

Abstract

Self-assembling peptides and oligonucleotides have given rise to synthetic materials with several applications in nanotechnology. Aggregation of synthetic oligosaccharides into well-defined architectures has not been reported even though natural polysaccharides, such as cellulose and chitin, are key structural components of biomaterials. Here, we report that six synthetic oligosaccharides, ranging from dimers to hexamers, self-assemble into nanostructures of varying morphologies and emit within the visible spectrum in an excitation-dependent manner. Well-defined differences in chain length, monomer modification, and aggregation methods yield glycomaterials with distinct shapes and properties. The excitation-dependent fluorescence in a broad range within the visible spectrum illustrates their potential for use in optical devices and imaging applications. We anticipate that our systematic approach of studying well-defined synthetic oligosaccharides will form the foundation of our understanding of carbohydrate interactions in nature.
Introduction
Simple peptides1 and nucleic acids2 can spontaneously self-assemble to form defined supramolecular patterns. These supramolecular architectures are the essence of modern bionanotechnology, with implications in the medical3 and energy1d,4 fields. The discovery that peptide dimers (i.e., diphenyl alanine) self-assemble provided fundamental insights into the progression of important diseases.5 The main limitation to the use of these systems is often associated with the modest quantities that can be produced. In contrast, natural polysaccharides comprise 80% of biomass, where they serve mainly structural roles.6 These materials, including cellulose7 and chitin,8 have a strong tendency to aggregate in well-defined architectures with different physical properties. Chemical modification tunes polysaccharide properties9 to serve as biocompatible,6,7c cheap, and renewable self-assembling materials for application in nanotechnology,10 optical components,7b,11 drug delivery systems,12 and tissue engineering.8a However, the use of polysaccharide materials is limited by poor quality control and reproducibility, owing to the polydispersity of chain length and modifications. While synthetic oligosaccharides should be able to self-assemble into tunable materials, this process has not been observed for structurally defined oligosaccharides, as access to pure glycans has been challenging. Chemical synthesis of oligosaccharides provides an attractive alternative to the modification of natural polysaccharides; however, it was extremely laborious prior to recent advances in automated synthesis.13 Rapid access to synthetic oligo- and polysaccharides provided material for systematic structural studies, showing that synthetic hexasaccharides may adopt defined shapes in solution.14
Here, we report that synthetic oligosaccharides self-assemble into defined structures. The systematic approach confirmed that differences in chain length and modification yield glycomaterials with distinct shapes and properties. This finding is particularly important in the prospective of creating novel carbohydrate materials with tunable properties. Moreover, these materials exhibit unexpected excitation-dependent intrinsic optical properties that can expand the applications of these materials even further and may result in new, cheap, and biocompatible optical devices. The dramatic differences in the aggregate morphologies stress the importance of a better knowledge of glycan presentation in biological systems, where carbohydrate–carbohydrate interactions regulate several cellular processes.15 We suggest that our approach, based on the study of the interaction of well-defined synthetic oligosaccharides, could shed light upon the rules that regulate cellular recognition and uptake.
Results and Discussion
Self-Assemby
Hexamer 4 (Figure 1A) proved so poorly soluble in many organic solvents that further chemical manipulations are impossible, while a similar compound that carries fewer benzyl ethers (i.e., hexamer 5) encountered fewer solubility and reactivity issues.14 These differences likely are a consequence of the formation of supramolecular structures due to strong intermolecular interactions, such as hydrogen bonding, together with π–π interactions of the benzyl ether modification. Exploiting such interactions to drive the self-assembly should give rise to novel oligosaccharide materials. Three dimers (1–3) as well as one additional hexasaccharide (6) were prepared to probe the influence of chain length, linkage, and modification on self-assembly (Figure 1A). Supramolecular aggregation was induced by slow dialysis (D) or fast solvent switch (S).16 Oligosaccharides dissolved in a dimethylacetamide (DMAc)/water mixture and dialyzed against water aggregated into nanoparticles with diameters of 40–60 nm (Figure 2A, a–f). These particles exist in solution, as confirmed by cryo-SEM of 2-D (Figure S1) and dynamic light scattering (DLS) measurements (Figure S2). Direct injection of water into a glycan solution in HFIP (fast solvent switch) results in faster mixing, higher oligosaccharide concentration, and altered self-assembly behavior (Figure 2A, g–l).17 Needle-like structures were found for 2-S-HFIP (5–10 μm length, 10–50 nm height, and 100–500 nm width, Figure 2A, h, and Figure S3) and a spheroidal architecture (1–2 μm diameter) for the hexamer 5-S-HFIP analogue (Figure 2A, k). These supramolecular structures were stable for 1 month under ambient conditions and resisted dilution and sonication (Figure S4). 1-S-HFIP assembled into a mixture of rods and toroid structures (Figure 2A, g), while 4-S-DMAc formed clusters of nanoparticles (Figure 2A, j). Differences in oligosaccharide structure such as linkage and modification patterns fundamentally affect the material morphology as 3-S-HFIP (Figure 2A, i) and 6-S-DMAc (Figure 2A, l) aggregated randomly and did not form any ordered supramolecular structure. Compounds 3 and 6 are based on a fairly rigid 1,4 glycosidic linkage (secondary alcohol) and therefore can adopt a limited number of conformations in solution. The flexibility of the 1,6 linkage allows for higher conformational diversity, permitting the formation of fundamentally different nanostructures.
Figure 1.

Chemical structure of well-defined oligosaccharides (A) and illustration of sample preparation methods (B). The sample names indicate the sugar oligomer (e.g., 2), the assembly method (D, S, or F), and the solvent used (e.g., HFIP). For example, 2-S-HFIP means compound 2 prepared by the solvent switch method with HFIP as good solvent.
Figure 2.

(A) Supramolecular structure formation with two different methods. (a–f) TEM images (scale bars: 100 nm) of samples prepared by the dialysis method (0.1 mg mL–1) for (a) 1-D, (b) 2-D, (c) 3-D, (d) 4-D*, (e) 5-D, and (f) 6-D*. *0.01 mg mL–1 due to poor solubility of the starting material. (g–l) SEM images (scale bars: 2 μm) of samples prepared by the solvent switch method for (g) 1-S-HFIP-low (0.1 mg mL–1), (h) 2-S-HFIP, (i) 3-S-HFIP, (j) 4-S-DMAc, (k) 5-S-HFIP, and (l) 6-S-DMAc. (B) Screening of assembly conditions for compound 2. (a) TEM image (scale bar: 500 nm) for 2-D-high (2 mg mL–1). (b–f) SEM images (scale bars: 2 μm) for (b) 2-S-HFIP-low (0.1 mg mL–1), (c) 2-S-HFIP-20%, (d) 2-S-iPrOH-20%, (e) 2-S-DMAc, and (f) 2-S-Ace-20%. If not mentioned, the standard concentration for the solvent switch method (S) is 2 mg mL–1 and the content of organic solvent is 2%.
The effects of assembly conditions on structure were studied in detail using dimer 2 that forms well-defined, disperse, and stable needle-like structures (Figure 2B). 2-S-HFIP presents an ordered morphology, as it showed intense birefringence under polarized light, typical of anisotropic materials (Figure S7). Moreover, staining with Congo red,18 a commonly used dye to detect highly ordered amyloid fibrils, gave intense gold-green birefringence (Figure S8). Dialysis using a higher concentration of 2 (2 mg mL–1) led to the formation of nanofibers (Figure 2B, a), likely due to the further association of the spherical particles existing in the diluted solution. The solvent exchange method generated longer needles when a lower concentration of compound 2 was employed (0.1 mg mL–1) (Figure 2B, b). A higher HFIP content (20%) did not change the shape or length of the supramolecular structures (Figure 2B, c). In this case, the selective solvation properties of HFIP, in a HFIP–H2O system,19 result in a similar local HFIP concentration, limiting aggregation diversity. A similar elongated morphology was obtained when isopropyl alcohol was used instead of HFIP (Figure 2B, d) and a gel-like microwire material was obtained in acetone (Figure 2B, f). The diversity observed is ascribed to the different conformations adopted by compound 2, when solvated by different solvents. In particular, the well-known ability of HFIP to cluster the hydrophobic regions of peptides and affect their folding (HFIP-induced enhancement of the hydrophobic effect)19,20 is responsible for the dramatic differences of the generated nanostructures.
Real-Time Measurement
The self-assembly of 2-S-HFIP was captured in real-time using bright-field microscopy (Figure 3, movie S1) by injecting a freshly prepared solution into a cell counting slide. Needle-like structures diffuse from the HFIP droplets containing the oligosaccharide into the surrounding water. The contact between the needles and a second HFIP droplet (Figure 3, time 06:52) disrupts the droplet to release the oligosaccharide and results in further needle growth. Surprisingly, glycan-containing HFIP droplets are intensely fluorescent. We believe that this phenomenon is the result of the formation of supramolecular chromophores within the material, as previously observed for self-assembled peptides, nucleic acids, and amino acids.17,21 An extended π-conjugation system and/or charge delocalization through a dense hydrogen-bonding network are generally responsible for this behavior.21b
Figure 3.
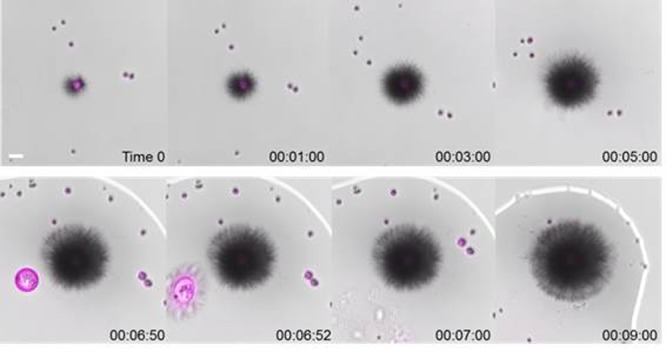
Real-time merged bright-field (scale of gray) and fluorescence (magenta) images illustrating the self-assembly process for 2-S-HFIP. Excitation wavelength at 405 nm and detection range 410–676 nm (scale bar: 20 μm).
Photophysical Characterization
Confocal microscopy analysis of different morphologies revealed that thin films prepared by direct evaporation of a glycan solution in HFIP on a slide glass (2-F-HFIP) emit strongly in four different channels (Figure 4A) upon visible light irradiation. Films prepared in other organic solvents showed a similar fluorescence behavior (Figure S10). Aggregates obtained via the solvent switch method are only weakly emissive (Figure 4A). This observation agrees with the supramolecular chromophore hypothesis, since emission intensity is strong in organic solvents, where a dense H-bonding network is favored and quenching occurs when the H-bonding pattern is disrupted by water. The morphology of these materials was further probed with X-ray powder diffraction (XRD) (Figure 4B). 2-S-HFIP exhibited sharp peaks, as typical for crystalline structures; in contrast, 2-F-HFIP shows broad peaks. This confirms the drastic change in morphology upon interaction with water (2-S-HFIP). To better evaluate the causes of this phenomenon, compounds 7–11 were prepared. To probe the importance of aromatic groups for the emissive behavior, compound 7 was synthesized. This amphiphilic, partially methylated analogue allows for the formation of a dense hydrogen bonding network, in the absence of aromatic groups. Upon film formation (7-F-HFIP), compound 7 showed a similar optical behavior, confirming that the optical properties are not merely a result of π–π stacking.
Figure 4.

(A) Confocal microscopy images of 2 prepared by HFIP film forming F (scale bars: 100 μm), solvent switch S (scale bars: 10 μm), and compounds 7–11 prepared by the film-forming method (scale bars: 100 μm) in four different channels (blue(ex/em), 405/451 nm; green, 488/529 nm; yellow, 561/597 nm; red, 633/709 nm). (B) XRD profiles of 2-F-HFIP (red) and 2-S-HFIP (black) and compounds 7–11.
Compounds 8–10 are fully functionalized, blocking the formation of a dense hydrogen bonding network within the material. Different substituents (Bn vs Me vs Ac) were tested. Surprisingly, confocal microscopy analysis showed emissive behavior for compounds 8-F-HFIP and 9-F-HFIP. We suspect that such compounds, even in the absence of a strong hydrogen bonding network, maintain a self-organization tendency. On the other hand, the per-acetylated analogue 10 as well as the fully deprotected compound 11 showed no emission. XRD analysis of all of the materials suggested a correlation between the broad XRD profile and the emissive behavior. Similarly, the appearance of sharp peaks in the XRD profiles, indicating high crystallinity, is associated with emission quenching.
Further photophysical characterization showed a broad absorption band for compound 2-F-HFIP, associated with the formation of new self-assembled entities upon film formation. The broadening of the absorption spectrum is not observed for compound 2 in solution, nor for the low emissive, crystalline sample 2-S-HFIP (Figure 5A). Excitation spectra (Figures S13 and S14) confirmed that the emissive species are linked to this spectral region (350–500 nm). Emission quantum yield was calculated for 2-F-HFIP (Φ(λex=360nm) = 0.85 ± 0.01%). Moreover, unlike commonly used dyes, where the emission peak position is independent of the excitation wavelength, the emission spectrum of 2-F-HFIP is drastically affected by the excitation wavelength (Figure 5B). A broad fluorescence emission profile was observed with maxima shifting from 410 to 490 nm as the excitation is changed from 340 to 410 nm. This red edge excitation shift (REES) is a common phenomenon observed in graphene oxide,21c ionic liquids,21d and highly ordered assemblies,17 suggesting potential applications of self-assembling oligosaccharides for optical devices, semiconductors, and nanotechnology.1d,17,21a,21b
Figure 5.

(A) Absorption spectra of 2-F-HFIP, 2-S-HFIP (recorded for the solid samples), and compound 2 in HFIP solution. (B) Normalized emission spectra of 2-F-HFIP at excitation wavelengths of 340, 350, 360, 370, 380, 390, 400, and 410, showing the red shifting of the emission maxima. Spectra acquired at RT.
Conclusions
In conclusion, we successfully generated supramolecular structures from fully synthetic well-defined oligosaccharides and demonstrated that the fine-tuning of the oligosaccharide structure has a tremendous effect on the material morphology. The three dimer and hexamer analogues with different glycosidic linkages and protective group patterns form similar nanospheres when generated by the slow dialysis method, whereas distinctive microstructures are obtained with the fast solvent switch method. These compounds show unique optical properties such as broad emission profiles and red edge excitation shift. Further studies to modulate the fluorescent properties of such materials are currently underway, with potential applications for optical devices and nanotechnology. These findings suggest that synthetic oligosaccharides are viable substrates for the fundamental study of the forces that guide the polysaccharide aggregation in nature. For example, tuning glycomaterial properties through the synthesis of well-defined structures will be relevant for drug delivery systems, where carbohydrate–carbohydrate interactions play a significant role in cellular uptake.
Acknowledgments
We thank the Max-Planck Society, the Minerva Fast Track Program, the MPG-FhG Cooperation Project Glyco3Dysplay, and the Clore Scholarship program for generous financial support. We thank Heike Runge, Dr. Oleksandr Savatieiev, Maria Hagen, Michaela Eder, Felix Fuchsberger, Eva Settels, Felix Hentschel, Olaf Niemeyer, Ingrid Zenke, Daniel Werner, Dr. Richard J. Fair, Alonso Pardo-Vargas, and Reinhild Dünnebacke for technical support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b11882.
Author Contributions
⊥ Y.Y., S.G.: These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- a Ghadiri M. R.; Granja J. R.; Milligan R. A.; McRee D. E.; Khazanovich N. Self-assembling organic nanotubes based on a cyclic peptide architecture. Nature 1993, 366, 324. 10.1038/366324a0. [DOI] [PubMed] [Google Scholar]; b Hartgerink J. D.; Beniash E.; Stupp S. I. Self-Assembly and Mineralization of Peptide-Amphiphile Nanofibers. Science 2001, 294, 1684–1688. 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]; c Bera S.; Mondal S.; Rencus-Lazar S.; Gazit E. Organization of Amino Acids into Layered Supramolecular Secondary Structures. Acc. Chem. Res. 2018, 51, 2187–2197. 10.1021/acs.accounts.8b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tao K.; Makam P.; Aizen R.; Gazit E. Self-assembling peptide semiconductors. Science 2017, 358, eaam9756. 10.1126/science.aam9756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rothemund P. W. K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]; b Dietz H.; Douglas S. M.; Shih W. M. Folding DNA into Twisted and Curved Nanoscale Shapes. Science 2009, 325, 725–730. 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ke Y.; Ong L. L.; Shih W. M.; Yin P. Three-Dimensional Structures Self-Assembled from DNA Bricks. Science 2012, 338, 1177–1183. 10.1126/science.1227268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Adler-Abramovich L.; Vaks L.; Carny O.; Trudler D.; Magno A.; Caflisch A.; Frenkel D.; Gazit E. Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat. Chem. Biol. 2012, 8, 701. 10.1038/nchembio.1002. [DOI] [PubMed] [Google Scholar]; b Webber M. J.; Appel E. A.; Meijer E. W.; Langer R. Supramolecular biomaterials. Nat. Mater. 2016, 15, 13. 10.1038/nmat4474. [DOI] [PubMed] [Google Scholar]; c Hu Q.; Li H.; Wang L.; Gu H.; Fan C. DNA Nanotechnology-Enabled Drug Delivery Systems. Chem. Rev. 2018, 10.1021/acs.chemrev.7b00663. [DOI] [PubMed] [Google Scholar]
- a Tao K.; Fan Z.; Sun L.; Makam P.; Tian Z.; Ruegsegger M.; Shaham-Niv S.; Hansford D.; Aizen R.; Pan Z.; Galster S.; Ma J.; Yuan F.; Si M.; Qu S.; Zhang M.; Gazit E.; Li J. Quantum confined peptide assemblies with tunable visible to near-infrared spectral range. Nat. Commun. 2018, 9, 3217. 10.1038/s41467-018-05568-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lan X.; Wang Q. DNA-programmed self-assembly of photonic nanoarchitectures. NPG Asia Mater. 2014, 6, e97. 10.1038/am.2014.16. [DOI] [Google Scholar]
- Reches M.; Gazit E. Casting Metal Nanowires Within Discrete Self-Assembled Peptide Nanotubes. Science 2003, 300, 625–627. 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- Klemm D.; Heublein B.; Fink H.-P.; Bohn A. Cellulose: Fascinating Biopolymer and Sustainable Raw Material. Angew. Chem., Int. Ed. 2005, 44, 3358–3393. 10.1002/anie.200460587. [DOI] [PubMed] [Google Scholar]
- a Habibi Y.; Lucia L. A.; Rojas O. J. Cellulose Nanocrystals: Chemistry, Self-Assembly, and Applications. Chem. Rev. 2010, 110, 3479–3500. 10.1021/cr900339w. [DOI] [PubMed] [Google Scholar]; b Parker R. M.; Guidetti G.; Williams C. A.; Zhao T.; Narkevicius A.; Vignolini S.; Frka-Petesic B. The Self-Assembly of Cellulose Nanocrystals: Hierarchical Design of Visual Appearance. Adv. Mater. 2018, 30, 1704477. 10.1002/adma.201704477. [DOI] [PubMed] [Google Scholar]; c Klemm D.; Kramer F.; Moritz S.; Lindström T.; Ankerfors M.; Gray D.; Dorris A. Nanocelluloses: A New Family of Nature-Based Materials. Angew. Chem., Int. Ed. 2011, 50, 5438–5466. 10.1002/anie.201001273. [DOI] [PubMed] [Google Scholar]; d Wang P.-X.; Hamad W. Y.; MacLachlan M. J. Structure and transformation of tactoids in cellulose nanocrystal suspensions. Nat. Commun. 2016, 7, 11515. 10.1038/ncomms11515. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wang P.-X.; Hamad W. Y.; MacLachlan M. J. Polymer and Mesoporous Silica Microspheres with Chiral Nematic Order from Cellulose Nanocrystals. Angew. Chem., Int. Ed. 2016, 55, 12460–12464. 10.1002/anie.201606283. [DOI] [PubMed] [Google Scholar]
- a Hassanzadeh P.; Kharaziha M.; Nikkhah M.; Shin S. R.; Jin J.; He S.; Sun W.; Zhong C.; Dokmeci M. R.; Khademhosseini A.; Rolandi M. Chitin nanofiber micropatterned flexible substrates for tissue engineering. J. Mater. Chem. B 2013, 1, 4217–4224. 10.1039/c3tb20782j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rolandi M.; Rolandi R. Self-assembled chitin nanofibers and applications. Adv. Colloid Interface Sci. 2014, 207, 216–222. 10.1016/j.cis.2014.01.019. [DOI] [PubMed] [Google Scholar]; c Zhong C.; Kapetanovic A.; Deng Y.; Rolandi M. A Chitin Nanofiber Ink for Airbrushing, Replica Molding, and Microcontact Printing of Self-assembled Macro-, Micro-, and Nanostructures. Adv. Mater. 2011, 23, 4776–4781. 10.1002/adma.201102639. [DOI] [PubMed] [Google Scholar]
- a Fox S. C.; Li B.; Xu D.; Edgar K. J. Regioselective Esterification and Etherification of Cellulose: A Review. Biomacromolecules 2011, 12, 1956–1972. 10.1021/bm200260d. [DOI] [PubMed] [Google Scholar]; b Liebert T.; Hornig S.; Hesse S.; Heinze T. Nanoparticles on the Basis of Highly Functionalized Dextrans. J. Am. Chem. Soc. 2005, 127, 10484–10485. 10.1021/ja052594h. [DOI] [PubMed] [Google Scholar]
- a Broaders K. E.; Grandhe S.; Fréchet J. M. J. A Biocompatible Oxidation-Triggered Carrier Polymer with Potential in Therapeutics. J. Am. Chem. Soc. 2011, 133, 756–758. 10.1021/ja110468v. [DOI] [PubMed] [Google Scholar]; b Bachelder E. M.; Beaudette T. T.; Broaders K. E.; Dashe J.; Fréchet J. M. J. Acetal-Derivatized Dextran: An Acid-Responsive Biodegradable Material for Therapeutic Applications. J. Am. Chem. Soc. 2008, 130, 10494–10495. 10.1021/ja803947s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerwall J. P. F.; Schütz C.; Salajkova M.; Noh J.; Hyun Park J.; Scalia G.; Bergström L. Cellulose nanocrystal-based materials: from liquid crystal self-assembly and glass formation to multifunctional thin films. NPG Asia Mater. 2014, 6, e80. 10.1038/am.2013.69. [DOI] [Google Scholar]
- Almeida I. F.; Pereira T.; Silva N. H. C. S.; Gomes F. P.; Silvestre A. J. D.; Freire C. S. R.; Sousa Lobo J. M.; Costa P. C. Bacterial cellulose membranes as drug delivery systems: An in vivo skin compatibility study. Eur. J. Pharm. Biopharm. 2014, 86, 332–336. 10.1016/j.ejpb.2013.08.008. [DOI] [PubMed] [Google Scholar]
- a Panza M.; Pistorio S. G.; Stine K. J.; Demchenko A. V. Automated Chemical Oligosaccharide Synthesis: Novel Approach to Traditional Challenges. Chem. Rev. 2018, 118, 8105–8150. 10.1021/acs.chemrev.8b00051. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pardo-Vargas A.; Delbianco M.; Seeberger P. H. Automated glycan assembly as an enabling technology. Curr. Opin. Chem. Biol. 2018, 46, 48–55. 10.1016/j.cbpa.2018.04.007. [DOI] [PubMed] [Google Scholar]
- Delbianco M.; Kononov A.; Poveda A.; Yu Y.; Diercks T.; Jiménez-Barbero J.; Seeberger P. H. Well-Defined Oligo- and Polysaccharides as Ideal Probes for Structural Studies. J. Am. Chem. Soc. 2018, 140, 5421–5426. 10.1021/jacs.8b00254. [DOI] [PubMed] [Google Scholar]
- a Bucior I.; Burger M. M. Carbohydrate–carbohydrate interactions in cell recognition. Curr. Opin. Struct. Biol. 2004, 14, 631–637. 10.1016/j.sbi.2004.08.006. [DOI] [PubMed] [Google Scholar]; b Rojo J.; Morales J. C.; Penadés S.. Carbohydrate-Carbohydrate Interactions in Biological and Model Systems. In Host-Guest Chemistry: Mimetic Approaches to Study Carbohydrate Recognition; Penadés S., Ed.; Springer: Berlin, Heidelberg, 2002; pp 45–92. [Google Scholar]
- Heinze T.; Hornig S.. Versatile Concept for the Structure Design of Polysaccharide-based Nanoparticles. Polysaccharide Materials: Performance by Design; American Chemical Society: Washington, DC, 2009; Vol. 1017, pp 169–183. [Google Scholar]
- Berger O.; Adler-Abramovich L.; Levy-Sakin M.; Grunwald A.; Liebes-Peer Y.; Bachar M.; Buzhansky L.; Mossou E.; Forsyth V. T.; Schwartz T.; Ebenstein Y.; Frolow F.; Shimon L. J. W.; Patolsky F.; Gazit E. Light-emitting self-assembled peptide nucleic acids exhibit both stacking interactions and Watson–Crick base pairing. Nat. Nanotechnol. 2015, 10, 353. 10.1038/nnano.2015.27. [DOI] [PubMed] [Google Scholar]
- Nilsson M. R. Techniques to study amyloid fibril formation in vitro. Methods 2004, 34, 151–160. 10.1016/j.ymeth.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Colomer I.; Chamberlain A. E. R.; Haughey M. B.; Donohoe T. J. Hexafluoroisopropanol as a highly versatile solvent. Nat. Rev. Chem. 2017, 1, 0088. 10.1038/s41570-017-0088. [DOI] [Google Scholar]
- Andersen N. H.; Dyer R. B.; Fesinmeyer R. M.; Gai F.; Liu Z.; Neidigh J. W.; Tong H. Effect of Hexafluoroisopropanol on the Thermodynamics of Peptide Secondary Structure Formation. J. Am. Chem. Soc. 1999, 121, 9879–9880. 10.1021/ja991829k. [DOI] [Google Scholar]
- a Pinotsi D.; Buell A. K.; Dobson C. M.; Kaminski Schierle G. S.; Kaminski C. F. A Label-Free, Quantitative Assay of Amyloid Fibril Growth Based on Intrinsic Fluorescence. ChemBioChem 2013, 14, 846–850. 10.1002/cbic.201300103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pinotsi D.; Grisanti L.; Mahou P.; Gebauer R.; Kaminski C. F.; Hassanali A.; Kaminski Schierle G. S. Proton Transfer and Structure-Specific Fluorescence in Hydrogen Bond-Rich Protein Structures. J. Am. Chem. Soc. 2016, 138, 3046–3057. 10.1021/jacs.5b11012. [DOI] [PubMed] [Google Scholar]; c Shang J.; Ma L.; Li J.; Ai W.; Yu T.; Gurzadyan G. G. The Origin of Fluorescence from Graphene Oxide. Sci. Rep. 2012, 2, 792. 10.1038/srep00792. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Samanta A. Dynamic Stokes Shift and Excitation Wavelength Dependent Fluorescence of Dipolar Molecules in Room Temperature Ionic Liquids. J. Phys. Chem. B 2006, 110, 13704–13716. 10.1021/jp060441q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.