

Abstract
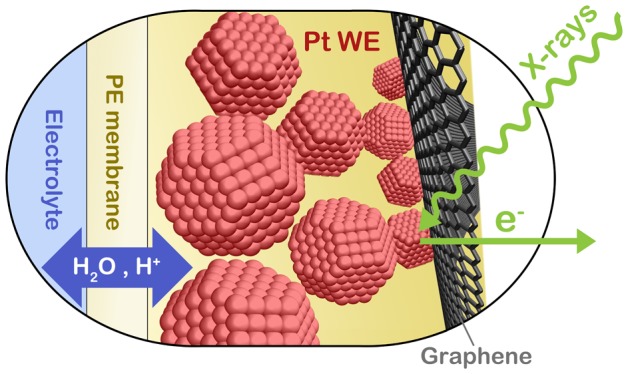
During the electrochemical reduction of oxygen, platinum catalysts are often (partially) oxidized. While these platinum oxides are thought to play a crucial role in fuel cell degradation, their nature remains unclear. Here, we studied the electrochemical oxidation of Pt nanoparticles using in situ XPS. When the particles were sandwiched between a graphene sheet and a proton exchange membrane that is wetted from the back, a confined electrolyte layer was formed, allowing us to probe the electrocatalyst under wet conditions. We show that the surface oxide formed at the onset of Pt oxidation has a mixed Ptδ+/Pt2+/Pt4+ composition. The formation of this surface oxide is suppressed when a Br-containing membrane is chosen due to adsorption of Br on Pt. Time-resolved measurements show that oxidation is fast for nanoparticles: even bulk PtO2·nH2O growth occurs on the subminute time scale. The fast formation of Pt4+ species in both surface and bulk oxide form suggests that Pt4+-oxides are likely formed (or reduced) even in the transient processes that dominate Pt electrode degradation.
1. Introduction
Fuel cells play a central role in the transition to a sustainable society, allowing us to use the energy stored in renewable carrier molecules such as hydrogen, methanol, or ammonia. With its high efficiency, compact design and modest operating temperature, the proton exchange membrane fuel cell (PEMFC) is of particular interest.1 As a catalyst for the multielectron reactions involved in the electrochemical fuel combustion, platinum has shown unique activity. To enable widespread implementation of PEMFCs with such a scarce metal, it is essential to minimize the Pt loading. A major hurdle toward this goal is catalyst degradation, which forces engineers to use a surplus of catalyst material to ensure long device lifetime.
Under the corrosive conditions present on the cathode side of PEMFCs, where the oxygen reduction reaction (ORR) takes place, platinum is prone to dissolution.2 In particular, transient electrode potentials3−8 and the combination of low pH and high oxygen pressure3,9 are detrimental. The importance of oxygen in the dissolution process suggests that platinum oxides are involved. Indeed, it was shown that the surface of Pt cathodes is (partially) oxidized at ORR potentials, particularly in the presence of oxygen.3,10−15
To understand the dissolution process, it is essential to identify which oxides are formed under ORR conditions. It has been established that Pt oxide formation starts with surface roughening at around 1.0 VRHE.13,16,17 The roughening originates from the place exchange between Pt atoms in the surface and O(H) adsorbates. The surface oxide saturates at 2 monolayers of oxygen (based on the passed charge density), from which it was assumed that an O–Pt–O–Pt layer is formed.17 However, the disorder in the surface structure precludes a clear assignment of the key structural elements and their oxidation state using common methods such as cyclic voltammetry, electrode mass analysis, scanning tunneling microscopy, and X-ray scattering. The onset of bulk oxidation is thermodynamically predicted around 1.0–1.2 VRHE,18,19 yet is usually observed only at significantly higher potentials due to kinetic limitations.17 A strongly hydrated and porous oxide evolves,17,20,21 which was shown to contain Pt4+ using ex situ X-ray photoelectron spectroscopy (XPS).21,22 A detailed spectroscopic characterization was precluded due to decomposition of the oxide in vacuum.22
To provide a detailed speciation of the electrochemically formed oxides on Pt, there is a clear need for in situ X-ray spectroscopy methods, which can identify the chemical state of the disordered structures in an element-specific manner. However, spectroscopy under electrochemical conditions constitutes a major challenge, particularly for the identification of surface species. Nonetheless, hard X-ray absorption studies have been able to monitor the amount of oxidized platinum formed on various Pt catalysts as a function of potential.10,14,15 X-ray photoelectron spectroscopy (XPS) allows for a more surface sensitive and detailed speciation of the oxide yet faces the technical challenge of dealing with the limited escape depth of electrons in liquid. Pioneering efforts toward electrochemical XPS have been made through the study of gas-phase electrochemistry on membrane-electrode assemblies,23−25 providing insight into the operation of high-temperature fuel cells. To enable studies under the more usual wet, low-temperature operation conditions, hard X-ray photoelectron spectroscopy has been employed.26−28 In another approach, the catalyst was probed through an array of microscopic graphene windows.29,30 While very promising, obtaining high-quality spectroscopy in these approaches remains challenging.
Inspired by Frevel et al.,31 we combine the methods above into a graphene-capped membrane-electrode assembly in order to obtain high-quality XPS and X-ray absorption spectroscopy in the soft energy range under wet electrochemical conditions. When Pt nanoparticles are sandwiched between a back-wetted proton exchange membrane and a graphene layer, a large-area confined electrolyte layer is formed. The graphene layer serves both as an electron transparent window and as a contact to the Pt nanoparticles, allowing for a single layer of separated particles to be investigated. Using this methodology, we study the reversible oxidation of Pt0 up to Pt4+ at potentials from 0.05 to 1.85 V vs RHE.
2. Methods
For this work, an in situ cell developed earlier25,32 has been modified with a graphene capping layer. As shown in Figure 1a, a monolayer of graphene is placed on top of the catalyst, which is supported by a proton exchange membrane. Behind the membrane is an electrolyte flow channel, which houses the Ag/AgCl (saturated KCl) reference electrode and the Pt counter electrode. The flow channel ensures wetting of the proton exchange membrane, which in turn leaks electrolyte to the catalyst side. The graphene capping layer provides a barrier against water evaporation from the catalyst surface into the vacuum of the ambient pressure XPS chamber. With the steady flow of water arriving to the catalyst layer through the membrane and the diminished evaporation rate from the catalyst layer due to the graphene, the partial pressure of water around the catalyst is pushed up. As will be shown in the section 3, this can lead to water condensation (see also ref (31)), so that the catalyst can be probed under wet electrochemical conditions. As shown in previous work,29,30,33,34 the graphene layer is transparent to X-rays and electrons (>300 eV kinetic energy), therefore allowing for spectroscopy to be conducted. In addition, the graphene provides electrical contact, so that isolated nanoparticles can be studied (see Figure 1b). The sample area, roughly 1 mm2, is much larger than the beam spot (100 μm × 200 μm). Therefore, the photon flux can be used more efficiently than in previous work with microscopic graphene windows,29,33,34 facilitating high quality spectroscopy with acquisition times down to the subminute scale. Finally, we should stress that the method is robust and inexpensive and can be used in conjunction with virtually any electrocatalyst. A deeper characterization of the cell will be published elsewhere.
Figure 1.

(a) Schematic representation of the electrochemical cell. (b) Transmission electron microscopy image of Pt nanoparticles sputtered on a Nafion film deposited on a Quantifoil TEM grid. The coverage shown here is the lowest used in this work (area coverage 37%), equivalent to a 1 nm layer. (c) High-resolution image of the sample in panel b.
The Pt working electrode is sputter-deposited on the vacuum side of the membrane. As shown in Figure 1b,c, the deposition resulted in crystalline, somewhat dendritic particles. Due to this asymmetric shape, the particles are best characterized by their typical surface-to-core distance rather than their equivalent radius. Our analysis indicates surface-to-core distances in the range of 1 to 2 nm. The catalyst loading, expressed as layer thickness, was varied between 1 and 16 nm. Nafion 117 (Ion Power) and Fumasep FAD55 (Fumatech) were used as proton exchange membranes. For Nafion 117, only water and protons can pass the membrane, whereas FAD55 also allows anions to diffuse to and from the working electrode. More details on the sample preparation and characterization can be found in ESI section S1.
The electrochemical cell was mounted on the near-ambient pressure XPS end station (NAPXPS1) of the ISISS beamline at the BESSY II/HZB synchrotron facility in Berlin, Germany. No gases were dosed to the cell chamber other than the leakage from the cell, leading to chamber pressures of 0.05 mbar to 0.15 mbar. For near-edge X-ray absorption fine structure (NEXAFS), electrons were collected on the analyzer nozzle to obtain the total electron yield (TEY) signal, while a partial electron yield (PEY) signal was simultaneously collected in the analyzer at a kinetic energy of 385 eV. This energy corresponds to the inelastically scattered background of the O KLL Auger peak, which combines good signal intensity with effective elimination of gas phase contributions from the PEY signal, in particular when a potential (90 V) is applied to the analyzer nozzle.35 To prevent beam damage effects, each spectrum was recorded on a fresh spot on the surface. All potentials were converted to the RHE scale. Details on the analysis procedure and beam effect studies can be found in ESI sections S2 and S3, respectively.
3. Results
As a first step, we established the degree of wetting on the Pt nanoparticles. Figure 2a shows O K-edge spectra for a 16 nm Pt layer on FAD55, with and without graphene cover layer. Both spectra are dominated by the contribution from the FAD membrane around 537.5 eV (see Figure S7a in the ESI for blank FAD). The most notable difference is the presence of a peak at 535 eV for the graphene-covered sample. A comparison of Figure 2a to the O K-edge spectrum of graphene deposited on gold via the same procedure indicates that this change is not due to oxygen functionalities on the graphene (see Figure S7b and its discussion in the ESI). Rather, the peak can be ascribed to the pre-edge feature of water with unsaturated hydrogen bonds.31,36−38 The relative intensity of this feature in the spectrum suggests that more than one layer of water has condensed on the catalyst, implying 100% relative humidity in the catalyst layer. Consistent with this, the O 1s spectrum (Figure S8 in the ESI) shows some increase around 533–534 eV, where multilayer H2O is expected to appear.36 Unfortunately, the many closely spaced contributions in the O 1s spectra and their attenuation by graphene preclude a more detailed analysis. Nonetheless, it is clear that the graphene cover layer enables proper wetting of the Pt working electrode.
Figure 2.

Effect of the graphene cover layer on electrolyte and oxidation properties of a 16 nm Pt layer on FAD55. (a) O K-edge spectra (PEY) recorded at open circuit potential (0.05 V). (b,c) Pt 4f spectra acquired during stepwise anodization of the same samples as shown in panel a, with an excitation energy of 580 eV and 0.1 M H2SO4 electrolyte. Each potential was held at least 5 min prior to acquisition. Potentials are shown versus RHE.
Figure 2b,c shows how wetting leads to a much more pronounced electrochemical response in the Pt 4f spectra. While the uncovered Pt layer only shows minor oxidation, even at 1.85 VRHE, the graphene-covered sample is clearly oxidized. The well-resolved peak at 76.7 eV indicates that a Pt(IV) oxyhydroxide layer is formed (hereafter referred to as PtO2·nH2O). Depth profiling (Figure S9 in the ESI) showed that the oxidation occurs at the outer surface of the electrode for the graphene-covered sample. In contrast, the uncovered sample is oxidized at the FAD55–Pt interface. These results highlight the importance of humidity for the electrochemical response in fuel cells. Indeed, it is well-documented that the power output of fuel cells is humidity-dependent.39,40 However, high humidity also leads to increased catalyst degradation.41,42 The comparison in Figure 2b,c suggests that this may be related to the increased degree of oxidation under wet conditions.
Further understanding of the electrolyte can be obtained from C 1s, N 1s, O 1s, and S 2p spectra (see Figure 3). Except for the oxygen species, one does not expect significant chemical changes. However, the observed binding energy shifts 1:1 with the electrostatic potential of the emitting atom. This can be exploited to identify the location of various electrolyte species. As shown schematically in Figure 3a, the electrostatic potential of the species belonging to or adsorbed on the (grounded) working electrode is zero versus the electron analyzer. If a nonconductive oxide is formed on the working electrode, a potential drop will occur in this layer. However, the constant Pt4+ peak position in Figure 2c suggests that this effect is negligible. In contrast, the electrostatic potential of electrolyte species will shift significantly depending on their proximity to the working electrode and the applied potential. As shown in Figure 3a, the electrostatic potential drops in the electrolyte. At the positive (anodic) potentials applied here, the electrolyte potential is negative with respect to the working electrode, leading to lower apparent binding energies.
Figure 3.

Influence of the local electrostatic potential on XPS peak positions. (a) Schematic representation of the electrostatic potential around the Pt particles. ΔU designates the difference in applied potential between the two depicted situations. The potential drop in the electrolyte can adopt a variety of functional forms but is shown as simple Gouy–Chapman exponential decay here. (b) Survey spectra of 16 nm graphene-covered Pt on FAD55 at open circuit potential (0.05 V) and 1.65 V, with an excitation energy of 1100 eV and 0.1 M H2SO4 electrolyte.
For instance, the N 1s peak belonging to the FAD membrane has shifted 1.7 eV at 1.65 VRHE with respect to open circuit potential (OCP, 0.05 VRHE), hence close to the expected bulk electrolyte shift. In contrast, the C 1s peak is shifted by only 0.5 eV, because it contains contributions from both FAD (electrolyte) and graphene (working electrode). Accordingly, the peak shape has clearly changed. Similarly, the O 1s peak shows an incomplete shift of 1.2 eV and a shape change, which can be explained as a mixed response of adsorbed water, graphene functionalities, bulk water, sulfate ions, and the FAD membrane. The S 2p peak also shows an incomplete shift (1.3 eV) and a clear broadening, suggesting that the sulfate anions are found in both the adsorbed and free form. Finally, we note that the shoulder around 69.3 eV in Figure 2c originates from Br 3d electrons from the FAD membrane. Similar to the case of the S 2p peak, the Br 3d shoulder does not shift as a whole but rather broadens toward lower binding energy. This suggests that Br species can be found both as adsorbates and free electrolyte species. Thus, we identify sulfate, Br-species, and O-species as the adsorbates in the Pt–FAD system and the N-functionalities and sulfate as bulk electrolyte species. An analogous analysis on Nafion is presented in Figure S10 in the ESI.
For both FAD and Nafion, we observe an intensity increase for membrane species during anodization (Br 3d peak in Figure 2c and S 2p peak in Figure S11 in the ESI), implying that the membranes redistribute within the catalyst layer. In agreement with this, the signal-to-noise ratio of the Pt 4f spectra decreases at elevated potential. When the potential is lowered back to 0.25 VRHE, the Br 3d peak disappears (not shown), yet the Pt signal loss is only partially reversed. This implies that some particles detach from the graphene during anodization and become completely encapsulated by the membrane. It has been suggested that the membrane redistribution occurs in order to screen the increasing electrode potential using membrane species43 (see section S7 in the ESI for further discussion). Such a rearrangement of charged species will require solvation, which explains the absence of a rising Br peak in the case of a poorly wetted catalyst (Figure 2b).
Having established the general behavior of our samples, we turn our attention to the detailed evolution of the Pt particles during anodization. To eliminate mass and charge transport limitations in the electrolyte, we investigated low-coverage samples with isolated nanoparticles (see Figure 1b), covered with graphene. We first discuss the oxidation of Pt on FAD55. To prevent any memory effects from air exposure, the working electrode was cycled 10 times between −0.15 VRHE and 1.25 VRHE at 50 mV/s. Subsequently, the sample was anodized in steps, while recording the spectra shown in Figure 4. The data were fitted using Pt0 (Pt 4f7/2 at 71.0 eV), Ptδ+ (71.8 eV), Pt2+ (72–72.7 eV) and Pt4+ (73.7 eV) doublets (details in ESI section S2).
Figure 4.

Stepwise anodization of graphene-covered, isolated Pt nanoparticles on FAD55 in 0.1 M H2SO4 electrolyte. (a) Pt 4f spectra acquired with an excitation energy of 600 eV and fit model using Pt0 (green), Ptδ+ (yellow), Pt2+ (blue), and Pt4+ (red) doublets. (b) The according O K-edge spectra.
At OCP (0.05 VRHE), a large Ptδ+ contribution is observed. Since the OCP lies well below the oxidation potential of platinum, the Ptδ+ peak must be adsorbate-induced. In agreement with this, hard X-ray absorption spectroscopy data10,15 showed broadening of the Pt L-edge white line in this potential range. At higher potentials, the L-edge white line was found to sharpen. Similarly, the Ptδ+ contribution decreases when moving to 0.85 VRHE and 1.05 VRHE in our measurements. These observations suggest a change in the adsorbate structure that reduces the adsorbate–Pt dipole. Although a detailed description of these adsorbate structures is beyond our measurement capabilities at this point, one could explain this as the result of an exchange between sulfate (2 charges per adsorbate) and bromine species (1 or fewer charges per adsorbate) on the surface, or as the result of hydrogen (co)adsorption at low potentials.
The O K-edges up to 1.05 VRHE in Figure 4b show little change with respect to OCP. This suggests that little to no adsorbed O(H)44,45 or surface oxide phases44 are formed, in contrast to literature for H2SO4 electrolytes.17,21 An explanation comes from the rise of the Br 3d peak (69.3 eV). As noted in our analysis of the electrolyte species, these Br species adsorb on the Pt particles. Studies on halide adsorption have shown that the Pt–Br interaction is sufficiently strong to outcompete O(H) adsorbates, thereby thermodynamically suppressing surface oxide formation.8,46,47 At sufficiently high potential, Pt will nonetheless oxidize. Indeed, an onset of oxide formation is observed at 1.25 V, through a minute rise in the Pt4+ component. The Pt4+ formation is accompanied by an increase in the O K-edge spectrum between 529.5 and 532 eV, consistent with PtO2.44 Some contribution may come from the oxidation of the graphene cover layer, although we expect this to occur at higher potentials (see ESI section S8 and ref (31).
Further increasing the potential to 1.45 V induces the formation of bulk PtO2·nH2O, as evidenced by the large Pt4+ contribution and the increased O K-edge peak at 531.5–532 eV. Since depth profiling implies a Pt0 core, Pt4+ shell situation (see Figure S9 in the ESI), the increase in the Ptδ+ contribution is attributed to the formation of a Pt–PtO2·nH2O interface. The PtO2·nH2O layer grows further at 1.85 V, almost completely consuming Pt0 (see also Figure S13 in the ESI for a Pt 4f spectrum recorded with higher excitation energy). Since the Pt–PtO2·nH2O interface is smaller in this situation, the Ptδ+ component shrank along with Pt0. Similar to Figure 2, the Pt4+ peak position and shape remain constant, suggesting that there is (almost) no potential drop over the oxide layer, even for the thick oxide generated here. This implies that the hydrous PtO2 generated under electrochemical conditions is more conductive than its crystalline anhydrous counterparts.48 Consistent with this, no charging was observed during XPS studies on thick PtO2·nH2O layers.21,22
The oxidation of Pt on Nafion shows striking differences to the case of Pt on FAD discussed above. Figure 5a shows the stepwise oxidation of a single layer of Nafion-supported nanoparticles. The onset potential for oxide formation lies between 0.9 VRHE and 1.15 VRHE (in agreement with the literature10,13−19), roughly 0.2 V lower than for Pt/FAD. Note that this shift in oxidation onset is also present for thicker Pt layers on Nafion. The initial oxidation of Nafion-supported Pt is evident in the Ptδ+, the Pt2+, and the Pt4+ contributions for the Nafion-supported particles, whereas Pt4+ growth was dominant for Pt/FAD. This observation shows that the Pt surface oxide is not uniform but rather contains Pt in various coordination environments, consistent with the rough surface morphology observed by X-ray scattering13 and scanning tunneling microscopy.16
Figure 5.

Pt oxidation on Nafion. (a) Stepwise oxidation of a single layer of Pt nanoparticles in 0.1 M H2SO4, with fit model using Pt0 (green), Ptδ+ (yellow), Pt2+ (blue), and Pt4+ (red) doublets. Excitation energy: 480 eV. (b) Graphene-covered 4 nm Pt layer at 1.85 VRHE with 0.1 M H2SO4. Excitation energy: 580 eV.
At potentials where bulk oxidation can occur (i.e., 1.25 V), the Pt4+ contribution becomes increasingly dominant, finally completely replacing Pt2+ at 1.85 V (Figure 5b). In agreement with bulk thermodynamics,18 this shows that bulk Pt2+ oxides are unstable with respect to PtO2·nH2O formation.
The absence of Pt2+ on our nanoparticles at high potentials differs from observations on planar bulk electrodes.22,26 This contrast can be attributed to kinetically hindered oxygen/H2O transport in planar electrodes. Indeed, creating a few nanometers of oxide on planar Pt can require hours.17,26 In our case, the response to applied potential is much more rapid. Figure 6a shows the evolution of the oxygen pre-edge feature of PtO2·nH2O during anodization, monitored using the PEY signal at an excitation energy of 531.8 eV (see ref (31) for technical details). The onset potential for oxidation is in good agreement with the static measurements from Figure 4b. The subsequent oxide growth nearly saturates in the following 2 min. Confirming this, a Pt 4f spectrum recorded roughly 2.5 min after reaching 1.85 V shows nearly completely oxidized particles (Figure 6c). Similar to oxide formation, complete reversal to the metallic situation can be achieved in a few minutes (also for the Nafion-supported samples). Again, such reversibility was not observed for planar electrodes.17,22,26
Figure 6.

Kinetics and reversibility of oxide formation on graphene-covered Pt nanoparticles on FAD55 in 0.1 M H2SO4 electrolyte. (a) Development of the PEY signal at 531.8 eV during anodization as shown in panel b. (c) Pt 4f spectra (hν = 600 eV) at OCP (0.05 VRHE) prior to the voltage sweep shown in panel b, ∼2.5 min after reaching 1.85 V, and after returning to OCP.
The facile bulk oxidation of platinum is quite specific to isolated nanoparticles and is not observed for thin films. Figure 7 shows a series of depth profiling experiments performed on catalyst layers of varying thickness. Already for a layer thickness of 4 nm, significantly less oxide is observed at 1.85 VRHE. Taking into account particle size effects using Shard’s method,49 we estimate that the oxide is about 1.5–2 times thinner than for the 1 nm sample. Conductivity measurements on 4 nm samples indicate that (most of) the particles are connected into a film. Clearly, this geometric difference from the 1 nm sample is already sufficient to change the oxidation behavior. To exclude that the film geometry hinders electrolyte mass transport to and from the Pt surface on the graphene side, we also investigated a 16 nm layer, for which this effect should be more pronounced. No further decrease in the oxide thickness is observed, indicating that electrolyte transport is not a limiting factor. Rather, we explain the deeper oxidation of the isolated nanoparticles as a result of their high surface-to-bulk ratio or structural flexibility,50 which facilitates easy penetration of oxygen or H2O into the particle core. Note that irrespective of the layer thickness, the oxidation kinetics were sufficient to oxidize any Pt2+ formed during the oxidation to Pt4+ (note the lack of Pt2+ in Figure 7).
Figure 7.

Depth profiling analysis of graphene-covered Pt nanoparticles/layers of various thickness on FAD55 in 0.1 M H2SO4 electrolyte, performed at 1.85 VRHE after a dwell period of at least 10 min. For fitting procedure, see section S2 in the ESI. The fraction of oxidic Pt was taken as the combined Pt2+ and Pt4+ contribution to the fitted spectra.
Our conclusions establish a clear framework for the discussion of Pt dissolution. Electrochemically generated Pt oxides range from mixed valence Ptδ+/Pt2+/Pt4+ surface oxide to pure Pt4+ bulk oxide (see Figure 8), with fast kinetics connecting these oxidic states with the metallic state for nanoparticles. Transient dissolution, which dominates Pt electrode degradation,3−8 can therefore occur during the formation and reduction of both surface and bulk oxide, even for short excursions to oxidative potentials. To relate our results to fuel cells, we should point out that while our sample geometry (nanoparticles on Nafion) and relative humidity (100%) are representative, we have not introduced O2 into the gas phase and our operating temperature is on the low side (23–120 °C for fuel cells). Since O2 is capable of oxidizing Pt even in the absence of applied potential,51 particularly at elevated temperature, we expect these differences to lead to a lower onset potential for oxide formation.
Figure 8.
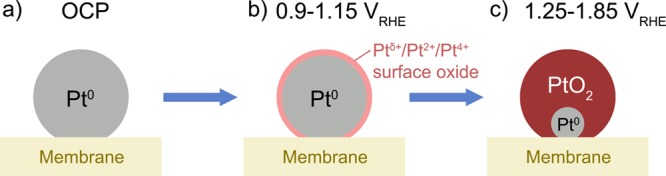
Schematic representation of the electrochemical oxidation of platinum. (a) Pristine metallic particles at open circuit potential. (b) Surface oxidation. This stage is absent for Pt on FAD due to Pt–Br interaction. (c) Bulk oxidation, which finally results in pure PtO2·nH2O.
4. Conclusion
We have studied the electrochemical oxidation of platinum nanoparticles using in situ XPS. Pt nanoparticles were sandwiched between a proton exchange membrane and a monolayer of graphene. While electrolyte can diffuse through the membrane from the backside, graphene provides a barrier for evaporation into the vacuum chamber. The appearance of a distinct H2O peak in the O K-edge spectra indicates that this geometry results in a well-wetted environment for the catalyst nanoparticles, opening the way for the study of wet electrochemistry using XPS and electron microscopy. We show that the wetting leads to much stronger Pt oxidation at high potentials, as compared to gas phase electro-oxidation.
Measurements conducted on a single layer of nanoparticles allowed us to probe the catalyst with minimal mass transport limitations in the electrolyte. We show that the initial oxidation of Pt on Nafion 117, between 0.9 VRHE and 1.15 VRHE, yields a surface oxide with mixed Ptδ+/Pt2+/Pt4+ oxidation state. On the alternative membrane, Fumasep FAD55, the formation of the surface oxide is suppressed due to the interaction between the Pt particles and Br species in the FAD membrane. For both membranes, anodization to higher potentials yields pure PtO2·nH2O (no PtO·nH2O) as thermodynamically expected.18 The oxidation on our nanoparticle catalysts is much faster than for (ultra)thin film or bulk electrodes, with significant bulk oxide growth on the subminute time-scale.
Based on these results, it is clear that the full range of Pt oxidation states (Pt0–Pt4+) should be considered in transient dissolution phenomena, even at modest oxidation potentials and subminute time scales. For transient potential excursions to high potentials, bulk oxide is a possible precursor to dissolution.
Acknowledgments
BESSY II/HZB is acknowledged for granting beam time under the proposal no. 17205966. R.M. was financially supported by the Bundesministerium für Bildung und Forschung through the InnoEMat consortium. The authors thank Eugen Stotz, Michael Hävecker, and Detre Teschner for their help in the cell construction, beamline optimization, and graphene transfer.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b12284.
Details on sample preparation and characterization, details on the analysis procedure for the Pt 4f and O K-edge spectra, an assessment of beam damage effects and prevention measures, O K-edge spectra of graphene on Au, in situ O 1s spectra, in situ depth profiling of the oxidized Pt samples with and without graphene cover, S 2p spectra recorded during anodization experiments on Pt/Nafion, and a Pt 4f spectrum of a Pt/FAD sample recorded at 1280 eV excitation energy (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Basualdo M. S.; Feroldi D.. PEM Fuel Cells with Bio-Ethanol Processor Systems: A Multidisciplinary Study of Modelling, Simulation, Fault Diagnosis and Advanced Control; Springer-Verlag, 2012. [Google Scholar]
- Shao-Horn Y.; Sheng W. C.; Chen S.; Ferreira P. J.; Holby E. F.; Morgan D. Instability of Supported Platinum Nanoparticles in Low-Temperature Fuel Cells. Top. Catal. 2007, 46 (3–4), 285–305. 10.1007/s11244-007-9000-0. [DOI] [Google Scholar]
- Matsumoto M.; Miyazaki T.; Imai H. Oxygen-Enhanced Dissolution of Platinum in Acidic Electrochemical Environments. J. Phys. Chem. C 2011, 115 (22), 11163–11169. 10.1021/jp201959h. [DOI] [Google Scholar]
- Xing L.; Jerkiewicz G.; Beauchemin D. Ion Exchange Chromatography Coupled to Inductively Coupled Plasma Mass Spectrometry for the Study of Pt Electro-Dissolution. Anal. Chim. Acta 2013, 785, 16–21. 10.1016/j.aca.2013.04.048. [DOI] [PubMed] [Google Scholar]
- Furuya Y.; Mashio T.; Ohma A.; Tian M.; Kaveh F.; Beauchemin D.; Jerkiewicz G. Influence of Electrolyte Composition and pH on Platinum Electrochemical And/or Chemical Dissolution in Aqueous Acidic Media. ACS Catal. 2015, 5 (4), 2605–2614. 10.1021/cs5016035. [DOI] [Google Scholar]
- Topalov A. A.; Cherevko S.; Zeradjanin A. R.; Meier J. C.; Katsounaros I.; Mayrhofer K. J. J. Towards a Comprehensive Understanding of Platinum Dissolution in Acidic Media. Chem. Sci. 2014, 5 (2), 631–638. 10.1039/C3SC52411F. [DOI] [Google Scholar]
- Cherevko S.; Zeradjanin A. R.; Keeley G. P.; Mayrhofer K. J. J. A Comparative Study on Gold and Platinum Dissolution in Acidic and Alkaline Media. J. Electrochem. Soc. 2014, 161 (12), H822–H830. 10.1149/2.0881412jes. [DOI] [Google Scholar]
- Lopes P. P.; Strmcnik D.; Tripkovic D.; Connell J. G.; Stamenkovic V.; Markovic N. M. Relationships between Atomic Level Surface Structure and Stability/Activity of Platinum Surface Atoms in Aqueous Environments. ACS Catal. 2016, 6 (4), 2536–2544. 10.1021/acscatal.5b02920. [DOI] [Google Scholar]
- Mitsushima S.; Koizumi Y.; Uzuka S.; Ota K.-I. Dissolution of Platinum in Acidic Media. Electrochim. Acta 2008, 54 (2), 455–460. 10.1016/j.electacta.2008.07.052. [DOI] [Google Scholar]
- Merte L. R.; Behafarid F.; Miller D. J.; Friebel D.; Cho S.; Mbuga F.; Sokaras D.; Alonso-Mori R.; Weng T.-C.; Nordlund D.; et al. Electrochemical Oxidation of Size-Selected Pt Nanoparticles Studied Using in Situ High-Energy-Resolution X-Ray Absorption Spectroscopy. ACS Catal. 2012, 2, 2371–2376. 10.1021/cs300494f. [DOI] [Google Scholar]
- Kongkanand A.; Ziegelbauer J. M. Surface Platinum Electrooxidation in the Presence of Oxygen. J. Phys. Chem. C 2012, 116 (5), 3684–3693. 10.1021/jp211490a. [DOI] [Google Scholar]
- Tang L.; Han B.; Persson K.; Friesen C.; He T.; Sieradzki K.; Ceder G. Electrochemical Stability of Nanometer-Scale Pt Particles in Acidic Environments. J. Am. Chem. Soc. 2010, 132 (2), 596–600. 10.1021/ja9071496. [DOI] [PubMed] [Google Scholar]
- Drnec J.; Ruge M.; Reikowski F.; Rahn B.; Carlà F.; Felici R.; Stettner J.; Magnussen O. M.; Harrington D. A. Pt Oxide and Oxygen Reduction at Pt(111) Studied by Surface X-Ray Diffraction. Electrochem. Commun. 2017, 84, 50–52. 10.1016/j.elecom.2017.10.002. [DOI] [Google Scholar]
- Friebel D.; Miller D. J.; Nordlund D.; Ogasawara H.; Nilsson A. Degradation of Bimetallic Model Electrocatalysts: An in Situ X-Ray Absorption Spectroscopy Study. Angew. Chem., Int. Ed. 2011, 50 (43), 10190–10192. 10.1002/anie.201101620. [DOI] [PubMed] [Google Scholar]
- Friebel D.; Viswanathan V.; Miller D. J.; Anniyev T.; Ogasawara H.; Larsen A. H.; O’grady C. P.; Nørskov J. K.; Nilsson A. Balance of Nanostructure and Bimetallic Interactions in Pt Model Fuel Cell Catalysts: In Situ XAS and DFT Study. J. Am. Chem. Soc. 2012, 134 (23), 9664–9671. 10.1021/ja3003765. [DOI] [PubMed] [Google Scholar]
- Wakisaka M.; Asizawa S.; Uchida H.; Watanabe M. In Situ STM Observation of Morphological Changes of the Pt(111) Electrode Surface during Potential Cycling in 10 mM HF Solution. Phys. Chem. Chem. Phys. 2010, 12 (16), 4184–4190. 10.1039/b923956a. [DOI] [PubMed] [Google Scholar]
- Conway B. E. Electrochemical Oxide Film Formation at Noble Metals as a Surface-Chemical Process. Prog. Surf. Sci. 1995, 49 (4), 331–452. 10.1016/0079-6816(95)00040-6. [DOI] [Google Scholar]
- Jacob T. Theoretical Investigations on the Potential-Induced Formation of Pt-Oxide Surfaces. J. Electroanal. Chem. 2007, 607 (1–2), 158–166. 10.1016/j.jelechem.2007.03.023. [DOI] [Google Scholar]
- Bard A. J.; Parsons R.; Jordan J.. Standard Potentials in Aqueous Solution; Dekker: New York, 1985. [Google Scholar]
- Zerbino J. O.; Perdriel C.; Arvia A. J. Ellipsometry of Hydrous Platinum Oxide Layers and Electrodispersed Platinum Surfaces Resulting from Their Electroreduction. Thin Solid Films 1993, 232, 63–67. 10.1016/0040-6090(93)90763-F. [DOI] [Google Scholar]
- Peuckert M.; Coenen F. P.; Bonzel H. P. XPS Study of the Electrochemical Surface Oxidation of Platinum in 1N H2SO4 Acid Electrolyte. Electrochim. Acta 1984, 29, 1305–1314. 10.1016/0013-4686(84)87002-4. [DOI] [Google Scholar]
- Johnson B.; Ranjan C.; Greiner M.; Arrigo R.; Schuster M. E.; Höpfner B.; Gorgoi M.; Lauermann I.; Willinger M.; Knop-Gericke A.; et al. Characterization of Platinum and Iridium Oxyhydrate Surface Layers from Platinum and Iridium Foils. ChemSusChem 2016, 9 (13), 1634–1646. 10.1002/cssc.201600143. [DOI] [PubMed] [Google Scholar]
- Saveleva V. A.; Papaefthimiou V.; Daletou M. K.; Doh W. H.; Ulhaq-Bouillet C.; Diebold M.; Zafeiratos S.; Savinova E. R. Operando Near Ambient Pressure XPS (NAP-XPS) Study of the Pt Electrochemical Oxidation in H2O and H2O/O2Ambients. J. Phys. Chem. C 2016, 120 (29), 15930–15940. 10.1021/acs.jpcc.5b12410. [DOI] [Google Scholar]
- Casalongue H. S.; Kaya S.; Viswanathan V.; Miller D. J.; Friebel D.; Hansen H. A.; Nørskov J. K.; Nilsson A.; Ogasawara H. Direct Observation of the Oxygenated Species during Oxygen Reduction on a Platinum Fuel Cell Cathode. Nat. Commun. 2013, 4, 2817. 10.1038/ncomms3817. [DOI] [Google Scholar]
- Arrigo R.; Hävecker M.; Schuster M. E.; Ranjan C.; Stotz E.; Knop-gericke A.; Schlögl R. In Situ Study of the Gas-Phase Electrolysis of Water on Platinum by. Angew. Chem., Int. Ed. 2013, 52, 11660–11664. 10.1002/anie.201304765. [DOI] [PubMed] [Google Scholar]
- Favaro M.; Valero-Vidal C.; Eichhorn J.; Toma F. M.; Ross P. N.; Yano J.; Liu Z.; Crumlin E. J. Elucidating the Alkaline Oxygen Evolution Reaction Mechanism on Platinum. J. Mater. Chem. A 2017, 5 (23), 11634–11643. 10.1039/C7TA00409E. [DOI] [Google Scholar]
- Takagi Y.; Wang H.; Uemura Y.; Nakamura T.; Yu L.; Sekizawa O.; Uruga T.; Tada M.; Samjeské G.; Iwasawa Y.; et al. In Situ Study of Oxidation States of Platinum Nanoparticles on a Polymer Electrolyte Fuel Cell Electrode by near Ambient Pressure Hard X-Ray Photoelectron Spectroscopy. Phys. Chem. Chem. Phys. 2017, 19 (8), 6013–6021. 10.1039/C6CP06634H. [DOI] [PubMed] [Google Scholar]
- Axnanda S.; Crumlin E. J.; Mao B.; Rani S.; Chang R.; Karlsson P. G.; Edwards M. O. M.; Lundqvist M.; Moberg R.; Ross P.; et al. Using “tender” X-Ray Ambient Pressure X-Ray Photoelectron Spectroscopy as A Direct Probe of Solid-Liquid Interface. Sci. Rep. 2015, 5, 9788. 10.1038/srep09788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-velez J. J.; Pfeifer V.; Hävecker M.; Weatherup R. S.; Arrigo R.; Chuang C.; Stotz E.; Weinberg G.; Salmeron M.; Schlögl R.; et al. Photoelectron Spectroscopy at the Graphene – Liquid Interface Reveals the Electronic Structure of an Electrodeposited Cobalt/Graphene Electrocatalyst. Angew. Chem., Int. Ed. 2015, 54, 14554–14558. 10.1002/anie.201506044. [DOI] [PubMed] [Google Scholar]
- Kolmakov A.; Dikin D. A.; Cote L. J.; Huang J.; Abyaneh M. K.; Amati M.; Gregoratti L.; Günther S.; Kiskinova M. Graphene Oxide Windows for in Situ Environmental Cell Photoelectron Spectroscopy. Nat. Nanotechnol. 2011, 6 (10), 651–657. 10.1038/nnano.2011.130. [DOI] [PubMed] [Google Scholar]
- Frevel L.; Mom R. V.; Velasco-velez J. J.; Plodinec M.; Knop-Gericke A.; Schlögl R.; Jones T. E. In Situ X-Ray Spectroscopy on the Electrochemical Development of Iridium Nanoparticles in Confined Electrolyte. J. Phys. Chem. C 2019, 10.1021/acs.jpcc.9b00731. [DOI] [Google Scholar]
- Pfeifer V.; Jones T. E.; Velasco-velez J. J.; Arrigo R.; Piccinin S.; Hävecker M.; Knop-gericke A.; Schlögl R. In Situ Observation of Reactive Oxygen Species Forming on Oxygen-Evolving Iridium Surfaces. Chem. Sci. 2017, 8, 2143–2149. 10.1039/C6SC04622C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherup R. S.; Eren B.; Hao Y.; Bluhm H.; Salmeron M. B. Graphene Membranes for Atmospheric Pressure Photoelectron Spectroscopy. J. Phys. Chem. Lett. 2016, 7 (9), 1622–1627. 10.1021/acs.jpclett.6b00640. [DOI] [PubMed] [Google Scholar]
- Velasco-velez J. J.; Pfeifer V.; Hävecker M.; Wang R.; Centeno A.; Zurutuza A.; Algara-Siller G.; Stotz E.; Skorupska K.; Teschner D.; et al. Atmospheric Pressure X-Ray Photoelectron Spectroscopy Apparatus: Bridging the Pressure Gap. Rev. Sci. Instrum. 2016, 87, 053121. 10.1063/1.4951724. [DOI] [PubMed] [Google Scholar]
- Hävecker M.; Cavalleri M.; Herbert R.; Follath R.; Knop-gericke A.; Hess C.; Hermann K.; Schlögl R. Methodology for the Structural Characterization of VxOy Species Supported on Silica under Reaction Conditions by Means of in Situ O K-Edge X-Ray Absorption Spectroscopy. Phys. Status Solidi B 2009, 246 (7), 1459–1469. 10.1002/pssb.200945067. [DOI] [Google Scholar]
- Nilsson A.; Nordlund D.; Waluyo I.; Huang N.; Ogasawara H.; Kaya S.; Bergmann U.; Näslund L.-Å.; Öström H.; Wernet P.; et al. X-Ray Absorption Spectroscopy and X-Ray Raman Scattering of Water and Ice; an Experimental View. J. Electron Spectrosc. Relat. Phenom. 2010, 177 (2–3), 99–129. 10.1016/j.elspec.2010.02.005. [DOI] [Google Scholar]
- Endo O.; Nakamura M.; Sumii R.; Amemiya K. 1D Hydrogen Bond Chain on Pt(211) Stepped Surface Observed by O K-NEXAFS Spectroscopy. J. Phys. Chem. C 2012, 116 (26), 13980–13984. 10.1021/jp302509k. [DOI] [Google Scholar]
- Bluhm H.; Ogletree F. D.; Fadley C. S.; Hussain Z.; Salmeron M. B. The Premelting of Ice Studied with Photoelectron Spectroscopy. J. Phys.: Condens. Matter 2002, 14, L227–233. 10.1088/0953-8984/14/8/108. [DOI] [Google Scholar]
- Zhang J.; Tang Y.; Song C.; Xia Z.; Li H.; Wang H.; Zhang J. PEM Fuel Cell Relative Humidity (RH) and Its Effect on Performance at High Temperatures. Electrochim. Acta 2008, 53, 5315–5321. 10.1016/j.electacta.2008.02.074. [DOI] [Google Scholar]
- Wang X.; Duan Y.; Yan W.; Weng F. Effect of Humidity of Reactants on the Cell Performance of PEM Fuel Cells with Parallel and Interdigitated Flow Field Designs. J. Power Sources 2008, 176, 247–258. 10.1016/j.jpowsour.2007.10.065. [DOI] [Google Scholar]
- Bi W.; Sun Q.; Deng Y.; Fuller T. F. The Effect of Humidity and Oxygen Partial Pressure on Degradation of Pt/C Catalyst in PEM Fuel Cell. Electrochim. Acta 2009, 54, 1826–1833. 10.1016/j.electacta.2008.10.008. [DOI] [Google Scholar]
- Xu H.; Kunz R.; Fenton J. M. Investigation of Platinum Oxidation in PEM Fuel Cells at Various Relative Humidities. Electrochem. Solid-State Lett. 2007, 10 (1), B1–B5. 10.1149/1.2372230. [DOI] [Google Scholar]
- Law Y. T.; Zafeiratos S.; Neophytides S. G.; Orfanidi A.; Costa D.; Dintzer T.; Arrigo R.; Knop-Gericke A.; Schlögl R.; Savinova E. R. In Situ Investigation of Dissociation and Migration Phenomena at the Pt/electrolyte Interface of an Electrochemical Cell. Chem. Sci. 2015, 6 (10), 5635–5642. 10.1039/C5SC01421B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. J.; Öberg H.; Kaya S.; Sanchez Casalongue H.; Friebel D.; Anniyev T.; Ogasawara H.; Bluhm H.; Pettersson L. G. M.; Nilsson A. Oxidation of Pt(111) under near-Ambient Conditions. Phys. Rev. Lett. 2011, 107 (19), 195502. 10.1103/PhysRevLett.107.195502. [DOI] [PubMed] [Google Scholar]
- Schiros T.; Näslund L.-Å.; Andersson K.; Gyllenpalm J.; Karlberg G. S.; Odelius M.; Ogasawara H.; Pettersson L. G. M.; Nilsson A. Structure and Bonding of the Water-Hydroxyl Mixed Phase on Pt(111). J. Phys. Chem. C 2007, 111 (41), 15003–15012. 10.1021/jp073405f. [DOI] [Google Scholar]
- Markovic N. M.; Ross P. N. Jr. Surface Science Studies of Model Fuel Cell Electrocatalysts. Surf. Sci. Rep. 2002, 45 (4–6), 117–229. 10.1016/S0167-5729(01)00022-X. [DOI] [Google Scholar]
- Minguzzi A.; Montagna L.; Falqui A.; Vertova A.; Rondinini S.; Ghigna P. Dynamics of Oxide Growth on Pt Nanoparticles Electrodes in the Presence of Competing Halides by Operando Energy Dispersive X-Ray Absorption Spectroscopy. Electrochim. Acta 2018, 270, 378–386. 10.1016/j.electacta.2018.03.092. [DOI] [Google Scholar]
- Seriani N.; Jin Z.; Pompe W.; Colombi Ciacchi L. Density Functional Theory Study of Platinum Oxides: From Infinite Crystals to Nanoscopic Particles. Phys. Rev. B: Condens. Matter Mater. Phys. 2007, 76, 155421. 10.1103/PhysRevB.76.155421. [DOI] [Google Scholar]
- Shard A. G. A Straightforward Method For Interpreting XPS Data From Core-Shell Nanoparticles. J. Phys. Chem. C 2012, 116, 16806–16813. 10.1021/jp305267d. [DOI] [Google Scholar]
- An W.; Liu P. Size and Shape Effects of Pd@Pt Core-Shell Nanoparticles: Unique Role of Surface Contraction and Local Structural Flexibility. J. Phys. Chem. C 2013, 117, 16144–16149. 10.1021/jp4057785. [DOI] [Google Scholar]
- Fantauzzi D.; Krick Calderón S.; Mueller J. E.; Grabau M.; Papp C.; Steinrück H. P.; Senftle T. P.; van Duin A. C. T.; Jacob T. Growth of Stable Surface Oxides on Pt(111) at Near-Ambient Pressures. Angew. Chem., Int. Ed. 2017, 56 (10), 2594–2598. 10.1002/anie.201609317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.