

Abstract
Modern mass spectrometry now permits genome-scale and quantitative measurements of biological proteomes. However, analysis of specific specimens is currently hindered by the incomplete representation of biological variability of protein sequences in canonical reference proteomes and the technical demands for their construction. Here, we report ProteomeGenerator, a framework for de novo and reference-assisted proteogenomic database construction and analysis based on sample-specific transcriptome sequencing and high-accuracy mass spectrometry proteomics. This enables the assembly of proteomes encoded by actively transcribed genes, including sample-specific protein isoforms resulting from non-canonical mRNA transcription, splicing, or editing. To improve the accuracy of protein isoform identification in non-canonical proteomes, ProteomeGenerator relies on statistical target–decoy database matching calibrated using sample-specific controls. Its current implementation includes automatic integration with MaxQuant mass spectrometry proteomics algorithms. We applied this method for the proteogenomic analysis of splicing factor SRSF2 mutant leukemia cells, demonstrating high-confidence identification of non-canonical protein isoforms arising from alternative transcriptional start sites, intron retention, and cryptic exon splicing as well as improved accuracy of genome-scale proteome discovery. Additionally, we report proteogenomic performance metrics for current state-of-the-art implementations of SEQUEST HT, MaxQuant, Byonic, and PEAKS mass spectral analysis algorithms. Finally, ProteomeGenerator is implemented as a Snakemake workflow within a Singularity container for one-step installation in diverse computing environments, thereby enabling open, scalable, and facile discovery of sample-specific, non-canonical, and neomorphic biological proteomes.
Keywords: proteogenomics, de novo database construction, transcriptomics, peptide fractionation, peptide–spectral matching, scoring function, protein isoform analysis
Graphical Abstarct
INTRODUCTION
Functional analysis of physiologic and pathologic cell activities requires accurate and complete identification and quantification of all involved effector molecules. Such global studies are principally based on the decoding and assembly of the human genome.1,2 Recent advances in messenger RNA (mRNA) sequencing and bioinformatics now enable the routine analysis of biological gene expression.3–5 However, direct and proteome-wide studies of proteins and their biological variation remain confined to specialized approaches.6–8
Modern mass spectrometry now permits genome-scale and quantitative measurements of cellular proteomes.9–13 This approach is based on mass spectrometric analysis of peptides, generated by proteolysis of proteomes, followed by matching their observed fragmentation spectra with the corresponding amino acid sequences.14,15 Generally, this is accomplished using statistical peptide–spectrum matching techniques that leverage scoring functions to assess the similarity of observed and theoretical mass spectra,16 with the corresponding confidence of spectral identification expressed as a global false discovery rate (FDR), estimated using target–decoy approaches.17
Peptide identification using peptide-spectrum matching (PSM) and target–decoy FDR estimation is based on the fundamental assumption that mass spectrometry search databases contain a complete and accurate list of all potential protein sequences. Consequently, the sensitivity and specificity of peptide identification depend on the fidelity of the target and decoy databases. Advances in genome analysis and assembly have led to the development of high-quality databases of consensus protein sequences such as UniProt and RefSeq.1,2,18,19 However, germline and somatic genetic variants, mRNA splicing, and other biological processes can diversify polypeptide sequences, thereby generating sequence variants that are not catalogued in the consensus or canonical databases.20,21 These sources of proteome variation are particularly prevalent in human cancers, which frequently present structural aberrations in genes and dysregulation of their expression,22–24 ultimately hindering the analysis of cancer proteomes based on reference consensus databases.
In principle, proteogenomic approaches that integrate genome and transcriptome sequencing data with mass spectrometric protein analysis can overcome this limitation by generating sample-specific target databases for proteomic analysis that more accurately reflect the expressed proteome.25–31 Such an approach was first introduced to support gene annotation using proteomic data32 and has since become a powerful tool for quantitative and integrative studies.30,33–38 In addition, related approaches were recently developed for cancer biology39–44 and immunology studies.45
Specifically, sequences of expressed gene transcripts obtained by high-throughput sequencing of mRNA (RNA-seq) are a convenient source for proteogenomic sample-specific database construction for two main reasons: (i) these measurements reflect sequence variability introduced by transcriptional and post-transcriptional processes, and (ii) restriction of the mass spectral match search space to the specifically expressed proteins can improve its sensitivity and accuracy, particularly as compared with proteins predicted from translation of all possible reading frames.46
Based on this rationale, several approaches have recently been developed to generate customized mass spectrometry search databases from RNA-seq data.28,44,47–49 However, sensitivity and accuracy of proteogenomic detection of neomorphic and non-canonical proteins remain limited by the under-sampling of rare peptides and challenges in the generation of sample-specific databases from non-strand-specific short-read RNA-seq data.25–27 Furthermore, while reference sequence databases such as UniProt are manually curated to improve accuracy,18 automated workflows are needed to enable facile sample-specific proteogenomic analyses at scale.
Here, we describe ProteomeGenerator, an open, modular, and scalable framework for de novo and referenced proteogenomic database construction and analysis written in the Snakemake workflow management system and implemented using Singularity for one-step installation in diverse computing environments. We controlled the accuracy of peptide–spectrum matching using sequence and spectral decoys and used this method to assess the performance of four state-of-the-art mass spectrometry search algorithms. Lastly, we used ProteomeGenerator for genome-scale proteomic analysis of splicing factor mutant leukemia cells based on the integration of deep mRNA sequencing and multidimensional high-capacity chromatography. This led to the high-confidence identification of non-canonical protein isoforms arising from alternative transcription start sites, intron retention, and cryptic exon splicing as well as improved accuracy of genome-scale proteome discovery as compared with conventional approaches.
EXPERIMENTAL SECTION
Reagents
Mass spectrometry grade (Optima liquid chromatography–mass spectrometry, LC–MS) water, acetonitrile (ACN), and methanol were purchased from Fisher Scientific (Fair Lawn, NJ). Formic acid of >99% purity (FA) was obtained from Thermo Scientific. All other reagents at MS-grade purity were obtained from Sigma-Aldrich (Saint Louis, MO).
Cell Culture
Human K052 cells were obtained from the Japanese Collection of Research Bioresources Cell Bank, identity confirmed using STR genotyping (Genetica DNA Laboratories, Burlington, NC), and cultured as described.50 Cells were collected while in the exponential growth phase, washed twice in ice-cold phosphate-buffered saline, snap-frozen, and stored at −80 °C.
mRNA Sequencing
RNA was extracted using QIAGEN RNeasy columns (Qiagen, Valencia, CA). Poly(A)-selected, unstranded Illumina libraries were prepared with a modified TruSeq protocol, and 0.5× AMPure XP beads (Beckman Coulter, Indianapolis IA) were added to the sample library to select for fragments of <400 base pairs (bp), followed by 1× beads to select for fragments of >100 bp. These fragments were then amplified via polymerase chain reaction (15 cycles) and sequenced on the Illumina HiSeq 2000 (100 000 000, 2 × 49 base-pair reads per sample).
ProteomeGenerator
ProteomeGenerator is written in Snakemake,51 a scalable, Python-based workflow management system (Figure 2). The entire workflow is available for download at https://github.com/jtpoirier/proteomegenerator. ProteomeGenerator ingests RNA-seq data, which is aligned to a reference genome by the STAR splice aware aligner (version 2.5.2a).52 Aligned reads are subsequently filtered to exclude low-quality and poorly mapping reads using Samtools (version 1.3).53 A sample-specific transcript model is then assembled either de novo or with assistance from reference transcript model if one is available using StringTie (version 1.3.3b),54 with simultaneous filtering for transcripts with coverage of ≥2.5, length of ≥300 base pairs, and abundance of ≥1% of expressed transcripts for a given gene. All resulting transcript models are then merged with StringTie using an expression threshold of one transcript per million with permissive intron inclusion. The resulting merged transcript model is then used to generate corresponding cDNA sequences using gffread (version 0.9.8). The longest uninterrupted open reading frame is detected within each cDNA using TransDecoder (version 2.1, https://github.com/TransDecoder).55,56 Shorter open reading frames with low expected values when searched against the UniProt database using BLAST (version 2.2.31) are retained.57 The predicted longest open reading frames are subsequently mapped back to genomic coordinates and translated to their respective unique peptide sequences. Peptides assigned to mass spectra are mapped back to their genomic coordinates using ProteomeGenerator for visualization in the Integrative Genomics Viewer.57 Unique tryptic peptide search space was calculated for each database using the EMBOSS58 tool pepdigest and filtered to include all peptides of at least 6 amino acids in length having a molecular weight between 600 and 4000 Da. ProteomeGenerator is distributed with a Singularity container,59 allowing for one-step installation in diverse computing environments. In addition, its current distribution automatically includes the MaxQuant mass spectrometry analysis algorithm.
Figure 2.

Schema for the ProteomeGenerator snakemake workflow. Sequencing reads are aligned using STAR followed by their de novo or referenced assembly intro transcriptomes using StringTie and processing to identify reading frames and protein isoforms. The resulting protein database is set as the target for peptide–mass spectral matching using MaxQuant.
Databases
Consensus protein sequences databases were downloaded from UniProt18 as of January 2016 (Homo sapiens SwissProt database including isoforms), September 2015 (Archaebacteria loki), and June 2014 (Escherichia coli). Contaminant sequences were retrieved from cRAP60 as of June 2014. For comparisons, proteogenomic databases were also generated using QUILTS27 using GRCh38 as reference genome, variant quality threshold of 0, and thresholds for supporting reads for novel splice junctions as 2 for “both boundaries annotated”, 3 for “left boundary annotated”, and 3 for “no boundary annotated.”
Proteome Extraction and Proteolysis
Protein extraction and proteolysis was performed as previously described.61 Briefly, frozen cell pellets were thawed on ice, resuspended in 6 M guanidinium hydrochloride and 100 mM ammonium bicarbonate at pH 7.6 (ABC), and lysed using the E210 adaptive focused sonicator (Covaris, Woburn, CA). The protein content in cell lysate was determined using the BCA assay according to the manufacturer’s instructions (Pierce, Rockford, IL). Upon reduction and alkylation, proteomes were digested using 1:100 w/w (protease:proteome) LysC endopeptidase (Wako Chemical, Richmond, VA) and 1:50 w/w MS sequencing-grade modified trypsin (Promega, Madison WI). Digestion was stopped by acidifying the reactions to pH 3 using formic acid (Thermo Scientific), and peptides were subsequently desalted using solid-phase extraction using C18 Macro Spin columns (Nest Group, Southborough, MA).
High-Resolution Peptide Chromatography
Peptide chromatographic fractionation was performed using the Alliance e2695 high-performance liquid chromatograph (Waters, Milford MA). High-pH reversed-phase separation was performed using the Xselect CSH 3.0 mm × 150 mm column (Waters, part no. 186006728) at a constant flow-rate of 250 μL/min. After an initial equilibration at 100% buffer A (50 mM ammonium hydroxide in water, pH 10) for 5 min, peptides were resolved by a 75 min 0–70% gradient of buffer B (80% ACN in water, pH 10), followed by 10 min at 100% buffer B. The eluate was collected in 0.5 mL aliquots using a fraction collector between 25 and 75 min, lyophilized to dryness in a vacuum centrifuge, and stored at −80 °C until analysis. Before LC–MS analysis, peptides were resuspended in 20 μL of 0.1% formic acid in water, and 2 μL were analyzed.
Strong cation exchange chromatography was performed using the Xselect Hi Res SP, 7 μm, 4.6 mm × 100 mm column (Waters, part no. 186004930) at a constant flow-rate of 500 μL/min. After an initial equilibration at 100% buffer A (0.1% formic acid, 5% ACN) for 5 min, peptides were resolved by a 80 min 0–30% gradient of buffer B (1 M KCl, 5% ACN), followed by 5 min of gradient 30–50% buffer B and a final hold for 5 min at 100% buffer B. The eluate was collected in 1 mL aliquots using a fraction collector between 25 and 85 min and lyophilized to dryness in a vacuum centrifuge. Pellets were solubilized in 0.3 mL of 0.1% aqueous formic acid, and peptides were desalted using solid-phase extraction with C18 Macro Spin columns (Nest Group). SPE eluates were lyophilized and stored at −80 °C until analysis. Before LC–MS analysis, peptides were resuspended in 20 μL of 0.1% formic acid in water, and 2 μL were analyzed.
Nanoscale Liquid Chromatography
Nanoscale liquid chromatography experiments were performed using the Ekspert NanoLC 425 chromatograph (Eksigent, Redwood City, CA), equipped with an autosampler module, 2 10-port and 1 6-port rotary valves, and 1 isocratic and 2 binary pumps. Column fabrication were performed as previously described.62 Briefly, samples were initially aspirated into a 10 μL PEEK sample loop. Chromatographic columns were fabricated by pressure filling the stationary phase into silica capillaries fritted with K-silicate. Reversed-phase columns were fabricated by packing Reprosil 1.9 μm silica C18 particles (Dr. Meisch, Ammerbuch-Entringen, Germany) into 75 μm × 40 cm fritted capillaries. Trap columns were fabricated by packing Poros R2 10 μm C18 particles (Life Technologies, Norwalk, CT) into 150 μm × 4 cm fritted capillaries. Vented trap-elute architecture was used for chromatography.63 Peptides were resolved by reversed-phase chromatography hyphenated to the nanoelectrospray ion source. Upon valve switching to connect the trap column in line with the analytical reverse-phase column and ion emitter, the pressure was equilibrated at a flow of 250 nL/min for 5 min in 5% buffer B (ACN, 0.1% FA) in buffer A (water, 0.1% FA). Subsequently, a 120 min (high-pH reverse-phase samples) or 180 min (SCX samples) linear gradient of 5–40% of buffer B was used to resolve peptides, followed by a 5 min 40–80% gradient prior to column wash at 80% buffer B for 30 min.
Nanoelectrospray Ionization and Orbitrap Mass Spectrometry
Electrospray emitters with terminal opening diameter of 10 μm were obtained from New Objective (Woburn, MA). The emitter was connected to the outlet of the reverse-phase column using a metal union that also served as the electrospray current electrode. Electrospray ionization was achieved using constant 1700 V voltage. During column loading, the electrospray emitter was washed with 50% aqueous methanol using the DPV-565 PicoView ion source (New Objective).
For all measurements, we used the Orbitrap Fusion mass spectrometer (Thermo Scientific, San Jose, CA). Precursor scans in the 400–2000 Th were performed in the orbitrap detector at 120 000 resolution, with 100 ms maximum injection time and automatic gain control set at 105 ions. Fragment spectra were recorded in the linear ion trap in rapid mode, with a maximum injection time of 75 ms and target of 104 ions and 1.2 Da quadrupolar precursor selection.
Data Analysis
Peptide–spectral matching calculations were performed using a custom-built computer server equipped with 4 Intel Xeon E5–4620 8-core CPUs operating at 2.2 GHz and 512 GB physical memory (Exxact Corporation, Freemont, CA). Peptide– spectral matching was performed using SEQUEST HT and Percolator16,64,65 as part of Proteome Discoverer version 2.1.0.81 (Thermo Scientific), Byonic version 2.7.84,66,67 MaxQuant version 1.5.4.1,68,69 and PEAKS version 8.0.70 The Linux-compatible version of MaxQuant71 was version 1.6.2.3. For all searches, MS1 and HCD MS2 mass tolerances were set to 10 ppm and 0.6 Da, respectively. Cysteine carbamidomethylation was set as fixed, while methionine oxidation and glutamine and asparagine deamidation were set as variable modifications, with a maximum of three variable modifications per peptide. Only peptides containing 7–35 residues and up to 2 missed trypsin cleavages were considered. Tryptic peptides observable by mass spectrometry were predicted based on proteome composition using the generate-peptides utility of Crux.72 ProteomeGenerator is openly available at https://github.com/jtpoirier/proteomegenerator.
RESULTS AND DISCUSSION
ProteomeGenerator for Deep Genome-Scale Transcriptomic and Proteomic Integration
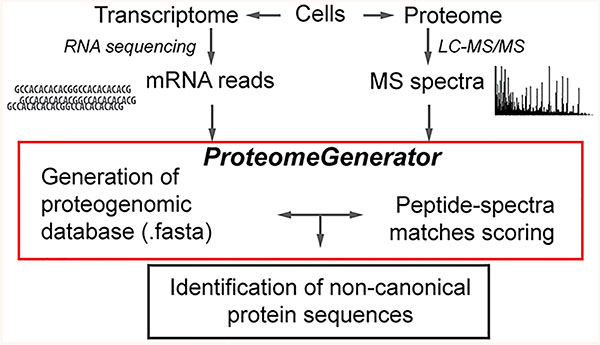
To facilitate proteogenomic analysis, we developed a computational framework, termed ProteomeGenerator, which automates proteomic analysis using sample-specific protein sequences databases. ProteomeGenerator is programmed in the Snake-make workflow management system for open, modular, and scalable analysis. The software is packaged using the Singularity software container to enable one-step installation in diverse computing environments.59 ProteomeGenerator first generates a sample-specific protein sequence database using high-coverage RNA-seq data based on de novo or referenced transcriptome assembly (Figure 1). This sample-specific proteome is then automatically set as the target database for peptide and protein isoform identification using statistical target–decoy database matching. In this work, identification specificity was also controlled by using spectrally calibrated sample-specific controls, and the sensitivity of mass spectrometric identification was enhanced by leveraging high-resolution nanoscale chromatography.
Figure 1.

ProteomeGenerator overview. Transcriptomes and proteomes from the same biologic sample are analyzed in parallel by high-coverage Illumina sequencing and high-resolution, high-accuracy mass spectrometry, respectively. ProteomeGenerator assembles fastq-for-matted mRNA sequencing reads into predicted transcripts, identifies reading frames and isoforms, and produces Fasta-formatted proteogenomic (PGX) databases containing canonical and non-canonical expressed protein isoforms for subsequent mass spectrometry searches.
We applied this method to analyze human K052 leukemia cells harboring splicing factor SRSF2 mutations, which were recently described to cause recurrent mRNA mis-splicing and are therefore expected to express non-canonical and neomorphic protein isoforms.50 First, we extracted mRNA and obtained high-coverage RNA-seq data of nearly 60 million reads by Illumina sequencing. ProteomeGenerator processed the raw sequencing reads in the following steps: (i) two-pass splice aware alignment to the user-supplied reference genome (in this case, GRCh38); (ii) assembly of possible transcript isoforms using StringTie assisted by the user-supplied transcript model (in this case, GENCODE version 20); (iii) prediction of the longest open reading frame for each possible transcript; and (iv) generation of FASTA-formatted proteogenomic database composed of unique protein sequences (Figure 2). Finally, we integrated the MaxQuant algorithm in the current distribution to automate proteomic analysis using the sample-specific database as the target for peptide–spectral matching.
The constructed K052 cells-specific proteogenomic database, to which we refer as PGX, consisted of 17 348 protein entries and was expected to produce 743 148 observable tryptic peptides with lengths between 7 and 35 residues, assuming 1 missed trypsin cleavage (Figure 3A,B). For comparison, the canonical reference human proteome in the UniProt database (SwissProt database including isoforms, as of March 2016) contained 42 123 proteins corresponding to 1 460 257 mass spectrometry observable tryptic peptides. The PGX database contained 37 158 peptides with no counterparts in UniProt, presumably originating from novel predicted protein isoforms specific for splicing factor mutant K052 cells and, consequently, not annotated in UniProt. The PGX database was approximately 51% smaller than UniProt with respect to mass spectrometry observable peptides, presumably because it contained only the expressed protein isoforms rather than the protein complement of all canonical protein isoforms as in the case of canonical reference proteomes.
Figure 3.

Comparison of the canonical and proteogenomic protein databases displaying (A) number of protein entries (B) and theoretical tryptic peptides amenable for mass spectrometry analysis specific for either UniProt, PGX, or both.
We compared the sample-specific database assembled by ProteomeGenerator to that produced using the QUILTS method.27 The K052 proteome predicted by QUILTS contained 201 718 protein sequence variants (using UniProt as reference), corresponding to 559 015 theoretically observable tryptic peptides (Figure S1). Considering only the predicted peptides not mapping in the UniProt database, QUILTS identified 544 331 non-canonical peptides as compared to 56 397 identified using ProteomeGenerator, with only 1440 (2.5%) peptides predicted by both methods. While accounting for canonical isoforms contained in UniProt, ProteomeGenerator exhibited a 63% decrease in variant sequences compared to QUILTS (Figure S1).
Genome-Scale Mass Spectrometry Proteomics
In parallel to transcriptome sequencing, the proteome of the same K052 cell population was analyzed by bottom-up mass spectrometry coupled to high-resolution, multidimensional chromatography to improve the detection of low abundance and rare peptides, whose selection for fragmentation was previously found to be limited by the finite sampling rate of current mass spectrometers.27 Tryptic peptides were generated using sequential LysC and trypsin proteolysis and fractionated off-line using high-pH reversed-phase (hRP) and strong-cation exchange (SCX) chromatography, leveraging the orthogonality of these separation modes to reverse-phase under acidic conditions.74 Each peptide fraction was then resolved by high-resolution nanoscale reverse-phase chromatography hyphenated via a nanoelectrospray ion source to the mass spectrometer. This strategy enabled the generation of a total of 2.8 million fragmentation spectra. To assess sampling efficiency, we used statistical database matching against UniProt to identify unique peptides and proteins at 1% FDR. Using a subsampling analysis, we observed that this strategy indeed maximized the sensitivity of detection of canonical proteins, though peptide sampling was apparently incompletely saturated (Figure S2A). Thus, high-resolution chromatography coupled with high-accuracy mass spectrometry and high-coverage transcriptome sequencing is suitable for genome-scale proteogenomics. Likewise, we found that high-coverage mRNA sequencing apparently saturated sampling of unique sequence reads obtained by transcriptomic analysis (Figure S2B).
Scoring Function Selection for Sensitive, Accurate, and Efficient Proteogenomic Discovery Using Spectral-Match Calibration
Having obtained genome-scale mass spectrometry data and the transcriptome-derived PGX database, we next sought to confirm accuracy of algorithms for scoring peptide–spectral matches and estimating FDR confidence. Such algorithms should ideally not only maximize sensitivity (i.e., the fraction of identified spectra) but also ensure high specificity, particularly when searching non-curated proteogenomic target databases that may contain erroneous sequences. To empirically assess sensitivity and specificity of these algorithms, we introduced two negative controls:75,76 (i) a set of mass spectra from a nonhuman proteome (E. coli) recorded under identical experimental parameters as the human K052 proteome, and (ii) a set of protein sequences from an evolutionarily divergent nonhuman species with minimal identity to the human proteome (in this case, the recently published proteome of A. loki archaebacteria).77 The negligible homology between the control E. coli and A. loki proteomes with the human one was confirmed by direct comparisons of the predicted tryptic peptides generated from each assembly (Figure 4A,B). Furthermore, we confirmed that the proteomes used as controls did not exhibit substantially different amino acid composition as compared to the human proteomes and were expected to produce tryptic peptides of similar lengths (based on frequency of K and R residues) and physicochemical properties (Figure S3).
Figure 4.

Sensitivity and specificity of mass spectrometry search algorithms. (A, B) Comparison of unique theoretical peptides in the experimental PGX proteome, canonical UniProt, and bacterial proteomes used as negative controls. (C) Sensitivity of tested algorithms expressed as the number of identified peptides. (D) Specificity of tested algorithms evaluated from the fraction of peptide–spectrum matches mapped to the negative controls. The PSM fraction mapped to A. loki is reported both in absolute terms (black) and normalized to take into account the relative sizes of the human and archaebacterial proteomes (shown in gray). Normalization was performed by multiplying the number of human peptides by the ratio of the A. loki and H. sapiens databases, expressed as the number of tryptic peptides.
For mass spectrometry search algorithms, we used four current state-of-the-art programs, chosen for their distinct methods for candidate sequence selection and FDR estimation: Sequest HT with Percolator as part of Proteome Discoverer,16,64,65 Byonic,66,67 MaxQuant,68,69 and PEAKS.70 For benchmarking purposes, we searched the experimental human K052 and negative control bacterial E. coli mass spectra against a concatenated database containing PGX and UniProt human databases, supplemented with sequences of negative control archaebacterial A. loki proteome and of common contaminants from cRAP.60 All searches were performed using identical search parameters at FDR < 0.01 at PSM, peptide, and protein level, as enabled by the specific algorithms. We assessed sensitivity based on the number of bona fide correct peptide identifications, mapping to either human or common contaminant proteins (Figure 4C and Table S1). We observed that Sequest HT, MaxQuant, and PEAKS had similar sensitivity, while Byonic showed superior sensitivity, as measured by the number of identified peptides mapping to the target database.
We estimated specificity based on the fraction of incorrect peptide identifications, corresponding to human sequences identified from E. coli spectra, or experimental human spectra matched to archaebacterial A. loki sequences (Figure 4D). Because of the difference in size between H. sapiens and A. loki proteomes, the latter could not be used directly as decoy database for conventional FDR calculation. However, to confirm accuracy of false-positive rate estimation by the target–decoy strategy, we empirically estimated the FDR by multiplying the number of observed incorrect PSMs by the ratio of the number of theoretical tryptic peptides in the H. sapiens and A. loki proteomes. This demonstrated that the empirical FDR deviated from the expected value of 1% using all tested mass spectrometry matching algorithms. In particular, the FDR appeared to be underestimated using Sequest HT, Byonic, and MaxQuant (all producing empirical FDR values in the 1.7–2.1 range), and over-estimated by PEAKS. In particular, Sequest and MaxQuant, which predominantly rely on accurate precursor mass measurements, were preferentially susceptible to in-correctly match human spectra to A. loki sequences, while Byonic and PEAKS, which prioritize candidate peptides based on de novo sequence tags, were more prone to incorrectly match decoy E. coli spectra to human sequences. Thus, in the context of proteogenomic analyses, in which the accuracy of the target database may be difficult to control, the latter approaches may be preferable. Despite its superior sensitivity and accuracy, the currently available implementation of PEAKS is not compatible with shared high-performance computing environments. Instead, the fully automated version of ProteomeGenerator uses the recently released Linux implementation of MaxQuant.71
Identification of Non-canonical Protein Isoforms Using ProteomeGenerator
Compelled by the superior accuracy of PEAKS, we next compared the peptides from K052 cells proteome identified using this algorithm against either PGX (i.e., the .fasta database file generated by ProteomeGenerator) or UniProt databases. All analyses were performed with identical search parameters, and global FDR < 0.01 at the PSM level. Based on the same set of 2 736 597 fragmentation spectra, we observed 611 275 and 621 960 peptide–spectrum matches (22% PSM rate) when searching PGX and UniProt databases, respectively (Figure 5A and Tables S2 and S3).
Figure 5.

Accurate proteome discovery using statistical target–decoy matching with spectral calibration. (A) Number of peptides identified (FDR < 0.01) based on matching spectra from K052 proteome against proteogenomic (PGX, red) and canonical (UniProt, gray) databases. (B) Overlap between the peptides identified in PGX (red) and UniProt (gray) databases. (C) Comparison of PEAKS scores for peptides identified in both PGX and UniProt databases. (D) PEAKS score distribution for peptides identified exclusively in PGX (red) and UniProt (gray) databases. (E) For peptides exclusively identified against the PGX database, PEAKS score distributions for peptides not mapping in UniProt (red) or present in the canonical database (gray). Boxes delimit the 25th and 75th percentiles, the middle line corresponds to the median, and whiskers correspond to the 5th and 95th percentiles. (F) PEAKS score distributions for peptides identified exclusively in PGX but also mapping in UniProt (gray) or exclusively mapping in the PGX database (red).
Most of these PSMs defined peptide sequences that were shared between the two databases (97% and 94% of the total PGX and UniProt identifications, respectively Figure 5B), and showed nearly identical confidence of identification (Figure 5C). A total of 7134 peptides were uniquely identified when searching against UniProt, although 999 (14%) of these sequences were encoded in the proteogenomic database. It is possible that the absence of these peptides was due to the limited sensitivity of mRNA sequencing, as previously described for analysis of HeLa and NCI-60 cells.13,78–81 The specific identification of the peptides encoded in the PGX database suggests that the greater diversity of sequences in UniProt may be due to the incorrect matching of homeometric peptides.82 This is consistent with the greater number of nearly isobaric theoretical peptides in UniProt, as compared to the proteogenomic database (Figure S4).
To compare the efficiency of ProteomeGenerator and QUILTS, we concatenated each database with the canonical UniProt human proteome and used it to score K052 spectra (Figure S5). ProteomeGenerator had a 3-fold higher number of discovered non-canonical peptides compared to QUILTS (2340 versus 752 peptides, respectively). In addition, 30% of the non-canonical peptides identified using QUILTS were also detected by ProteomeGenerator, indicating that, at least with respect to this comparison, ProteomeGenerator has similar sensitivity and superior specificity as QUILTS (Figure S5B).
The 3445 identified peptides unique to the proteogenomic database originated from inclusion of sample-specific protein sequences as well as increased statistical power of database spectral matching produced by the reduced search space,7 as was recently reported in proteogenomic context.80,81 In particular, we observed that 30% of peptide sequences specifically identified against the PGX database had no apparent counterparts in UniProt. However, 70% (2411 out of 3445) of PGX-specific identified peptides were apparently the result of increased statistical power. In particular, we observed that the reduced size of the sample-specific PGX database led to a lower PSM score threshold as compared with searches against Uniprot, as the score cutoff corresponding to FDR < 0.01 was 18.5 and 18.7 for searches against PGX and UniProt, respectively (Figure 5D–F). Supporting this idea, we noticed that searches against individual proteogenomic and canonical databases had superior sensitivity compared with searches against concatenated databases (Figure S5C).
To identify non-canonical protein isoforms, we analyzed the subset of peptides with sequences not mapping in UniProt, as prioritized based on the apparent statistical confidence of their identifications (PEAKS PSM score of >50). Analysis of these sequences using BLAST indicated that the majority (94%) mapped to isoforms annotated in the non-reviewed section of UniProt or RefSeq (Table S4). Notably, we also identified peptides corresponding to previously un-annotated isoforms of APH (APEH), YB-1 (YBX1), and MUNC13D (UNC13D). These isoforms were found to be consistent with alternative splicing and intron retention for APEH and MUNC13D (Figures 6A,B and S4), and peptide identification was confirmed by manual inspection of the mass fragmentation spectra (Figures 6C and S2).
Figure 6.

Identification of non-canonical protein isoforms using ProteomeGenerator. (A) Genome tracks of non-canonical APEH isoform generation via inclusion of an intronic sequence normally spliced in the canonical APEH isoform. (B) The K052-specific isoform of APEH contains a novel N-terminal sequence, with the splicing junction encompassed by peptide AGPDPGVSPAQVLLSEPEEAAALYR. Residues 35–276 of the protein sequences defined by ProteomeGenerator are identical to residues 4–245 of the canonical UniProt protein sequence. (C) Fragmentation spectrum of the peptide encompassing the novel splice junction, with diagnostic fragment ions and amino acid residues labeled. Italicized ion labels indicate ions with relative intensity above 25% of the maximum. Asterisks denote internal ions. (D) Peptide abundance as based on total fragment ion current for all identified APEH peptides (red: peptides from K052-specific sequence; gray: peptides from canonical sequence).
In the case of APEH, the non-canonical N-terminal domain produced by apparent intron retention was detected by mass spectrometry from two unique and independent peptides, including one spanning the non-canonical splice junction. Because no peptides corresponding to this region were detected from the canonical isoform, we used the total ion current of the fragmentation spectrum associated with this PSM to estimate the differential abundance of the novel and canonical APEH protein isoforms (Figure 6D). This analysis revealed that the identified non-canonical APEH isoform represents the majority of cellular APEH, identified specifically by ProteomeGenerator.
CONCLUSIONS
Here, we introduced an analytical framework for scalable de novo and reference-guided assembly of sample-specific proteomic databases based on mRNA sequencing and proteogenomic peptide–spectral matching. We designed ProteomeGenerator to produce sample-specific databases containing only proteins that are predicted to be expressed based on transcriptomic sequencing. As a result, PGX databases have markedly reduced search space as compared with canonical reference databases, such as UniProt, and workflows requiring the use of concatenated databases (such as QUILTS).27 As a result, the ProteomeGenerator workflow exhibits enhanced sensitivity using global FDR as confidence metric for statistical database matching.
Because no methods currently exist for an a priori definition of complete transcriptomes and proteomes, we confirmed the accuracy of ProteomeGenerator based on three measures: (i) the set of peptides identified using ProteomeGenerator largely overlaps with that obtained by conventional matching against consensus human proteomes; (ii) the identification of novel non-canonical peptides identified by ProteomeGenerator was supported by robust experimental evidence both at proteomic (i.e., complete fragmentation spectra) and transcriptomic (i.e., multiple sequencing reads) levels and were annotated in other non-reviewed or provisional databases; and (iii) the accuracy of peptide–spectral matching was confirmed by stringent bench-marking of scoring functions and their calibration using spectral and sequence negative controls.
ProteomeGenerator and related approaches are limited by the accuracy of transcriptome assembly, which is dependent on mRNA sequencing quality, depth, length, and strand specificity. We anticipate that ProteomeGenerator will benefit from increasing adoption of long-read and strand-specific RNA sequencing technologies. To this end, it will also be important to further improve the sensitivity of proteome sampling by peptide mass spectrometry, such as optimizing proteome proteolysis, its chromatographic resolution, and methods for de novo mass spectral identification.83,84 Lastly, ProteomeGenerator is implemented as a Snakemake workflow, enabling open, scalable, and facile discovery of sample-specific, non-canonical, and neomorphic biological proteomes. In addition, ProteomeGenerator is currently distributed as a Singularity container with automatic MaxQuant integration, enabling its one-step installation and execution in diverse computing environments. This should facilitate the discovery of natural variation in cellular and tissue proteomes, which can contribute to normal tissue development and its dysregulation in human disease such as cancer.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Fenyö and Kelly Ruggles for help with QUILTS. We also thank John Phillips for constructive discussion, Nathaniel Kwok for assistance with computer scripting, Wilfred Tang for assistance with Byonic searches, and Robert Holmes for computer server support.
Funding
This work was supported by the NIH R21 CA188881, R01 CA204396, P30 CA008748, the Burroughs Wellcome Fund, the Josie Robertson Investigator Program, the Rita Allen Foundation, the Alex’s Lemonade Stand Foundation, the American Society of Hematology, and the Gabrielle’s Angel Foundation (A.K.) and the American Italian Cancer Foundation (P.C.). A.Y. is supported by grants from the Aplastic Anemia and MDS International Foundation and the Lauri Strauss Leukemia Foundation. A.K. is the Damon Runyon-Richard Lumsden Foundation Clinical Investigator.
ABBREVIATIONS
- BCA
bicinchoninic acid
- FDR
false discovery rate
- LC
liquid chromatography
- MS
mass spectrometry
- PGX
proteogenomic
- PSM
peptide–spectral match
- RNA-seq
RNA sequencing
- RP
reversed-phase
- SCX
strong cation exchange
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00295.
Figures showing a comparison of proteogenomic databases, subsampling analysis of mass spectrometric and RNA sequencing data, a composition of target and negative control protein databases, the frequency of isobaric peptides generated by the PGX and UniProt databases, a comparison of peptides identified using as target database UniProt human proteome concatenated to ProteomeGenerator and QUILTS, and raw spectra for peptide AGPDPGVSPAQVLLSEPEEAAALYR (PDF)
Tables showing the calibration of peptide-spectral matching using negative controls, peptides matched to the PGX and UniProt databases, and identified peptides with PEAKS score higher than 50 and not mapping the reference UniProt database (ZIP)
The authors declare no competing financial interest.
RNA sequencing data are available from the NCBI BioProject repository with the accession code PRJNA427716. Mass spectrometry raw files and search results are publicly available through the ProteomeXchange data repository with identifiers PXD008381 and PXD008463.73
REFERENCES
- (1).International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409 (6822), 860–921. [DOI] [PubMed] [Google Scholar]
- (2).International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431 (7011), 931–945. [DOI] [PubMed] [Google Scholar]
- (3).Mortazavi A; Williams BA; McCue K; Schaeffer L; Wold B Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5 (7), 621–28. [DOI] [PubMed] [Google Scholar]
- (4).ICGC Breast Cancer Working Group, Oslo Breast Cancer Research Consortium. Direct Transcriptional Consequences of Somatic Mutation in Breast Cancer. Cell Rep. 2016, 16 (7), 2032–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Engström PG; Steijger T; Sipos B; Grant GR; Kahles A; Rätsch G; Goldman N; Hubbard TJ; Harrow J; Guigó R; Bertone P RGASP Consortium. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 2013, 10 (12), 1185–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Edwards AM; Isserlin R; Bader GD; Frye SV; Willson TM; Yu FH Too many roads not taken. Nature 2011, 470 (7333), 163–5. [DOI] [PubMed] [Google Scholar]
- (7).Nesvizhskii AI Proteogenomics: concepts, applications and computational strategies. Nat. Methods 2014, 11 (11), 1114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ruggles KV; Krug K; Wang X; Clauser KR; Wang J; Payne SH; Fenyo D; Zhang B; Mani DR Methods, Tools and Current Perspectives in Proteogenomics. Mol. Cell. Proteomics 2017, 16 (6), 959–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Aebersold R; Mann M Mass spectrometry-based proteomics. Nature 2003, 422 (6928), 198–207. [DOI] [PubMed] [Google Scholar]
- (10).Wilhelm M; Schlegl J; Hahne H; Gholami AM; Lieberenz M; Savitski MM; Ziegler E; Butzmann L; Gessulat S; Marx H; Mathieson T; Lemeer S; Schnatbaum K; Reimer U; Wenschuh H; Mollenhauer M; Slotta-Huspenina J; Boese J-H; Bantscheff M; Gerstmair A; Faerber F; Kuster B Mass spectrometry-based draft of the human proteome. Nature 2014, 509 (7502), 582–7. [DOI] [PubMed] [Google Scholar]
- (11).Hebert AS; Richards AL; Bailey DJ; Ulbrich A; Coughlin EE; Westphall MS; Coon JJ The one hour yeast proteome. Mol. Cell. Proteomics 2014, 13 (1), 339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).de Godoy LMF; Olsen JV; Cox J; Nielsen ML; Hubner NC; Fröhlich F; Walther TC; Mann M Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 2008, 455 (7217), 1251–4. [DOI] [PubMed] [Google Scholar]
- (13).Nagaraj N; Wiśniewski JR; Geiger T; Cox J; Kircher M; Kelso J; Pääbo S; Mann M Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol 2011, 7 (1), 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mann M; Wilm M Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem 1994, 66 (24), 4390–9. [DOI] [PubMed] [Google Scholar]
- (15).Shevchenko A; Wilm M; Vorm O; Mann M Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem 1996, 68 (5), 850–8. [DOI] [PubMed] [Google Scholar]
- (16).Eng JK; McCormack AL; Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 1994, 5 (11), 976–89. [DOI] [PubMed] [Google Scholar]
- (17).Elias JE; Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4 (3), 207–14. [DOI] [PubMed] [Google Scholar]
- (18).The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017, 45 (1), D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Pruitt KD; Harrow J; Harte RA; Wallin C; et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009, 19 (7), 1316–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Samandi SA; Roy V; Delcourt V; Lucier J-F; Gagnon J; Beaudoin MC; Vanderperre B; Breton M-A; Motard J; Jacques J-F; Brunelle M; Gagnon-Arsenault I; Fournier I; Ouangraoua A; Hunting DJ; Cohen AA; Landry CR; Scott MS; Roucou X Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. eLife 2017, 6, 27860DOI: 10.7554/eLife.27860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Couso J-P; Patraquim P Classification and function of small open reading frames. Nat. Rev. Mol. Cell Biol 2017, 18 (9), 575–89. [DOI] [PubMed] [Google Scholar]
- (22).Dvinge H; Bradley RK Widespread intron retention diversifies most cancer transcriptomes. Genome Med. 2015, 7 (1), 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Negrini S; Gorgoulis VG; Halazonetis TD Genomic instability-an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol 2010, 11 (3), 220–8. [DOI] [PubMed] [Google Scholar]
- (24).DeBoever C; Ghia EM; Shepard PJ; Rassenti L; Barrett CL; Jepsen K; Jamieson CHM; Carson D; Kipps TJ; Frazer KA Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput. Biol 2015, 11 (3), e1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Krug K; Popic S; Carpy A; Taumer C; Macek B Construction and assessment of individualized proteogenomic databases for large-scale analysis of nonsynonymous single nucleotide variants. Proteomics 2014, 14 (23), 2699–708. [DOI] [PubMed] [Google Scholar]
- (26).Li Y; Wang X; Cho J-H; Shaw TI; Wu Z; Bai B; Wang H; Zhou S; Beach TG; Wu G; Zhang J; Peng J JUMPg: An Integrative Proteogenomics Pipeline Identifying Unannotated Proteins in Human Brain and Cancer Cells. J. Proteome Res 2016, 15 (7), 2309–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ruggles KV; Tang Z; Wang X; Grover H; Askenazi M; Teubl J; Cao S; McLellan MD; Clauser KR; Tabb DL; Mertins P; Slebos R; Erdmann-Gilmore P; Li S; Gunawardena HP; Xie L; Liu T; Zhou J-Y; Sun S; Hoadley KA; Perou CM; Chen X; Davies SR; Maher CA; Kinsinger CR; Rodland KD; Zhang H; Zhang Z; Ding L; Townsend RR; Rodriguez H; Chan D; Smith RD; Liebler DC; Carr SA; Payne S; Ellis MJ; Fenyo D An Analysis of the Sensitivity of Proteogenomic Mapping of Somatic Mutations and Novel Splicing Events in Cancer. Mol. Cell. Proteomics 2016, 15 (3), 1060–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wang X; Zhang B customProDB: an R package to generate customized protein databases from RNA-Seq data for proteomics search. Bioinformatics 2013, 29 (24), 3235–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wen B; Xu S; Zhou R; Zhang B; Wang X; Liu X; Xu X; Liu S PGA: an R/Bioconductor package for identification of novel peptides using a customized database derived from RNA-Seq. BMC Bioinf. 2016, 17 (1), 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Woo S; Cha SW; Merrihew G; He Y; Castellana N; Guest C; MacCoss M; Bafna V Proteogenomic database construction driven from large scale RNA-seq data. J. Proteome Res. 2014, 13 (1), 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zickmann F; Renard BY MSProGene: integrative proteogenomics beyond six-frames and single nucleotide polymorphisms. Bioinformatics 2015, 31 (12), 106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Jaffe JD; Berg HC; Church GM Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics 2004, 4 (1), 59–77. [DOI] [PubMed] [Google Scholar]
- (33).Evans VC; Barker G; Heesom KJ; Fan J; Bessant C; Matthews DA De novo derivation of proteomes from transcriptomes for transcript and protein identification. Nat. Methods 2012, 9 (12), 1207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Liu Y; Gonzàlez-Porta M; Santos S; Brazma A; Marioni JC; Aebersold R; Venkitaraman AR; Wickramasinghe VO Impact of Alternative Splicing on the Human Proteome., Cell Rep. 2017, 20 (5), 1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Castellana NE; Payne SH; Shen Z; Stanke M; Bafna V; Briggs SP Discovery and revision of Arabidopsis genes by proteogenomics. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (52), 21034–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Omasits U; Quebatte M; Stekhoven DJ; Fortes C; Roschitzki B; Robinson MD; Dehio C; Ahrens CH Directed shotgun proteomics guided by saturated RNA-seq identifies a complete expressed prokaryotic proteome. Genome Res. 2013, 23 (11), 1916–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Menschaert G; Van Criekinge W; Notelaers T; Koch A; Crappé J; Gevaert K; Van Damme P Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol. Cell. Proteomics 2013, 12 (7), 1780–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Khatun J; Yu Y; Wrobel JA; Risk BA; Gunawardena HP; Secrest A; Spitzer WJ; Xie L; Wang L; Chen X; Giddings MC Whole human genome proteogenomic mapping for ENCODE cell line data: identifying protein-coding regions. BMC Genomics 2013, 14 (1), 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhang H; Liu T; Zhang Z; Payne SH; Zhang B; McDermott JE; Zhou J-Y; Petyuk VA; et al. CPTAC Investigators. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166 (3), 755–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhang B; Wang J; Wang X; Zhu J; Liu Q; Shi Z; Chambers MC; Zimmerman LJ; Shaddox KF; Kim S; Davies SR; Wang S; Wang P; Kinsinger CR; Rivers RC; Rodriguez H; Townsend RR; Ellis MJC; Carr SA; Tabb DL; Coffey RJ; Slebos RJC; Liebler DC NCI CPTAC. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513 (7518), 382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Mertins P; Mani DR; Ruggles KV; Gillette MA; Clauser KR; Wang P; et al. NCI CPTAC, “Proteogenomics connects somatic mutations to signalling in breast cancer.,. Nature 2016, 534 (7605), 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Rolland DCM; Basrur V; Jeon Y-K; McNeil-Schwalm C; Fermin D; Conlon KP; Zhou Y; Ng SY; Tsou C-C; Brown NA; Thomas DG; Bailey NG; Omenn GS; Nesvizhskii AI; Root DE; Weinstock DM; Faryabi RB; Lim MS; Elenitoba-Johnson KSJ Functional proteogenomics reveals biomarkers and therapeutic targets in lymphomas. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (25), 6581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Branca RMM; Orre LM; Johansson HJ; Granholm V; Huss M; Pérez-Bercoff Å; Forshed J; Kall L; Lehtiö J HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat. Methods 2014, 11 (1), 59–62. [DOI] [PubMed] [Google Scholar]
- (44).Komor MA; Pham T; Hiemstra AC; Piersma SR; Bolijn AS; Schelfhorst T; Delis-van Diemen PM; Tijssen M; Sebra RP; Ashby M; Meijer GA; Jimenez CR; Fijneman RJA Identification of differentially expressed splice variants by the proteogenomic pipeline Splicify. Mol. Cell. Proteomics 2017, 16 (10), 1850–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Laumont CM; Daouda T; Laverdure J-P; Bonneil É ; Caron-Lizotte O; Hardy M-P; Granados DP; Durette C; Lemieux S; Thibault P; Perreault C Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun 2016, 7, 10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Soares NC; Spät P; Krug K; Macek B Global dynamics of the Escherichia coli proteome and phosphoproteome during growth in minimal medium. J. Proteome Res 2013, 12 (6), 2611–21. [DOI] [PubMed] [Google Scholar]
- (47).Park H; Bae J; Kim H; Kim S; Kim H; Mun D-G; Joh Y; Lee W; Chae S; Lee S; Kim HK; Hwang D; Lee S-W; Paek E Compact variant-rich customized sequence database and a fast and sensitive database search for efficient proteogenomic analyses. Proteomics 2014, 14 (23), 2742–9. [DOI] [PubMed] [Google Scholar]
- (48).Wang X; Slebos RJC; Wang D; Halvey PJ; Tabb DL; Liebler DC; Zhang B Protein identification using customized protein sequence databases derived from RNA-Seq data. J. Proteome Res 2012, 11 (2), 1009–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Cesnik AJ; Shortreed MR; Sheynkman GM; Frey BL; Smith LM Human Proteomic Variation Revealed by Combining RNA-Seq Proteogenomics and Global Post-Translational Modification (G-PTM) Search Strategy. J. Proteome Res. 2016, 15 (3), 800–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kim E; Ilagan JO; Liang Y; Daubner GM; Lee SC-W; Ramakrishnan A; et al. SRSF2Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27 (5), 617–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Köster J; Rahmann S Snakemake-a scalable bioinformatics workflow engine. Bioinformatics 2012, 28 (19), 2520–2. [DOI] [PubMed] [Google Scholar]
- (52).Dobin A; Davis CA; Schlesinger F; Drenkow J; Zaleski C; Jha S; Batut P; Chaisson M; Gingeras TR STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29 (1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Li H; Handsaker B; Wysoker A; Fennell T; Ruan J; Homer N; Marth G; Abecasis G; Durbin R 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25 (16), 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Pertea M; Pertea GM; Antonescu CM; Chang T-C; Mendell JT; Salzberg SL StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol 2015, 33 (3), 290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Haas BJ; Papanicolaou A; Yassour M; Grabherr M; Blood PD; Bowden J; Couger MB; Eccles D; Li B; Lieber M; MacManes MD; Ott M; Orvis J; Pochet N; Strozzi F; Weeks N; Westerman R; William T; Dewey CN; Henschel R; LeDuc RD; Friedman N; Regev A De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc 2013, 8 (8), 1494–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Altschul SF; Gish W; Miller W; Myers EW; Lipman DJ Basic local alignment search tool. J. Mol. Biol 1990, 215 (3), 403–10. [DOI] [PubMed] [Google Scholar]
- (57).Robinson JT; Thorvaldsdóttir H; Winckler W; Guttman M; Lander ES; Getz G; Mesirov JP Integrative genomics viewer. Nat. Biotechnol 2011, 29 (1), 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Rice P; Longden I; Bleasby A EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000, 16 (6), 276–7. [DOI] [PubMed] [Google Scholar]
- (59).Kurtzer GM; Sochat V; Bauer MW Singularity: Scientific containers for mobility of compute. PLoS One 2017, 12 (5), e0177459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Mellacheruvu D; Wright Z; Couzens AL; Lambert J-P; St-Denis NA; Li T; Miteva YV; Hauri S; et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10 (8), 730–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Dhabaria A; Cifani P; Reed C; Steen H; Kentsis A A High-Efficiency Cellular Extraction System for Biological Proteomics. J. Proteome Res 2015, 14 (8), 3403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Cifani P; Kentsis A High sensitivity quantitative proteomics using automated multidimensional nano-flow chromatography and accumulated ion monitoring on quadrupole-Orbitrap-linear ion trap mass spectrometer. Mol. Cell. Proteomics 2017, 16 (11), 2006–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ficarro SB; Zhang Y; Carrasco-Alfonso MJ; Garg B; Adelmant G; Webber JT; Luckey CJ; Marto JA Online nanoflow multidimensional fractionation for high efficiency phosphopeptide analysis. Mol. Cell. Proteomics 2011, 10 (11), O111011064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Käll L; Canterbury JD; Weston J; Noble WS; MacCoss MJ Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4 (11), 923–5. [DOI] [PubMed] [Google Scholar]
- (65).Spivak M; Weston J; Bottou L; Käll L; Noble WS Improvements to the percolator algorithm for Peptide identification from shotgun proteomics data sets. J. Proteome Res. 2009, 8 (7), 3737–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Bern M; Cai Y; Goldberg D Lookup peaks: a hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry. Anal. Chem 2007, 79 (4), 1393–400. [DOI] [PubMed] [Google Scholar]
- (67).Bern MW; Kil YJ Two-dimensional target decoy strategy for shotgun proteomics. J. Proteome Res 2011, 10 (12), 5296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26 (12), 1367–72. [DOI] [PubMed] [Google Scholar]
- (69).Cox J; Neuhauser N; Michalski A; Scheltema RA; Olsen JV; Mann M Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res 2011, 10 (4), 1794–805. [DOI] [PubMed] [Google Scholar]
- (70).Zhang J; Xin L; Shan B; Chen W; Xie M; Yuen D; Zhang W; Zhang Z; Lajoie GA; Ma B PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 2012, 11 (4), M111010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Sinitcyn P; Tiwary S; Rudolph J; Gutenbrunner P; Wichmann C; Yılmaz S; Hamzeiy H; Salinas F; Cox J MaxQuant goes Linux. Nat. Methods 2018, 15 (6), 401. [DOI] [PubMed] [Google Scholar]
- (72).McIlwain S; Tamura K; Kertesz-Farkas A; Grant CE; Diament B; Frewen B; Howbert JJ; Hoopmann MR; Käll L; Eng JK; MacCoss MJ; Noble WS Crux: rapid open source protein tandem mass spectrometry analysis. J. Proteome Res 2014, 13 (10), 4488–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Vizcaíno JA; Côté RG; Csordas A; Dianes JA; Fabregat A; Foster JM; Griss J; Alpi E; Birim M; Contell J; O’Kelly G; Schoenegger A; Ovelleiro D; Pérez-Riverol Y; Reisinger F; Ríos D; Wang R; Hermjakob H The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2012, 41, D1063–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Gilar M; Olivova P; Daly AE; Gebler JC Orthogonality of separation in two-dimensional liquid chromatography. Anal. Chem 2005, 77 (19), 6426–34. [DOI] [PubMed] [Google Scholar]
- (75).Granholm V; Noble WS; Käll L On using samples of known protein content to assess the statistical calibration of scores assigned to peptide-spectrum matches in shotgun proteomics. J. Proteome Res 2011, 10 (5), 2671–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Keich U; Noble WS On the importance of well-calibrated scores for identifying shotgun proteomics spectra. J. Proteome Res 2015, 14 (2), 1147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Spang A; Saw JH; Jørgensen SL; Zaremba-Niedzwiedzka K; Martijn J; Lind AE; van Eijk R; Schleper C; Guy L; Ettema TJG Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 2015, 521 (7551), 173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Helmy M; Sugiyama N; Tomita M; Ishihama Y Oncoproteogenomics: a novel approach to identify cancer-specific mutations combining proteomics and transcriptome deep sequencing. Genome Biol. 2010, 11 (1), P17. [Google Scholar]
- (79).Gholami AM; Hahne H; Wu Z; Auer FJ; Meng C; Wilhelm M; Kuster B Global proteome analysis of the NCI-60 cell line panel. Cell Rep. 2013, 4 (3), 609–20. [DOI] [PubMed] [Google Scholar]
- (80).Karpova MA; Karpov DS; Ivanov MV; Pyatnitskiy ML; Chernobrovkin AL; Lobas AA; Lisitsa AV; Archakov AI; Gorshkov MV; Moshkovskii SA Exome-Driven Characterization of the Cancer Cell Lines at the Proteome Level: The NCI-60 Case Study. J. Proteome Res 2014, 13 (12), 5551–60. [DOI] [PubMed] [Google Scholar]
- (81).Alfaro JA; Ignatchenko A; Ignatchenko V; Sinha A; Boutros PC; Kislinger T Detecting protein variants by mass spectrometry: a comprehensive study in cancer cell-lines. Genome Med. 2017, 9 (62), 1DOI: 10.1186/s13073-017-0454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Frank AM; Savitski MM; Nielsen ML; Zubarev RA; Pevzner PA De novo peptide sequencing and identification with precision mass spectrometry. J. Proteome Res 2007, 6 (1), 114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Wang X; Codreanu SG; Wen B; Li K; Chambers M; Liebler DC; Zhang B Detection of proteome diversity resulted from alternative splicing is limited by trypsin cleavage specificity. Mol. Cell. Proteomics 2018, 17, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Zhou F; Lu Y; Ficarro SB; Adelmant G; Jiang W; Luckey CJ; Marto JA Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat. Commun 2013, 4, 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.