

Abstract
Myeloperoxidase (MPO) is a pro-inflammatory enzyme abundantly secreted by activated myeloid cells after stroke. We show that when MPO activity is either blocked by the specific inhibitor 4-aminobenzoic acid hydrazide (ABAH) in wildtype (WT) mice or congenitally absent (MPO−/−), there was decreased cell loss, including degenerating neurons and oligodendrocytes, in the ischemic brains compared to vehicle-treated WT mice after stroke. MPO inhibition also reduced the number of activated myeloid cells after ischemia. MPO inhibition increased cytoprotective heat shock protein 70 (Hsp70) by 70% and p-Akt by 60%, while decreased the apoptotic marker p53 level by 62%, compared to vehicle-treated mice after ischemia. Similarly, MPO inhibition increased the number of Hsp70+/NeuN+ cells after stroke by 60%. Notably, MPO inhibition significantly improved neurological outcome compared with the vehicle-treated group after stroke. We further found longer treatment periods resulted in larger reduction of infarct size and greater neurobehavioral improvement from MPO inhibition, even when given days after stroke. Therefore, MPO inhibition with ABAH or MPO deficiency creates a protective environment that decreased inflammatory cell recruitment and increased expression of survival factors to improve functional outcome. MPO inhibition may represent a promising therapeutic target for stroke therapy, possibly even days after stroke has occurred.
Keywords: Inflammation, ischemia, myeloperoxidase, neuroprotection, stroke
Introduction
Stroke is an acute cerebrovascular disease that remains the fourth leading cause of mortality and permanent disability affecting 800,000 individuals each year with many survivors experiencing persistent difficulty with daily tasks in the United States.1,2 Ischemic stroke mainly occurs due to interruption of blood flow caused by thrombosis or embolism.3,4 Currently, recombinant tissue-plasminogen activator (t-PA) is the only FDA-approved agent for clinical use in acute stroke, but has a limited therapeutic window and carries a risk of hemorrhage.5,6 However, previous neuroprotective agents aimed at reducing or preventing neuronal damage and neurobehavioral dysfunction have not been successful in clinical trials.7 Thus, new and effective approaches for stroke therapy are urgently needed.
Myeloperoxidase (MPO) is an inflammatory and oxidative enzyme secreted by activated polymorphonuclear neutrophils (PMN), monocytes, microglia, and macrophage. Elevated MPO activity had been found to increase infarct size.8 MPO forms highly reactive species, including hypochlorous acid (HOCl), tyrosyl radicals, and aldehydes that can damage tissue and increase pro-inflammatory cytokine generation.9,10 MPO-H2O2-chloride system also promotes endothelial cell dysfunction, inducible nitric oxide synthase (iNOS) production, and lipid peroxidation that can exacerbate inflammation.9,11,12 MPO consumes endothelial-derived NO, thereby attenuating NO bioavailability and impairing its vasodilating and anti-inflammatory properties.13 MPO−/− mice exhibit less atrial fibrosis, reduced MMP-9 activity and lowered 3-chlorotyrosine.14 MPO also delays neutrophil apoptosis.15 In human stroke, high MPO levels were correlated with larger infarct volume and MPO has been implicated in cardiovascular diseases.16,17
4-aminobenzoic acid hydrazide (ABAH) is an irreversible specific MPO inhibitor that has been found to improve multiple sclerosis (MS) and stroke in experimental models.8,18 Post-insult ABAH treatment (40 mg/kg, i.p.) or MPO−/− mice markedly reduced infarct size.8 Additionally, ABAH treatment in wildtype (WT) mice or MPO−/− mice stimulated neurogenesis after stroke.19 These findings suggest that MPO may emerge to be a key molecule for stroke therapy. Whether MPO-targeted therapy has cellular neuroprotective effects in stroke has not been investigated. In this study, we show that MPO inhibition exerts cellular neuroprotective effects after experimental stroke by decreasing inflammatory cell recruitment and increasing factors that promote cell survival to improve functional outcome.
Material and methods
Animal care
All animal study experiments were approved by the Massachusetts General Hospital Animal Care and Use Committee in accordance with National Institute of Health guidelines and with the ARRIVE guidelines. Animals were housed in a facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care International. WT C57BL/6J male mice (8–10 weeks, 23–25 g) (n = 166) were obtained from Charles River (Charles River Laboratory, MA, USA) and MPO−/− mice (8–10 weeks, n = 22, 13th generation backcross on the C57BL/6J background) were obtained from Jackson Laboratory (Bar Harbor, ME, USA), and randomly assigned to one of the following three groups: (i) sham-operated control animals (WT: n = 43, MPO−/−: n = 6), (ii) transient middle cerebral artery occlusion (tMCAO)-induced ischemia followed by saline-treatment (WT: n = 53, MPO−/−: n = 8) and (iii) tMCAO-induced ischemia followed by ABAH-treatment (WT: n = 54, MPO−/−: n = 8). Additional C57BL/6J mice were randomly assigned to (iv) tMCAO-induced ischemia followed by acute ABAH treatment (n = 8) and (v) tMCAO-induced ischemia followed by subacute ABAH treatment (n = 8). Food and water were freely accessible to the mice and the holding rooms were maintained on a 12-h light/dark cycle. Mice were sacrificed at 3 days for Western blotting and for neurobehavioral tests up to day 21 days after focal ischemia. Animals were also sacrificed on day 7 or 21 after stroke for immunohistochemistry.
Mouse cerebral ischemia model
Focal cerebral ischemia was induced by transiently occluding the right middle cerebral artery (tMCAO) for 30 min followed by reperfusion in mice as described previously.20 The mice were anesthetized by using 1.5–2.0% isoflurane (Baxter health care corporation, Deerfield, IL, USA). Under a dissecting microscope, a ventral midline neck incision was performed and the right carotid bifurcation was exposed. The common carotid artery (CCA), internal carotid artery (ICA) and external carotid artery (ECA) were carefully isolated from surrounding connective tissues. A small nick was made in the ECA stump and a 7–0 fine surgical nylon monofilament (diameter 0.19–0.20 mm) coated with silicon (Cat # 7019PK5Re, Doccol Co., Sharon, MA, USA) was inserted into the right ICA lumen through the ECA stump and advanced up to the MCA origin at the Circle of Willis.
Regional cerebral blood flow (rCBF) to the middle cerebral artery (MCA) territory was continuously monitored using laser Doppler flowmetry ((LDF), AD instruments, model: ML191, Colorado Springs, CO, USA) at the coordinates of 1 mm posterior and 5 mm right lateral to the bregma, as previously described.21 Ischemia was induced successfully when rCBF decreased to 10–30% of baseline and returned to > 70% of baseline after reperfusion. After 30-min occlusion, the suture was gently withdrawn for reperfusion and then ECA was ligated. During the surgery, mice were maintained at a body temperature of 37℃ by using a heating pad (FHC Bowdoinham, ME, USA). After that, mice were kept in an incubator for recovery for 2 h at 35℃. Sham-operated animals were treated identically except suture was not inserted into the MCA.
Drug administration protocol
C57BL/6J (wild type) mice were intraperitoneally treated with either 400 µl of saline vehicle (controls) or 400 µL of the specific MPO inhibitor 4-aminobenzoic acid hydrazide (ABAH, Sigma-Aldrich, St Louis, MO, USA) at a dose of 40 mg/kg with twice daily up to days 3, 7 or 21 following stroke (Supplementary Figure 1). ABAH is a small molecule with likely a short blood half-life and 40 mg/kg regimen was found empirically to be most clinically effective.18 For acute and subacute treatments, the mice were subjected to two different injections. For the acute treatment group, the mice were given ABAH (40 mg/kg, i.p.) 30 min after tMCAO followed by another injection 8 h later on the first day (day 0) (two doses). The subacute treatment group received daily injections of the same dose of ABAH starting on day 2, continuing to day 21 after tMCAO (i.e. days 2–21). For Western blotting, we administrated ABAH or saline to mice from day 0 to day 3 following tMCAO and mice were euthanized on day 3.
Immunohistochemistry
We performed immunostaining according to previous methods with some modifications.22 Mice were transcardially perfused with phosphate-buffered saline (PBS, pH 7.4). Brain tissues were quickly sinked in dry ice-precooled 2-methyl butane (Fisher Scientific, New Jersey, USA) and stored at −80℃. Coronal brain sections (20 µm) corresponding to bregma 1.18 to −0.10 mm (SVZ) and bregma −1.22 to −2.80 (DG) were cut with a cryostat (Microm International GmbH, Model: HM 505 E, Walldorf, Germany) and stored at −80℃. We collected approximately 36 sections per mouse.
Tissues were fixed with 4% paraformaldehyde (PFA) for 15 min and washed three times for 10 min with PBS 0.1 M (pH 7.4). After that, brain sections were incubated with blocking solutions including 0.1% Triton X-100 and 5% normal goat serum in PBS for 1 h at room temperature. Immunostaining was performed on day 7 or 21 following stroke, as previously described.22 Primary antibodies were incubated overnight at 4℃ and were used as follows: mouse anti-heat shock protein 70 (Hsp70, 1:100, (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), mouse-anti-NeuN (a mature neuronal marker, 1:200, Millipore, Billerica, MA, USA), rat anti-CD11b (1: 200, Bio-Rad Lab, Inc., USA), and mouse anti-myelin basic protein (MBP, 1:300, Covance, MD, USA). Brain sections were washed three times for 10 min with PBST solutions, and then secondary antibodies against the appropriate species were incubated for 2 h in a humidified chamber at room temperature. All secondary antibody combinations were carefully tested to ensure that there was no cross talk between fluorescent dyes or cross-reactivity between secondary antibodies, especially anti-rat and anti-mouse secondary antibodies. The secondary antibodies were Alexa flour 555 and Alexa flour 488 conjugated with goat anti-mouse (1:500) and goat anti-rat (1:3000) (Invitrogen, Carlsbad, CA, USA). Other secondary antibodies were mouse anti-FITC, rat anti-FITC (1:200) and mouse anti-Cy3-conjugated antibody (1:250) (Jackson Immunoresearch, West Grove, PA, USA). Brain sections were rinsed three times for 10 min with PBS, mounted onto super frost slides, and cover slips were applied with Vectashield Fluorescent Mounting Medium (Vector Laboratories, Inc., Burlingame, CA, USA). Control experiments were performed for brain coronal tissue sections as outlined above, but did not use the primary antibodies. We detected fluorescence signals by using a fluorescence microscope (Nikon, Nikon Eclipse 80i, and Japan). Images captured from selective objectives using imaging software and transferred images into the ImageJ software. The number of positive cells in the ipsilateral brains after stroke was counted using the 40 × objective lens.
Fluoro-Jade B staining
Fluoro-Jade (Millipore, Billerica, MA, USA) is one of polyanionic fluorescein derivatives and detects dying neurons. The slides were treated with a solution containing 1% sodium hydroxide in 80% ethanol (20 mL of 5% NaOH added to 80 mL alcohol) for 5 min and coronal brain sections were immersed in 0.06% potassium permanganate (KMnO4) for 10 min. The sections were washed with distilled water and placed in a 0.0004% FJ-B solution made by adding 4 mL of a 0.01% stock solution of FJ-B to 96 mL of 0.1% acetic acid. After 20 min in the staining solution, brain slides were washed and dehydrated using alcohol and xylene. The dry slides were cleared by immersion in xylene before cover slips were applied with DPX (Fluka, Milwaukee WI), a mounting medium. For visualizing FJ-B, which shows excitation peak at 480 nm and emission peak at 525 nm, we used a fluorescein/FITC filter.23 FJ-B-positive cells were imaged with a fluorescence microscope (Nikon, Nikon Eclipse 80i, and Japan) and counted by 40 × objectives in the ipsilateral hippocampus, CA1, striatum and parietal cortex against corresponding region of control mice.
Quantification of immunoreactive cells
Brain tissues were cut into 20 µm-thick coronal sections. We first performed hematoxylin and eosin (H&E) staining. All assessments were done by an experimenter blinded to the group assignment. We selected the sections to image for each mouse by examining the brain anatomy on H&E staining at 2 × compared to a mouse atlas (Supplementary Figure 2). We then progressively zoomed in (10 × , 20 × , and then 40 × magnification) to different areas within the structure of interest up to 40 × . At 40 × , images were captured for analysis. Adjacent sections were used for immunostaining as described above. Six random fields per structure of interest per mouse were captured at 40 × for analysis for each staining. Double immunostaining of Hsp70+/NeuN+ cells was performed on day 7 after stroke. H&E staining was investigated in the ischemic striatum and parietal cortex on day 7 after stroke. We counted immuno-positive cell numbers in the ipsilateral brains including striatum, white matter (WM), hippocampal CA1 and parietal cortex (Pcox) on days 7 and 21 following cerebral ischemia. We quantified MBP loss according to the MBP scoring system24 as follows: 0: no abnormality, 1: minimal loss of MBP staining, 2: more MBP loss of processes, 3: loss of branch processes and fragment capsule, 4: ragged appearing capsule and thinning, 5: near total loss of capsule and process. Immunostained cells were counted in three to five animals in each group on a fluorescence microscope (Nikon, Nikon Eclipse 80i, and Japan). Images were analyzed by the ImageJ software. Results were determined by the number of cells per high-power field (HPF).
Western blotting
Western blotting was performed according to the previously described method.22 Briefly, ischemic and non-ischemic hemispheres dissected from mouse brain. We further separated the striatum and cortex. Brain tissues were homogenized with the RIPA lysis buffer containing 0.5 M Tris-HCl (pH 7.4), 1.5 M NaCl, 2.5% deoxycholic acid, 10% NP-40, and 10 mM EDTA (Cat # 20-188, Millipore, USA) with protein inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany) by using one tablet per 10 mL buffer. Tissue lysates were sonicated for 30 s and centrifuged at 12,000 g for 15 min at 4℃. Protein concentration in the supernatants was measured using BCA protein assay kits (Pierce Biotechnology Inc., Rockford, IL, USA). Sample lysates were boiled for 10 min at 100℃ and centrifuged for 5 min at 12,000 g. Equal protein amount (20 µg–30 µg) was loaded in the 4–12% SDS bis/tris gel (Invitrogen Corporation, Carlsbad, CA, USA) and protein was transferred into polyvinylidene fluoride (PVDF) membrane and membrane was blocked with 5% non-fat milk for 1 h at room temp. Subsequently, the membrane was incubated overnight at 4℃ following primary antibodies: The primary antibodies were as follows: mouse anti-heat shock protein 70 (Hsp70) (1:500, Santa Cruz Biotechnology Inc., Dallas, Texas, USA), rabbit-phospho Akt (Ser 473) (1:300, Millipore), mouse-anti p53 (1:250, Santa Cruz Biotechnology), mouse -anti Hsp27 (1:300, Santa Cruz Biotechnology), mouse-anti-Bcl-2 (1:250, Santa Cruz Biotechnology), and mouse-anti β-actin (1:4000, Sigma Aldrich, St Louis, MO, USA). Membranes were three times rinsed with PBS containing 0.1% tween 20 (PBST) for 10 min and incubated with horseradish peroxidase-(HRP) conjugated anti-mouse, anti-rabbit, or anti-goat secondary antibodies (Santa Cruz Biotechnology, 1:2000) for 1 h at room temperature. After washing (3×), membranes were treated with an enhanced chemiluminescence (ECL) kit (GE Healthcare, Amersham ECL kit, Buckinghamshire, U.K) and analyzed by normalizing to the same well actin control.
Functional behavioral assessment
The grid-walk (foot fault) test, adhesive tape removal test and forelimb use asymmetry (cylinder) test were performed to assess stroke-induced deficits and subsequent functional recovery after ABAH treatment (sham: n = 12, vehicle: n = 15, ABAH: n = 14). All behavioral tests were performed one day prior to surgery and continued on days −1, 1, 3, 7, 10, 14 and 21 after stroke by a blinded investigator. Data were recorded by video recorder and were analyzed in a blinded manner without knowledge of the treatment group assignment. To further characterize the effects of ABAH were specific to MPO, we performed the 8-point test as follows: sham MPO−/− mice treated with vehicle or ABAH (three mice in each group) and stroke MPO−/− mice treated with vehicle or ABAH (four mice in each group) on day 14 after stroke. Another 10 MPO−/− mice were used for histopathological analysis and neurobehavioral tests.
Grid walk test (foot-fault)
Grid walk test assesses for motor, coordination and movement impairment by evaluating limb function during locomotion in rodents. Stroke-induced mice generally exhibited more contralateral (impaired limb) foot faults and normal animals observed few or no foot faults.25 Mice were placed on an elevated wire grid measuring 37 cm (H) × 48 cm (W) with 1.5 cm × 1.5 cm square holes, and allowed to walk for 5–10 min to get 100 steps. Precise grip and placement of forelimb are required to complete this test and measured foot fault of the affected forelimbs. With each weight-bearing step, the forelimbs may fall or slip between the wire. This was recorded as a foot fault. The video camera was located in front of the grid panel in the bottom to track mice movement with the appropriate angle. All assessments were done by an experimenter blinded to the group assignment. Sham, vehicle and ABAH-treated mice were tested on days −1, 1, 3, 7, 10, 14, and 21 days after tMCAO. The number of both contralateral and ipsilateral foot fault for each limb is assessed by total number of steps and showed foot fault value. The equation for grid walk score was calculated by (left foot fault-right foot fault)/number of steps × 100.26
Adhesive removal test
This test can assess stroke-induced impairments of tactile extinction as well as somatosensory and motor function.27 Ipsilateral forelimb paws (unimpaired limb) will demonstrate decreased contact/removal time of adhesives compared with contralateral forelimb paws (impaired limb) in tMCAO. Mice were placed on the acrylic transparent cylinder and small colorful-adhesive tapes (0.3 cm × 0.4 cm) were applied as bilateral tactile stimuli on the dorsal side of forelimb paws on both sides. Two strips of sticky tapes of equal size were applied with equal pressure to the forepaws. Normal mice very quickly remove the adhesive tapes from their forelimb paws by grooming. We assessed time-to-contact and time-to-removal for both forelimbs (left, right) three times per day.28 We performed pre-training and baseline tests to identify any pre-operative asymmetries. The time-to-contact for each paw and to remove the adhesive was measured with a maximum of 180 s. The tape removal time was recorded and test was performed on days −1, 1, 3, 7, 10, 14 and 21 days after ischemia. Time-to-contact and time-to-remove separated out sensory and motor functions. The sensory time was counted starting from the time of mouse placement into the chamber to when the mouse contacted the tape with its mouth. The equation was (Lc − Rc)/(Lc + Rc; L, left, R, right. c, contact) × 100. The motor tests were assessed by measuring the time for the mouse to bring its forepaws to mouth and completely removing the tape. The equation was (Lr − Rr)/(Lr + Rr, r, removal) × 100.
Forelimb use asymmetry test (cylinder test)
The purpose of this test was to evaluate spontaneous forelimb use including locomotors asymmetry of forelimb activity between ischemic and control mice by measuring touch times of the cylinder wall.29 The mice were allowed to freely explore the clear plexiglass cylinder (18 cm (H) × 10 cm (W) for 30 min and we measured the number of wall touches using left, right and both forelimb simultaneously. The cylinder was high enough so that the animal could not reach the top margin of the apparatus by rearing. Animals were video recorded from a lateral perspective. We calculated the forelimb asymmetry score by using (number of right-left)/(number of both sides) × 100 (%).
8-point neurological behavioral test
We performed the 8-point score test, which assesses motor, sensory and reflex test, to form a global assessment of the neurobehavioral outcome.22 To characterize acute and subacute ABAH treatment effects for stroke, we assessed sham (n = 8), vehicle (n = 11), acute (n = 5), subacute (n = 8) and continuous ABAH-treated group (n = 10) from days −1 to 21 post tMCAO in C57BL/6 J mice. Another experiment compared 8-point behavioral scores between C57BL/6 J mice and MPO−/− mice with or without ABAH treatment. These animal groups were divided into five groups: sham (no stroke, WT, n = 8), stroke WT vehicle-treated (WT, n = 11), stroke WT ABAH-treated (n = 10), stroke vehicle-treated MPO−/− (n = 4), and stroke MPO−/− ABAH-treated (n = 3) on day 7 after cerebral ischemia. Behavioral test was performed one day before tMCAO for baseline study and on days 1, 3, 7, 10, 14 and 21 following stroke.
MRI and determination of infarct volumes
MR imaging was performed up to 21 days following stroke induction, including days 1 and 21, on a 4.7 T animal MRI scanner (Bruker, Billerica, MA) including a dedicated mouse head coil. Coronal diffusion-weighted images (DWI) were acquired on day 1 and coronal T1w and T2w images were obtained at later time points. Mice were anesthetized with isoflurane (0.5–2.5 %), which was adjusted during the imaging procedure to keep a consistent respiratory rate while being observed by the Model 1025 MRI compatible small animal watching and gating system (SA instruments, Stony Brook, NY). Rectal temperature was also checked and maintained at 37℃ by adjusting an MR-compatible small animal heating system (SA instruments) throughout the imaging process. After scanning, the mice were heated using the Air-Therm ATX (World Precision Instruments, Sarasota, FL) until they were conscious.
Statistical analysis
Data are expressed as the mean ± SEM. For groups with non-Gaussian distributions, comparison was made with the nonparametric Mann–Whitney test. For groups with a Gaussian distribution, means were compared using the Student’s t-test. For comparison among groups, we performed ANOVA followed by Bonferroni’s post hoc testing. p < 0.05 was considered statistically significant. We used Graphpad Prism version 6.00 for statistical analysis (GraphPad Software, La Jolla California USA, www. graphpad. com).
The number of mice used for each group was calculated to achieve a power of 90% (30% difference in means and standard deviation of 15%) based on our prior experience using this model,8,19 resulting in at least n = 3 per group.
Results
ABAH-treated WT and MPO−/− mice decreased cell loss on day 7 after stroke
We investigated whether ABAH treatment or MPO-deficiency can decrease cell death compared with stroke-induced control animals on day 7 after stroke. On H&E staining, we confirmed that sham-operated mice showed intact cell morphology but vehicle-treated stroke mice exhibited a shrunken cell morphology and increased cell loss in the ischemic brains on day 7 after stroke in C57BL/6 J (WT) mice (Figure 1(a)). In contrast, post-insult ABAH administration demonstrated less morphological change and cell loss compared with vehicle-treated stroke mice. In addition, vehicle-treated stroke MPO−/− mice were similarly resistant to cell death. However, ABAH-treated MPO−/− mice did not show further effect compared to vehicle-treated group in MPO−/− mice, consistent with ABAH being specific and redundant in these MPO-deficient mice (Figure 1(a)). Quantified result showed that vehicle-treated wild type stroke mice had significantly lower number of H&E-positive cells compared to MPO-inhibited mice in the ipsilateral striatum on day 7 after tMCAO (78.1 ± 2.4; 142.3 ± 1.3; p < 0.001, Vehicle WT vs. ABAH WT, n = 10) (Figure 1(b)). A similar effect was found in ipsilateral parietal cortex (81.0 ± 2.4; 123.3 ± 2.6, p < 0.001, Vehicle WT vs. ABAH WT, n = 10) (Figure 1(c)). Vehicle-treated MPO−/−, ABAH-treated C57BL/6 and ABAH-treated MPO−/− stroke mice did not show a significant difference in the number of H&E-positive cells. There was no significant difference between the animal groups in the mean laser Doppler flow (LDF) values during stroke induction (Supplementary Figure 3, 11.6 ± 0.9 versus (vs.) 11.8 ± 0.9 in the vehicle (n = 30) vs. ABAH (n = 30); p = 0.90).
Figure 1.

MPO inhibition and MPO−/− mice decreased cell loss on day 7 after tMCAO. (a) H&E staining in the ipsilateral striatum (Str) and parietal cortex (pcox). In striatum: Sham (WT), vehicle (WT), note that the cell morphology reveals cell shrinkage and loss. Vehicle (MPO−/−), ABAH (WT) and ABAH (MPO−/−). In parietal cortex: Sham (WT),vehicle (WT), vehicle (MPO−/−), ABAH (WT), and ABAH (MPO−/−) mice. Arrows indicate damaged cells. Quantified result of H&E staining in the ipsilateral striatum (b) and parietal cortex (c) on day 7 after tMCAO. Data are mean ± SEM. ANOVA followed by Bonferroni’s post hoc test. ns: no significant. ** p < 0.01, *** p < 0.001 between indicated groups. Data analyzed from 10 animals per group, except MPO−/− mice where three animals were analyzed.
ABAH treatment and MPO-deficiency decreased ischemia-induced neuronal death
Next, we investigated whether MPO inhibition affects neuronal survival by FJ-B+ staining (a degenerating neuronal marker) on day 7 after tMCAO. We examined the FJ-B+ cells in the ipsilateral hippocampus and striatum regions for each group. As expected, vehicle-treated tMCAO mice showed increased labeling of FJ-B+ cells in the ipsilateral hippocampus CA1 and striatum on day 7 after ischemia compared with the labeling in the corresponding brain regions of the sham-operated animals (Figure 2(a)). On the other hand, ABAH treatment markedly decreased the number of FJ-B+ cells. Vehicle-treated MPO−/− mice also showed a reduction in the number of FJ-B+ cells compared to vehicle-treated wild type mice, underscoring a detrimental role of MPO in ischemic brain injury. ABAH-treated MPO−/− mice showed similar labeling of FJ-B+ cells compared to vehicle-treated MPO−/− mice in the ischemic hemispheres on day 7 after stroke (Figure 2(a)). Quantified results for FJ-B+ cells in the ipsilateral hippocampus CA1 are shown in Figure 2(b) (34.0 ± 3.1; 2.3 ± 0.88, p < 0.001, Vehicle WT vs. ABAH WT). We also confirmed similar quantified analysis for FJ-B+ cells in the ipsilateral striatum (Figure 2(c), 40.0 ± 4.8; 9.3 ± 2.2, p < 0.05, Vehicle WT vs. ABAH WT) on day 7 after stroke.
Figure 2.

MPO inhibition decreased Fluoro-Jade B staining after tMCAO. (a) Fluoro-Jade B (FJ-B) effects on ipsilateral hippocampus (HP) CA1 and striatum (Str). In HP (CA1): sham (WT), vehicle (WT), vehicle (MPO−/−) mice, ABAH (WT), and ABAH (MPO−/−) mice. Arrows identify FJ-B+ cells. In striatum: sham (WT), vehicle (WT), vehicle (MPO−/−) mice, ABAH (WT) and ABAH (MPO−/−) mice. Magnification × 40. Scale bar, 50 µm. Quantified results of FJ-B+ cells in hippocampus CA1 (b) and striatum (c) on day 7 after tMCAO. Data are mean ± SEM. ANOVA followed by Bonferroni’s post hoc test. * p < 0.05, *** p < 0.001 between indicated groups. Data analyzed from three animals per group.
ABAH treatment and MPO-deficiency prevent MBP loss after stroke
To address the question whether ABAH or MPO-deficiency could protect against the death of MBP-expressing oligodendrocytes in the ischemic brains, we assessed for MBP staining in the ipsilateral WM regions of sham-operated (WT), vehicle (WT), ABAH (WT), vehicle (MPO−/−), and ABAH (MPO−/−)-treated stroke mice. Vehicle-treated animals showed marked fragmentation of the MBP staining in the ipsilateral WM seven days post-stroke compared with sham-operated animals (Figure 3(a)). Notably, ABAH treatment decreased MBP staining fragmentation in the ischemic WM on day 7 after tMCAO. Similarly, vehicle-treated MPO−/− mice had more intact MBP staining compared to vehicle-treated WT mice. However, ABAH treatment in MPO−/− mice did not reveal additional effects on MBP staining in the ischemic brain regions compared with the vehicle-treated MPO−/− mice (Figure 3(a)). Quantified results showed that vehicle-treated stroke animals exhibited significantly worse (higher) MBP scores compared to sham control. When MPO was either blocked or congenitally absent, the MBP scores improved (lower compared to vehicle-treated C57BL/6 stroke mice). Note that MPO−/− stroke mice, whether vehicle-treated or ABAH-treated, and ABAH-treated C57BL/6 stroke mice did not show a significant difference in the MBP score (Figure 3(b)).
Figure 3.

MPO inhibition and MPO−/− mice decreased MBP loss on day 7 after tMCAO. (a) Ipsilateral white matter area with MBP+/DAPI+ images. Sham (C57BL/6J, WT), vehicle (C57BL/6J), ABAH (C57BL/6J), vehicle (MPO−/−), and ABAH (MPO−/−) mice. Note the marked fragmentation of the MBP staining in the vehicle-treated C57BL/6J image. Representative images from three animals per group. Magnification, × 40. Scale bar, 50 µm. Lat: lateral corpus callosum; WM: white matter; Str: striatum. (b) Quantified myelin basic protein (MBP) score in ipsilateral white matter lateral region on day 7 after stroke. Vehicle-treated wild type mice significantly increased MBP score compared to sham-treated control. Data are mean ± SEM. ANOVA followed by Bonferroni’s post hoc test. ns; no significant. *** p < 0.001 between indicated groups. Data analyzed from three animals per group.
ABAH attenuates the inflammatory response in the ischemic brain
We found that there were more cells with activated microglia morphology with strong staining of the myeloid cell marker CD11b+ in the ipsilateral striatum and corpus callosum seven days post stroke (Figure 4(a) and (b)). Post-insult ABAH treatment decreased the number of these cells compared to vehicle-treated control mice. Sham-operated animals showed very few of these cells. Quantified result in the striatum is shown in Figure 4(b) on day 7 after stroke (p < 0.001). Similar effect in the ischemic corpus callosum was confirmed on day 7 after stroke (42.0 ± 0.59 vs. 18 ± 0.58 cells/HPF, vehicle (n = 3) vs. ABAH (n = 3); p < 0.001). On day 21 after stroke, the number of CD11b+ cells continued to be decreased after ABAH treatment in the ipsilateral striatum and corpus callosum (Figure 4(a)) striatum, quantified in 4 C, p < 0.001). Similar results were found in the ischemic corpus callosum (42 ± 1.7 vs. 18.7 ± 2.9 cells/HPF, vehicle (n = 3) vs. ABAH (n = 3); p < 0.001). Additionally, ED1 (CD68), a macrophage/microglia marker, was increased by stroke but ABAH treatment decreased the number of ED1-positive cells in the ischemic striatum on day 14 after stroke (Supplementary Figure 4(a)). Quantified result confirmed that post-stroke ABAH treatment significantly attenuated the number of ED1-positive cells (Supplementary Figure 4(b)).
Figure 4.

MPO inhibition decreased CD11b+ cells after stroke. (a) Immunostaining for CD11b+ cells on day 7 after tMCAO. Striatum (Str): Sham, CD11b+ (green)/DAPI+ (blue), vehicle, and ABAH. Arrows show CD11b+ cells. (b) Corpus callosum (CC): Sham, vehicle, and ABAH. Immunostaining for CD 11 b+ cells on day 21 after tMCAO. Striatum: Sham, CD11b+ (green)/DAPI+ (blue), vehicle and ABAH. Arrows show CD11b+ cells. Corpus callosum: Sham, vehicle, and ABAH. Magnification, × 40. Scale bar, 50 µm. Quantification of ipsilateral striatum on day 7 (b) and day 21 (c), respectively. Data represent mean ± SEM. *** p < 0.001. ANOVA followed by Bonferroni’s post hoc test. Data analyzed from three animals per group.
MPO inhibition increased cytoprotective proteins and attenuated pro-apoptotic protein
Heat shock proteins (Hsps) have multiple functions including neuroprotective, anti-apoptotic, and anti-inflammatory effects in stroke.30,31 We examined whether post-insult MPO inhibition affects Hsp70 levels on day 3 after tMCAO. Compared to vehicle treatment, ABAH treatment increased Hsp70 level in the ipsilateral striatum on day 3 post stroke. Akt functions are a major downstream target for phosphatidylinositol 3-kinase (PI3-K) and are involved in cell survival through phosphorylation and activation of mTOR and CREB pathways.32 These events modulate neural stem cell differentiation and neuroprotective effects. Post-insult ABAH treatment increased phosphor-Akt (Ser473) levels in the ipsilateral striatum and cortex compared with vehicle-treated mice on day 3 after cerebral ischemia (Figure 5(a), Supplementary Figure 5). p53 is a pro-apoptotic protein and is involved in the neuronal cell death signaling cascade. Cerebral ischemia up-regulated p53 but ABAH treatment inhibited p53 protein levels in the ischemic striatum after cerebral ischemia (Figure 5(a), Supplementary Figure 5). Quantified results showed that post-insult ABAH treatment increased Hsp70 levels by 70% compared to vehicle-treated stroke mice (p < 0.001) (Figure 5(b)). ABAH also increased p-Akt levels compared to vehicle-treated stroke animals (60%, p < 0.001, Figure 5(c)). ABAH-treatment decreased the pro-apoptotic p53 levels compared to vehicle-treated stroke animals (62%, p < 0.001, Figure 5(d)).
Figure 5.
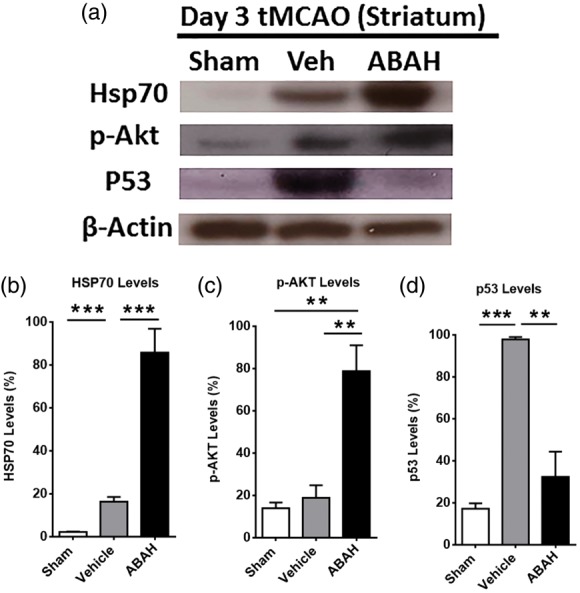
Effect of MPO inhibition on neuroprotective and apoptotic factors. (a) Western blots for Hsp70, pAkt and p53 levels in the ipsilateral striatum on day 3 after tMCAO in mice. β-actin was used for loading controls. Data shown are representative from each group. (b–d) Quantified result of Hsp70, p-Akt (Ser 473) and p53 levels, respectively. Data are mean ± SEM of percentage of protein levels with three to four animals in each group. ** p < 0.05, *** p < 0.001. ANOVA followed by Bonferroni’s post hoc test.
To further validate our findings, we examined whether Hsp70 colocalized with specific cell types. Double immunostaining for Hsp70+/NeuN+ cells was markedly increased in the ipsilateral striatum after ABAH treatment compared to that of vehicle-treated mice on day 7 after ischemic injury (Figure 6(a) and (b)). We also found that ABAH treatment significantly increased the number of NeuN+ cells in the striatum in the ipsilateral striatum on day 7 after stroke compared with vehicle-treated animals (Figure 6(a)). Quantified results for Hsp70+ cells in the ipsilateral striatum are shown in Figure 6(b) (p < 0.001), for NeuN+ cells in Figure 6(c) (p < 0.001) and Hsp70+/NeuN+ cells in Figure 6(d) (p < 0.001). The majority of Hsp70+ cells colocalized with NeuN+ cells. Similar to the above, both ABAH- and vehicle-treated stroke MPO−/− mice showed a similar increase in the number of Hsp70+/NeuN+ cells in the ipsilateral striatum, parietal cortex, and CA1 of the hippocampus compared to WT vehicle-treated stroke mice (Supplementary Figure 6), indicating ABAH does not exhibit additional effect other than MPO inhibition (Supplementary Figure 6(a) to (c)). Quantified results showed no significant difference in Hsp70+, NeuN+, Hsp70+/NeuN+ cells in vehicle- and ABAH-treated MPO−/− mice (Supplementary Figure 5(d) to (f)) and parietal cortex (Supplementary Figure 6(g) to (i)) on day 7 after stroke. While Hsp70 can be found in both nucleus and cytoplasm, in stress conditions, heat shock proteins such as Hsp70 translocate to the nucleus.33–36 Correspondingly, we found that Hsp70 mostly colocalized with DAPI nucleus staining after stroke.
Figure 6.

Double immunostaining for Hsp70+/NeuN+ cells in the ipsilateral striatum on day 7 after tMCAO in mice. (a) Hsp70 (red) in vehicle- and ABAH-treated mice, NeuN (green), DAPI (blue), and merge. Note that the majority of Hsp70+ cells were localized in NeuN+ cells. Short arrows show Hsp70+ cells, long arrows identify NeuN+ cells. Dotted arrows identify DAPI+ cells. Str: striatum, triangles show colocalized cells. Magnification, × 40. Scale bar, 50 µm. Quantified data of (b) Hsp70+cells, (c) NeuN+ cells, and (d) Hsp70+/NeuN+ cells. Data are mean ± SEM. ANOVA followed by Bonferroni’s post hoc test. *** p < 0.001 between indicated groups. Representative data analyzed from three animals per group.
MPO inhibition improved function after stroke
The grid walk tests were performed to assess motor coordination impairments of limb functioning and rehabilitation effects after stroke.27 We found that sham-treated mice performed consistently well and maintained baseline scores (0.56, n = 8). Vehicle-treated stroke mice showed increased number of foot faults (9.8, n = 11), but ABAH-treated stroke mice showed significantly improved grid walk scores (3.4, n = 10, p < 0.0001) on day 21 after stroke (Figure 7(a)). In the adhesive motor test, vehicle-treated stroke mice similarly showed significantly worse scores than ABAH-treated stroke mice (Figure 7(b), n = 10, p < 0.0001). In the adhesive sensory test, the ABAH-treated mice also demonstrated improved adhesive sensory deficits compared with vehicle-treated animals on days 1 and 7 after stroke (Figure 7(c), n = 10, p < 0.05). Forelimb asymmetry test (cylinder test) was also performed to investigate forelimb deficits. On day 21 after tMCAO, ABAH-treatment significantly reduced forelimb asymmetry deficits compared with vehicle-treatment (Figure 7(d), p < 0.05).
Figure 7.

MPO inhibition with ABAH markedly improved functions up to on day 21 after stroke. (a) Grid walk test. Sham (n = 8), vehicle (n = 12), and ABAH (n = 12). Data represent mean ± SEM. ANOVA followed by Bonferroni’s post hoc test. *** p < 0.001, vehicle vs. ABAH-treated groups. (b) Adhesive motor test. Scores were analyzed on days 1, 1, 3, 7, 10, 14 and 21 after tMCAO. Sham (n = 8), vehicle (n = 12), and ABAH (n = 12). Data represent mean ± SEM. ANOVA followed by Bonferroni’s post hoc test. * p < 0.05, vehicle vs. ABAH-treated groups. (c) Adhesive sensory test. Scores were analyzed on days 1, 1, 3,7,10, 14 and 21 after tMCAO. Sham (n = 8), vehicle (n = 12), and ABAH (n = 12). Data represent mean ± SEM. ANOVA followed by Bonferroni’s post hoc test. * p < 0.05, vehicle vs. ABAH-treated groups. (d) forelimb use asymmetry test, day 21 following tMCAO. Sham (n = 8), vehicle (n = 14) and ABAH (n = 15). Data represent mean ± SEM. ANOVA followed by Bonferroni’s post hoc test. ** p < 0.01, vehicle vs. ABAH-treated groups.
Length of MPO inhibition and neurobehavioral response
We had previously shown that delayed ABAH treatment (initiated during the subacute phase of stroke) captured most of the functional benefit of treatment throughout the entire stroke period when assessed on day 21.8 In this study, we first confirmed that delayed (susbacute) treatment resulted in smaller day 21 lesion size (Supplementary Figure 7(a) and (b)). In addition, we studied the evolution of neurobehavioral functions. On day 1 after stroke, both vehicle-treated and subacute treated stroke mice showed similar 8-point scores, while stroke mice treated only on day 0 (acute group) demonstrated a slight improved score. Interestingly, the continuous treatment group, which had received an additional day of treatment by day 1, exhibited a further improvement, consistent with a dose-response. Following the withdrawal of the treatment, the acute group demonstrated a similar course as that of the vehicle-treated group. However, the subacute treatment group started to improve after the initiation of treatment, achieving a level similar to that of the continuous treatment group by day 7 and maintained that level to the end of the observation period (up to day 21) (Supplementary Figure 7(c)).
Discussion
In this study, we found that MPO, a key enzyme secreted in inflammation, plays a detrimental role in the ischemic brain, which could be reversed by MPO inhibition. MPO inhibition with the specific irreversible MPO inhibitor ABAH and MPO-deficiency decreased cell loss following stroke, including FJ-B+ degenerating neurons and MBP+ oligodendrocytes. We further found that ABAH treatment attenuated inflammatory cell recruitment, including CD11b+and ED1+ myeloid cells in the ischemic striatum. Moreover, post-insult ABAH treatment increased the levels of cytoprotective proteins such as Hsp70, and p-Akt but decreased pro-apoptotic p53 in the ischemic brain. We found a dose-dependent response to ABAH treatment after stroke, with longer period of treatment conferring greater benefit. Early treatment was also beneficial, but the effect did not persist once the treatment stopped. In neurobehavioral assessments, post-insult ABAH treatment improved sensory and motor functions after stroke. Together, these beneficial effects underscore a protective role for MPO inhibition after cerebral ischemia.
Elevated MPO activity in the infarct has been found to come from both early infiltrating neutrophils as well as later infiltrating macrophages after stroke,37,38 making it an attractive therapeutic target. MPO itself has pro-inflammatory and cell activation properties through polymorphonuclear neutrophil (PMN) attraction driven by physical forces.38 Furthermore, MPO binding to the neutrophil integrin Mac-1 can stimulate PMN adhesion and inflammatory cell recruitment.39 MPO also inhibits neutrophil apoptosis to prolong acute inflammation.15 Additionally, acting as a catalytic enzyme, MPO depletes NO consumption and makes neurotoxic oxidants such as HOCl, tyrosyl radicals and lipid oxidation. Inflammatory response from ischemic injury triggers pro-inflammatory cytokines, leukocytes, chemokines, microglial activation and adhesion molecule recruitment in the vascular endothelial cells.40–42 By inhibiting MPO activity, these deleterious effects are reversed, thus decreasing inflammation. These result in decreased overall inflammatory burden that served to further decrease inflammatory cell recruitment and response. As neuroprotection is associated with decreased recruitment of inflammatory cells,42 we correspondingly found decreased number of CD11b+ and ED1+cells with MPO inhibition.
At 40 mg/kg i.p. twice daily, ABAH was found to suppress MPO activity in the infarct by 30%.8 However, this degree of suppression was sufficient to achieve > 60% decrease in the number of CD11b+ myeloid cells recruited to or activated in the brain. Correspondingly, we observed 60–70% improved cell loss as well as day 21 lesion size. Similarly, neuroprotective factors such as Hsp70 and p-AKT increased by > 70%, while apoptotic factors such as p53 decreased by > 60%. While these changes (e.g. increase in Hsp70) may be due to more surviving cells after treatment, other factors such as Hsp27 and Bcl-2 did not increase with ABAH treatment (Supplementary Figure 8), suggesting that these changes are likely due to MPO inhibition rather than as a artifact of increased cell survival. The degree of the changes appears to be concordant with the degree of inflammatory myeloid cells changes, suggesting that suppressing MPO activity also affects the recruitment of MPO-containing myeloid cells. MPO activity likely also acts as a positive feedback signal to exacerbate inflammation, and may serve to recruit/activate these myeloid cells, including microglia.
Thus, there are multiple benefits to inhibiting MPO activity after stroke, including improved neurogenesis,19 decreased infarct size,8 improved neurobehavioral outcome, decreased inflammatory cell recruitment, increased expression of cytoprotective proteins, and decreased cell loss. It is important to note that while MPO’s key biological role is in fighting infection, this role does not appear to be limiting to develop MPO inhibition as a potential therapy, given that MPO-deficient humans do not exhibit increased infection rate.43 Thus, it is not surprising that multiple pharmaceutical companies are developing and translating MPO inhibitors.44,45
Conclusion
MPO inhibition with ABAH and MPO deficiency is neuroprotective and improved functions in cerebral ischemia in mouse models in a dose-dependent manner. Therefore, MPO inhibition is a potentially promising therapeutic strategy for stroke.
Supplemental Material
Supplemental material for Reducing myeloperoxidase activity decreases inflammation and increases cellular protection in ischemic stroke by Hyeon J Kim, Ying Wei, Gregory R Wojtkiewicz, Ji Y Lee, Michael A Moskowitz and John W Chen in Journal of Cerebral Blood Flow & Metabolism
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health grants R01 NS070835 and R01 NS0721267.
Declaration of conflicting interests
The author(s) declared no potential conflict of interest with respect to the research, authorship, and/or publication of this articles.
Authors’ contributions
All authors reviewed and edited the manuscript. HJK designed research and performed experiments, data analysis, and wrote the manuscript. YW performed experiments and data analysis. GW conducted experiments (imaging) and data analysis. JL assisted with experiment and data analysis. MAM, designed research and performed data analysis. JWC designed research, performed data analysis, and wrote the manuscript with HJK.
Supplementary material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb
References
- 1.Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation 2011; 123: 933–944. [DOI] [PubMed] [Google Scholar]
- 2.Winstein CJ, Stein J, Arena R, et al. Guidelines for adult stroke rehabilitation and recovery: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2016; 47: e98–e169. [DOI] [PubMed] [Google Scholar]
- 3.Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med 2009; 7: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron 2010; 67: 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furlan AJ, Katzan IL, Caplan LR. Thrombolytic therapy in acute ischemic stroke. Curr Treat Options Cardiovasc Med 2003; 5: 171–180. [DOI] [PubMed] [Google Scholar]
- 6.Shuaib A, Hussain MS. The past and future of neuroprotection in cerebral ischaemic stroke. Eur Neurol 2008; 59: 4–14. [DOI] [PubMed] [Google Scholar]
- 7.O'Collins VE, Macleod MR, Donnan GA, et al. 1,026 experimental treatments in acute stroke. Ann Neurol 2006; 59: 467–477. [DOI] [PubMed] [Google Scholar]
- 8.Forghani R, Kim HJ, Wojtkiewicz GR, et al. Myeloperoxidase propagates damage and is a potential therapeutic target for subacute stroke. J Cereb Blood Flow Metab 2015; 35: 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang R, Brennan ML, Shen Z, et al. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J Biol Chem 2002; 277: 46116–46122. [DOI] [PubMed] [Google Scholar]
- 10.Nussbaum C, Klinke A, Adam M, Baldus S, et al. Myeloperoxidase: a leukocyte-derived protagonist of inflammation and cardiovascular disease. Antioxid Redox Signal 2013; 18: 692–713. [DOI] [PubMed] [Google Scholar]
- 11.Galijasevic S, Saed GM, Diamond MP, et al. Myeloperoxidase up-regulates the catalytic activity of inducible nitric oxide synthase by preventing nitric oxide feedback inhibition. Proc Natl Acad Sci U S A 2003; 100: 14766–14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malle E, Marsche G, Panzenboeck U, et al. Myeloperoxidase-mediated oxidation of high-density lipoproteins: fingerprints of newly recognized potential proatherogenic lipoproteins. Arch Biochem Biophys 2006; 445: 245–255. [DOI] [PubMed] [Google Scholar]
- 13.Loria V, Dato I, Graziani F, et al. Myeloperoxidase: a new biomarker of inflammation in ischemic heart disease and acute coronary syndromes. Mediators Inflamm 2008; 2008: 135625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rudolph V, Andrie RP, Rudolph TK, et al. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat Med 2010; 16: 470–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El Kebir D, Jozsef L, Pan W, et al. Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circ Res 2008; 103: 352–359. [DOI] [PubMed] [Google Scholar]
- 16.Luo Y, Yin W, Signore AP, et al. Neuroprotection against focal ischemic brain injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. J Neurochem 2006; 97: 435–448. [DOI] [PubMed] [Google Scholar]
- 17.Tang WH, Brennan ML, Philip K, et al. Plasma myeloperoxidase levels in patients with chronic heart failure. Am J Cardiol 2006; 98: 796–799. [DOI] [PubMed] [Google Scholar]
- 18.Forghani R, Wojtkiewicz GR, Zhang Y, et al. Demyelinating diseases: myeloperoxidase as an imaging biomarker and therapeutic target. Radiology 2012; 263: 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim H, Wei Y, Lee JY, et al. Myeloperoxidase inhibition increases neurogenesis after ischemic stroke. J Pharmacol Exp Ther 2016; 359: 262–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breckwoldt MO, Chen JW, Stangenberg L, et al. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci U S A 2008; 105: 18584–18589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taninishi H, Jung JY, Izutsu M, et al. A blinded randomized assessment of laser Doppler flowmetry efficacy in standardizing outcome from intraluminal filament MCAO in the rat. J Neurosci Methods 2015; 241: 111–120. [DOI] [PubMed] [Google Scholar]
- 22.Kim HJ, Leeds P, Chuang DM. The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J Neurochem 2009; 110: 1226–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 2000; 874: 123–130. [DOI] [PubMed] [Google Scholar]
- 24.Liu XB, Shen Y, Plane JM, et al. Vulnerability of premyelinating oligodendrocytes to white-matter damage in neonatal brain injury. Neurosci Bull 2013; 29: 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang L, Schallert T, Zhang ZG, et al. A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J Neurosci Methods 2002; 117: 207–214. [DOI] [PubMed] [Google Scholar]
- 26.Bland ST, Schallert T, Strong R, et al. Early exclusive use of the affected forelimb after moderate transient focal ischemia in rats: functional and anatomic outcome. Stroke 2000; 31: 1144–1152. [DOI] [PubMed] [Google Scholar]
- 27.Schaar KL, Brenneman MM, Savitz SI. Functional assessments in the rodent stroke model. Exp Transl Stroke Med 2010; 2: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Modo M, Stroemer RP, Tang E, et al. Neurological sequelae and long-term behavioural assessment of rats with transient middle cerebral artery occlusion. J Neurosci Methods 2000; 104: 99–109. [DOI] [PubMed] [Google Scholar]
- 29.Schallert T. Behavioral tests for preclinical intervention assessment. NeuroRx. 2006; 3: 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yenari MA, Liu J, Zheng Z, et al. Antiapoptotic and anti-inflammatory mechanisms of heat-shock protein protection. Ann N Y Acad Sci 2005; 1053: 74–83. [DOI] [PubMed] [Google Scholar]
- 31.Zheng Z, Kim JY, Ma H, et al. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab 2008; 28: 53–63. [DOI] [PubMed] [Google Scholar]
- 32.Hers I, Vincent EE, Tavare JM. Akt signalling in health and disease. Cell Signal 2011; 23: 1515–1527. [DOI] [PubMed] [Google Scholar]
- 33.Knowlton AA, Salfity M. Nuclear localization and the heat shock proteins. J Biosci 1996; 21: 123–132. [Google Scholar]
- 34.Adhikari AS, Sridhar Rao K, et al. Heat stress-induced localization of small heat shock proteins in mouse myoblasts: intranuclear lamin A/C speckles as target for alphaB-crystallin and Hsp25. Exp Cell Res 2004; 299: 393–403. [DOI] [PubMed] [Google Scholar]
- 35.Dastoor Z, Dreyer J. Nuclear translocation and aggregate formation of heat shock cognate protein 70 (Hsc70) in oxidative stress and apoptosis. J Cell Sci 2000; 113(Pt 16): 2845–2854. [DOI] [PubMed] [Google Scholar]
- 36.Manzerra P, Brown IR. The neuronal stress response: nuclear translocation of heat shock proteins as an indicator of hyperthermic stress. Exp Cell Res 1996; 229: 35–47. [DOI] [PubMed] [Google Scholar]
- 37.Barone FC, Hillegass LM, Price WJ, et al. Polymorphonuclear leukocyte infiltration into cerebral focal ischemic tissue: myeloperoxidase activity assay and histologic verification. J Neurosci Res 1991; 29: 336–345. [DOI] [PubMed] [Google Scholar]
- 38.Barone FC, Hillegass LM, Tzimas MN, et al. Time-related changes in myeloperoxidase activity and leukotriene B4 receptor binding reflect leukocyte influx in cerebral focal stroke. Mol Chem Neuropathol. 1995; 24: 13–30. [DOI] [PubMed] [Google Scholar]
- 39.Lau D, Mollnau H, Eiserich JP, et al. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci U S A 2005; 102: 431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol 2006; 66: 232–245. [DOI] [PubMed] [Google Scholar]
- 41.Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab 2012; 32: 1677–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, Hallenbeck JM, Ruetzler C, et al. Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab 2003; 23: 748–755. [DOI] [PubMed] [Google Scholar]
- 43.Lanza F. Clinical manifestation of myeloperoxidase deficiency. J Mol Med 1998; 76: 676–681. [DOI] [PubMed] [Google Scholar]
- 44.Churg A, Marshall CV, Sin DD, et al. Late intervention with a myeloperoxidase inhibitor stops progression of experimental chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 185: 34–43. [DOI] [PubMed] [Google Scholar]
- 45.Ali M, Pulli B, Courties G, et al. Myeloperoxidase inhibition improves ventricular function and remodeling after experimental myocardial infarction. JACC Basic to Transl Sci 2017; 1: 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material for Reducing myeloperoxidase activity decreases inflammation and increases cellular protection in ischemic stroke by Hyeon J Kim, Ying Wei, Gregory R Wojtkiewicz, Ji Y Lee, Michael A Moskowitz and John W Chen in Journal of Cerebral Blood Flow & Metabolism