells through the transwell chamber coated with gelatine and endothelial cells. Data presented as mean + SD or median and interquartile range; mini-max ranges marked as whiskers; n=6. MDA-MB-231 cells were incubated with PDI inhibitors and free thiol blockers following their prior stimulation with 5 mg/ml LPS and allowed to migrate through the transwell chamber coated with a gelatine and the monolayer of either HMEC-1 or EA.hy926 endothelial cells. Significance of differences was analysed with one-way ANOVA and the post-hoc multiple comparisons Tukey’s test and planned comparisons were verified the bootstrap-boosted unpaired student’s t test (10000 iterations) with the Bonferroni’s correction for multiple comparisons.
Collagen gel contraction
Figure 7A presents the differences in collagen contraction promoted by different cell lines. MCF-10A cells are unable to initiate collagen contraction, whereas MDA-MB-231 cells exhibit higher collagen contraction properties than non-invasive MCF-7 cells. Cystamine and Q3Rut inhibit collagen contraction initiated by MCF-7 cells only at the point of 12 hr and their blocking effects diminish with time (Figure 7B) meanwhile DTNB, PCMBS, 16F16 and PACMA-31 inhibit MCF-7-dependent collagen contraction at all tested time points. MDA-MB-231-mediated collagen contraction can be blocked by cystamine, PCMBS, 16F16 and PACMA-31 with only slightly inhibitory effect observed after incubation with Q3Rut. Solvents, like DMSO and ethanol demonstrate no significant effect on collagen contraction (Supplementary Figure 6).
Figure 7.

Effects of PDI inhibitors and free thiol blockers on the collagen gel contraction by MCF-10A, MCF-7 and MDA-MB-231 cells. Data presented as mean + SD, mini-max ranges marked as whiskers; n=12. NIE SEM! Collagen floating gels supplemented with the MCF-10A, MCF-7 and MDA-MB-231 cells in the NaOH-buffered medium containing DMEM, collagen type I, glutamine and antibiotics, were allowed to pomymerize (A) Two hours after polymerization gels were supplemented with PDI inhibitors and free thiol blockers, pictured and incubated for up to 72 hr (B) Significance of differences was analysed with one-way ANOVA and the post-hoc multiple comparisons Tukey’s test and planned comparisons were verified the bootstrap-boosted unpaired student’s t test (10000 iterations) with the Bonferroni’s correction for multiple comparisons.
Immunostaining
Figure 8A shows that after during MCF-10A adhesion to endothelium most of integrin β1 molecules on Eahy.926 cells remains in inactivated form (antibodies anti-β1 clone P4C10), when integrin β1 on MCF-10 became activated (antibodies anti-β1 clone 12G10 and antibodies anti-β1 clone HUTS-4). In contrast integrin αVβ3 on endothelial cells can be found both active (antibodies anti-αVβ3 clone LM609) or inactive state (antibodies anti-αVβ3 clone CBL544), whereas αVβ3 on MCF-10A remains mostly in inactive form. Since MCF-7 cells displays very low level of both β1 and αVβ3, neither active nor inactive form of integrins can be observed on MCF-7 surface during adhesion to endothelium monolayer (Figure 8B). In response to MDA-MB-231 adhesion to endothelium integrin β1 on cancer cells became activated (antibodies clone 12G10 and clone HUTS-4) and integrin αVβ3 stays mostly in inactive form (clone CBL544), meanwhile αVβ3 on endothelial cells can be found in active form (clone LM609) (Figure 8C).
Figure 8.

Fluorescent microscopic images of of MCF-10A, MCF-7 and MDA-MB-231 cells adhering to EA.926 endothelial cells. MCF-10A (A), MCF-7 (B) and MDA-MB-231 cells (C) Adhering to endothelial EA.hy926 cells were fixed and immunostained with specific antibodies for inactive (clone P4C10) or active (clone 12G10, HUTS-4) form of integrin β1 and inactive (cloneCBL544) or active (clone LM609) form of integrin αVβ3. Immunostaining with secondary antibodies conjugated with Alexa Fluor served as a negative controls. All images were taken by using ZOE Fluorescent Cell Imager (Bio-Rad, magnification 700 ×).
Western blot and labelling of free thiol groups
Figure 9A presents the differences in protein expression of integrin β1, integrin β3 and PDI. MDA-MB-231 exhibits high levels of integrin β1 and integrin β3 as compared to MCF-10A. MCF-7 may be characterized by a very poor expression of the two tested integrins. Figure 9B shows the presence of surface proteins containing free thiols on all of the tested cell lines. Not-labelled EA.hy926 lysate was used as a negative control of MPB-labelling and avidin-HRP blotting. Based on Western blot analysis, it is possible to evaluate the prevalence of thiol-containing proteins, however, signals from different proteins with a similar molecular mass may overlap. To give an insight into the thiols pattern on specific proteins, MPB-labelled lysates were precipitated with neutravidin-agarose resin which binds only labelled proteins through avidin-biotin interaction. Figure 9C demonstrates the presence of free-thiol containing integrin β1 and β3 subunits on the surface of MCF-10A and MDA-MB-231, but MDA-MB-231 exhibits a higher number of free-thiol containing β3 molecules. PDI molecules labelled with MPB can be found on the surface of all the tested cell lines with a higher exposition on the surface of MCF-7.
Figure 9.

Contents of integrins and PDI proteins, overall content of free thiol groups and free thiol groups in specific integrins and PDI protein in MCF-10A, MCF-7 and MDA-MB-231 cells. Data presented as mean + SD; mini-max ranges marked as whiskers; n=10. A. Western blot analysis of integrin β1, integrin β3 and PDI protein in different cell lines. B. Free thiol groups in different cell lines. C. Free thiol groups in the molecules of integrin β1, integrin β3 and PDI protein. Precipitation of non-labelled lysate and immunoblotting with β-actin were used as a negative control of MPB labelling and precipitation. Significance of differences was analysed with one-way ANOVA and the post-hoc multiple comparisons Tukey’s test and planned comparisons were verified the bootstrap-boosted unpaired student’s t test (10000 iterations) with the Bonferroni’s correction for multiple comparisons.
Blocking of free thiols on integrin molecules
Using free thiol blockers and PDI inhibitors during MDA-MB-231 adhesion to integrin ligands leads to diminished exposition of thiol groups on integrin β1 and integrin β3 (Figure 10). Effectiveness of tested agents in blocking free thiol exposition may be correlated with effectiveness in blocking the adhesion itself since i.e. cystamine and PCMBS are very potent blockers of free thiols on integrins, meanwhile DTNB that exhibits no blocking effect on adhesion to collagen or vitronectin shows the weakest effectiveness in blocking free thiol exposition on integrins molecules. Also 16F16 seems to be ineffective in blocking free thiols on integrins, which corresponds with other experiments, where 16F16 was the weakest of all tested PDI inhibitors. Q3Rut that exhibits very low blocking effect on free thiols exposition on surface PDI molecules effectively blocks free thiols on integrins molecules, which may suggest that blocking the ligand binding pocket on PDI by Q3Rut does not affect free thiols exposition on PDI but as results blocks integrin activation via thiol-disulfide exchanges dependent on PDI binding to integrins. Western blot analysis of proteins not precipitated by neutravidin-agarose resin serves as an indicator of equal amount of protein content in examined samples.
Figure 10.

Exposure of free thiols on integrin receptors and PDI molecules of MDA-MB-231 cells after incubation with free thiol blockers and PDI inhibitors. Immunoblotting of non-precipitated lysate with β-actin was used as a negative control of MPB labelling and precipitation. Data presented as mean + SD or median and interquartile range; mini-max ranges marked as whiskers, n=5.
Using blocking antibodies for integrin β1 and integrin αVβ3 during cell-cell adhesion (Figure 11A) and migration through endothelial monolayer (Figure 11B) proves that inactivation of integrin receptors leads to inhibition of cells movement. Adhesion of MDA-MB-231 cells to endothelium seems to be dependent merely on β1 activation since anti-αVβ3 antibodies desbite atenuation of cancer cells attachment, does not block cell-cell interaction in statistically significant matter. Meanwhile migration of cancer cells through endothelium layer can be blocked by both: anti-β1 or anti-αVβ3 antibodies irrespective of type of endothelial cells.
Figure 11.
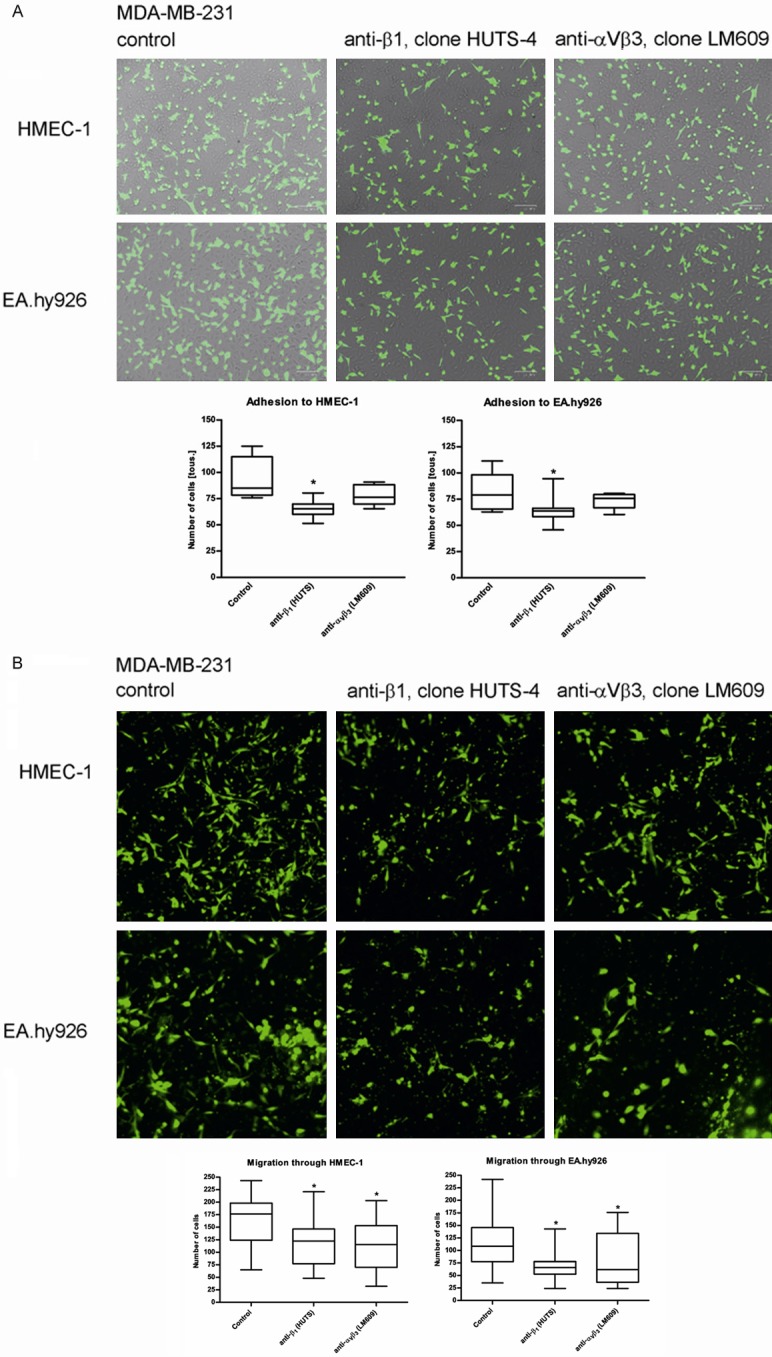
Adhesion (A) and migration (B) of MDA-MB-231 cells to endothelial cells HMEC-1 and EA.hy926 in the presence of anti-β1 (clone HUTS-4) or anti-αVβ3 (clone LM609) blocking antibodies used at concentration 1:50. Data presented as mean + SD or median and interquartile range; mini-max ranges marked as whiskers; n=3.
Discussion
The interaction of tumour cells with the endothelium is an important step in the metastatic process. Metastasis is especially dangerous for breast cancer patients. Only 12% of patients in Stage I/II of breast cancer dies during the first 10 years following the diagnosis, whereas, 60% of Stage III patients, with invaded nearby lymph nodes, and over 90% of Stage IV patients, with invaded distant organs, die within 10 years following the diagnosis [43].
Integrin receptors involved in breast cancer cell invasiveness include β1 and β3 [10,11,15,16,44]. Thiol-disulfide exchanges are important for integrin activation, which was illustrated by experiments involving the substitution of a serine residue at a site of one or both of the cysteine residues that form the initial disulfide bond. Substitution of a serine may lead to four different results: 1) It may have no effect on integrin activity, 2) It may lead to reduced expression of integrin molecules on the cell surface 3) It may maintain integrin receptor in an inactive state or 4) it may initiate a constitutively active state of integrin [56,57]. Since activation of β1 and β3 integrins can be promoted by modifications of extracellular disulfide bonds on integrin extracellular domains [17-19], blocking of integrin-mediated adhesion and migration of cancer cells through the endothelium by factors inhibiting thiol-disulfide exchanges, seems to be a logical suggestion for improving chemotherapy.
We provide evidence which supports the study showing that cancer MCF-7 cell line exhibits low level of integrin β1 and β3 expression, which may explain its low efficiency in adhesion and migration on integrin ligands. On the other hand, MDA-MB-231, highly metastatic breast cancer cells express high level of β1 and β3, which corresponds to high efficiency of adhesion and migration on collagen type I and vitronectin. Other researchers suggest that β1 is necessary for cancer-endothelium interaction and lack of β1 expression is the cause of MCF-7 non-invasive character [10,11]. We agree with this assumption but integrin expression cannot be the only factor responsible for effectiveness of cancer cell migration and adhesion since both cell lines expressing β1 and β3, i.e. non-invasive MCF-10A and highly invasive MDA-MB-231, adhere and migrate with different effectiveness. If we agree with scientific reports about MCF-10A sharing features of mesenchymal cancer cell lines, we have to assume that the total level of integrin expression seems to be an important though not the decisive factor for cancer cells adhesion and migration on integrin ligands, since MDA-MB-231 cells adhere and migrate with high efficiency as compared to MCF-10A despite similar level of integrins expression.
LPS-induced upregulation of αVβ3 promotes αVβ3-mediated adhesion and invasiveness of metastatic breast cancer [16]. However, in our study we do not find the active form of integrin αVβ3 present on cancer cell surface during adhesion to the endothelial monolayer. The active form of αVβ3 was rather found on endothelial cells, which suggests that during interaction between cancer cells and the endothelium, activation of αVβ3 may occur not only on cancer cells but rather on both type of cells. We demonstrate that during adhesion of MDA-MB-231 to endothelial cells, the active form of integrin β1 is present mainly on cancer cells meanwhile on endothelial cells β1 remains in inactive form. This observation stays in agreement with scientific reports suggesting that β1 is one of numerous features responsible for the highly-metastatic character of MDA-MB-231 and low level of β1 expression is the cause of MCF-7 non-invasive character [10,11]. Activation of β1 may lead to β1-dependent phosphorylation and dissociation of β-catenin from the VE-cadherin complex, which occurs during an interaction between cancer cells and endothelial cells [10]. Based on that observation, we hypothesize that activation of β1 integrin on cancer cells is necessary for the first steps of cancer cells migration through the endothelium which involves adhesion of cancer cells to endothelial cells. Then the following steps may be mediated by the activated integrin αVβ3 since overexpression of αVβ3 correlates with the invasive character of cancer cells [12-14]. Blocking of MDA-MB-231 adhesion and migration through endothelium by anti-β1 antibodies and blocking only migration step by anti-αVβ3 antibodies without significant effect on adhesion itself, seems to confirm this assumption.
We prove that the mechanism mediated by β1 and β3 like adhesion and migration to integrin ligands can be attenuated by factors blocking free thiol groups, thus blocking thiol-disulfide exchanges inside and between proteins. The most potent agent blocking cell adhesion to integrin ligands is PCMBS which, according to its molecular mass and electric charge, cannot be transported through the cellular membrane and blocks only surface thiols. Since other tested thiol blockers, like DTNB and cystamine, are membrane-permeable, we may assume that blocking surface proteins is essential for inhibiting adhesion of cancer cells to ECM proteins. The inhibitory effects of thiol blockers may be caused by: 1) Blocking free thiols on PDI molecules or other thiol isomerases, which are used for reduction of disulfide bonds in integrins, 2) Blocking freshly introduced free thiols on integrin molecules, since the active state of integrin may be related not to the disruption of disulfide bonds sensu stricte but with the exposition of free thiol group in specific position of aminoacid chain [56,57].
Other factors employed in our study include PDI inhibitors: 16F16, PACMA-31 and Q3Rut. 16F16 is a small molecule suppressor of polyglutamine-induced apoptosis with two PDI isoforms as specific targets. PACMA-31, which similarly to 16F16 is irreversible but more specific, covalently binds to active site cysteines of PDI and shows promising effects in therapy of ovarian cancer [45-48]. In our study we demonstrate that PDI inhibitor PACMA-31 displays tendencies to inhibit proliferation of breast cancer MCF-7 and MDA-MB-231 cells. The anti-proliferating activity of PACMA-31 increases when the concentration exceeds 10 μM, which corresponds to the results obtained by the discoverers of PACMA-31 suggesting that PACMA-31 can be cytotoxic for human breast cancer cells even at micromolar concentrations [36] without any significant impact on normal cells [36,45].
Q3Rut was chosen due to its therapeutic implications in thrombotic disease and a lack of a negative influence on the human organism [25,49]. Q3Rut is also one of the most specific PDI inhibitors among all the rutin derivatives and in contrast to 16F16 and PACMA-31 it cannot be transported through the cellular membrane [28]. Q3Rut blocks the functions of PDI in a thiol-independent matter by binding to the b or x’ domain of PDI, which is a substrate docking domain and not a catalytic domain [49].
From all the tested PDI inhibitors the lowest efficiency in blocking adhesion to integrin ligands can be correlated with activity of 16F16 and the highest one with PACMA-31. Lower effectiveness of PDI inhibitors comparing with thiol blockers may be the result of targeting only PDI and not other proteins like integrins. Moreover in response to PDI inhibition other oxidoreductases present on cell surface may probably act as PDI replacement [58,59]. It is also possible that secretion of fresh PDI molecules in response to PDI inhibition may still leads to integrin activation during adhesion or migration.
Among all the tested cell lines MDA-MB-231 exhibits the highest efficiency of adhesion and migration on β1 and β3 ligands. The non-invasive MCF-10A most commonly used as a normal breast cell model, expresses β1 integrin but displays low efficiency in adhesion and migration on ECM proteins. The non-invasive MCF-7 cells that do not express β1 and β3 can be characterised by poor adhesion and migration, the same as non-cancer cell line MCF-10A. MDA-MB-231 more effectively adheres not only to ECM proteins but also to different endothelial cells. Adhesion of MDA-MB-231 to the endothelium, analogously to adhesion to collagen and vitronectin, can be attenuated by thiol blockers and PDI inhibitors. Measured with thiols labelling and neutravidin precipitation diminished exposition of free thiols on integrin molecules in response to incubation with tested factors, proves that integrins are targets of thiol-blocking properties of cystamine, PCMBS, DTNB or PDI inhibitors. Similarities in blocking ECM adhesion and cell-cell adhesion by different thiol blockers or PDI inhibitors suggest that both of these processes are, at some point at least, mediated by integrins. This assumption was proved by using anti integrins antibodies that blocks adhesion of MDA-MB-231 cells to endothelium and inhibits cancer cell migration through endothelial layer.
The highly metastatic MDA-MB-231 is the only tested cell line that demonstrates the ability to migrate through a transwell chamber coated with gelatine or through the endothelial monolayer. Blocking the invasiveness of MDA-MB-231 on the endothelium by thiol blockers and PDI inhibitors proves the involvement of integrins in cancer progression and metastasis. The first step of the transendothelial migration involves adhesion of cancer cells to the endothelial monolayer, and staining with specific anti-integrins antibodies shows that integrin β1 on cancer cells became activated during adhesion to HMEC-1 or EA.hy926. However, the non-invasive MCF-10A that also expresses β1 are unable to migrate through the endothelium despite their ability to attach to endothelial cells.
MCF-10A is the only tested cell line unable to promote collagen contraction, whereas, both cancer cell lines: MCF-7 and MDA-MB-231 can promote collagen contraction. The highly metastatic MDA-MB-231 initiates this process more effectively than the non-invasive MCF-7. Collagen and its modifications are crucial for cancer cells, since remodelling of the ECM and penetration of cancer cells through tissue barriers is necessary for metastasis. Collagen type I seems to be important for breast cancer progression since it is widely distributed in breast tissue and increased stromal collagen can contribute to mammary tumor formation and metastasis [50]. Enhanced collagen I-α2β1 cross-talking promotes tumor metastasis to the bone matrix which contains abundant amounts of collagen type I [51]. Integrin α2β1 regulates expression of collagen type I and main metalloproteinase responsible for collagen degradation-MMP-1 [52]. Collagen contraction can be promoted by integrins such as integrin α11 [19] which was proposed as a novel biomarker for breast cancer [60]. Blocking the collagen contraction by thiol blockers or PDI inhibitors may shed a light on another integrin-dependent process involved in metastasis of cancer cells.
Our findings demonstrate that blocking free thiols on cell surface can inhibit cell-cell adhesion and transendothelial migration of the highly metastatic MDA-MB-231 cell line. We are aware that sulfhydryl reagents like free thiol blockers would be predicted to affect dozens of proteins. However integrins must belong to this group since tested factors affects mechanisms mediated directly by integrins like adhesion and migration on ECM proteins. What is more important we show that all of tested agents, with various effectiveness, affects free thiol exposition on integrin molecules present on cell surface, which may lead to blocking of integrin activation since exposition of free thiols, as a consequence of breakage of disulfide bonds, can initiate a constitutively active state of integrin [56,57].
Other researchers showed that the suppression of cell adhesion, spreading and invasion of MDA-MB-231 can be related to inhibition of β1 by blocking surface PDI using MNS (3,4-methylenedioxy-β-nitrostyrene) [20]. We provide evidences that also other PDI inhibitors can affect cancer cell migration, but we are aware that specificity of these factors may be debatable. 16F16 and PACMA-31 were constructed as permeable factors and may be easily sequestered by endogenous thiols. Moreover in the cytosol there are many different disulfide isomerases with a range of abundances and all of them may be susceptible to alkylation at their redox-active cysteine residues [60], so the anti-adhesion ad anti-migration effect of 16F16 and PACMA-31 may not be related only to blocking surface PDI. Q3Rut due to the lack of negative effects on human organism and impermeable properties seems to be very promising for further studies, but even Q3Rut high specificity to PDI and not other isomerases was questioned [61].
Unfortunately, we did not manage to find any thiol blocker or PDI inhibitor that would be specific for invasive cancer cells. Despite the huge differences in adhesion, migration and cell-cell adhesion, all the tested cell lines including normal MCF-10A, cancer but non-invasive MCF7 and highly invasive MDA-MB-231 respond to incubation with thiol blockers and PDI inhibitors in a similar manner. The factors characterised by the highest effectiveness, i.e. PACMA-31 and PCMBS, influence the activity of all the tested cell lines in a similar way. Additionally, all the tested cell lines exhibit similar levels of PDI including surface PDI molecules exposing free thiols. The most promising observations involve an increased number of free thiols on αVβ3 on MDA-MB-231, which may be correlated with a high number of activated integrin molecules on the surface of invasive cancer cells. Our observation that a non-toxic, specific inhibitor of extracellular PDI Q3Rut displays potency in blocking cancer cells adhesion and migration may be a very good starting point for a further analysis of the role thiol isomerases in cell migration. As a conclusion, this paper shows that blocking thiol isomerases like PDI or blocking activation of integrin receptors by stopping thiol-disulfide exchanges can be a promising target for PDI-dependent anti-cancer therapy not only depending on accumulation of misfolded proteins but also on attenuating integrin-mediated cancer invasiveness.
Acknowledgements
This work was supported by the grant “Prostacyclin, nitric oxide and carbon monoxide-based pharmacotherapy of endothelial dysfunction and platelet activation-a novel strategy to inhibit cancer metastasis” (acronym: METENDOPHA) funded by the National Center for Research and Development (Cracow, Poland) under the Polish Strategic Framework Program STRATEGMED “Prevention practices and treatment of civilization” (the grant coordinated by the Jagiellonian Center for Experimental Therapeutics-Jagiellonian University, Cracow, Poland), No. STRATEGMED1/233226/11/NCBR/2015.
Disclosure of conflict of interest
None.
Abbreviations
- 16F16
2-(2-Chloroacetyl)-2,3,4,9-tetrahydro-1-methyl-1H-pyrido[3,4-b]indole-1-carboxylic acid methyl ester)
- DTNB
5,5’-dithiobis-(2-nitrobenzoic acid)
- MPB
3-(N-maleimido-propinyl)biocytin
- NEMN
ethylmaleimide
- Q3Rut
quercetin-3-rutinoside
- PACMA 31
N-(2,4-Dimethoxyphenyl)-N-(1-oxo-2-propyn-1-yl)-2-(2-thienyl)glycyl-glycine ethyl ester
- PCMBS
p-chloromercuribenzene sulfonate
Supporting Information
References
- 1.Läubli H, Borsig L. Selectins promote tumor metastasis. Semin Cancer Biol. 2010;20:169–177. doi: 10.1016/j.semcancer.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Desgrosellier JS, Cheresh DA. Integrins in cancer: Biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. J Biol Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 4.Hegerfeldt Y, Tusch M, Bröcker EB, Friedl P. Collective cell movement in primary melanoma explants: plasticity of cell-cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 2002;62:2125–2130. [PubMed] [Google Scholar]
- 5.Dardik R, Savion N, Kaufmann Y, Varon D. Thrombin promotes platelet-mediated melanoma cell adhesion to endothelial cells under flow conditions: role of platelet glycoproteins P-selectin and GPIIb-IIIA. Br J Cancer. 1998;77:2069–2075. doi: 10.1038/bjc.1998.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramirez NE, Zhang Z, Madamanchi A, Boyd KL, O’Rear LD, Nashabi A, Li Z, Dupont WD, Zijlstra A, Zutter MM. The α2β1 integrin is a metastasis suppressor in mouse models and human cancer. J Clin Invest. 2011;121:226–237. doi: 10.1172/JCI42328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho WC, Heinemann C, Hangan D, Uniyal S, Morris VL, Chan BM. Modulation of in vivo migratory function of alpha 2 beta 1 integrin in mouse liver. Mol Biol Cell. 1997;8:1863–1875. doi: 10.1091/mbc.8.10.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang C, Zeisberg M, Lively JC, Nyberg P, Afdhal N, Kalluri R. Integrin alpha 1 beta 1 and alpha 2 beta 1 are the key regulators of hepatocarcinoma cell invasion across the fibrotic matrix microenvironment. Cancer Res. 2003;63:8312–8317. [PubMed] [Google Scholar]
- 9.Yoshimura K, Meckel KF, Laird LS, Chia CY, Park JJ, Olino KL, Tsunedomi R, Harada T, Iizuka N, Hazama S, Kato Y, Keller JW, Thompson JM, Chang F, Romer LH, Jain A, Iacobuzio-Donahue C, Oka M, Pardoll DM, Schulick RD. Integrin alpha 2 mediates selective metastasis to the liver. Cancer Res. 2009;69:7320–7332. doi: 10.1158/0008-5472.CAN-09-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haidari M, Zhang W, Caivano A, Chen Z, Ganjehei L, Mortazavi A, Stroud C, Woodside DG, Willerson JT, Dixon RA. Integrin α2β1 mediates tyrosine phosphorylation of vascular endothelial cadherin induced by invasive breast cancer cells. J Biol Chem. 2012;287:32981–32992. doi: 10.1074/jbc.M112.395905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taherian A, Li X, Liu Y, Haas TA. Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer. 2011;11:293. doi: 10.1186/1471-2407-11-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Filardo EJ, Brooks PC, Deming SL, Damsky C, Cheresh DA. Requirement of the NPXY motif in the integrin beta (3) subunit cytoplasmic tail for melanoma cell migration in vitro and in vivo. J Cell Biol. 1995;130:441–450. doi: 10.1083/jcb.130.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Felding-Habermann B, O’Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH, Hughes PE, Pampori N, Shattil SJ, Saven A, Mueller BM. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci U S A. 2001;98:1853–1868. doi: 10.1073/pnas.98.4.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manes T, Zheng DQ, Tognin S, Woodard AS, Marchisio PC, Languino LR. Alpha (v) beta (3) integrin expression up-regulates cdc2, which modulates cel migration. J Cell Biol. 2003;161:817–826. doi: 10.1083/jcb.200212172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao F, Li L, Guan L, Yang H, Wu C, Liu Y. Roles for GP IIb/IIIa and αvβ3 integrins in MDA-MB-231 cell invasion and shear flow-induced cancer cell mechanotransduction. Cancer Lett. 2014;344:62–73. doi: 10.1016/j.canlet.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 16.Yang H, Wang B, Wang T, Xu L, He C, Wen H, Yan J, Su H, Zhu X. Toll-like receptor 4 prompts human breast cancer cells invasiveness via lipopolysaccharide stimulation and is overexpressed in patients with lymph node metastasis. PLoS One. 2014;9:e109980. doi: 10.1371/journal.pone.0109980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swiatkowska M, Szymański J, Padula G, Cierniewski CS. Interaction and functional association of protein disulfide isomerase with alpha V beta 3 integrin on endothelial cells. FEBS J. 2008;275:1813–1823. doi: 10.1111/j.1742-4658.2008.06339.x. [DOI] [PubMed] [Google Scholar]
- 18.Swiatkowska M, Padula G, Michalec L, Stasiak M, Skurzynski S, Cierniewski CS. Ero1alpha is expressed on blood platelets in association with Protein-Disulfide Isomerase and contributes to redox-controlled remodeling of alpha IIb beta 3. J Biol Chem. 2010;285:29874–29883. doi: 10.1074/jbc.M109.092486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Popielarski M, Ponamarczuk H, Stasiak M, Michalec L, Bednarek R, Studzian M, Pulaski L, Swiatkowska M. The role of Protein Disulfide Isomerase and thiol bonds modifications in activation of integrin subunit alpha 11. Biochem Biophys Res Commun. 2018;495:1635–1641. doi: 10.1016/j.bbrc.2017.11.186. [DOI] [PubMed] [Google Scholar]
- 20.Chen IH, Chang FR, Wu YC, Kung PH, Wu CC. 3,4-Methylenedioxy-β-nitrostyrene inhibits adhesion and migration of human triple-negative breast cancer cells by suppressing β1 integrin function and surface protein disulfide isomerase. Biochimie. 2015;110:81–92. doi: 10.1016/j.biochi.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 21.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 22.Hua D, Liu MY, Cheng ZD, Qin XJ, Zhang HM, Chen Y, Qin GJ, Liang G, Li JN, Han XF, Liu DX. Small interfering RNA-directed targeting of Toll-like receptor 4 inhibits human prostate cancer cell invasion, survival, and tumorigenicity. Mol Immunol. 2009;46:2876–2884. doi: 10.1016/j.molimm.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 23.Jasuja R, Furie B, Furie BC. Endothelium-derived but not platelet-derived Protein Disulfide Isomerase is required for thrombus formation in vivo. Blood. 2010;116:4665–4674. doi: 10.1182/blood-2010-04-278184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Essex DW, Chen K, Swiatkowska M. Localization of protein disulfide isomerase to the external surface of the platelet plasma membrane. Blood. 1995;86:2168–2173. [PubMed] [Google Scholar]
- 25.Jasuja R, Passam FH, Kennedy DR, Kim SH, Van Hessem L, Lin L, Bowley SR, Joshi SS, Dilks JR, Furie B, Furie BC, Flaumenhaft R. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest. 2012;122:2104–2113. doi: 10.1172/JCI61228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett TA, Edwards BS, Sklar LA, Rogelj S. Sulfhydryl regulation of L-selectin shedding: phenylarsine oxide promotes activation-independent L-selectin shedding from leukocytes. J Immunol. 2000;164:4120–4129. doi: 10.4049/jimmunol.164.8.4120. [DOI] [PubMed] [Google Scholar]
- 27.Bi S, Hong P, Lee W, Baum LG. Galectin-9 binding to cell surface Protein disulfide isomerase regulates the redox environment to enhance T-cell migration and HIV entry. Proc Natl Acad Sci U S A. 2011;108:10650–10655. doi: 10.1073/pnas.1017954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willems SH, Tape CJ, Stanley PL, Taylor NA, Mills IG, Neal DE, McCafferty J, Murphy G. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochem J. 2010;428:439–450. doi: 10.1042/BJ20100179. [DOI] [PubMed] [Google Scholar]
- 29.Lovat PE, Corazzari M, Armstrong JL, Martin S, Pagliarini V, Hill D, Brown AM, Piacentini M, Birch-Machin MA, Redfern CP. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res. 2008;68:5363–5369. doi: 10.1158/0008-5472.CAN-08-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonome T, Levine DA, Shih J, Randonovich M, Pise-Masison CA, Bogomolniy F, Ozbun L, Brady J, Barrett JC, Boyd J, Birrer MJ. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008;68:5478–5486. doi: 10.1158/0008-5472.CAN-07-6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Welsh JB, Sapinoso LM, Su AI, Kern SG, Wang-Rodriguez J, Moskaluk CA, Frierson HF Jr, Hampton GM. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–5978. [PubMed] [Google Scholar]
- 32.Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, Taylor JM, Iannettoni MD, Orringer MB, Hanash S. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 33.Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nat Genet. 2005;37:382–390. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- 34.Goplen D, Wang J, Enger PØ, Tysnes BB, Terzis AJ, Laerum OD, Bjerkvig R. Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res. 2006;66:9895–9902. doi: 10.1158/0008-5472.CAN-05-4589. [DOI] [PubMed] [Google Scholar]
- 35.Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, Wang Y. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 2005;11:7234–7242. doi: 10.1158/1078-0432.CCR-05-0683. [DOI] [PubMed] [Google Scholar]
- 36.Hashida T, Kotake Y, Ohta S. Protein disulfide isomerase knockdown-induced cell death is cell-line-dependent and involves apoptosis in MCF-7 cells. J Toxicol Sci. 2011;36:1–7. doi: 10.2131/jts.36.1. [DOI] [PubMed] [Google Scholar]
- 37.Yamada R, Cao X, Butkevich AN, Millard M, Odde S, Mordwinkin N, Gundla R, Zandi E, Louie SG, Petasis NA, Neamati N. Discovery and preclinical evaluation of a novel class of cytotoxic propynoic acid carbamoyl methyl amides (PACMAs) J Med Chem. 2011;54:2902–2914. doi: 10.1021/jm101655d. [DOI] [PubMed] [Google Scholar]
- 38.Sun S, Lee D, Ho AS, Pu JK, Zhang XQ, Lee NP, Day PJ, Lui WM, Fung CF, Leung GK. Inhibition of prolyl 4-hydroxylase, beta polypeptide (P4HB) attenuates temozolomide resistance in malignant glioma via the endoplasmic reticulum stress response (ERSR) pathways. Neuro Oncol. 2013;15:562–577. doi: 10.1093/neuonc/not005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soule HD, Maloney TM, Wolman SR, Peterson WD Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- 40.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imbalzano KM, Tatarkova I, Imbalzano AN, Nickerson JA. Increasingly transformed MCF-10A cells have a progressively tumor-like phenotype in three-dimensional basement membrane culture. Cancer Cell Int. 2009;9:7. doi: 10.1186/1475-2867-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat. 2004;83:249–289. doi: 10.1023/B:BREA.0000014042.54925.cc. [DOI] [PubMed] [Google Scholar]
- 43.Singletary SE, Connolly JL. Breast cancer staging: working with the sixth edition of the AJCC cancer staging manual. CA Cancer J Clin. 2006;56:37–47. doi: 10.3322/canjclin.56.1.37. [DOI] [PubMed] [Google Scholar]
- 44.Liao SJ, Zhou YH, Yuan Y, Li D, Wu FH, Wang Q, Zhu JH, Yan B, Wei JJ, Zhang GM, Feng ZH. Triggering of Toll-like receptor 4 on metastatic breast cancer cells promotes αvβ3-mediated adhesion and invasive migration. Breast Cancer Res Treat. 2012;133:853–863. doi: 10.1007/s10549-011-1844-0. [DOI] [PubMed] [Google Scholar]
- 45.Xu S, Butkevich AN, Yamada R, Zhou Y, Debnath B, Duncan R, Zandi E, Petasis NA, Neamati N. Discovery of an orally active small-molecule irreversible inhibitor of Protein Disulfide Isomerase for ovarian cancer treatment. Proc Natl Acad Sci U S A. 2012;109:16348–16353. doi: 10.1073/pnas.1205226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaplan A, Stockwell BR. Structural elucidation of a small molecule inhibitor of protein disulfide isomerase. ACS Med Chem Lett. 2015;6:966–971. doi: 10.1021/acsmedchemlett.5b00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaplan A, Gaschler MM, Dunn DE, Colligan R, Brown LM, Palmer AG, Lo DC, Stockwell BR. Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc Natl Acad Sci U S A. 2015;112:2245–2252. doi: 10.1073/pnas.1500439112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vatolin S, Phillips JG, Jha BK, Govindgari S, Hu J, Grabowski D, Parker Y, Lindner DJ, Zhong F, Distelhorst CW, Smith MR, Cotta C, Xu Y, Chilakala S, Kuang RR, Tall S, Reu FJ. Novel Protein Disulfide Isomerase inhibitor with anticancer activity in multiple myeloma. Cancer Cancer Res. 2016;76:3340–3350. doi: 10.1158/0008-5472.CAN-15-3099. [DOI] [PubMed] [Google Scholar]
- 49.Lin L, Gopal S, Sharda A, Passam F, Bowley SR, Stopa J, Xue G, Yuan C, Furie BC, Flaumenhaft R, Huang M, Furie B. Quercetin-3-rutinoside inhibits Protein Disulfide Isomerase by binding to its b’x Domain. J Biol Chem. 2015;290:23543–23552. doi: 10.1074/jbc.M115.666180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hall CL, Dai J, Van Golen KL, Keller ET, Long MW. Type I collagen receptor (alpha 2 beta 1) signaling promotes the growth of human prostate cancer cells within the bone. Cancer Res. 2006;66:8648–54. doi: 10.1158/0008-5472.CAN-06-1544. [DOI] [PubMed] [Google Scholar]
- 52.Riikonen T, Westermarck J, Koivisto L, Broberg A, Kahari VM, Heino J. Integrin alpha 2 beta 1 is a positive regulator of collagenase (MMP-1) and collagen alpha 1 (I) gene expression. J Biol Chem. 1995;270:13548–13552. doi: 10.1074/jbc.270.22.13548. [DOI] [PubMed] [Google Scholar]
- 53.Pan Y, Liu G, Yuan Y, Zhao J, Yang Y, Li Y. Analysis of differential gene expression profile identifies novel biomarkers for breast cancer. Oncotarget. 2017;8:114613–114625. doi: 10.18632/oncotarget.23061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Neill S, Robinson A, Deering A, Ryan M, Fitzgerald DJ, Moran N. The platelet integrin αIIbβ3 has an endogenous thiol isomerase activity. J Biol Chem. 2000;275:36984–36990. doi: 10.1074/jbc.M003279200. [DOI] [PubMed] [Google Scholar]
- 55.Zhu G, Zhang Q, Reddy EC, Carrim N, Chen Y, Xu XR, Xu M, Wang Y, Hou Y, Ma L, Li Y, Rui M, Petruzziello-Pellegrini TN, Lavalle C, Stratton TW, Lei X, Adili R, Chen P, Zhu C, Wilkins JA, Hynes RO, Freedman J, Ni H. The integrin PSI domain has an endogenous thiol isomerase function and is a novel target for antiplatelet therapy. Blood. 2017;129:1840–1854. doi: 10.1182/blood-2016-07-729400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mor-Cohen R, Rosenberg N, Einav Y, Zelzion E, Landau M, Mansour W, Averbukh Y, Seligsohn U. Unique disulphide bonds in epidermal growth factor (EGF) domains of beta3 affect structure and function of αIIbβ3 and αvβ3 integrins in different manner. J Biol Chem. 2012;287:8879–8891. doi: 10.1074/jbc.M111.311043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruiz C, Liu CY, Sun QH, Sigaud-Fiks M, Fressinaud E, Muller JY, Nurden P, Nurden AT, Newman PJ, Valentin N. A point mutation in the cysteine-rich domain of glycoprotein (GP) IIIa results in the expression of a GPIIb-IIIa (alpha IIb beta 3) integrin receptor locked in a high-affinity state and a Glanzmann thrombasthenia-like phenotype. Blood. 2001;98:2432–2441. doi: 10.1182/blood.v98.8.2432. [DOI] [PubMed] [Google Scholar]
- 58.Schulman S, Bendapudi P, Sharda A, Chen V, Bellido-Martin L, Jasuja R, Furie BC, Flaumenhaft R, Furie B. Extracellular thiol isomerases and their role in thrombus formation. Antioxid Redox Signal. 2016;24:1–15. doi: 10.1089/ars.2015.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dihazi H, Dihazi GH, Bibi A, Eltoweissy M, Mueller CA, Asif AR, Rubel D, Vasko R, Mueller GA. Secretion of ERP57 is important for extracellular matrix accumulation and progression of renal fibrosis, and is an early sign of disease onset. J Cell Sci. 2013;126:3649–3663. doi: 10.1242/jcs.125088. [DOI] [PubMed] [Google Scholar]
- 60.Foster CK, Thorpe C. Challenges in the evaluation of thiol-reactive inhibitors of human Protein Disulfide Isomerase. Free Radic Biol Med. 2017;108:741–749. doi: 10.1016/j.freeradbiomed.2017.04.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bekendam RH, Bendapudi PK, Lin L, Nag PP, Pu J, Kennedy DR, Feldenzer A, Chiu J, Cook KM, Furie B, Huang M, Hogg PJ, Flaumenhaft R. A substrate-driven allosteric switch that enhances PDI catalytic activity. Nat Commun. 2016;7:12579. doi: 10.1038/ncomms12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.