

Abstract
Graphene quantum dots (GQDs) are emerging as environmentally friendly, low-cost, and highly tunable building blocks in solar energy conversion architectures, such as solar (fuel) cells. Specifically, GQDs constitute a promising alternative for organometallic dyes in sensitized oxide systems. Current sensitized solar cells employing atomically precise GQDs are based on physisorbed sensitizers, with typically limited efficiencies. Chemisorption has been pointed out as a solution to boost photoconversion efficiencies, by allowing improved control over sensitizer surface coverage and sensitizer-oxide coupling strength. Here, employing time-resolved THz spectroscopy, we demonstrate that chemisorption of atomically precise C42-GQDs (hexa-peri-hexabenzocoronene derivatives consisting of 42 sp2 carbon atoms) onto mesoporous metal oxides, enabled by their functionalization with a carboxylate group, enhances electron transfer (ET) rates by almost 2 orders of magnitude when compared with physisorbed sensitizers. Density functional theory (DFT) calculations, absorption spectroscopy and valence band X-ray photoelectron spectroscopy reveal that the enhanced ET rates can be traced to stronger donor–acceptor coupling strength enabled by chemisorption.
Graphene quantum dots (GQDs) are nanosized graphene fragments, which have nonzero, size-dependent band-gaps due to quantum confinement effects.1−3 Moreover, GQDs are metal-free and hence potentially low-cost and environmentally friendly. These features have motivated researchers to apply GQDs in solar energy conversion schemes (e.g., solar cells4−7 and photocatalytic devices8,9). GQDs are typically prepared by hydrothermal treatment of graphene or small molecules;1−3 although certain control of GQD size has been achieved following this synthesis protocol,2 samples are generally defined by broad absorption features induced by inhomogeneous broadening (i.e., samples does not consist on a narrow distribution of chemical structures), an aspect that is detrimental for optoelectronic applications.
Alternatively, large polycyclic aromatic hydrocarbons (PAHs) have been synthesized in the field of organic chemistry over the last decades, with hexa-peri-hexabenzocoronene (HBC), consisting of 42 sp2 carbon atoms, being a representative example.10,11 Recently, Yan et al. reported the synthesis of large PAHs consisting of 132, 168, and 170 sp2 carbon atoms and referred to them as colloidal GQDs.12,13 Such PAHs, sometimes also called nanographenes, can indeed serve as atomically precise zero dimensional GQDs, owing to their well-defined size- and shape-dependent optoelectronic properties as predicted by theoretical predictions.14−16 Yan et al. also reported the use of such well-defined GQDs as absorbers in a sensitized solar cell geometry.17 However, these initial solar cell devices revealed low photoconversion efficiencies, mainly linked with low short-circuit currents. The poor photocurrent produced in the cells was tentatively correlated with the low affinity of the employed GQDs sensitizers, which were physisorbed onto the mesoporous oxide surface. In a follow up report from the same group, functionalization of the colloidal GQDs by carboxylate groups was reported to allow better control on the interfacial bonding geometry on polar surfaces (specifically on mica) by chemisorption.18 Furthermore, theoretical studies have shown that chemisorption, instead of physisorption, of sensitizers should favor donor–acceptor coupling and hence boost electron transfer at GQD/oxide interfaces.19,20 Although all these works have suggested that chemisorption of GQDs onto metal oxides might improve photoconversion efficiencies in sensitized systems, there is at present no experimental evidence to support that claim. Here, we quantify interfacial electron transfer (ET) rates for atomically precise GQDC42 (HBC with 42 sp2 carbon atoms) chemisorbed and physisorbed on mesoporous SnO2 by optical pump-terahertz probe (OPTP) spectroscopy. We demonstrate that sensitizer chemisorption onto the oxide electrode substantially improves ET rates induced by strong overlap (hybridization) between donor and acceptor wave functions, which is triggered by the functionalization of the GQDC42 by a carboxylate group. This claim is directly evident from the OPTP data and is further supported by absorption spectroscopy, valence band X-ray photoelectron spectroscopy (VBXPS) and density functional theory (DFT) calculations.
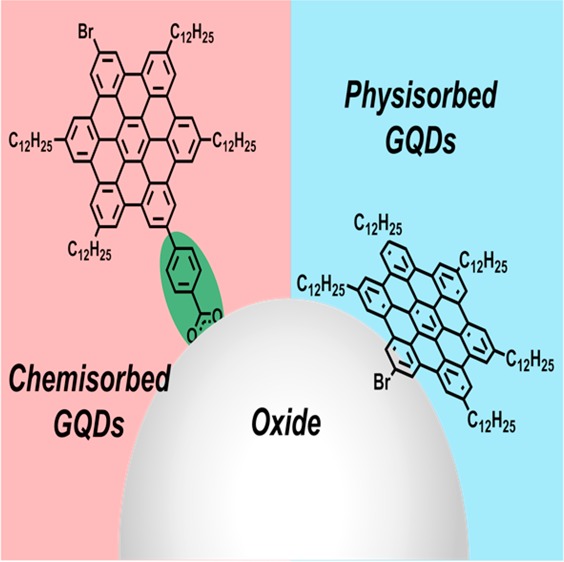
The molecular structures of the two GQDC42 samples used in this study are shown in Figure 1. For clarity, we name our samples as GQDC42 and GQDC42-PhCOOH for the sensitizer without and with a phenyl carboxylic acid functional group, respectively. The GQDC42 sample was prepared following a protocol described previously21 and the synthesis of GQDC42-PhCOOH is described in the Supporting Information (SI). In brief, GQDC42-PhCOOH was synthesized starting from a hexaphenylbenzene derivative bearing bromo- and iodo-groups through a Suzuki-coupling reaction to selectively introduce a phenyl ester moiety and subsequent cyclodehydrogenation reaction to convert the hexaphenylbenzene core into HBC. Finally, the ester group was hydrolyzed under basic conditions to the desired carboxylic acid anchoring group. The synthesized samples were diluted in toluene, and the obtained suspensions were employed for sensitizing the mesoporous oxide films (see the SI for sample preparation details). The functionalized films were subsequently characterized by OPTP spectroscopy; the measurements were performed under nitrogen conditions to prevent any potential sensitizer photo-oxidation.
Figure 1.

Chemical structures of the graphene quantum dots, GQDC42 and GQDC42-PhCOOH, analyzed in this study; they differ on the presence of a carboxylate functional group which is expected to serve as a covalent link to the oxide electrode.
OPTP spectroscopy is a powerful tool to investigate ultrafast interfacial dynamics for dye-22−26 and QD-27−29 sensitized oxide systems. As the employed THz probe (∼1.5 THz bandwidth) is primarily sensitive to free carrier motion (i.e., photoconductivity), an OPTP measurement in a sensitized oxide neatly probes the emergence of photoconductivity in the oxide electrode after selective excitation of the sensitizer. As such, it resolves, in time, the arrival of electrons from the sensitizer’s populated molecular orbitals (e.g., LUMO) into the oxide’s conduction band (CB). Figure 2a shows normalized OPTP dynamics for GQDC42 and GQDC42-PhCOOH sensiti-zing SnO2 mesoporous films (blue diamonds and red circles respectively; 400 nm pump excitation, 0.6 mJ/cm2); these dynamics were collected in the linear single-exciton regime (see Figure S1). The lack of response under 400 nm pump excitation of a bare SnO2 mesoporous oxide film is also presented in Figure 2a (black triangles). As evident from Figure 2a, the sensitization of mesoporous SnO2 by the sensitizer functionalized with a phenyl carboxylate group (GQDC42-PhCOOH, Figure 1), which is expected to chemisorb at the oxide surface, results in faster ET rates when compared with the sensitizer lacking functionalization (GQDC42 in Figure 1), which is expected to physisorb at the oxide surface. Both traces can be well described phenomenologically by a biphasic exponential model (solid red lines in Figure 2), providing time constants of τ1 = 12 ± 1 ps and τ2 = 2025 ± 450 ps for GQDC42 sensitized SnO2 films; and τ1 = 0.2 ± 0.1 ps and τ2 = 51 ± 3 ps for GQDC42-PhCOOH sensitized SnO2 films. Biphasic ET dynamics are a common observable for dye- and QD- sensitized oxide systems that have been generally explained in terms of two distinct transfer channels toward the oxide electrode. They might be linked with “hot” and “cold” ET channels30,31 or, alternatively, to “cold” ET channels induced by two donor–acceptor interfacial conformations (i.e., providing distinct donor–acceptor energetics).32 Biphasic dynamics have been also rationalized by considering effects induced by molecules loosely attached (physisorbed) to the surface or those present in form of the aggregates.29,33,34 Even though it is difficult to rule out any of these scenarios from the current data, the fact that both ET components become faster when QDs are chemisorbed onto the oxide matrix support the view that two ET channels define our interfacial dynamics. A deeper analysis of the nature of the biphasic signals is underway and will be reported elsewhere.
Figure 2.

(a) OPTP dynamics of GQDC42 (open blue diamonds) and GQDC42-PhCOOH (open red circles) sensitizing SnO2 films (400 nm pump excitation, 0.6 mJ cm–2). Traces are normalized to the plateau of the biexponential fits (solid red lines). Black open triangles are OPTP dynamics for a bare SnO2 film. (b) Frequency-resolved complex photoconductivity for both sensitized systems (1 ns after photoexcitation); solid and dashed lines correspond to Drude–Smith fits for the real and imaginary components of the frequency dependent conductivity.
To validate the conclusion of faster ET rates for the chemisorbed sensitizers inferred from the OPTP data, we analyze the frequency-resolved complex photoconductivity in the samples. This approach can tell whether OPTP dynamics indeed refer uniquely to electrons populating the oxide conduction band. Figure 2b presents the real and imaginary (closed and open symbols) components of the complex conductivity for GQDC42 (blue) and GQDC42-PhCOOH (red) sensitized oxide films (1 ns after excitation, 1.5 THz bandwidth). As evident from Figure 2b, the complex photoconductivity spectra for chemisorbed and physisorbed dots onto the oxide overlap quite well, indicating that the nature of the monitored photoconductivity for both samples is identical, as expected for electrons in SnO2. The resolved complex spectra can be well described by the phenomenological Drude–Smith (DS) model:35
| 1 |
where ωp, ε0, τs, and c refer to the plasma frequency, vacuum permittivity, scattering time and localization factor, respectively. Within the DS model, the measurable frequency resolved photoconductivity can be attributed to free carriers experiencing preferential backscattering at the boundaries of the nanocrystalline mesoporous oxide electrode.22,36 The strength of the localization is parametrized by c, which ranges between 0 (free carrier, Drude reponse) and −1 (representing full localization of charges). Best fit to the data using eq 1 for both samples provides the same fitting parameters within error; a scattering rate of τs = 46 ± 2 fs and localization factor of c = −0.69 ± 0.1. These results are in good agreement with previous values inferred for nanostructured SnO2 films37−39 and, hence, demonstrate that the monitored signal in Figure 2a refers uniquely to electrons populating the oxide conduction band. These results support our conclusion that the changes in the monitored interfacial ET rates can be traced uniquely to the presence of the phenyl carboxylate group functionalizing the GQDC42-PhCOOH, allowing for chemisorption. Furthermore, these results reveal that electron transport within the analyzed SnO2 electrode is unaffected by the type of functionalization (chemisorption or physisorption).
To check the generality of our observation that ET is accelerated for chemisorbed sensitizers, we also measured interfacial ET dynamics for both sensitizers onto mesoporous ZnO and TiO2 films (Figure S3). For ZnO samples, we observed as well faster ET rates for the chemisorbed sensitizers, analogous to the case of SnO2 based samples (Figure 2). For titania, ET occurs faster than our experimental sub-picosecond time resolution for both sensitizers, so we are unable to determine changes in ET rates for this set of samples. The overall slower ET rates observed for ZnO and SnO2 electrodes when compared to titania, generally explained in terms of larger density of states for the latter improving donor–acceptor coupling strength, agree qualitatively with those reported for organometallic dye sensitizers onto the same electrodes.40 Overall, the results support the view that the presence of a phenyl carboxylate group facilitates electron transfer toward the electrode independently of its nature.
To rationalize our finding of faster ET for chemisorbed GQDC42, we investigate the nature of the interaction between sensitizer donor and oxide acceptor. Figure 3 presents the relative energy level alignments. For the SnO2 electrode, the energy positions are obtained from ultraviolet photoelectron spectroscopy (UPS, see Figure S5) and absorbance. For the two molecules, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were obtained from gas-phase DFT calculations. When the interaction of the metal oxide with the molecules is not taken into account, upon sensitization, the slightly larger ΔG (the energy difference between the LUMO of the sensitizer and the CB of the electron-accepting oxide, see Figure 3) for the GQDC42 lacking a carboxylate group should lead to faster ET for the physisorbed sensitizers.29 From the OPTP data shown in Figure 2a, it is clear that this is not the case, which shows that the chemical interaction between chemisorbed GQDC42-PhCOOH and the oxide electrode affects donor–acceptor energetics (and hence coupling strength). The frontier orbitals obtained from DFT calculations offer some insight on this aspect. As shown in Figure 3, the sensitizer expected to chemisorb to the oxide surface (GQDC42-PhCOOH) reveals a substantial LUMO distribution on the carboxylate group, which is expected to deprotonate and bond to the oxide surface. The localization of the LUMO nearer the oxide surface will provide enhanced donor–acceptor coupling strength (donor–acceptor wave function overlap) when compared with the physisorbed case, in line with the OPTP data shown in Figure 2a. Faster ET rates as a function of shorter donor–acceptor orbital distances between sensitizers and metal oxides have been demonstrated in quantum dot–oxide41 and dye–oxide40 systems.
Figure 3.
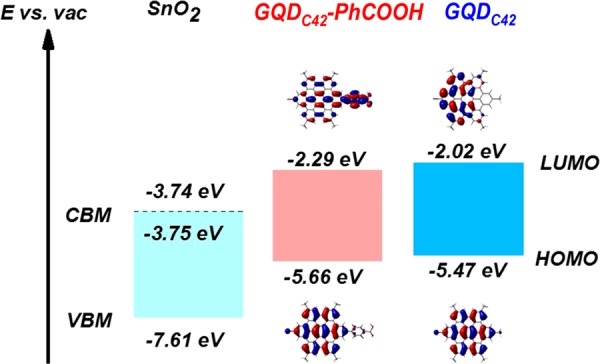
Interfacial donor–acceptor energetics derived from gas-phase DFT calculations for GQDC42 and GQDC42-PhCOOH sensitizers and from UPS for tin oxide. Frontier orbital distributions of HOMO and LUMO for both sensitizers are also presented. The Fermi level for the tin oxide sample is within the oxide CB, indicating strong n-type character.
To reveal a stronger interaction between chemisorbed GQDC42 orbitals and the oxide conduction band, we analyzed the samples by optical absorption and valence band X-ray photoelectron spectroscopy. In Figure 4a, we present the UV–vis absorbance spectrum of both sensitized systems (solid red and blue lines for chemisorbed and physisorbed GQDC42, respectively) along with the bare oxide film (black line). Figure 4a also provides the spectra of the toluene solution of both sensitizers (dashed red and blue lines for chemisorbed and physisorbed GQDC42 respectively); the absorption profiles peak at 3.45 and 3.39 eV for GQDC42 and GQDC42-PhCOOHs, respectively. Whereas the peak absorbance for GQDC42 remains essentially identical upon sensitization, a clear blue shift is resolved for the peak absorbance of the GQDC42-PhCOOH sensitizers after binding to the oxide electrode (dotted and solid red lines in Figure 4a). A similar blue shift in the absorbance after sensitization has been previously reported for dye-sensitized films and explained in terms of the strong coupling induced by deprotonation of the dyes42−45 induced by chemisorption; in good agreement with our expectations.
Figure 4.

(a) UV–vis absorbance spectrum of GQDC42 (blue) and GQDC42-PhCOOH (red) sensitized SnO2 films and for sensitizers dispersed in solution (dashed lines). The spectrum of bare SnO2 film is also shown (solid black line). (b) Valence band X-ray spectra of GQDC42 (blue) and GQDC42-PhCOOH (red) sensitized and bare (black) SnO2 electrodes. The dashed lines are linear fits enabling binding energy estimates for the analyzed systems.
In Figure 4b, we present valence band X-ray photoelectron spectroscopy (VBXPS) data for a bare SnO2 mesoporous film (solid black line) and oxide electrodes functionalized by GQDC42-PhCOOH and GQDC42 sensitizers (red and blue line respectively). As evident from the plot, the physisorbed GQDC42 sensitized and the bare tin oxide electrodes reveal identical electron binding energies. However, the electron binding energy is reduced by approximately 2 eV upon chemisorption of GQDC42-PhCOOHs, indicating significant/efficient modification/decoration of the surface of the oxide, which was absent upon the physisorption (the results were reproducible within ∼50 meV for samples produced in three different batches). The observed change in electron binding energy can be explained by chemical modification at the oxide interface (e.g., deprotonation of COOH groups able to modify the Fermi energy at the oxide) and/or by dipolar effects (modifying vacuum levels induced by the presence of a surface electric field induced by sensitization).46 In any case, it reveals a strong donor–acceptor interaction for the chemisorbed GQDC42, in qualitative agreement with absorbance spectra shown in Figure 4a and the expected closer proximity of frontier orbitals inferred by DFT calculation (Figure 3). All these results support qualitatively the experimentally resolved effect on interfacial OPTP dynamics, i.e. the main conclusion of this work: chemisorption of GQDC42, enabled by their functionalization with a carboxylate group, substantially enhances ET rates.
In summary, we investigated the rates of electron transfer from physisorbed and chemisorbed graphene quantum dot (GQD) sensitizers to metal oxides, using hexa-peri-hexabenzocoronene derivatives as atomically precise GQDs. An increase of ET rates as large as 2 orders of magnitude is observed for chemisorbed sensitizers when compared with physisorbed ones. Accelerated electron transfer is correlated with enhanced donor–acceptor coupling (i.e., wave function hybridization). These results demonstrate that functionalization of GQDs with anchoring head groups represents a potential path for improved photoconversion efficiencies in carbon-based sensitizer/oxide electrodes employed in solar energy conversion schemes.
Acknowledgments
This work has been financially supported by the Max Planck Society. E. C. acknowledges financial support from the Max Planck Graduate Center and the regional government of Comunidad de Madrid under project (2017-T1/AMB-5207).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.9b00399.
Materials and methods; OPTP dynamics of GQDs sensitized SnO2 in linear single-exciton regime; measurement artifact in frequency-resolved photoconductivity; OPTP dynamics of GQDs Sensitized ZnO/TiO2; frequency-resolved photoconductivity of GQDs sensitized ZnO/TiO2; UPS and absorption spectrum of mesoporous SnO2 Film (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Zheng P.; Wu N. Fluorescence and Sensing Applications of Graphene Oxide and Graphene Quantum Dots: A Review. Chem. - Asian J. 2017, 12 (18), 2343–2353. 10.1002/asia.201700814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Rui M.; Song J.; Shen Z.; Zeng H. Carbon and Graphene Quantum Dots for Optoelectronic and Energy Devices: A Review. Adv. Funct. Mater. 2015, 25 (31), 4929–4947. 10.1002/adfm.201501250. [DOI] [Google Scholar]
- Li L.; Wu G.; Yang G.; Peng J.; Zhao J.; Zhu J.-J. Focusing on Luminescent Graphene Quantum Dots: Current Status and Future Perspectives. Nanoscale 2013, 5 (10), 4015. 10.1039/c3nr33849e. [DOI] [PubMed] [Google Scholar]
- Zamiri G.; Bagheri S. Fabrication of Green Dye-Sensitized Solar Cell Based on ZnO Nanoparticles as a Photoanode and Graphene Quantum Dots as a Photo-Sensitizer. J. Colloid Interface Sci. 2018, 511, 318–324. 10.1016/j.jcis.2017.10.026. [DOI] [PubMed] [Google Scholar]
- Chen L.; Guo C. X.; Zhang Q.; Lei Y.; Xie J.; Ee S.; Guai G.; Song Q.; Li C. M. Graphene Quantum-Dot-Doped Polypyrrole Counter Electrode for High-Performance Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2013, 5 (6), 2047–2052. 10.1021/am302938a. [DOI] [PubMed] [Google Scholar]
- Dinari M.; Momeni M. M.; Goudarzirad M. Dye-Sensitized Solar Cells Based on Nanocomposite of Polyaniline/Graphene Quantum Dots. J. Mater. Sci. 2016, 51 (6), 2964–2971. 10.1007/s10853-015-9605-9. [DOI] [Google Scholar]
- Mihalache I.; Radoi A.; Mihaila M.; Munteanu C.; Marin A.; Danila M.; Kusko M.; Kusko C. Charge and Energy Transfer Interplay in Hybrid Sensitized Solar Cells Mediated by Graphene Quantum Dots. Electrochim. Acta 2015, 153, 306–315. 10.1016/j.electacta.2014.11.200. [DOI] [Google Scholar]
- Bayat A.; Saievar-Iranizad E. Graphene Quantum Dots Decorated Rutile TiO2 Nanoflowers for Water Splitting Application. J. Energy Chem. 2018, 27 (1), 306–310. 10.1016/j.jechem.2017.09.036. [DOI] [Google Scholar]
- Rajender G.; Kumar J.; Giri P. K. Interfacial Charge Transfer in Oxygen Deficient TiO2-Graphene Quantum Dot Hybrid and Its Influence on the Enhanced Visible Light Photocatalysis. Appl. Catal., B 2018, 224, 960–972. 10.1016/j.apcatb.2017.11.042. [DOI] [Google Scholar]
- Narita A.; Wang X.-Y.; Feng X.; Müllen K. New Advances in Nanographene Chemistry. Chem. Soc. Rev. 2015, 44 (18), 6616–6643. 10.1039/C5CS00183H. [DOI] [PubMed] [Google Scholar]
- Wu J.; Pisula W.; Müllen K. Graphenes as Potential Material for Electronics. Chem. Rev. 2007, 107 (3), 718–747. 10.1021/cr068010r. [DOI] [PubMed] [Google Scholar]
- Yan X.; Li B.; Li L.-S. Colloidal Graphene Quantum Dots with Well-Defined Structures. Acc. Chem. Res. 2013, 46 (10), 2254–2262. 10.1021/ar300137p. [DOI] [PubMed] [Google Scholar]
- Yan X.; Cui X.; Li L.-S. Synthesis of Large, Stable Colloidal Graphene Quantum Dots with Tunable Size. J. Am. Chem. Soc. 2010, 132 (17), 5944–5945. 10.1021/ja1009376. [DOI] [PubMed] [Google Scholar]
- Rieger R.; Müllen K. Forever Young: Polycyclic Aromatic Hydrocarbons as Model Cases for Structural and Optical Studies. J. Phys. Org. Chem. 2010, 23 (4), 315–325. 10.1002/poc.1644. [DOI] [Google Scholar]
- Tan Y.-Z.; Yang B.; Parvez K.; Narita A.; Osella S.; Beljonne D.; Feng X.; Müllen K. Atomically Precise Edge Chlorination of Nanographenes and Its Application in Graphene Nanoribbons. Nat. Commun. 2013, 4 (1), 2646. 10.1038/ncomms3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.-Y.; Narita A.; Müllen K. Precision Synthesis versus Bulk-Scale Fabrication of Graphenes. Nat. Rev. Chem. 2017, 2 (1), 0100. 10.1038/s41570-017-0100. [DOI] [Google Scholar]
- Yan X.; Cui X.; Li B.; Li L.-S. Large, Solution-Processable Graphene Quantum Dots as Light Absorbers for Photovoltaics. Nano Lett. 2010, 10 (5), 1869–1873. 10.1021/nl101060h. [DOI] [PubMed] [Google Scholar]
- Hamilton I. P.; Li B.; Yan X.; Li L. S. Alignment of Colloidal Graphene Quantum Dots on Polar Surfaces. Nano Lett. 2011, 11 (4), 1524–1529. 10.1021/nl200298c. [DOI] [PubMed] [Google Scholar]
- Seyed-Talebi S. M.; Beheshtian J.; Neek-amal M. Doping Effect on the Adsorption of NH3 Molecule onto Graphene Quantum Dot: From the Physisorption to the Chemisorption. J. Appl. Phys. 2013, 114 (12), 124307. 10.1063/1.4822165. [DOI] [Google Scholar]
- Long R. Understanding the Electronic Structures of Graphene Quantum Dot Physisorption and Chemisorption onto the TiO2 (110) Surface: A First-Principles Calculation. ChemPhysChem 2013, 14 (3), 579–582. 10.1002/cphc.201200882. [DOI] [PubMed] [Google Scholar]
- Ito S.; Wehmeier M.; Brand J. D.; Kübel C.; Epsch R.; Rabe J. P.; Müllen K. Synthesis and Self-Assembly of Functionalized Hexa-Peri-Hexabenzocoronenes. Chem. - Eur. J. 2000, 6 (23), 4327–4342. . [DOI] [PubMed] [Google Scholar]
- Turner G. M.; Beard M. C.; Schmuttenmaer C. A. Carrier Localization and Cooling in Dye-Sensitized Nanocrystalline Titanium Dioxide. J. Phys. Chem. B 2002, 106 (45), 11716–11719. 10.1021/jp025844e. [DOI] [Google Scholar]
- Tiwana P.; Docampo P.; Johnston M. B.; Snaith H. J.; Herz L. M. Electron Mobility and Injection Dynamics in Mesoporous ZnO, SnO2, and TiO2 Films Used in Dye-Sensitized Solar Cells. ACS Nano 2011, 5 (6), 5158–5166. 10.1021/nn201243y. [DOI] [PubMed] [Google Scholar]
- Karakus M.; Zhang W.; Räder H. J.; Bonn M.; Cánovas E. Electron Transfer from Bi-Isonicotinic Acid Emerges upon Photodegradation of N3-Sensitized TiO2 Electrodes. ACS Appl. Mater. Interfaces 2017, 9 (40), 35376–35382. 10.1021/acsami.7b08986. [DOI] [PubMed] [Google Scholar]
- Tiwana P.; Parkinson P.; Johnston M. B.; Snaith H. J.; Herz L. M. Ultrafast Terahertz Conductivity Dynamics in Mesoporous TiO 2: Influence of Dye Sensitization and Surface Treatment in Solid-State Dye-Sensitized Solar Cells. J. Phys. Chem. C 2010, 114 (2), 1365–1371. 10.1021/jp908760r. [DOI] [Google Scholar]
- Brauer J. C.; Moser J.-E. Transient Photoconductivity of Dye-Sensitized TiO2 Nanocrystalline Films Probed by Optical Pump-THz Probe Spectroscopy. Ultrafast Phenom. XVII 2011, 358–360. [Google Scholar]
- Wang H.; Barceló I.; Lana-Villarreal T.; Gómez R.; Bonn M.; Cánovas E. Interplay Between Structure, Stoichiometry, and Electron Transfer Dynamics in SILAR-Based Quantum Dot-Sensitized Oxides. Nano Lett. 2014, 14 (10), 5780–5786. 10.1021/nl5026634. [DOI] [PubMed] [Google Scholar]
- Zhao K.; Pan Z.; Mora-Sero I.; Canovas E.; Wang H.; Song Y.; Gong X.; Wang J.; Bonn M.; Bisquert J.; et al. Boosting Power Conversion Efficiencies of Quantum-Dot-Sensitized Solar Cells Beyond 8% by Recombination Control. J. Am. Chem. Soc. 2015, 137 (16), 5602–5609. 10.1021/jacs.5b01946. [DOI] [PubMed] [Google Scholar]
- Cánovas E.; Moll P.; Jensen S. A.; Gao Y.; Houtepen A. J.; Siebbeles L. D. A.; Kinge S.; Bonn M. Size-Dependent Electron Transfer from PbSe Quantum Dots to SnO2 Monitored by Picosecond Terahertz Spectroscopy. Nano Lett. 2011, 11 (12), 5234–5239. 10.1021/nl202550v. [DOI] [PubMed] [Google Scholar]
- Wang H. I.; Infante I.; Brinck S. t.; Cánovas E.; Bonn M. Efficient Hot Electron Transfer in Quantum Dot-Sensitized Mesoporous Oxides at Room Temperature. Nano Lett. 2018, 18 (8), 5111–5115. 10.1021/acs.nanolett.8b01981. [DOI] [PubMed] [Google Scholar]
- Benkö G.; Kallioinen J.; Korppi-Tommola J. E. I.; Yartsev A. P.; Sundström V. Photoinduced Ultrafast Dye-to-Semiconductor Electron Injection from Nonthermalized and Thermalized Donor States. J. Am. Chem. Soc. 2002, 124 (3), 489–493. 10.1021/ja016561n. [DOI] [PubMed] [Google Scholar]
- Furube A.; Katoh R.; Yoshihara T.; Hara K.; Murata S.; Arakawa H.; Tachiya M. Ultrafast Direct and Indirect Electron-Injection Processes in a Photoexcited Dye-Sensitized Nanocrystalline Zinc Oxide Film: The Importance of Exciplex Intermediates at the Surface. J. Phys. Chem. B 2004, 108 (33), 12583–12592. 10.1021/jp0487713. [DOI] [Google Scholar]
- Wenger B.; Grätzel M.; Moser J.-E. Rationale for Kinetic Heterogeneity of Ultrafast Light-Induced Electron Transfer from Ru(II) Complex Sensitizers to Nanocrystalline TiO2. J. Am. Chem. Soc. 2005, 127 (35), 12150–12151. 10.1021/ja042141x. [DOI] [PubMed] [Google Scholar]
- Cánovas E.; Wang H.; Karakus M.; Bonn M. Hot Electron Transfer from PbSe Quantum Dots Molecularly Bridged to Mesoporous Tin and Titanium Oxide Films. Chem. Phys. 2016, 471, 54–58. 10.1016/j.chemphys.2015.11.005. [DOI] [Google Scholar]
- Smith N. Classical Generalization of the Drude Formula for the Optical Conductivity. Phys. Rev. B: Condens. Matter Mater. Phys. 2001, 64 (15), 155106. 10.1103/PhysRevB.64.155106. [DOI] [Google Scholar]
- Ulbricht R.; Hendry E.; Shan J.; Heinz T. F.; Bonn M. Carrier Dynamics in Semiconductors Studied with Time-Resolved Terahertz Spectroscopy. Rev. Mod. Phys. 2011, 83 (2), 543–586. 10.1103/RevModPhys.83.543. [DOI] [Google Scholar]
- Tsokkou D.; Othonos A.; Zervos M. Carrier Dynamics and Conductivity of SnO2 Nanowires Investigated by Time-Resolved Terahertz Spectroscopy. Appl. Phys. Lett. 2012, 100 (13), 133101. 10.1063/1.3698097. [DOI] [Google Scholar]
- Regan K. P.; Swierk J. R.; Neu J.; Schmuttenmaer C. A. Frequency-Dependent Terahertz Transient Photoconductivity of Mesoporous SnO2 Films. J. Phys. Chem. C 2017, 121 (29), 15949–15956. 10.1021/acs.jpcc.7b02318. [DOI] [Google Scholar]
- Zou X.; Luo J.; Lee D.; Cheng C.; Springer D.; Nair S. K.; Cheong S. A.; Fan H. J.; Chia E. E. M. Temperature-Dependent Terahertz Conductivity of Tin Oxide Nanowire Films. J. Phys. D: Appl. Phys. 2012, 45 (46), 465101. 10.1088/0022-3727/45/46/465101. [DOI] [Google Scholar]
- Asbury J. B.; Hao E.; Wang Y. Q.; Ghosh H. N.; Lian T. Q. Ultrafast Electron Transfer Dynamics from Molecular Adsorbates to Semiconductor Nanocrystalline Thin Films. J. Phys. Chem. B 2001, 105 (20), 4545. 10.1021/jp003485m. [DOI] [Google Scholar]
- Wang H.; McNellis E. R.; Kinge S.; Bonn M.; Cánovas E. Tuning Electron Transfer Rates through Molecular Bridges in Quantum Dot Sensitized Oxides. Nano Lett. 2013, 13 (11), 5311–5315. 10.1021/nl402820v. [DOI] [PubMed] [Google Scholar]
- Hagberg D. P.; Edvinsson T.; Marinado T.; Boschloo G.; Hagfeldt A.; Sun L. A Novel Organic Chromophore for Dye-Sensitized Nanostructured Solar Cells. Chem. Commun. 2006, (21), 2245. 10.1039/b603002e. [DOI] [PubMed] [Google Scholar]
- Wang Z.-S.; Sugihara H. N3-Sensitized TiO2 Films: In Situ Proton Exchange toward Open-Circuit Photovoltage Enhancement. Langmuir 2006, 22 (23), 9718–9722. 10.1021/la061315g. [DOI] [PubMed] [Google Scholar]
- Lin L.-Y.; Tsai C.-H.; Wong K.-T.; Huang T.-W.; Hsieh L.; Liu S.-H.; Lin H.-W.; Wu C.-C.; Chou S.-H.; Chen S.-H.; et al. Organic Dyes Containing Coplanar Diphenyl-Substituted Dithienosilole Core for Efficient Dye-Sensitized Solar Cells. J. Org. Chem. 2010, 75 (14), 4778–4785. 10.1021/jo100762t. [DOI] [PubMed] [Google Scholar]
- Nazeeruddin M. K.; Humphry-Baker R.; Liska P.; Grätzel M. Investigation of Sensitizer Adsorption and the Influence of Protons on Current and Voltage of a Dye-Sensitized Nanocrystalline TiO2 Solar Cell. J. Phys. Chem. B 2003, 107 (34), 8981–8987. 10.1021/jp022656f. [DOI] [Google Scholar]
- Cahen D.; Kahn A. Electron Energetics at Surfaces and Interfaces: Concepts and Experiments. Adv. Mater. 2003, 15 (4), 271–277. 10.1002/adma.200390065. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.