

Abstract
Background/Aim: Recent research highlights the role of cancer-associated adipocytes (CAA) in promoting breast cancer cell migration, invasion and resistance to therapy. This study aimed at identifying cellular proteins differentially regulated in breast cancer cells co-cultured with CAA. Materials and Methods: Adipocytes isolated from human breast adipose tissue were co-cultured with hormone receptor-positive (MCF-7) or -negative (MDA-MB-231) breast cancer cells using a transwell co-culture system. Proteomes of co-cultured and control breast cancer cells were compared quantitatively using iTRAQ labelling and tandem mass spectrometry, and the results were validated by western blotting. Results: A total of 1,126 and 1,218 proteins were identified in MCF-7 and MDA-MB-231 cells, respectively. Among these, 85 (MCF-7) and 63 (MDA-MB-231) had an average fold change >1.5 following co-culture. Pathway analysis revealed that CAA-induced enrichment of proteins involved in metabolism, the ubiquitin proteasome, and purine synthesis. Conclusion: This study provides a proteomic platform for investigating the paracrine role of CAA in promoting breast cancer cell metastasis and resistance to therapy.
Keywords: Cancer-associated adipocytes, CAA, iTRAQ, breast cancer, metabolism, tumour microenvironment
Breast tissue, particularly in postmenopausal women, is rich in adipocytes that function as endocrine cells with the ability to exert biological effects through the secretion of growth factors, hormones and adipokines (1). Adipocytes at the invasive edge of human breast tumours exhibit a modified phenotype, in which they become delipidated with enlarged interstitial space, display a decrease in late adipocyte differentiation markers and overexpress inflammatory cytokines including IL-6 and IL-1β (2). Consequently, these adipocytes have been named cancer-associated adipocytes (CAA), and this modified CAA phenotype is similarly exhibited by mature adipocytes co-cultured with breast cancer cells in vitro (2).
Direct stimulation of breast cancer cells with factors known to be secreted by mature adipocytes, such as leptin and IL-6, is capable of supporting and promoting breast tumorigenicity in both oestrogen responsive and unresponsive breast cancer cells (3,4). However, cell–cell interactions in vivo are complex and adipocytes are unlikely to obligate breast cancer cell behaviour through a single secreted molecule. In vitro studies using conditioned media from 3T3-L1 derived murine adipocytes showed up-regulation of anti-apoptotic transcriptional programs in MCF-7 breast cancer cells (5). Similarly, conditioned media from breast adipocytes induced migration in non-cancerous normal breast epithelial cells (6), and increased migration, proliferation, viability and invasion in a variety of oestrogen receptor (ER)-positive and ER-negative breast cancer cell lines (7-13).
In vitro studies, using transwell (non-contact) co-culture of mature adipocytes with breast cancer cells, have established that CAA promote breast cancer cell proliferation, viability, migration and invasion (2,8,13-18). Recent evidence suggests that cross talk with CAA induces breast cancer cell invasiveness, in part, through metabolic remodelling of the cancer cell, promoting a shift towards increased mitochondrial fatty acid oxidation (17,19). In addition, breast cancer cells co-cultured with CAA undergo a partial epithelial-to-mesenchymal transition (EMT) (2), and become more resistant to radiotherapy (20) and other breast cancer therapies (21,22). These findings are further supported by in vivo xenograft studies, where human breast cancer cells co-cultured with CAA and subsequently implanted in mice show an increased tumour growth and metastasis (16,18,23).
The majority of these co-culture studies have focused on the altered production of specific adipocyte derived factors, such as CCL5, IL-6, IGF-1 and IGFBP-2, that promote breast cancer cell pro-tumour behaviour via paracrine actions (2,8,14-16). In addition to altered expression of adipocyte secreted factors, many of these studies have observed modified expression of targeted genes and proteins in breast cancer cells following co-culture with CAA, including IL-6, IL-8, CCL5, MCT2, CPT1A and MMP-2. A number of genes and pathways have been identified by cDNA microarray analysis to be differentially expressed in breast cancer cells after co-culture with CAA (13,18) or CAA-conditioned media (5,10), yet the expression of translated proteins are not necessarily predicted by this mRNA expression data. To date, the global regulation of cell protein in breast cancer cells following co-culture with CAA has not been assessed.
Therefore, the aim of the current study was to identify key proteins and cellular pathways that are differentially regulated in breast cancer cells co-cultured with CAA. This study performed proteomic profiling on ER positive (MCF-7) and hormone receptor negative (MDA-MB-231) human breast cancer cells cultured alone or co-cultured with CAA isolated from human breast adipose tissue, and identified a number of differentially regulated molecules and enriched pathways, each of which may be playing an important role in the relationship between CAA and breast cancer cells in the tumor microenvironment.
Materials and Methods
Breast cancer cell lines. Human breast cancer cell lines (MCF7: Luminal A, oestrogen receptor (ER)+, progesterone receptor (PR)+, HER2–; and MDA-MB-231: Basal, ER–/PR–/HER2–) (24) were purchased from American Tissue Culture Collection (ATCC; Manassas, VA, USA) and used for experiments within the first 30 passages. All cells used were tested using PCR and found to be mycoplasma free. Cells were cultured in Dulbecco’s modified Eagle medium (DMEM) (Thermo Fisher Scientific, Rockford, IL, USA) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin and 100 μg/ml streptomycin (Life Technologies, Carlsbad, CA, USA), in a 37˚C incubator with 5% CO2.
Breast adipose tissue samples. Breast adipose tissue samples were collected from female patients (mean age=63.3 years; age range=35-81 years) undergoing surgery at Christchurch Hospital for therapeutic or prophylactic mastectomy between March 2016 and March 2017. All samples collected from patients with breast tumours were characterised as invasive ductal carcinoma (IDC). Adipose tissue was sampled by a pathologist from macroscopically normal breast tissue. Ethical approval for the study was obtained from the University of Otago Ethics Committee (reference number 12/319). All patients gave informed written consent, and samples were collected by the Cancer Society Tissue Bank, Christchurch using standard operating procedures (25).
In vitro growth and differentiation of breast adipocytes. Breast adipose tissue was processed within 1 h of surgical resection to isolate the stromal vascular fraction (SVF) and differentiate stromal cells into mature adipocytes, as previously described (26). Briefly, fibrous and vascular tissue was cut away and the remaining adipose tissue was minced and enzymatically dissociated using collagenase, (type I from Clostridium histolyticum; 1 mg/ml in Hanks Balanced Salt Solution (HBSS); Sigma Aldrich, St. Louis, MA, USA) for 2 h at 37˚C with gentle agitation. Stromal vascular cells were re-suspended in DMEM/F12 nutrient media with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin, and plated onto 12 well CellBIND plates (In Vitro Technologies, Melbourne, Australia) at the equivalent of 0.6 g of adipose tissue per well. After 24 h, adherent cells were washed with phosphate buffered saline (PBS) (Sigma-Aldrich) and replenished with fresh DMEM/F12 with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin. Two days after reaching full confluence, cells were supplemented for 5 days with serum free complete differentiation DMEM/F12 media consisting of 0.5 mmol/l 3-isobutyl-1-methylxanthine (IBMX) (Sapphire Biosciences, Redfern, NSW, Australia), 100 nmol/l insulin (Life Technologies), 100 nmol/l dexamethasone (Sapphire Biosciences), 2 nmol/l triiodothyronine (Sigma-Aldrich,), 10 ng/ml transferrin (Sigma-Aldrich), 1 μmol/l rosiglitazone (Sapphire Biosciences), 33 μmol/l biotin (Sigma-Aldrich) and 17 μmol/l pantothenic acid (Sigma-Aldrich). Following the initial induction of differentiation, cells were replenished with serum free differentiation media DMEM/F12, without 0.5 mmol/l IBMX, for a further 5 days. Following differentiation, the adipocytes were maintained in DMEM/F12 with insulin (0.05 mmol/l) and dexamethasone (10 nmol/l) until fully mature and lipid filled (approximately 14-21 days). Differentiation of the pre-adipocytes to mature adipocytes was determined by light microscopy, and only wells with >70% differentiated adipocytes were used in experiments. All cell culture was performed at 37˚C incubation with 5% CO2.
Transwell co-culture of human breast adipocytes with breast cancer cell lines. Transwell inserts (Transwell® permeable supports 0.4 μm polyester membrane 12 mm insert, costar®, Corning) were soaked in DMEM/F12, 10% FBS media at 37˚C for ≥1 h. Differentiated adipocytes (on the bottom of each well in the 12-well plates) were replenished immediately before co-culture with fresh DMEM/F12 with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin. The transwell inserts were placed into the wells suspended above the differentiated adipocytes, and 500 μl of media containing breast cancer cells (MCF7=2.4×105 cells/ml; MDA-MB-231=2×105 cells/ml) was added to each insert. Transwell co-culture plates were then incubated at 37˚C with 5% CO2 for 3 days. Following transwell co-culture, media was removed from the inserts and breast cancer cells were collected for cell proteome analysis (n=2 biological replicates for CAA-MCF-7 and CAA-MDA-MB-231; each replicate uses adipose tissue from a different patient). Control MCF-7 (n=2) and MDA-MB-231 (n=2) cells were placed in transwell inserts and cultured alone (i.e. without adipocytes) in DMEM/F12 with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin for 3 days.
Sample preparation and mass spectrometry analysis of the breast cancer cell proteome. Sample preparation of the control (MCF-7 and MDA-MB-231) and transwell co-cultured (CAA-MCF-7 and CAA-MDA-MB-231) breast cancer cells was carried out following the filter-aided sample preparation (FASP) procedure (27). Briefly, cells were homogenised in lysis buffer (20 mM Hepes pH 7.5, 1 mM EDTA, 1 mM EGTA, 0.2% SDS, 5 mM tris(2-carboxyethyl)phosphine (TCEP) and protease inhibitor (complete mini EDTA free) (Roche, Basel, Switzerland) and then sonicated in short intervals for a total period of 2 min. Homogenates were centrifuged for 60 min at 16,000 × g to remove insoluble material. The supernatant was loaded onto Ultra-0.5 ml centrifugal filter units (Merck Millipore, Billerica, MA, USA) with a 3-kDa cutoff for two detergent depletion steps in 0.2 M triethylammonium bicarbonate (TEAB) containing 8 M urea and subsequent buffer exchanged into 0.2 M triethylammonium bicarbonate (TEAB). Proteins were reduced and alkylated on filter according to the instructions of the 8plex iTRAQ (isobaric Tags for Relative and Absolute Quantification) kit (AB Sciex, Foster City, CA, USA), and then buffer exchanged into 0.5 M TEAB. Total protein concentrations were normalised using a Bradford assay and protein was digested overnight using mass spectrometry grade trypsin (Promega Corporation, Madison, WI, USA). Prepared peptides from control (MCF-7 and MDA-MB-231) and transwell co-cultured (CAA-MCF-7, CAA-MDA-MB-231) breast cancer cells were labelled with isopropanol activated iTRAQ labels (Figure 1) from the iTRAQ® Reagents Kit, following manufacturer’s instructions. The labelled samples were further normalised according to the total iTRAQ labelling efficiency, measured by LC-coupled LTQ-Orbitrap tandem MS (as outlined below) in a test sample of pooled aliquots of all samples. Samples were then pooled accordingly and desalted by C-18 solid phase extraction. To reduce sample complexity and improve peptide identification, the iTRAQ labelled peptides were pre-fractionated by OFFGEL isoelectric focusing over a pH gradient (pH 3-10) into 12 individual fractions, using the 3100 OFFGEL fractionator (Agilent Technologies, CA, USA), following the manufacturer’s instructions. Individual fractions were then desalted by C-18 solid-phase extraction, vacuum concentrated to dryness and reconstituted in mass spectrometry loading buffer (5% acetonitrile in aqueous 0.2% formic acid). Each fraction was analysed by liquid chromatography coupled tandem mass spectrometry (LC-MS/MS) using an Ultimate 3000 RSLC-system fitted inline to the nanospray ionisation source of an LTQ-Orbitrap XL Mass Spectrometer (Thermo Fisher Scientific). The reversed-phase high performance liquid chromatography (RP-HPLC) separation of peptides was achieved on an in-house fused silica emitter tip column (15 cm in length with 75 μm inner diameter) packed with C-18 beads (3 μm diameter, 100 Å pore size), using a flow rate of 400 nl/min. To improve identification accuracy and quantification counts, three distinct reversed phase LC gradients were used to analyse each sample in technical triplicates. Gradients were established in three linear stages, 5 to 25%, 25 to 45% and 45 to 99% acetonitrile in aqueous 0.2% formic acid. The length of each gradient run varied across sample replicates between 40, 65 and 90 min for stage 1, 9, 10 and 9 min for stage 2, and 6, 5 and 6 min for stage 3. The column was washed and re-equilibrated between each sample. Full precursor ion scans were performed in the orbitrap analyser in a mass range of m/z 400-2000 and at a resolution of 60,000 at m/z 400. The highest four peptide signals for replicate one, and highest five peptide signals for replicate two and three, were selected for both CID (collision induced dissociation) at a normalised collision energy of 35% in the LTQ ion trap for protein identification, and HCD (high-energy collision induced dissociation) at a normalised collision energy of 55% for reporter ion detection and quantification in the Orbitrap at a resolution of 15,000 at m/z 400. For replicate one, dynamic exclusion allowed for two repeated MS/MS measurements of the same precursor ion before a 90 sec exclusion duration, and for replicate two and three, dynamic exclusion allowed for two repeated measurements before a 150 second exclusion duration. The raw data were processed by the Proteome Discoverer software (version 1.4, Thermo Fisher Scientific) under default peak processing settings. Using Sequest HT (Thermo Fisher Scientific) the lists of peaks were searched against the human reference amino acid sequence database. iTRAQ labels on lysines, tyrosines and the peptide N-terminus were selected as variable modifications. Other variable modifications included oxidised methionine, methylthio cysteine and deamidation of asparagine and glutamine. To reduce false positive identifications, the score threshold was adjusted to an estimated false discovery rate (FDR) of less than 1% using the Percolator node in Proteome Discoverer 1.4. Based on iTRAQ reporter ion intensities, the iTRAQ quantification node was used to determine the relative protein abundances. These ratios were normalised against the control protein ratio median, and only protein identifications with two or more significant peptide hits were accepted for quantification. To be considered for further analysis, proteins had to have two or more unique peptide hits, three or more ratio counts, control/control ratios less than 1.3 and match against the reviewed Swiss-Prot database. Identified proteins with a fold change of 1.5 or greater between control and transwell co-cultured breast cancer cells were considered as differentially regulated.
Figure 1. Experimental workflow. Schematic of the proteomic analysis and relative protein abundance validation workflow [n=number of biological replicates performed as independent experiments (expts)].

Bioinformatic analysis. The iTRAQ ratios were averaged and log transformed for each identified protein. Two different lists were uploaded into the PANTHER (protein annotation through evolutionary relationship) classification system (Version 12.0; http://www.pantherdb.org/) for cellular component and pathway analysis in each cell line using gene list analysis (28,29). The first list was analysed for overrepresented cellular components (PANTHER GO-Slim cellular components) (30), and included all the proteins that were identified by proteomic profiling in MCF-7 (n=1126) and MDA-MB-231 (n=1218) breast cancer cells. The second list was analysed for over-represented PANTHER pathways (31), and included only those proteins that had an average fold change ≥1.1 compared to controls, to indicate potentially meaningful pathways enriched in CAA-MCF-7 (n=576) and CAA-MDA-MB-231 (n=554). The statistical over-representation test was performed using the human genome as the reference list, and the binomial statistic p-values were corrected for multiple testing using the Bonferroni correction method.
Validation of mass spectrometry results using western analysis. To validate iTRAQ-based quantifications, western blotting was used to measure the relative protein abundances of three selected proteins (NDRG1, N-myc downstream-regulated gene 1 protein; PGK1, Phosphoglycerate kinase 1; and TFF1, Trefoil factor 1, Breast cancer oestrogen-inducible protein) in CAA-MCF-7 and CAA-MDA-MB-231 cells or control breast cancer cells cultured alone. Additional transwell co-culture experiments (n=3) were performed to obtain total protein samples for western analysis, and the CAA-MCF-7 and CAA-MDA-MB-231 cells used for western analysis were co-cultured with mature adipocytes isolated from three separate patients (Figure 1). Total protein was extracted from control and transwell co-cultured breast cancer cells, and protein concentration was determined by the BCA assay. A total of 5 μg of sample protein was mixed with BoltTM LDS sample buffer (Thermo Fisher Scientific) and dithiothreitol (DTT), denatured and resolved by BoltTM 4-12% and 12% Bis-Tris Gels with MOPS and MES SDS running buffer (Life Technologies), respectively. Proteins were transferred onto PVDF (polyvinylidene fluoride) membrane using BoltTM Transfer buffer (Life Technologies) with 10% methanol. Membranes were blocked with 5% w/v skimmed milk powder in tris buffered saline-tween 20 (TBS-T) (Sigma-Aldrich) for a minimum of 1 h before incubation with primary antibodies overnight at 4˚C. Primary antibodies were optimised at the following concentrations: anti-NDRG1 rabbit monoclonal antibody [EPR5593] at 1/5,000; anti-PGK1 rabbit polyclonal antibody (Abcam, Cambridge, MA, USA) at 1/1,000; anti-oestrogen inducible protein pS2 (TFF1) rabbit monoclonal antibody [EPR3972] (Abcam) at 1/1,000 and anti-β-actin mouse monoclonal antibody at 1/10,000 (Sigma-Aldrich). Each western analysis was repeated in triplicate for control (MCF-7, MDA-MB-231) and co-cultured (CAA-MCF-7, CAA-MDA-MB-231) breast cancer cell samples. The secondary antibodies used included polyclonal goat anti-rabbit immunoglobulins/HRP (Dako, Glostrup, Copenhagen, Denmark) at 1/5,000 for NDRG1, PGK1 and TFF1, and goat anti-mouse polyclonal immunoglobulins/HRP (Dako) used for β-actin at 1/10,000. Secondary antibodies were diluted in TBS-T and incubated at room temperature for 1 h. PVDF membranes were imaged using enhanced chemiluminescence (ECL) on an Alliance 4.7 (Uvitec Cambridge, Cambridge, UK) and were quantified using ImageJ software. The NDRG1, PGK1 and TFF1 densitometry values were compared to their respective β-actin bands to calculate relative expression values, and the relative expression for CAA-MCF-7 and CAA-MDA-MB-231 were compared to their respective controls to generate fold change values.
Statistical analysis. Western analysis results are presented as mean fold-change±SEM for all co-culture compared to control experiments (n=3, transwell co-culture experiments using adipose tissue from separate patients). Using GraphPad Prism 5 western blot ratios were analysed first by Kolmogrov-Smirnov to test data distribution. For normally distributed data one sample two-tailed t-tests were used to compare sample means to 1, and for non-normal data Wilcoxon signed-rank tests were performed to compare sample medians to 1. p-Values <0.05 were considered statistically significant. Statistical testing and graphical formatting were performed using GraphPad Prism 5.
Results
iTRAQ and LC-MS/MS identified and quantified relative protein abundance differences between control and transwell co-cultured breast cancer cells. By combining iTRAQ labelling, peptide fractionation by off gel isoelectric focusing and LC-MS/MS, a total of 1,126 and 1,218 proteins were identified from MCF-7 and MDA-MB-231 breast cancer cells, respectively (Figure 1). For each protein all co-culture/control iTRAQ ratio (fold change) combinations were used for quantification. Similar fold changes were reported from transwell co-culture experiments using adipocytes isolated from two individual breast cancer patients, suggesting that the CAA activity was not likely to be patient-specific.
The distribution of the average protein fold changes for CAA-MCF-7 and CAA-MDA-MB-231 illustrates that the majority of the proteins identified are similarly expressed in CAA co-cultured cells compared with the cells cultured alone (Figure 2A and B). The median fold change values for MCF-7 and MDA-MB-231 were –0.029 and –0.003, respectively. On average, 85 proteins in CAA-MCF-7 and 63 proteins in CAA-MDA-MB-231 were differentially regulated by 1.5-fold or greater compared to control cell lines (Figure 2A and B). Of the proteins that were changed by 1.5-fold or greater, more were down-regulated (n=53) than up-regulated (n=32) in CAA-MCF-7, whereas more proteins were up-regulated (n=51) than down-regulated (n=12) in CAA-MDA-MB-231. Three proteins, PGK1 (phosphoglycerate kinase 1), PPGB (lysosomal protective protein) and VPS35 (vacuolar protein sorting-associated protein 35) were differentially regulated in co-cultured versus control cells by 1.5-fold or greater in both cell lines. However, PGK1 was the only up-regulated protein in both cell lines, whereas PPGB and VPS35 were down-regulated in CAA-MCF-7 but up-regulated in CAA-MDA-MB-231 (Figure 2C).
Figure 2. Distribution of iTRAQ ratios identified and quantified in co-culture compared to control. A and B: Waterfall plots showing the distribution of the average co-culture (CC) versus control ratios (log2) for all the proteins identified in CAA-MCF-7 cells (1,126 proteins) and CAA-MDA-MB-231 cells (1,218 proteins), respectively. Vertical black bars represent different proteins and horizontal dotted lines represent a fold change (FC) of 1.5 (log2). In MCF-7 cells n=85 and in MDA-MB-231 n=63 proteins were enriched by greater than 1.5-fold. Of these enriched proteins, n=53/85 were down-regulated in MCF-7, whereas n=51/63 proteins were up-regulated by more than 1.5-fold in MDA-MB-231 cells. C: The three proteins identified and quantified to be differentially abundant by 1.5-fold or greater in both CAA-MCF-7 and CAA-MDA-MB-231 cells after co-culture with CAA. *FC values are the log of the average CC/control ratios.

Lists of the top 20 most differentially regulated proteins (excluding keratin proteins) in MCF-7 and MDA-MB-231 cells following transwell co-culture with CAA are presented in Table I and Table II. In line with the abovementioned trend, 17 of the top 20 differentially regulated proteins in CAA-MCF-7 were down-regulated, and 16 out of the top 20 were up-regulated in CAA-MDA-MB-231.
Table I. The top 20 proteins organised by greatest fold change (FC) in MCF-7 cells co-cultured with mature breast adipocytes.

aAmino acid residues; bNumber of unique peptides identified based on a FDR of <1%; cLog of the average co-culture/control ratios (fold change); dVariability of the peptide ratios as a percentage (normalised measure of the peptide ratio spread).
Table II. The top 20 proteins organised by greatest fold change (FC) in MDA-MB-231 cells co-cultured with mature breast adipocytes.

aAmino acid residues; bNumber of unique peptides identified based on a FDR of <1%; cLog of the average co-culture/control ratios (fold change); dVariability of the peptide ratios as a percentage (normalised measure of the peptide ratio spread).
Overall, this global and quantitative view shows that MCF-7 and MDA-MB-231 breast cancer cells exhibit unique alterations in protein expression profiles after co-culture with CAA, and furthermore, that majority of the proteins identified by discovery proteomics in breast cancer cells remain unchanged by cross-talk with CAA.
Bioinformatic analysis identified cellular components and pathways overrepresented by differentially regulated proteins following co-culture in breast cancer cells. Cellular components and pathways enriched in CAA-MCF-7 and CAA-MDA-MB-231 breast cancer cells were identified by statistical over-representation testing in PANTHER using gene list analysis, on the GO-Slim cellular components and PANTHER pathways (p<0.05) (Table III and Table IV). When analysis included all identified proteins, similar cellular components were enriched in MCF-7 and MDA-MB-231 cells, mostly pertaining to intracellular cytoplasmic, organelle and macromolecular complexes. More specifically, the enriched components were proteins from the nucleus and nuclear structures, as well as cell cytoskeleton, ribosomal and protein complexes. The enrichment of pathways relating to proteins that were differentially regulated in CAA-MCF-7 and CAA-MDA-MB-231 cells following co-culture, the direction of regulation and the corresponding proteins associated with these pathways are shown in Tables III and IV. The enriched tricarboxylic acid (TCA) cycle in CAA-MCF-7 and glycolysis pathway in CAA-MDA-MB-231 were the only pathways with all associated proteins up-regulated during co-culture. The ubiquitin proteasome pathway was commonly enriched in both CAA-MCF-7 and CAA-MDA-MB-231 cells, in which the majority of the associated proteins were 26S proteasome subunit proteins, and were down-regulated during co-culture. The CAA-MCF-7 cells predominantly down-regulated the 19S regulatory particle subunit proteins, whereas CAA-MDA-MB-231 decreased expression of both 19S and 20S subunit proteins. Differentially regulated proteins associated with Huntington’s and Parkinson’s disease pathways were significantly enriched in CAA-MCF-7 and CAA-MDA-MB-231, respectively. A component of the eukaryotic translation initiation factor 3 complex (EIF3F) required for initiation of protein synthesis was down-regulated in both cell types. Finally, proteins involved in de novo purine biosynthesis were differentially regulated in CAA-MDA-MB-231 cells, with up- and down-regulation occurring within this pathway (Table IV).
Table III. Significantly enriched (p<0.05) cellular pathways in the complement of proteins that were found differentially regulated in CAA-MCF-7.

aProteins increased by 1.1-fold or greater in MCF-7 and MDA-MB-231 cells following transwell co-culture with mature adipocytes; bProteins decreased by 1.1-fold or greater in MCF-7 and MDA-MB-231 cells following transwell co-culture with mature adipocytes; *Proteins are represented by gene names as reported by UniProtKB.
Table IV. Significantly enriched (p<0.05) cellular pathways in the complement of proteins that were found differentially regulated in CAA-MDAMB-231.

aProteins increased by 1.1-fold or greater in MCF-7 and MDA-MB-231 cells following transwell co-culture with mature adipocytes; bProteins decreased by 1.1-fold or greater in MCF-7 and MDA-MB-231 cells following transwell co-culture with mature adipocytes; *Proteins are represented by gene names as reported by UniProtKB.
Validation of mass spectrometry results by Western analysis. To validate mass spectrometry results, western analysis was used to determine the protein abundance of NDRG1 (N-myc downstream regulated 1), PGK1 (Phosphoglycerate kinase 1) and TFF1 (Trefoil factor 1; Breast cancer oestrogen-inducible protein) in CAA-MCF-7 and CAA-MDA-MB-231 cells or control breast cancer cells cultured alone (Figure 3). These three candidate proteins were selected based on differential abundance in either or both CAA-MCF-7 and CAA-MDA-MB-231, their relevance to breast cancer and the availability of well-characterised primary antibodies. In concordance with the iTRAQ quantification, PGK1 and TFF1 showed increased expression in CAA-MCF-7 cells (n=3 experiments), when measured by western blotting (fold change: PGK1=1.551 and TFF1=2.856) (Figure 3A and C). Similarly, NDRG1 and PGK1 were increased in CAA-MDA-MB-231 cells, validating the iTRAQ results (fold change: NDRG1=4.632 and PGK1=1.547) (Figure 3B and D). However, the NDRG1 up-regulation was more than two-fold higher when measured by western analysis compared to the iTRAQ quantification (NDRG1=2.137) (Figure 3B and D). As expected, mass spectrometry did not detect TFF1 expression in hormone receptor negative MDA-MB-231 cells, and this was validated by western analysis (Figure 3B and D). The only discordance was NDRG1 fold change measured in CAA-MCF-7 cells, showing an up-regulation by western analysis (2.193) but no change in expression by iTRAQ quantification (0.952) (Figure 3A and C). Furthermore, in accordance with the findings of the iTRAQ measurements, the fold changes measured by western analysis for PGK1 and TFF1 candidate proteins remained consistent across the replicates, whereas the fold changes for NDRG1 were more variable, as shown by the large error bars (Figure 3A and B).
Figure 3. Validation of candidate protein abundance by western blot analysis. A and B: The average fold change (FC) in protein abundance in coculture (CC) compared to control (Con) MCF-7 and MDA-MB-231 cells, for each candidate protein (NDRG1, PGK1 and TFF1) as measured by iTRAQ LC-MS/MS (white) and western analysis (grey). Data is presented as mean±SEM. C and D: Representative western blots for the abundance measurement of PGK1, TFF1 and NDRG1 in MCF-7 (Con), CAA-MCF-7 (CC), MDA-MB-231 (Con) and CAA-MDA-MB-231 (CC).

Discussion
The complex reciprocal communication between breast cancer cells and mature breast adipocytes has pro-tumorigenic consequences (2,20). However, the cellular processes causing CAA-related breast cancer pathogenesis are not well understood. Thus, studies investigating proteomic and phenotypic changes in breast cancer cells are clinically important. Previously, the differential regulation of rationally targeted proteins has been investigated following co-culture with CAA (14,17,18). In this study, we identified global protein abundance and associated pathways that are differentially regulated in two well-characterised breast cancer cell lines, representing key breast cancer molecular subtypes (hormone receptor positive, MCF7; triple negative, MDA-MB-231), following 3-day transwell co-culture with human breast adipocytes.
In this study, differential protein abundance of 1.5-fold or greater was quantified for a relatively small number of the proteins identified in both MCF-7 (n=85/1126) and MDA-MB-231 (n=63/1218) breast cancer cell lines. The oestrogen and progesterone responsive MCF-7 cells, categorised as having luminal A phenotype (ER+, PR+ and HER2–) (24), showed predominant down-regulation of highly differentially regulated proteins (n=53/85; 62%). In contrast, MDA-MB-231 hormone receptor negative (ER–, PR– and HER2–) cells, which are categorized as mesenchymal stem-like or basal B (24), mainly demonstrated up-regulation of highly differentially regulated proteins (n=51/63; 81%). Phosphoglycerate kinase-1 (PGK1) was the only protein to be similarly differentially regulated by more than 1.5-fold in both cell lines after adipocyte co-culture (i.e. up-regulated in both cell lines), highlighting a potential common mechanism by which CAA may promote breast cancer cell migration, invasion and resistance to therapy.
PGK1 is overexpressed in a variety of cancers (32-37), and high PGK1 protein expression in tumours is associated with increased metastatic dissemination (38,39), a multidrug, resistant phenotype (36,40,41) and poor patient survival outcomes (35-37). In breast cancer, PGK1 mRNA and protein are more highly expressed in breast tumour than in normal breast tissue (36), and elevated tumour PGK1 protein expression is associated with high histologic grade, positive ER, HER-2 and p53 status, and poor overall survival in patients treated with paclitaxel (36). PGK1 protein is highly expressed in HER-2/neu-positive tumours and breast cancer cell lines (42), but is not significantly associated with any particular breast cancer molecular subtype (36).
PGK1 catalyzes the reversible conversion of 1,3-diphosphoglycerate to 3-phosphoglycerate, and is the first ATP generating enzyme in the glycolytic pathway (43). Recent evidence shows that, in addition to its function as a glycolytic enzyme, PGK1 has a number of other mechanistically distinct functions that are determined by post-translational modification and subcellular localisation (44-47). For example, PGK1 can function as a protein kinase to reduce mitochondrial pyruvate metabolism and initiate cellular stress-induced autophagy. Therefore, PGK1 has the potential to co-ordinate aerobic glycolysis, mitochondrial metabolism and autophagy, to maintain cellular homeostasis and promote proliferation during tumourigenesis (45,47). The current study did not address the metabolic phenotypes of breast cancer cells after transwell co-culture with CAA. Therefore, future studies are required to better understand the subcellular localisation and function of PGK1 in breast cancer cells exposed to CAA, and its role in co-ordinating metabolic pathways with other cellular processes critical to breast cancer cell survival, growth and metastasis.
Three proteins identified by mass spectrometry (NDRG1, PGK1 and TFF1) were selected for validation by western analysis. These three candidate proteins were chosen because they appeared in the top 20 most differentially regulated proteins for either MCF-7 (Table I) or MDA-MB-231 (Table II) cells. In addition, expression of all three candidates has been associated with poor breast cancer outcomes and, in some cases, increased resistance to anti-tumour therapeutics (36,48,49); underlining the clinical relevance of the current findings. Western analysis validated the iTRAQ results for all but NDRG1 protein abundance in MCF-7 cells. The potential for iTRAQ measurements to underestimate fold change may explain the smaller fold change values reported here by iTRAQ compared to western analysis, particularly for NDRG1 expression in MCF-7 cells (50). Since the aim of the current study was to obtain proteomic profiles from two disparate breast cancer cell lines after exposure to CAA for lead generation purposes, the subsequent application of additional independent methods to confirm protein fold change values is required as the next step in any further investigation of candidate protein(s).
Pathway analysis showed that transwell co-culture with CAA induced both MCF7 and MDA-MB-231 breast cancer cell lines to up-regulate a number of proteins involved in glycolysis and the tricarboxylic acid (TCA) cycle (Figure 4). CAA-MCF-7 cells predominantly up-regulated proteins involved in the tricarboxylic acid (TCA) cycle (Table III), whereas CAA-MDA-MB-231 cells primarily up-regulated proteins involved in glycolysis (Table IV). In line with a metabolic shift towards enhanced glycolysis, CAA-MDA-MB-231 cells also showed up-regulated abundance of lactate dehydrogenase subunits LDHA and LDHB, and monocarboxylate transporter 4 (MCT4) (see supplementary material), which are involved in the interconversion of pyruvate and lactate (51,52), and transport of cellular lactate (53), in and from breast tumour cells, respectively.
Figure 4. MCF-7 and MDA-MB-231 breast cancer cells up-regulate enzymes of glycolysis and TCA cycle after co-culture with CAA. iTRAQ LC-MS/MS protein abundance data and bioinformatic analysis indicates an enriched up-regulation of TCA cycle and glycolysis pathway proteins in MCF-7 and MDA-MB-231 breast cancer cells after co-culture with CAA. Up and down arrows represent enzymes with abundance changes greater than 10%, and the direction of regulation (increased or decreased) compared to control breast cancer cells. White circles show enzymes that were detected, but not differentially regulated following co-culture. White circles with black lines are enzymes undetected in this study. *Complex comprising three subunits: OGDH, DLST and DLD, respectively. **Complex comprising four subunits: SDHAF2, SDHB, SDHC and SDHD, respectively. Complex subunits are gene names as reported by UniProtKB.
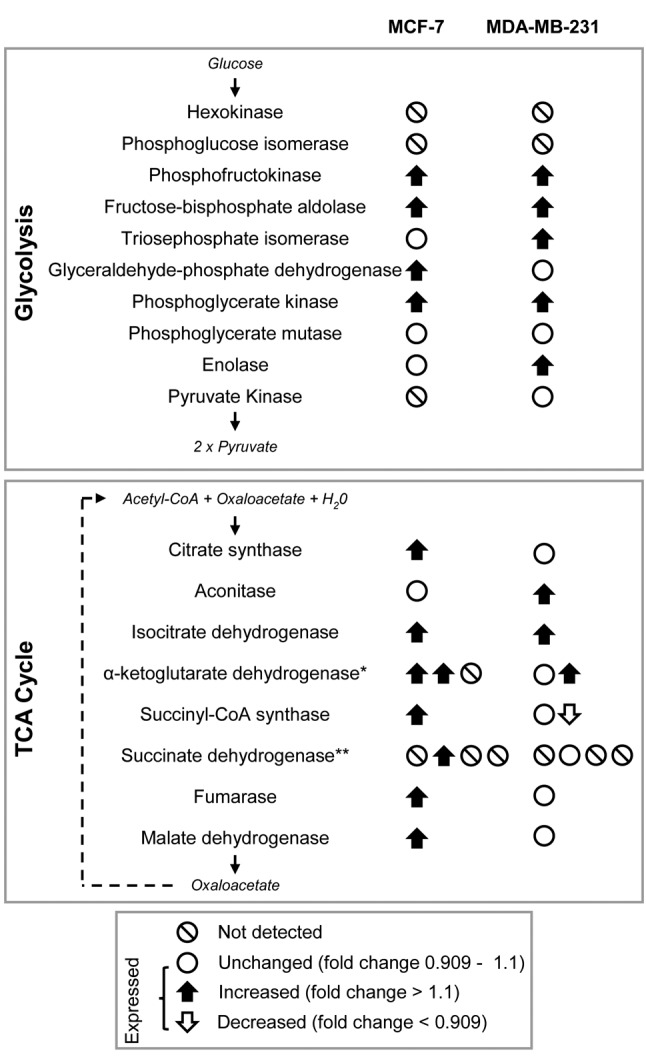
The ‘Warburg effect’ describes the metabolic shift to aerobic glycolysis cancer cells undergo to fulfil their energetic requirements, even under conditions of adequate oxygen supply (54), and triple negative breast cancer cells and tumours are characterised by elevated glycolysis (55). More recently, the two-compartment energy model has been proposed to explain the unique metabolic relationships occurring between cancer cells and stromal cells within the tumour microenvironment. According to this model, cancer cells act as metabolic parasites that stimulate catabolic pathways in proximal stromal cells to gain an energetic advantage that fuels cancer cell metabolism and tumour progression (56). Previous studies have shown that ovarian (57), prostate (58) and breast cancer (17,19) cells induce lipolysis in stromal adipocytes, and that the resulting metabolites (glycerol, free fatty acids) are secreted and transferred to cancer cells where they induce proliferation, migration and invasion. Furthermore, these studies showed that adipocyte co-culture enhances breast cancer cell progression by stimulating fatty acid uptake, storage and mitochondrial fatty acid oxidation in both MCF7 and MDA-MB-231 breast cancer cells (17,19). Carnitine palmitoyltransferase 1 (CPT1) is the rate-limiting enzyme in mitochondrial fatty acid oxidation (59), and basal levels of CPT1 and fatty acid oxidation are higher in MCF-7 than MDA-MB-231 breast cancer cells (17). In the current study, global proteome analysis identified an up-regulation of CPT1A in both CAA-MCF-7 and CAA-MDA-MB-231 breast cancer cells (see supplementary material). Together with the observed enrichment for increased expression of TCA cycle proteins in CAA-MCF-7 breast cancer cells, these results suggest that mitochondrial fatty acid oxidation is an important source of metabolites for the TCA cycle in MCF-7 cells with an adipocyte rich environment.
Differences in metabolic responses in CAA may be driven in part by oncogenic mutations characteristic to each breast cancer cell line (60). For example, MCF-7 cells are PIK3CA mutant but carry a wildtype TP53 gene, whereas the more invasive MDA-MB-231 cells have a mutant TP53 gene but are wildtype for PIK3CA (60). Both of these genes are recognized as prognostic markers in breast cancer (61,62), and may also influence the unique protein expression profiles exhibited by MCF-7 and MDA-MB-231 breast cancer cells following co-culture with CAA. It has been shown that by inhibiting PDH kinase 2 (PDK2), functional p53 up-regulates the activity of pyruvate dehydrogenase (PDH) (63). This promotes the conversion of pyruvate to acetyl-CoA, which subsequently enters the TCA cycle, and may contribute to the up-regulation of TCA cycle proteins in MCF-7 cells during co-culture.
In addition to metabolic pathways, this study identified down-regulation of ubiquitin proteasome pathway proteins in both MCF-7 and MDA-MB-231 breast cancer cells, up-regulation of proteins involved in de novo purine biosynthesis in MDA-MB-231, and down-regulation of a component of the eukaryotic translation initiation factor 3 complex (EIF3F) in both cell types. These adipocyte co-culture induced changes may represent shifts in protein degradation and DNA synthesis that aid in promoting breast cancer cell proliferation and migration. Taken together, these data demonstrate the complex nature of breast cancer cell proteomic heterogeneity and the interplay between breast cancer cells and stromal CAA.
This study used a 2D transwell co-culture model that physically separates CAA and breast cancer cells, yet allows reciprocal cross-talk between the two cell populations via secreted factors (2,8,14,17,18). A major strength of the current study was that it used adipocytes isolated and differentiated from human mammary adipose tissue, whereas most previous studies have used murine pre-adipocyte cell lines or human abdominal adipose tissue pre-adipocytes (2,8,14,17). Breast tumours in vivo have potential for direct CAA and breast cancer cell contact (2), and previous studies using 3D collagen gel matrixes have all observed similar but more pronounced effects of CAA on breast cancer cell growth, migration and invasion than those observed using 2D models (9,12,64). Furthermore, this study measured a snapshot of protein expression in a population of breast cancer cells after a three-day adipocyte co-culture, whereas the expression of proteins in vivo is likely to be dynamic. Thus, further studies are required to better understand how CAA affect breast cancer cell phenotype and function by altering abundance of proteins and their associated pathways over time.
Conclusion
This study identified a number of relative protein abundance changes in breast cancer cells co-cultured with human mature breast adipocytes in an in vitro transwell system. Hormone receptor-positive (MCF7) and triple-negative (MDA-MB-231) breast cancer cells showed predominant down- and up-regulation of highly differentially regulated proteins, respectively, supporting the concept that reciprocal communication between breast cancer cells and CAA is heterogeneous, and likely breast cancer cell type specific. Analysis showed that the regulated molecules participate in pathways related to metabolism, protein ubiquitination and purine synthesis, and identified PGK1 as the only commonly up-regulated protein in both cells. Overall, this investigation aids in better understanding the molecular mechanisms by which CAA regulate breast cancer cell phenotype and function, and provides a platform for future research to identify novel protein targets involved in breast cancer cell-CAA crosstalk.
Conflicts of Interest
The Authors declare that they have no conflict of interest regarding this study.
Authors’ Contributions
R.C.: Designed and performed experimental work, collected and analysed data, and was the primary author of the manuscript; E.P.: Contributed substantially to the conception and design of the study and the analysis and interpretation of data, supervised research, and provided critical revision of the manuscript; T.K.: Contributed substantially to design, collection, analysis and interpretation of mass spectrometry data, and provided critical revision of the manuscript; M.C.: Contributed substantially to the conception and design of the study and interpretation of data, supervised research, and co-wrote the manuscript.
Supplementary Material
Lists of proteins that were identified in MCF-7 and MDA-MB-231 breast cancer cells co-cultured with cancer associated adipocytes (CAA), measured by iTRAQ LC-MS/MS. Lists include fold change values that signify the difference in protein abundance between co-culture and control. Available at: https://www.otago.ac.nz/mackenzie-cancer/research/otago715163.html
Acknowledgements
The Authors would like to thank the Cancer Society Tissue Bank Christchurch (Curator Helen Morrin) for collecting and providing human breast adipose tissue samples.
Funding: This work was supported by The Mackenzie Charitable Foundation, The Cancer Society of New Zealand (CSNZ) (CSNZ 17.06) and the Canterbury Medical Research Foundation (CMRF) (PRO2016-001).
References
- 1.Rajala MW, Scherer PE. Minireview: The Adipocyte—At the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–3773. doi: 10.1210/en.2003-0580. PMID: 12933646. DOI: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- 2.Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, Wang YY, Meulle A, Salles B, Le Gonidec S, Garrido I, Escourrou G, Valet P, Muller C. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011;71:2455–2465. doi: 10.1158/0008-5472.CAN-10-3323. PMID: 21459803. DOI: 10.1158/0008-5472.CAN-10-3323. [DOI] [PubMed] [Google Scholar]
- 3.Walter M, Liang S, Ghosh S, Hornsby PJ, Li R. Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene. 2009;28:2745–2755. doi: 10.1038/onc.2009.130. PMID: 19483720. DOI: 10.1038/onc.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strong AL, Ohlstein JF, Biagas BA, Rhodes LV, Pei DT, Tucker HA, Llamas C, Bowles AC, Dutreil MF, Zhang S, Gimble JM, Burow ME, Bunnell BA. Leptin produced by obese adipose stromal/stem cells enhances proliferation and metastasis of estrogen receptor positive breast cancers. Breast Cancer Res. 2015;17:112. doi: 10.1186/s13058-015-0622-z. PMID: 26286584. DOI: 10.1186/s13058-015-0622-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iyengar P, Combs TP, Shah SJ, Gouon-Evans V, Pollard JW, Albanese C, Flanagan L, Tenniswood MP, Guha C, Lisanti MP, Pestell RG, Scherer PE. Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene. 2003;22:6408–6423. doi: 10.1038/sj.onc.1206737. PMID: 14508521. DOI: 10.1038/sj.onc.1206737. [DOI] [PubMed] [Google Scholar]
- 6.Carter JC, Church FC. Mature breast adipocytes promote breast cancer cell motility. Exp Mol Pathol. 2012;92:312–317. doi: 10.1016/j.yexmp.2012.03.005. PMID: 22445926. DOI: 10.1016/j.yexmp.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Grisouard J, Dembinski K, Mayer D, Keller U, Müller B, Christ-Crain M. Targeting AMP-activated protein kinase in adipocytes to modulate obesity-related adipokine production associated with insulin resistance and breast cancer cell proliferation. Diabetol Metab Syndr. 2011;3:16. doi: 10.1186/1758-5996-3-16. PMID: 21774820. DOI: 10.1186/1758-5996-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Esposito V, Passaretti F, Hammarstedt A, Liguoro D, Terracciano D, Molea G, Canta L, Miele C, Smith U, Beguinot F, Formisano P, Smith U, Canta L. Adipocyte-released insulin-like growth factor-1 is regulated by glucose and fatty acids and controls breast cancer cell growth in vitro. Diabetologia. 2012;55:2811–2822. doi: 10.1007/s00125-012-2629-7. PMID: 22798065. DOI: 10.1007/s00125-012-2629-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, Dray C, Guiet R, Maridonneau-Parini I, Le Gonidec S. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013;73:5657–5668. doi: 10.1158/0008-5472.CAN-13-0530. PMID: 23903958. DOI: 10.1158/00 08-5472.CAN-13-0530. [DOI] [PubMed] [Google Scholar]
- 10.Lapeire L, Hendrix A, Lambein K, Van Bockstal M, Braems G, Van Den Broecke R, Limame R, Mestdagh P, Vandesompele J, Vanhove C, Maynard D, Lehuédé C, Muller C, Valet P, Gespach CP, Bracke M, Cocquyt V, Denys H, De Wever O. Cancer-associated adipose tissue promotes breast cancer progression by paracrine oncostatin M and Jak/STAT3 signaling. Cancer Res. 2014;74:6806–6819. doi: 10.1158/0008-5472.CAN-14-0160. PMID: 25252914. DOI: 10.1158/0008-5472.CAN-14-0160. [DOI] [PubMed] [Google Scholar]
- 11.Drew BG, Hamidi H, Zhou Z, Villanueva CJ, Krum SA, Calkin AC, Parks BW, Ribas V, Kalajian NY, Phun J, Daraei P, Christofk HR, Hewitt SC, Korach KS, Tontonoz P, Lusis AJ, Slamon DJ, Hurvitz SA, Hevener AL. Estrogen receptor (ER)α-regulated lipocalin 2 expression in adipose tissue links obesity with breast cancer progression. J Biol Chem. 2015;290:5566–5581. doi: 10.1074/jbc.M114.606459. PMID: 25468909. DOI: 10.1074/jbc.M114.606459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujisaki K, Fujimoto H, Sangai T, Nagashima T, Sakakibara M, Shiina N, Kuroda M, Aoyagi Y, Miyazaki M. Cancer-mediated adipose reversion promotes cancer cell migration via IL-6 and MCP-1. Breast Cancer Res Treat. 2015;150:255–263. doi: 10.1007/s10549-015-3318-2. PMID: 25721605. DOI: 10.1007/s10549-015-3318-2. [DOI] [PubMed] [Google Scholar]
- 13.Sakurai M, Miki Y, Takagi K, Suzuki T, Ishida T, Ohuchi N, Sasano H. Interaction with adipocyte stromal cells induces breast cancer malignancy via S100A7 upregulation in breast cancer microenvironment. Breast Cancer Res. 2017;19:70. doi: 10.1186/s13058-017-0863-0. PMID: 28629450. DOI: 10.1186/s13058-017-0863-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C, Gao C, Meng K, Qiao H, Wang Y. Human adipocytes stimulate invasion of breast cancer MCF-7 cells by secreting IGFBP-2. PLoS One. 2015;10:e0119348. doi: 10.1371/journal.pone.0119348. PMID: 25747684. DOI: 10.1371/journal.pone.0119348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Esposito V, Liguoro D, Ambrosio MR, Collina F, Cantile M, Spinelli R, Raciti GA, Miele C, Valentino R, Campiglia P, De Laurentiis M, Di Bonito M, Botti G, Franco R, Beguinot F, Formisano P, D’Esposito V, Liguoro D, Ambrosio MR, Collina F, Cantile M, Spinelli R, Raciti GA, Miele C, Valentino R, Campiglia P, De Laurentiis M, Di Bonito M, Botti G, Franco R, Beguinot F, Formisano P. Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget. 2016;7:24495–24509. doi: 10.18632/oncotarget.8336. PMID: 27027351. DOI: 10.18632/oncotarget.8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Picon-Ruiz M, Pan C, Drews-Elger K, Jang K, Besser AH, Zhao D, Morata-Tarifa C, Kim M, Ince TA, Azzam DJ, Wander SA, Wang B, Ergonul B, Datar RH, Cote RJ, Howard GA, El-Ashry D, Torn e-Poyatos P, Marchal JA, Slingerland JM. Tumor and stem cell biology interactions between adipocytes and breast cancer cells stimulate cytokine production and drive src/sox2/mir-302b–mediated malignant progression. Cancer Res. 2016;76:491–504. doi: 10.1158/0008-5472.CAN-15-0927. PMID: 26744520. DOI: 10.1158/0008-5472.CAN-15-0927. [DOI] [PubMed] [Google Scholar]
- 17.Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, Cairns R, Thomas KC, Fazakerley DJ, Grewal T, Holst J, Saunders DN, Hoy AJ. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017;5 doi: 10.1186/s40170-016-0163-7. PMID: 28101337. DOI: 10.1186/s40170-016-0163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang CK, Chang PH, Kuo WH, Chen CL, Jeng YM, Chang KJ, Shew JY, Hu CM, Lee WH. Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via β-hydroxybutyrate. Nat Commun. 2017;8:14706. doi: 10.1038/ncomms14706. PMID: 28281525. DOI: 10.1038/ncomms14706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang YY, Attané C, Milhas D, Dirat B, Dauvillier S, Guerard A, Gilhodes J, Lazar I, Alet N, Laurent V, Le Gonidec S, Biard D, Hervé C, Bost F, Ren GS, Bono F, Escourrou G, Prentki M, Nieto L, Valet P, Muller C. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI insight. 2017;2:e87489. doi: 10.1172/jci.insight.87489. PMID: 28239646. DOI: 10.1172/jci.insight.87489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bochet L, Meulle A, Imbert S, Salles B, Valet P, Muller C. Cancer-associated adipocytes promotes breast tumor radioresistance. Biochem Biophys Res Commun. 2011;411:102–106. doi: 10.1016/j.bbrc.2011.06.101. PMID: 21712027. DOI: 10.1016/j.bbrc.2011.06.101. [DOI] [PubMed] [Google Scholar]
- 21.De Angel RE, Blando JM, Hogan MG, Sandoval MA, Lansakara-P DS, Dunlap SM, Hursting SD, Cui Z. Stearoyl gemcitabine nanoparticles overcome obesity-induced cancer cell resistance to gemcitabine in a mouse postmenopausal breast cancer model. Cancer Biol Ther. 2013;14:357–364. doi: 10.4161/cbt.23623. PMID: 23358472. DOI: 10.4161/cbt.23623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duong MN, Cleret A, Matera E-L, Chettab K, Mathé D, Valsesia-Wittmann S, Clémenceau B, Dumontet C. Adipose cells promote resistance of breast cancer cells to trastuzumab-mediated antibody-dependent cellular cytotoxicity. Breast Cancer Res. 2015;17:57. doi: 10.1186/s13058-015-0569-0. PMID: 25908175. DOI: 10.1186/s13058-015-0569-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rowan BG, Gimble JM, Sheng M, Anbalagan M, Jones RK, Frazier TP, Asher M, Lacayo EA, Friedlander PL, Kutner R, Chiu ES. Human adipose tissue-derived stromal/stem cells promote migration and early metastasis of triple negative breast cancer xenografts. PLoS One. 2014;9:e89595. doi: 10.1371/journal.pone.0089595. PMID: 24586900. DOI: 10.1371/journal.pone.0089595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe J-P, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo W-L, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. PMID: 17157791. DOI: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrin H, Gunningham S, Currie M, Dachs G, Fox S, Robinson B. The Christchurch Tissue Bank to support cancer research. N Z Med J. 2005;118:U1735. PMID: 16286947. [PubMed] [Google Scholar]
- 26.Lee M-J, Wu Y, Fried SK. A modified protocol to maximize differentiation of human preadipocytes and improve metabolic phenotypes. Obesity. 2012;20:2334–2340. doi: 10.1038/oby.2012.116. PMID: 22627913. DOI: 10.1038/oby.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. PMID: 19377485. DOI: 10.1038/NMETH. 1322. [DOI] [PubMed] [Google Scholar]
- 28.Consortium GO. Gene ontology annotations and resources. Nucleic Acids Res. 2013;41:D530–D535. doi: 10.1093/nar/gks1050. PMID: 23161678. DOI: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–86. doi: 10.1093/nar/gks1118. PMID: 23193289. DOI: 10.1093/nar/ gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. PMID: 23868073. DOI: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mi H, Thomas P. PANTHER Pathway: An ontology-based pathway database coupled with data analysis tools. Methods Mol Biol. 2009;563 doi: 10.1007/978-1-60761-175-2_7. PMID: 19597783. DOI: 10.1007/978-1-60761-175-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang TL, Liang Y, Chien KY, Yu JS. Overexpression and elevated serum levels of phosphoglycerate kinase 1 in pancreatic ductal adenocarcinoma. Proteomics. 2006;6:2259–2272. doi: 10.1002/pmic.200500345. PMID: 16493704. DOI: 10.1002/pmic.200500345. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Ying G, Wang J, Jung Y, Lu J, Zhu J, Pienta KJ, Taichman RS. Characterization of phosphoglycerate kinase-1 expression of stromal cells derived from tumor micro-environment in prostate cancer progression. Cancer Res. 2010;70:471–480. doi: 10.1158/0008-5472.CAN-09-2863. PMID: 20068185. DOI: 10.1158/0008-5472.CAN-09-2863. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Yan H, Yang K, Xiao H, Zou YJ, Zhang W Bin, Liu HY. Over-expression of cofilin-1 and phosphoglycerate kinase 1 in astrocytomas involved in pathogenesis of radioresistance. CNS Neurosci Ther. 2012;18:729–736. doi: 10.1111/j.1755-5949.2012.00353.x. PMID: 22742733. DOI: 10.1111/j.1755-5949.2012.00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmad SS, Glatzle J, Bajaeifer K, Bühler S, Lehmann T, Königsrainer I, Vollmer JP, Sipos B, Ahmad SS, Northoff H, Königsrainerand A, Zieker D. Phosphoglycerate kinase 1 as a promoter of metastasis in colon cancer. Int J Oncol. 2013;43:586–590. doi: 10.3892/ijo.2013.1971. PMID: 23727790. DOI: 10.3892/ijo.2013.1971. [DOI] [PubMed] [Google Scholar]
- 36.Sun S, Liang X, Zhang X, Liu T, Shi Q, Song Y, Jiang Y, Wu H, Jiang Y, Lu X, Pang D. Phosphoglycerate kinase-1 is a predictor of poor survival and a novel prognostic biomarker of chemoresistance to paclitaxel treatment in breast cancer. Br J Cancer. 2015;112:1332–1339. doi: 10.1038/bjc.2015.114. PMID: 25867275. DOI: 10.1038/ bjc.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie H, Tong G, Zhang Y, Liang S, Tang K, Yang Q. PGK1 Drives hepatocellular carcinoma metastasis by enhancing metabolic process. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18081630. PMID: 28749413. DOI: 10.3390/ijms18081630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zieker D, Königsrainer I, Tritschler I, Löffler M, Beckert S, Traub F, Nieselt K, Bühler S, Weller M, Gaedcke J, Taichman RS, Northoff H, Brücher BLDM, Königsrainer A. Phosphoglycerate kinase 1 a promoting enzyme for peritoneal dissemination in gastric cancer. Int J Cancer. 2010;126:1513–1520. doi: 10.1002/ijc.24835. PMID: 19688824. DOI: 10.1002/ijc.24835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ameis HM, Drenckhan A, Von Loga K, Escherich G, Wenke K, Izbicki JR, Reinshagen K, Gros SJ. PGK1 as predictor of CXCR4 expression, bone marrow metastases and survival in neuroblastoma. PLoS One. 2013;8 doi: 10.1371/journal.pone.0083701. PMID: 24376734. DOI: 10.1371/journal.pone.0083701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duan Z, Lamendola DE, Yusuf RZ, Penson RT, Preffer FI, Seiden MV. Overexpression of human phosphoglycerate kinase 1 (PKG1) induces a multidrug resistance phenotype. Anticancer Res. 2002;22:1933–1941. PMID: 12174867. [PubMed] [Google Scholar]
- 41.Schneider CC, Archid R, Fischer N, Bühler S, Venturelli S, Berger A, Burkard M, Kirschniak A, Bachmann R, Königsrainer A, Glatzle J, Zieker D. Metabolic alteration – Overcoming therapy resistance in gastric cancer via PGK-1 inhibition in a combined therapy with standard chemotherapeutics. Int J Surg. 2015;22:92–98. doi: 10.1016/j.ijsu.2015.08.020. PMID: 26298000. DOI: 10.1016/J.IJSU.2015. 08.020. [DOI] [PubMed] [Google Scholar]
- 42.Zhang D, Tai LK, Wong LL, Chiu L-L, Sethi SK, Koay ESC. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Mol Cell Proteomics. 2005;4:1686–1696. doi: 10.1074/mcp.M400221-MCP200. PMID: 16048908. DOI: 10.1074/mcp.M400221-MCP200. [DOI] [PubMed] [Google Scholar]
- 43.Bernstein BE, Hol WGJ. Crystal structures of substrates and products bound to the phosphoglycerate kinase active site reveal the catalytic mechanism. Biochemistry. 1998;37:4429–4436. doi: 10.1021/bi9724117. PMID: 9521762. DOI: 10.1021/bi9724117. [DOI] [PubMed] [Google Scholar]
- 44.Lay AJ, Jiang XM, Kisker O, Flynn E, Underwood A, Condron R, Hogg PJ. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature. 2000;408:869–873. doi: 10.1038/35048596. PMID: 11130727. DOI: 10.1038/35048596. [DOI] [PubMed] [Google Scholar]
- 45.Li X, Jiang Y, Meisenhelder J, Yang W, Hawke DH, Zheng Y, Xia Y, Aldape K, He J, Hunter T, Wang L, Lu Z. Mitochondria-Translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol Cell. 2016;61 doi: 10.1016/j.molcel.2016.02.009. PMID: 26942675. DOI: 10.1016/j.molcel. 2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qian X, Li X, Lu Z. Protein kinase activity of the glycolytic enzyme PGK1 regulates autophagy to promote tumorigenesis. Autophagy. 2017;13:1246–1247. doi: 10.1080/15548627.2017.1313945. PMID: 28486006. DOI: 10.1080/15548627.2017.1313945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu Z, Hunter T. Metabolic kinases moonlighting as protein kinases. Trends Biochem Sci. 2018;43:301–310. doi: 10.1016/j.tibs.2018.01.006. PMID: 29463470. DOI: 10.1016/J.TIBS.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Z, Liu Q, Chen Q, Zhu R, Zhu H. Overexpression of NDRG1: relationship with proliferative activity and invasiveness of breast cancer cell line and breast cancer metastasis. Zhonghua Bing Li Xue Za Zhi. 2006;35:333–338. PMID: 16834906. [PubMed] [Google Scholar]
- 49.Pelden S, Insawang T, Thuwajit C, Thuwajit P. The trefoil factor 1 (TFF1) protein involved in doxorubicin-induced apoptosis resistance is upregulated by estrogen in breast cancer cells. Oncol Rep. 2013;30:1518–1526. doi: 10.3892/or.2013.2593. PMID: 23836323. DOI: 10.3892/or.2013.2593. [DOI] [PubMed] [Google Scholar]
- 50.Evans C, Noirel J, Ow SY, Salim M, Pereira-Medrano AG, Couto N, Pandhal J, Smith D, Pham TK, Karunakaran E, Zou X, Biggs CA, Wright PC. An insight into iTRAQ: where do we stand now. Anal Bioanal Chem. 2012;404:1011–1027. doi: 10.1007/s00216-012-5918-6. PMID: 22451173. DOI: 10.1007/s00216-012-5918-6. [DOI] [PubMed] [Google Scholar]
- 51.Fantin V, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. PMID: 16766262. DOI: 10.1016/J.CCR.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 52.Mack N, Mazzio EA, Bauer D, Flores-Rozas H, Soliman KFA. Stable shRNA silencing of lactate dehydrogenase a (LDHA) in human MDA-MB-231 breast cancer cells fails to alter lactic acid production, glycolytic activity, ATP or survival. Anticancer Res. 2017;37:1205–1212. doi: 10.21873/anticanres.11435. PMID: 28314283. DOI: 10.21873/anticanres.11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baenke F, Dubuis S, Brault C, Weigelt B, Dankworth B, Griffiths B, Jiang M, Mackay A, Saunders B, Spencer-Dene B, Ros S, Stamp G, Reis-Filho JS, Howell M, Zamboni N, Schulze A. Functional screening identifies MCT4 as a key regulator of breast cancer cell metabolism and survival. J Pathol. 2015;237:152–165. doi: 10.1002/path.4562. PMID: 25965974. DOI: 10.1002/path.4562. [DOI] [PubMed] [Google Scholar]
- 54.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. PMID: 21508971. DOI: 10.1038/ nrc3038. [DOI] [PubMed] [Google Scholar]
- 55.Lanning NJ, Castle JP, Singh SJ, Leon AN, Tovar EA, Sanghera A, MacKeigan JP, Filipp FV, Graveel CR. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017;5:6. doi: 10.1186/s40170-017-0168-x. PMID: 28852500. DOI: 10.1186/s40170-017-0168-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, Tanowitz HB, Sotgia F, Lisanti MP. Energy transfer in “parasitic” cancer metabolism Mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle. 2011;10:4208–4216. doi: 10.4161/cc.10.24.18487. PMID: 22033146. DOI: 10.4161/cc.10.24.18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nieman KM, Kenny HA, Penicka C V, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, Lengyel E. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–1503. doi: 10.1038/nm.2492. PMID: 22037646. DOI: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Diedrich JD, Rajagurubandara E, Herroon MK, Mahapatra G, Hüttemann M, Podgorski I, Diedrich JD, Rajagurubandara E, Herroon MK, Mahapatra G, Hüttemann M, Podgorski I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF-1a activation. Oncotarget. 2016;7:64854–64877. doi: 10.18632/oncotarget.11712. PMID: 27588494. DOI: 10.18632/oncotarget.11712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bruce CR, Hoy AJ, Turner N, Watt MJ, Allen TL, Carpenter K, Cooney GJ, Febbraio MA, Kraegen EW. Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat diet-induced insulin resistance. Diabetes. 2009;58:550–558. doi: 10.2337/db08-1078. PMID: 19073774. DOI: 10.2337/db08-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kao J, Salari K, Bocanegra M, Choi Y-L, Girard L, Gandhi J, Kwei K a, Hernandez-Boussard T, Wang P, Gazdar AF, Minna JD, Pollack JR. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One. 2009;4:e6146. doi: 10.1371/journal.pone.0006146. PMID: 19582160. DOI: 10.1371/journal.pone.0006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gasco M, Shami S, Crook T. The p53 pathway in breast cancer. Breast Cancer Res. 2002;4:70–76. doi: 10.1186/bcr426. PMID: 11879567. DOI: 10.1186/bcr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mukohara T. PI3K mutations in breast cancer: prognostic and therapeutic implications. Breast Cancer Targets Ther. 2015;111 doi: 10.2147/BCTT.S60696. PMID: 26028978. DOI: 10.2147/BCTT.S60696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012;72:560–567. doi: 10.1158/0008-5472.CAN-11-1215. PMID: 22123926. DOI: 10.1158/0008-5472.CAN-11-1215. [DOI] [PubMed] [Google Scholar]
- 64.Manabe Y, Toda S, Miyazaki K, Sugihara H. Mature adipocytes, but not preadipocytes, promote the growth of breast carcinoma cells in collagen gel matrix culture through cancer-stromal cell interactions. J Pathol. 2003;201:221–228. doi: 10.1002/path.1430. PMID: 14517839. DOI: 10.1002/path.1430. [DOI] [PubMed] [Google Scholar]