

Abstract
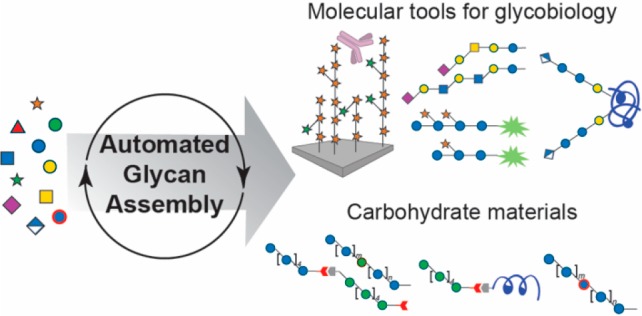
The intrinsic complexity of carbohydrate structures has hampered access to pure glycans and hence impeded progress in the glycosciences. Automated Glycan Assembly (AGA) has facilitated the procurement of synthetic glycans, to be used in diagnostics, vaccine development, enzyme characterization and structure–function relationship studies. A general approach for obtaining complex glycans from mammalian, bacterial, fungal and plant classes provides molecular tools for glycobiology research. Recent advances in AGA technology pave the way for the production of novel carbohydrate materials. This perspective describes the state-of-the art of AGA and aspects of the technology where additional improvements are needed.
Introduction
Carbohydrates, the most abundant biopolymers in nature, are a part of most living organisms, where they fulfill structural functions or play roles in diverse biological processes such as cell adhesion, pathogen-host interaction and numerous cell-signaling processes.1 Compared to polynucleotides and polypeptides that are linear polymers, polysaccharides are more diverse, complex and often branched. Each monosaccharide unit contains multiple hydroxyl groups that can serve as attachment points for further chain growth. Furthermore, in contrast to the phosphate diesters that connect nucleotides or amide linkages in peptides, each glycosidic linkage is a stereogenic center (Figure 1).
Figure 1.

Solid-phase synthetic strategy depends on biopolymer structure.
Carbohydrate complexity has slowed progress in the glycosciences when compared to molecular biology,2 as access to pure glycans has been a bottleneck for investigations into glycan function. Isolation from natural sources is difficult as carbohydrates are typically obtained in small amounts and as microheterogeneous mixtures.3 Access to defined structures in useful quantities without contamination4 relies on synthetic glycans as essential tools to study glycan function.
In principle, glycan synthesis is straightforward considering that glycans are of perceived complexity. Only one type of chemical bond, the glycosidic linkage, has to be constructed in a stereoselective manner. This conceptual simplicity stands in stark contrast to the practical challenges that the synthesis of complex glycans pose. Protecting group manipulations are required to ensure the desired product regio- and stereochemistry and translate into several months of work for traditional solution-phase approaches.5 Strategies aiming to accelerate oligosaccharide synthesis include convergent, one-pot, solid-supported and tag-assisted syntheses6 in combination with chemical, enzymatic or chemoenzymatic glycosylations.
Enzymatic Synthesis
Enzymatic approaches use unprotected sugars as substrates, thus avoiding protecting group manipulations. The variety of structures accessible via enzymatic synthesis is limited by enzyme availability and substrate specificities. The portfolio of enzymes for complex oligosaccharide synthesis keeps expanding.7
Methods to reduce the number of manual manipulations and purification steps during enzymatic syntheses have been reported.8−11 Approaches where the growing oligosaccharide is bound to a tag or solid support can be potentially combined with an automated process for expeditious glycan synthesis. However, tag methods often fail for oligosaccharides that are larger than their tag due to purification difficulties, or when large tags negatively influence synthesis efficiencies.9,12,13
Efficient enzymatic reactions on matrices are difficult and rendered solid-phase enzymatic synthesis elusive.9 Automated enzymatic glycan synthesis using the HPLC-based glycan synthesizer “Golgi” yielded the sialyl Lewisx (SLex) antigen.14 A dendrimer solid support improved the synthesis efficiency, but yields were reduced by a significant loss of material. Recently, a CEM Liberty Blue peptide synthesizer was used for the fully automated enzymatic synthesis of a series of glycan antigens.15 A thermoresponsive solid support polymer ensured efficient enzymatic glycosylations while minimizing product loss. Automated enzymatic synthesis is a promising avenue, but with just few examples to date, the scope of the method remains to be illustrated.
Streamlined Chemical Synthesis
One-pot strategies rely on performing multiple sequential glycosylations without intermediate protecting group manipulation or product isolation. In this way, a range of glycans has been synthesized.16 One-pot iterative glycosylations were used to procure the pieces that were later condensed to prepare an arabinogalactan 92-mer.17
The systematic exploitation of anomeric reactivity differences between glycosyl donors for their sequential glycosylation (‘programmable one-pot synthesis’) is based on the quantification of relative reactivity values (RRVs). RRVs guide the selection of building blocks according to their reactivity.18 Recently, an extended library of RRVs for building blocks, including virtual values predicted through machine learning, were incorporated into an updated software. The “Auto-CHO” software assists hierarchical one-pot syntheses by guiding the selection of building blocks including fragments generated via one-pot synthesis.19 RRV application is limited as it disregards other parameters such as acceptor or solvent influence.16 Reactivity-based protocols are difficult to generalize as minor protecting group changes can greatly influence reactivity. Solution-phase one-pot methodologies suffer from difficulties associated with the removal of reagents and side products.
Automated Chemical Synthesis
Automated Glycan Assembly (AGA) has expedited access to synthetic glycans up to 50-mers,20 while other automated platforms based on electrochemical assembly,21 fluorous-assisted solution-phase,22 and HPLC-assisted synthesis23 have been limited to few examples not exceeding hexasaccharides.6 From the proof-of-concept using a modified peptide synthesizer in 2001 to the first commercial Glyconeer 2.1 synthesizer,24 AGA has been developed using the syntheses of glycans of mammalian, bacterial, and plant origin as challenge.2,25 Here, we focus on AGA as a method for fast and reliable oligosaccharide synthesis by reviewing recent advances, pinpointing the remaining bottlenecks, and future perspectives.
AGA Approach
In solid-phase synthesis, a solid support equipped with a linker is used to successively couple building blocks and assemble a growing oligomer chain. Monomers carry a temporary protecting group (tPG) that is removed from the resin-bound oligomer to allow for subsequent chain growth in the next coupling cycle. For oligosaccharide assembly, regio- and stereocontrol of the coupling is ensured by the appropriate selection of orthogonally protected monosaccharide building blocks that carry a combination of temporary and permanent protecting groups (Figure 2).
Figure 2.

(A) Segment of a branched oligosaccharide represented with (a) chemical structure, (b) symbol representation according to the Symbol Nomenclature for Glycans (SNFG) and (c) SNFG with linkage presentation following the Oxford system.26 (B) Steps and building blocks required for the assembly of the branched oligosaccharide segment using solid-phase synthesis. For details on leaving groups, protecting groups and reaction conditions, see Figure 4.
The AGA oligosaccharide synthesis workflow is designed to minimize the number of purification steps and manipulations (Figure 3). Inside the synthesizer’s reaction vessel, a resin-bound linker serves as an anchor to successively attach monosaccharide building blocks. In this way, excess reagents can be washed away and time-consuming intermediate purification steps can be avoided. After completion of the synthesis, the resin-bound oligosaccharide is removed from the synthesizer and the oligosaccharide is cleaved from the solid support. Analytical normal-phase high performance liquid chromatography (NP-HPLC) and MALDI analysis of the crude product after cleavage are used to qualitatively assess the synthesis success (‘Control point 1’, Figure 3). The protected glycan is purified using preparative NP-HPLC. Global deprotection removes all permanent protecting groups (PGs) and after reverse-phase HPLC (RP-HPLC) the unprotected glycan is obtained. The final product is characterized typically by 1H, 13C, 2D NMR, and HRMS (‘Control point 2’, Figure 3).
Figure 3.

AGA oligosaccharide synthesis workflow.
AGA syntheses require careful selection of a compatible set of linker-functionalized solid support and building blocks (Figure 4). Merrifield polystyrene resin, a common solid support for peptide and oligonucleotide assembly, is used for its swelling and mechanical properties and chemical stability.27,28 The linker has to be readily and effectively cleaved at the end of the synthesis, but has to withstand all reaction conditions including acidic glycosylation and basic deprotection conditions. Cleavage of the linker and global deprotection should render the glycan reducing end in a useful form. “Approved building blocks” for AGA are those that can be easily produced on large scale, are stable over long periods of time but upon activation react with high yield and stereoselectivity, and bear protecting groups that can be selectively and effectively removed. An increasing number of these “approved building blocks” are now becoming commercially available.
Figure 4.

Summary of reactions and conditions commonly used for oligosaccharide synthesis. Transformations indicated with “*” are either of limited utility or have been tested only for a small number of glycans such that the scope remains to be fully determined.
Building Blocks
Building block selection is critical for AGA. The anomeric leaving group and protecting groups influence reactivity, stereoselectivity and regioselectivity of the building block as glycosyl donor and subsequently as nucleophile (glycosyl acceptor). Thioglycosides,24,29 glycosyl phosphates30,31 and glycosyl imidates32,33 are commonly used in AGA (Figure 4). Stock solutions for the activation of these glycosyl donors (NIS/TfOH for thioglycosides, or TMSOTf for glycosyl phosphates and imidates) remain stable for several days when kept under argon on the synthesizer. Thioglycosides are particularly attractive for commercial use as they are bench stable over long periods of time.34 In addition, thioglycosides react with reduced formation of hydrolyzed donor side product at temperatures (generally around 0 °C) that are significantly higher than those used for glycosyl phosphate and imidate building blocks. Building blocks that require very low glycosylation temperatures are inconvenient as they pose challenges to instrumentation and prolonged cycle times are required for cooling and warming.
The selection of protecting groups in the glycosyl donor determines glycosylation stereoselectivity (Figure 2B). Control over 1,2-trans glycosidic linkage formation is excercised by anchimeric assistance of participating protecting groups at the C2 hydroxyl. As nonparticparting groups at C2 do not allow for complete stereocontrol, 1,2-cis glycosides are installed with the help of remote participating groups and careful control over solvent and glycosylation temperature.5,35,36
The regioselectivity of the glycosylation reaction is controlled by protecting group selection in the acceptor. Permanent protecting groups are installed on hydroxyl groups that are present as free hydroxyls in the target molecule, and are removed by global deprotection after automated assembly. Temporary protecting groups (tPGs) mask hydroxyl groups that are a part of glycosidic linkages in the target molecule. Orthogonal tPGs are used for branching. Positions where modifications such as sulfation are present in the target molecule are protected with orthogonal tPGs as well.
The “approved building block” concept minimizes the number of PGs that are used during AGA (Figure 4). Benzyl (Bn) ether groups serve as permanent nonparticipating PGs and benzoyl (Bz) esters as permanent participating PGs. Acetate (Ac) esters are occasionally used for remote participation or to tune building block reactivity.29 Recently, cyanopivalolyl (PivCN) was introduced as a participating group for the AGA of oligorhamnans.32 Azido and trichloroacetyl (TCA) protecting groups perform well as nitrogen nonparticipating and participating protecting groups, respectively. Permanent protecting groups are removed after AGA by methanolysis or hydrogenolysis.
Naphthyl (Nap) ether (removed by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone, DDQ), levulinoyl (Lev) ester (removed using hydrazine), and 9-fluorenylmethyloxycarbonyl (Fmoc) carbonate (removed by piperidine) serve as tPGs that can be cleaved orthogonally. The Fmoc protecting group is preferentially installed at positions marked for chain elongation, as it can be cleaved quickly (5 min) and its removal can be monitored by UV as is the case routinely in oligopeptide synthesis.37 Levulinoyl ester is the most commonly used orthogonal tPG to mask branching points. Syntheses including Nap tPG need to be carefully designed, since occasional cleavage of primary Bn groups was observed during Nap cleavage.38p-Methoxybenzyl (PMB) ether groups can be used instead of Nap as a nonparticipating tPG.38 Milder treatment with DDQ used for PMB cleavage does not interfere primary Bn groups. Recently, 2-(azidomethyl)benzoyl (Azmb) ester served as participating tPG,30 and was chemoselectively removed with tributylphosphine to add one degree of orthogonality.
Linker
Linkers are support-bound protecting groups that have been developed in the context of different protecting and anomeric leaving group schemes. Upon cleavage, linkers can reveal various forms of the oligosaccharide reducing end. Metathesis-labile linker 1 is cleaved under conditions that are chemo-orthogonal to the cleavage of temporary and permanent protecting groups, and furnishes an n-pentenyl glycoside that can serve as glycosylating agent (Figure 5). Linker 1 is less used as it is not compatible with the electrophilic reagents required for thioglycoside activation.39,40 Base-labile linker 2, after treatment with sodium methoxide, affords conjugation-ready glycans to be printed on glycan arrays or for use in glycoconjugates. Photolabile linker 3, now commonly used in AGA, is fast, easily and chemoselectively removed by photocleavage in a commercial flow photoreactor, and affords conjugation-ready glycans after global deprotection.41,42 Traceless linker 4, a modified version of linker 3, was developed to obtain glycans with a free reducing end.43 The use of chemo-orthogonal methods for cleavage from the solid support produces fully protected oligosaccharides that are easier to purify using NP-HPLC conditions than the partially protected glycans obtained after cleavage of a base-labile linker. Semiprotected glycans greatly vary in terms of solubility and polarity such that finding appropriate conditions for chromatographic separation is time-consuming and difficult to generalize. Photocleavable linkers offer strategic and practical advantages, but cleavage efficiency is affected by photochemical side-reactions.44 The development of modified linkers with higher cleavage efficiency is an important goal in order to improve AGA.
Figure 5.

Linkers used for AGA. LG: -OP(O)(OR)2, -OC(NR)CR′3 for 1 or -SR, -OP(O)(OR)2, -OC(NR)CR′3 for 2–4. Cleavage and deprotection conditions: (a) Grubbs’s catalyst, CH2CH2; (b) NaOMe/MeOH; (c) flow photoreactor (UV 305 nm); (d) H2, Pd/C.
Automated Synthesis
The retrosynthetic analysis that precedes AGA is straightforward as it dissects the target glycan to identify building blocks based on monosaccharide identity (glucose, mannose, etc.), connectivity (1→4, 1→6, branching, etc.) and glycosidic linkage stereochemistry (α or β). An increasing number of “approved building blocks” and linker-functionalized resins are commercially available.45 The operator adds the linker-functionalized resin to the reaction vessel and attaches the bottles that contain the dissolved building blocks to the instrument. Building block solutions are freshly prepared with anhydrous solvent, but all other reagents and solvents can be used for several automated syntheses. After the operator selects a program for coupling the building blocks, according to the target sequence, a fully automated assembly process is executed.
AGAs are performed using the Glyconeer 2.1 or home-built synthesizers. Conceptionally, these instruments are similar to peptide synthesizers, but the temperature in the reactor can be controlled from −50 to +50 °C. Syntheses are currently performed at 12.5–45 μmol scales.32,41,46 For a scale-up, the potential influence of mixing effects inside the reaction vessel could be overcome by using a larger reaction vessel. The addition of each monomer relies on a coupling cycle that consists of glycosylation, capping and cleavage of a tPG (Figure 4) as well as intermediate washing steps to remove excess reagents. Inside the reaction vessel, the first monosaccharide is attached via its reducing end to the resin-bound linker. Then, a temporary protecting group is removed, to unmask a hydroxyl group on the resin-bound oligosaccharide that will act as a nucleophile in the subsequent glycosylation step. A capping step between the glycosylation and deprotection steps minimizes the formation of side-products by preventing further reaction of deletion sequences that are the product of incomplete glycosylations. For each step, the automated synthesizer controls reagent delivery, temperature and time. The output line from the reaction vessel can be directed to a fraction collector, to recover the excess building block used to drive glycosylation reactions to completion. This setup is particularly useful in homopolymer syntheses. Coupling efficiency can be tracked by UV-monitoring of dibenzofulvene, the product of Fmoc release.24
Until recently, capping was used rarely, to avoid further prolongation of already long synthesis times.25 A fast, mild, and quantitative capping protocol based on Ac2O/MsOH now allows for capping to be performed in every coupling cycle and can be incorporated in most AGA syntheses.47 A more time-consuming capping method that incorporates benzoate esters offers an alternative when acetyl caps are not suitable due to acetyl cleavage or migration in subsequent steps.48,49
Postautomation Operations
After automated assembly, cleavage from the solid support is performed according to the linker used (see above), and the protected oligosaccharide is purified using NP-HPLC. If no further modifications are required, global deprotection will remove all permanent protecting groups. A combination of methanolysis followed by hydrogenolysis is suitable for the removal of all permanent PGs commonly used during AGA. After hydrogenolysis, RP-HPLC purification furnishes the final oligosaccharide. Difficulties in deprotection due to solubility issues50 were overcome by using novel solvent mixtures or by changing the global deprotection regimen to Birch reduction followed by methanolysis.
Scope of AGA
Constant improvement in AGA methods and synthesis protocols paved the way to produce many glycans representing the major classes of mammalian carbohydrates (Figure 6). Poly N-acetyllactosamine assembly required efficient methods to incorporate GlcNAc,51 a challenging monosaccharide both as glycosyl donor and acceptor.52 Glycosylations involving uronic acids and sulfation strategies were implemented for the syntheses of glycosaminoglycans (GAGs) such as keratan sulfates,51 dermatan sulfates53 and hyaluronan.54 Methods for the installation of multiple cis-glycosidic linkages were key to the synthesis of globoside oligosaccharides and α-galactosyl epitopes.36 Efficient branching strategies facilitated access to lactoside oligosaccharides, including blood-group related Lewis antigens and tumor-associated carbohydrate antigens.29,55 Many glycans found on microorganisms were obtained by AGA: Polyglucosides such as α-, β-glucans and dextran;36,50 GlcNAc oligomers like chitin and β-1,6-poly-N-acetylglucosamine (PNAG);50 mycobacterial arabinofuranosides;56 α-oligorhamnans;57 and α-mannans.25,50 Moreover, AGA was used to synthesize defined portions of the capsular polysaccharides (CPSs) of Streptococcus pneumoniae serotypes 3 and 8.4,58 β-Mannosidic linkages were implemented in the syntheses of mannuronic acid alginates.59 To date, highly stereoselective β-mannosylation have proven elusive for AGA60 as neither neighboring group participation nor the anomeric effect can be used to obtain the desired anomer.
Figure 6.

Representative oligosaccharides synthesized using AGA. Structures are represented following SNFG.26 The stereochemistry of the glycosidic linkage is β for pyranoses with gluco configuration and α for pyranoses with manno configuration at C2, unless indicated otherwise. Glc, glucose; GlcNAc, glucosamine; Gal, galactose; GalNAc, galactosamine; Man, mannose; IdoA, iduronic acid; ManA, mannuronic acid; GlcA, glucuronic acid; Araf, arabinofuranose; Xylp, xylopyranose; Rha, rhamnose; Fuc, fucose; 13C-Glc, 13C-labeled glucose.
Plant carbohydrates such as polyglucosides amylose, cellulose, and mixed-linkage glucans are also readily accessible as molecular tools via AGA.31,36,50 Libraries of type-I and type-II arabinogalactans as well as arabinoxylans were produced.30,46,48,49 Arabinogalactans feature multiple challenging α-(1–4)-Gal linkages and arabinoxylans have diverse branching patterns, including disubstituted xylose residues. AGA is versatile in generating linkages involving hydroxyl groups that are poorly nucleophilic or hindered with high yield and stereoselectivity.
AGA makes it possible to create a variety of glycans by combining monosaccharides in a different order or connectivity and to generate unnatural sequences by introducing unnatural monosaccharides. Oligosaccharide probes with strategic single-site substitutions such as 13C-labeled building blocks and glycans carrying ester and amino moieties at the termini for further functionalization are examples for such glycans.50
Ever longer sequences such as 30- and 50-mer polymannosides were synthesized to assess the efficiency and reproducibility of AGA.25 Process improvements including optimized coupling cycles that require less time and solvent,50 as well as a Ac2O/MsOH capping procedure were tested in the context of the 50-mer synthesis.47 Shortened coupling cycles (90 min instead of 300 min including capping) facilitated capping during each of the 50 iterative coupling cycles with mannose 6 (Figure 7). Target glycan 7 was assembled in 22% yield within 75 h, a considerable improvement over 4% in 250 h obtained with previous protocols. Capping reduced the overall building block consumption by one-third and facilitated the purification of 7 from deletion sequences. The improved yield can be partly attributed to a better interaction between the glycosyl donor and the bulky resin-bound nucleophile, as capping minimizes the amount of large deletion sequences bound to the resin. Thus, AGA of long glycans is beginning to move past the mere proof of principle stage.
Figure 7.
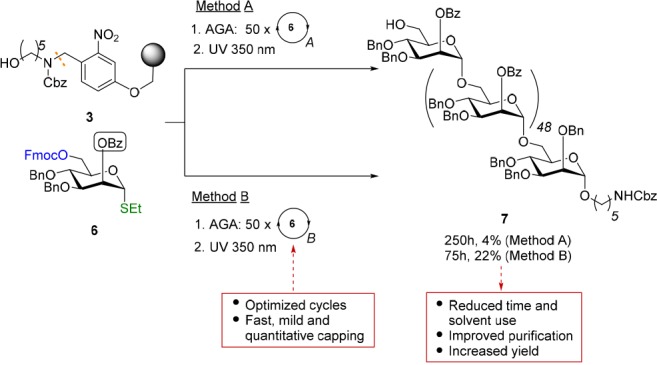
Synthesis of a 50-mer polymannoside to test the limits of AGA. A new capping procedure enables rapid access to longer oligosaccharide sequences. Reaction conditions 1. AGA: Method A for each coupling cycle i. acid wash: TMSOTf, DCM, −20 °C; ii. glycosylation: 6.4 equiv of 6 and NIS/TfOH in DCM/dioxane for −40 °C (1 min) → −20 °C (20 min) (repeated twice for cycles 46 to 50); iii. capping: py, Ac2O at 25 °C (30 min), repeated three times. iv. Fmoc deprotection: TEA (20% in DMF) at 25 °C (5 min), twice. Method B: for each coupling cycle i. acid wash: TMSOTf, DCM, −20 °C; ii. glycosylation: 6.5 equiv of 6 and NIS/TfOH in DCM/dioxane for −20 °C (5 min) → 0 °C (20 min); iii. capping: MsOH (2%) and Ac2O (10%) in anhydrous DCM at 25 °C (20 min). iv. Fmoc deprotection: piperidine (20% in DMF) at 25 °C (5 min). 2. Photocleavage: hν (305 nm).
High yields and stereoselectivities for glycosylations are key to the success of AGA and the construction of certain linkages remains challenging. In those cases, synthetic strategies that combine AGA to accelerate construction of a glycan backbone or a glycan segment, together with other techniques to install the challenging glycosidic linkage can be employed (Figure 8). Difficulties resulting from poor stereoselectivity during α-xyloside formation were bypassed by using a disaccharide building block for the AGA of xyloglucans and galactosylated xyloglucans (Figure 8A).38,42
Figure 8.

Selected oligosaccharides assembled using AGA in combination with other techniques. Structures are represented following the SNFG nomenclature.26 The stereochemistry of the glycosidic linkage is β for pyranoses with gluco configuration and α for pyranoses with manno configuration at C2, unless indicated otherwise. Neu5Ac, N-acetylneuramic acid. Linkages that remain a challenge for AGA are indicated by a pink arrow. (A) Solution phase glycosylation is used to install a challenging linkage in a disaccharide that will serve as a building block for AGA. (B) AGA is used to generate a variety of structures that serve as substrates for enzymatic sialylation. (C) AGA provides rapid access to a tetrasaccharide donor, which is then coupled in solution phase to a myo-inositol-containing acceptor. (D) Fragments obtained as AGA are used as scaffolds for the syntheses of carbohydrate materials using block coupling.
Sialosides are important mammalian glycans that mediate pathogen host-interactions, cell-signaling processes and immune response.1 Sialic acids are nine-carbon monosaccharides bearing a carboxylic acid at C1. High-yielding, stereoselective chemical sialylation is troublesome since the anomeric center is an unreactive quaternary carbon adjacent to the C1 carboxyl electron-withdrawing group. Moreover, no participating group can be placed at C3 to favor the desired α-stereoselectivity. Different AGA methods were tested for the syntheses of sialosides. Sialyl α-(2,3) and α-(2,6) galactosyl imidates were employed as disaccharide building blocks for the AGA of sialosides including tetrasaccharide sialyl Lewisx (Figure 8A).33 Protected sialyl α-(2,3) and α-(2,6) di- and trisaccharides were obtained through an AGA-only glycosylation strategy using 4O,5N-oxazolidinone N-acetylneuramic acid (Neu5Ac) derivatives as sialyl donors.61 Satisfactory results were achieved for the AGA of some α-(2,6) sialosides, but for α-(2,6) sialosides with GlcNAc in the backbone and for α-(2,3) linkages the glycosylation efficiencies remain significantly lower than those obtained for other couplings. Those target structures are among the less reactive acceptors, as the TCA protecting group in GlcNAc is electron-withdrawing and the C3 OH in Gal (in α-(2,3) sialosides) is less reactive than the primary C6 OH. An alternative approach combines AGA with enzymatic sialylation (Figure 8B). Linear oligosaccharides obtained by AGA, bearing a C5-aminolinker at the reducing end, served as substrates for enzymatic sialylation with α-(2,3)-sialyltransferase and cytodine monophosphate (CMP)-Neu5Ac.62
Fast access to glycosylphosphatidylinositol (GPI) glycans found on the surface of parasites is important to the development of diagnostics and vaccines.63 Methods to install the α-linkage between inositol and glucosamine by AGA remain elusive. A tetra-mannosyl fragment prepared by AGA was converted into a glycosyl donor for solution-phase coupling to an inositol-containing disaccharide (Figure 8C).64
Oligosaccharides obtained via AGA can be combined through block-couplings to create tailor-made carbohydrate materials (Figure 8D).50 The oligomer blocks were covalently linked by coupling the amino and carboxylic acid groups placed at the termini of each block. This strategy also produced glycan-peptide hybrid materials that differ from the glycopeptides prepared on a dedicated synthesizer.65 The fully automated assembly of hybrid materials holds great potential beyond the proof-of-concept realized to date.
Applications of Glycans Obtained by AGA
Glycan Arrays
Immobilization of diverse glycans in a spatially defined arrangement on a slide surface allows for high-throughput screening of carbohydrate-binding macromolecules.66 Binding of soluble proteins, whole viruses, bacteria, yeast or mammalian cells can be screened.67 AGA is ideally suited to generate glycan collections to populate arrays, since entire carbohydrate families of different lengths and substitution patterns can be generated using a set of monosaccharide building blocks (Figure 9). Glycans produced by AGA bear a C5-aminolinker for covalent immobilization on slides that are functionalized with N-hydroxysuccinimide (NHS) esters.68 A glycan array containing keratan sulfate GAGs served to identify keratan sulfate as a receptor candidate for a viral gene-therapy vector.51 Synthetic arabinoxylans, xyloglucans and galactosylated xyloglucans with custom-made substitution patterns helped to determine the binding specificities of several monoclonal antibodies (mAbs) commonly used for immunolabeling studies of plant cell walls.38,46 Four frameshifts of native S. pneumoniae ST8 CPS synthesized by AGA were placed on glycan arrays for mAb epitope mapping, en route to the discovery of protective glycotopes.4
Figure 9.

Applications of oligosaccharides synthesized by AGA. Structures are represented following SNFG nomenclature.26
Glycoconjugates
Oligosaccharides obtained by AGA using linkers 2 or 3 are readily conjugated in solution-phase to carrier proteins (Figure 9). Semisynthetic vaccine candidates may confer protective immune responses against infectious diseases. Based on microarray glycotope screening, a S. pneumoniae ST8 CPS sequence was selected for conjugation to CRM197 carrier protein, immunization studies, and mAb production for the identification of protective glycotopes.4 Combination of the glycoconjugate with the pneumococcal vaccine Prevnar 13 resulted in a 14-valent coformulation that generated a robust antibacterial immune response against ST8 without undermining the immunogenicity of Prevnar 13. AGA was used to synthesize fragments of S. pneumoniae ST3 CPS that are tested as vaccine candidates.58
Enzymatic Assays
Synthetic glycans are useful tools for active site mapping and to determine the substrate specificity of enzymes such as hydrolases and transglycosylases. Arabinoxylans, arabinogalactans and mixed-linkage glucans obtained through AGA were applied for determining the substrate specificity of xylan-deconstructing enzymes,30 endogalactanases49 and lichenase.31 To this end, tailor-made carbohydrates with specific substitution patterns were used as enzyme substrates and time-course experiments and HPLC analysis of digestion products were performed.
HPLC analysis of the products of the enzymatic reaction of xyloglucan sequences were used to probe the acceptor-substrate specificity of a xylosyltransferase. In addition, the synthetic, conjugation-ready xyloglucans were coupled to fluorescein (FC) to evaluate the activity of plant xyloglucan endotransglycosylases on glycan arrays.69 Synthetic FC-labeled xyloglucans were incorporated into plant sections.
Chemoenzymatic Synthesis
Sialylated glycans bearing a C5-aminolinker were obtained by combining AGA with α-(2,3) enzymatic sialylation (see above).62 The methodology is currently being expanded to branched fucosylated oligosaccharides and α-(2,6) sialosides.
Labeled Carbohydrates
Linear β-(1–6) glucose hexasaccharides were prepared using a standard building block and its 13C-labeled analogue by placing the 13C-labeled monosaccharide at different positions in the sequence.50 Thereby, chemical shifts corresponding to specific monosaccharides provided structural information from the coupling constants 1JH1C1 and 3JH1H2. Rapid access to labeled glycans by AGA offers new tools to gain conformational and geometric information from synthetic glycans.
Carbohydrate Standards
Synthetic glycans served as standards for developing ion mobility spectrometry-mass spectrometry (IM-MS) as a glycan characterization technique.70 In IM-MS, molecules are separated according to their mass, charge, size and shape. The analysis of synthetic trisaccharide standards showed that IM-MS can differentiate carbohydrate connectivity and anomeric stereoisomers, a feat beyond the capability of simple MS techniques (Figure 9). IM-MS detects as little as 0.1% of a minor isomer in a mixture quickly, while requiring minute amounts of sample without prior derivatization. Therefore, IM-MS has the potential to replace time-consuming and sample-demanding NMR experiments for the full characterization of oligosaccharides.
Carbohydrate Materials
Oligosaccharides were combined through block-coupling to create tailor-made carbohydrate materials and glycan-peptide hybrids (Figure 9).50 Structural studies revealed that single-site substitutions on homooligomer chains can dramatically impact their conformation. The production of novel carbohydrate materials based on changes in monomer substitution and the combination of different blocks is currently being investigated.
Current Challenges and Future Perspectives
Over the past two decades, AGA has evolved from an idea to a technology that produces glycans as molecular tools for various applications. Oligosaccharide assembly is now a streamlined process where potential bottlenecks have been addressed systematically. The automated synthesizer, as well as linker-functionalized resins and monosaccharide building blocks are now commercially available to facilitate access to this methodology to more laboratories. Reliable conditions have been developed for the rapid synthesis of ever more complex oligosaccharides. The bottleneck that once resided with oligosaccharide synthesis moved downstream to the global deprotection and final purification of deprotected glycans. Longer oligosaccharides sometimes aggregate and become insoluble, a phenomenon well-known from peptide chemistry.50 Faster techniques for final product characterization will reduce analysis times and thus the procurement time before the glycan use.
Establishing a reliable set of orthogonal protecting groups and based thereupon the selection of “approved building blocks” was the basis to the assembly of a wide variety of mammalian, bacterial, fungal and plant glycans. Protocols to install multiple cis-glycosidic linkages, sulfates, or poorly reactive building blocks such as uronic acids, Gal 4-OH nucleophiles or GlcNAc were developed. Some linkages, such as β-mannosides, cannot yet be stereoselectively installed using AGA.
While several reliable orthogonal participating temporary PGs are available, just two nonparticipating temporary PGs (Nap or PMB) are currently used. The construction of glycans containing 1,2-cis linkages with branching at C2 found, e.g. in antigenic glycans of schistosome parasites,71 will depend on the development of new, nonparticipating tPGs. The field would also benefit from more work in this area of installing multiple α-galactosides, glucosamines and glucuronic acids in one molecule. The extension of AGA to areas of glycospace not yet explored with this method will push its development forward, to meet new synthetic challenges that cannot yet be foreseen.
The complexity of carbohydrates renders NMR characterization of limited samples time-consuming. Often, more glycan is needed for characterization than for the actual experiments (e.g., glycan arrays). The combination of mass spectrometry with other spectroscopic techniques is a fast alternative that requires minimal sample amounts without derivatization. IM-MS or infrared multiple photon dissociation (IRPMD)-MS are complementary techniques to overcome MS limitations related to carbohydrate mass isomerisms and may distinguish monosaccharide content, anomeric configuration, regiochemistry, and glycosidic linkage stereochemistry.70,72 Synthetic carbohydrate standards will accelerate the development of analytical technologies for carbohydrate sequencing and for the full characterization of synthetic glycans.
AGA facilitates access to a host of glycans for biological applications such as vaccine development, epitope mapping and enzyme characterization as well as molecular tools for fundamental glycobiology investigations. Currently, AGA is paving the way to material sciences based on synthetic rather than isolated glycans. Gaining a detailed understanding of structure–property relationships is crucial for the development of novel carbohydrate-based materials.
Acknowledgments
We thank the Max-Planck Society for generous financial support. Thanks to all current and former co-workers who have made AGA a reality through their hard work and creativity.
The authors declare no competing financial interest.
References
- Essentials of Glycobiology, 2nd ed.; Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, 2009. [PubMed] [Google Scholar]
- Seeberger P. H. The Logic of Automated Glycan Assembly. Acc. Chem. Res. 2015, 48, 1450–1463. 10.1021/ar5004362. [DOI] [PubMed] [Google Scholar]
- Nedra D.; Castro S. P. M.; Paulín E. G. L.; Kwiatkowski S.; Edgar S.; Reyes R. E.; González C. R.; Jiménez R. C.; Herrera O.; Andrade A. A.; et al. The Complex World of Polysaccharides; Karunaratne D. N., Ed.; InTech, 2012. [Google Scholar]
- Schumann B.; Hahm H. S.; Parameswarappa S. G.; Reppe K.; Wahlbrink A.; Govindan S.; Kaplonek P.; Pirofski L.; Witzenrath M.; Anish C.; et al. A Semisynthetic Streptococcus Pneumoniae Serotype 8 Glycoconjugate Vaccine. Sci. Transl. Med. 2017, 9, eaaf5347. 10.1126/scitranslmed.aaf5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Schmidt R. R. New Principles for Glycoside-Bond Formation. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- Panza M.; Pistorio S. G.; Stine K. J.; Demchenko A. V. Automated Chemical Oligosaccharide Synthesis: Novel Approach to Traditional Challenges. Chem. Rev. 2018, 118, 8105–8150. 10.1021/acs.chemrev.8b00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moremen K. W.; Ramiah A.; Stuart M.; Steel J.; Meng L.; Forouhar F.; Moniz H. A.; Gahlay G.; Gao Z.; Chapla D.; et al. Expression System for Structural and Functional Studies of Human Glycosylation Enzymes. Nat. Chem. Biol. 2018, 14, 156–162. 10.1038/nchembio.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; Chen X. One-Pot Multienzyme (OPME) Systems for Chemoenzymatic Synthesis of Carbohydrates. Org. Biomol. Chem. 2016, 14, 2809–2818. 10.1039/C6OB00058D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L.; Edmunds G.; Gibbons C.; Zhang J.; Gadi M. R.; Zhu H.; Fang J.; Liu X.; Kong Y.; Wang P. G. Toward Automated Enzymatic Synthesis of Oligosaccharides. Chem. Rev. 2018, 118, 8151–8187. 10.1021/acs.chemrev.8b00066. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Wu Z.; Gadi M. R.; Wang S.; Guo Y.; Edmunds G.; Guan W.; Fang J. Cation Exchange Assisted Binding-Elution Strategy for Enzymatic Synthesis of Human Milk Oligosaccharides (HMOs). Bioorg. Med. Chem. Lett. 2017, 27, 4285–4287. 10.1016/j.bmcl.2017.08.041. [DOI] [PubMed] [Google Scholar]
- Prudden A. R.; Liu L.; Capicciotti C. J.; Wolfert M. A.; Wang S.; Gao Z.; Meng L.; Moremen K. W.; Boons G.-J. Synthesis of Asymmetrical Multiantennary Human Milk Oligosaccharides. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 6954–6959. 10.1073/pnas.1701785114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra A.; Li Y.; Yu H.; Slack T. J.; Wang P. G.; Chen X. Highly Efficient Chemoenzymatic Synthesis and Facile Purification of α-Gal Pentasaccharyl Ceramide Galα3nLc4βCer. Chem. Commun. 2017, 53, 8280–8283. 10.1039/C7CC04090C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J.; Yu H.; Malekan H.; Sugiarto G.; Li Y.; Qu J.; Nguyen V.; Wu D.; Chen X. Highly Efficient One-Pot Multienzyme (OPME) Synthesis of Glycans with Fluorous-Tag Assisted Purification. Chem. Commun. 2014, 50, 3159–3162. 10.1039/C4CC00070F. [DOI] [PubMed] [Google Scholar]
- Matsushita T.; Nagashima I.; Fumoto M.; Ohta T.; Yamada K.; Shimizu H.; Hinou H.; Naruchi K.; Ito T.; Kondo H.; et al. Artificial Golgi Apparatus: Globular Protein-like Dendrimer Facilitates Fully Automated Enzymatic Glycan Synthesis. J. Am. Chem. Soc. 2010, 132, 16651–16656. 10.1021/ja106955j. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chen C.; Gadi M. R.; Gibbons C.; Guo Y.; Cao X.; Edmunds G.; Wang S.; Liu D.; Yu J.; et al. Machine-Driven Enzymatic Oligosaccharide Synthesis by Using a Peptide Synthesizer. Angew. Chem., Int. Ed. 2018, 57, 16638–16642. 10.1002/anie.201810661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S. S.; Wang C. C.; Sabbavarapu N. M.; Podilapu A. R.; Liao P. H.; Hung S. C. One-Pot” Protection, Glycosylation, and Protection-Glycosylation Strategies of Carbohydrates. Chem. Rev. 2018, 118, 8025–8104. 10.1021/acs.chemrev.8b00036. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Xiong D. C.; Chen S. C.; Wang Y. S.; Ye X. S. Total Synthesis of Mycobacterial Arabinogalactan Containing 92 Monosaccharide Units. Nat. Commun. 2017, 8, 14851. 10.1038/ncomms14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Ollmann I. R.; Ye X. S.; Wischnat R.; Baasov T.; Wong C. H. Programmable One-Pot Oligosaccharide Synthesis. J. Am. Chem. Soc. 1999, 121, 734–753. 10.1021/ja982232s. [DOI] [Google Scholar]
- Cheng C.-W.; Zhou Y.; Pan W.-H.; Dey S.; Wu C.-Y.; Hsu W.-L.; Wong C.-H. Hierarchical and Programmable One-Pot Synthesis of Oligosaccharides. Nat. Commun. 2018, 9, 5202. 10.1038/s41467-018-07618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plante O. J.; Palmacci E. R.; Seeberger P. H. Automated Solid-Phase Synthesis of Oligosaccharides. Science 2001, 291, 1523–1527. 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]
- Nokami T.; Hayashi R.; Saigusa Y.; Shimizu A.; Liu C.-Y.; Mong K.-K. T.; Yoshida J. Automated Solution-Phase Synthesis of Oligosaccharides via Iterative Electrochemical Assembly of Thioglycosides. Org. Lett. 2013, 15, 4520–4523. 10.1021/ol402034g. [DOI] [PubMed] [Google Scholar]
- Tang S. L.; Linz L. B.; Bonning B. C.; Pohl N. L. B. Automated Solution-Phase Synthesis of Insect Glycans to Probe the Binding Affinity of Pea Enation Mosaic Virus. J. Org. Chem. 2015, 80, 10482–10489. 10.1021/acs.joc.5b01428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh N. V.; Fujikawa K.; Tan Y. H.; Stine K. J.; Demchenko A. V. HPLC-Assisted Automated Oligosaccharide Synthesis. Org. Lett. 2012, 14, 3036–3039. 10.1021/ol301105y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm H. S.; Schlegel M. K.; Hurevich M.; Eller S.; Schuhmacher F.; Hofmann J.; Pagel K.; Seeberger P. H. Automated Glycan Assembly Using the Glyconeer 2.1 Synthesizer. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E3385–E3389. 10.1073/pnas.1700141114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naresh K.; Schumacher F.; Hahm H. S.; Seeberger P. H. Pushing the Limits of Automated Glycan Assembly: Synthesis of a 50mer Polymannoside. Chem. Commun. 2017, 53, 9085–9088. 10.1039/C7CC04380E. [DOI] [PubMed] [Google Scholar]
- Stanley P.; Hart G.; Schnaar R. L.; Darvill A.; Prestegard J. J.; Kinoshita T.; Varki A.; Cummings R. D.; Aebi M.; Packer N. H.; et al. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 2015, 25, 1323–1324. 10.1093/glycob/cwv091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield R. B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. 10.1021/ja00897a025. [DOI] [Google Scholar]
- Collot M.; Eller S.; Weishaupt M.; Seeberger P. H. Glycosylation Efficiencies on Different Solid Supports Using a Hydrogenolysis-Labile Linker. Beilstein J. Org. Chem. 2013, 9, 97–105. 10.3762/bjoc.9.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm H. S.; Liang C.-Fu; Lai C.-H.; Fair R. J.; Schuhmacher F.; Seeberger P. H. Automated Glycan Assembly of Complex Oligosaccharides Related to Blood Group Determinants. J. Org. Chem. 2016, 81, 5866–5877. 10.1021/acs.joc.6b00554. [DOI] [PubMed] [Google Scholar]
- Senf D.; Ruprecht C.; de Kruijff G. H. M.; Simonetti S. O.; Schuhmacher F.; Seeberger P. H.; Pfrengle F. Active Site Mapping of Xylan-Deconstructing Enzymes with Arabinoxylan Oligosaccharides Produced by Automated Glycan Assembly. Chem. - Eur. J. 2017, 23, 3197–3205. 10.1002/chem.201605902. [DOI] [PubMed] [Google Scholar]
- Dallabernardina P.; Schuhmacher F.; Seeberger P. H.; Pfrengle F. Mixed-Linkage Glucan Oligosaccharides Produced by Automated Glycan Assembly Serve as Tools To Determine the Substrate Specificity of Lichenase. Chem. - Eur. J. 2017, 23, 3191–3196. 10.1002/chem.201605479. [DOI] [PubMed] [Google Scholar]
- Geert Volbeda A.; Van Mechelen J.; Meeuwenoord N.; Overkleeft H. S.; Van Der Marel G. A.; Codée J. D. C. Cyanopivaloyl Ester in the Automated Solid-Phase Synthesis of Oligorhamnans. J. Org. Chem. 2017, 82, 12992–13002. 10.1021/acs.joc.7b02511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D.; Hurevich M.; Castagner B.; Wang C. C.; Seeberger P. H. Automated Synthesis of Sialylated Oligosaccharides. Beilstein J. Org. Chem. 2012, 8, 1601–1609. 10.3762/bjoc.8.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeberger P. H.; Haase W. C. Solid-Phase Oligosaccharide Synthesis and Combinatorial Carbohydrate Libraries. Chem. Rev. 2000, 100, 4349–4393. 10.1021/cr9903104. [DOI] [PubMed] [Google Scholar]
- Chatterjee S.; Moon S.; Hentschel F.; Gilmore K.; Seeberger P. H. An Empirical Understanding of the Glycosylation Reaction. J. Am. Chem. Soc. 2018, 140, 11942–11953. 10.1021/jacs.8b04525. [DOI] [PubMed] [Google Scholar]
- Hahm H. S.; Hurevich M.; Seeberger P. H. Automated Assembly of Oligosaccharides Containing Multiple Cis-Glycosidic Linkages. Nat. Commun. 2016, 7, 1–8. 10.1038/ncomms12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. -D; Meienhofer J. Solid-Phase Peptide Synthesis Using Mild Base Cleavage of Nα-Fluorenylmethyloxycarbonylamino Acids, Exemplified By a Synthesis of Dihydrosomatostatin. Int. J. Pept. Protein Res. 1978, 11, 246–249. 10.1111/j.1399-3011.1978.tb02845.x. [DOI] [PubMed] [Google Scholar]
- Dallabernardina P.; Ruprecht C.; Smith P. J.; Hahn M. G.; Urbanowicz B. R.; Pfrengle F. Automated Glycan Assembly of Galactosylated Xyloglucan Oligosaccharides and Their Recognition by Plant Cell Wall Glycan-Directed Antibodies. Org. Biomol. Chem. 2017, 15, 9996–10000. 10.1039/C7OB02605F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade R. B.; Plante O. J.; Melean L. G.; Seeberger P. H. Solid-Phase Oligosaccharide Synthesis: Preparation of Complex Structures Using a Novel Linker and Different Glycosylating Agents. Org. Lett. 1999, 1, 1811–1814. 10.1021/ol991071+. [DOI] [PubMed] [Google Scholar]
- Fraser-Reid B.; Udodong U. E.; Wu Z.; Ottosson H.; Merritt J. R.; Rao C. S.; Roberts C.; Madsen R. n-Pentenyl Glycosides in Organic Chemistry: A Contemporary Example of Serendipity. Synlett 1992, 1992, 927–942. 10.1055/s-1992-21543. [DOI] [Google Scholar]
- Eller S.; Collot M.; Yin J.; Hahm H. S.; Seeberger P. H. Automated Solid-Phase Synthesis of Chondroitin Sulfate Glycosaminoglycans. Angew. Chem., Int. Ed. 2013, 52, 5858–5861. 10.1002/anie.201210132. [DOI] [PubMed] [Google Scholar]
- Dallabernardina P.; Schuhmacher F.; Seeberger P. H.; Pfrengle F. Automated Glycan Assembly of Xyloglucan Oligosaccharides. Org. Biomol. Chem. 2016, 14, 309–313. 10.1039/C5OB02226F. [DOI] [PubMed] [Google Scholar]
- Wilsdorf M.; Schmidt D.; Bartetzko M. P.; Dallabernardina P.; Schuhmacher F.; Seeberger P. H.; Pfrengle F. A Traceless Photocleavable Linker for the Automated Glycan Assembly of Carbohydrates with Free Reducing Ends. Chem. Commun. 2016, 52, 10187–10189. 10.1039/C6CC04954K. [DOI] [PubMed] [Google Scholar]
- Guillier F.; Orain D.; Bradley M. Linkers and Cleavage Strategies in Solid-Phase Organic Synthesis and Combinatorial Chemistry. Chem. Rev. 2000, 100, 2091–2158. 10.1021/cr980040+. [DOI] [PubMed] [Google Scholar]
- www.glycouniverse.com.
- Schmidt D.; Schuhmacher F.; Geissner A.; Seeberger P. H.; Pfrengle F. Automated Synthesis of Arabinoxylan-Oligosaccharides Enables Characterization of Antibodies That Recognize Plant Cell Wall Glycans. Chem. - Eur. J. 2015, 21, 5709–5713. 10.1002/chem.201500065. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Kononov A.; Delbianco M.; Seeberger P. H. A Capping Step During Automated Glycan Assembly Enables Access to Complex Glycans in High Yield. Chem. - Eur. J. 2018, 24, 6075–6078. 10.1002/chem.201801023. [DOI] [PubMed] [Google Scholar]
- Bartetzko M. P.; Schuhmacher F.; Hahm H. S.; Seeberger P. H.; Pfrengle F. Automated Glycan Assembly of Oligosaccharides Related to Arabinogalactan Proteins. Org. Lett. 2015, 17, 4344–4347. 10.1021/acs.orglett.5b02185. [DOI] [PubMed] [Google Scholar]
- Bartetzko M. P.; Schuhmacher F.; Seeberger P. H.; Pfrengle F. Determining Substrate Specificities of β1,4-Endogalactanases Using Plant Arabinogalactan Oligosaccharides Synthesized by Automated Glycan Assembly. J. Org. Chem. 2017, 82, 1842–1850. 10.1021/acs.joc.6b02745. [DOI] [PubMed] [Google Scholar]
- Delbianco M.; Kononov A.; Poveda A.; Yu Y.; Diercks T.; Jiménez-Barbero J.; Seeberger P. H. Well-Defined Oligo- and Polysaccharides as Ideal Probes for Structural Studies. J. Am. Chem. Soc. 2018, 140, 5421–5426. 10.1021/jacs.8b00254. [DOI] [PubMed] [Google Scholar]
- Hahm H. S.; Broecker F.; Kawasaki F.; Mietzsch M.; Heilbronn R.; Fukuda M.; Seeberger P. H. Automated Glycan Assembly of Oligo- N-Acetyllactosamine and Keratan Sulfate Probes to Study Virus-Glycan Interactions. Chem. 2017, 2, 114–124. 10.1016/j.chempr.2016.12.004. [DOI] [Google Scholar]
- Crich D.; Dudkin V. Why Are the Hydroxy Groups of Partially Protected N-Acetylglucosamine Derivatives Such Poor Glycosyl Acceptors, and What Can. Be Done about It? A Comparative Study of the Reactivity of N-Acetyl-, N-Phthalimido-, and 2-Azido-2-Deoxy-Glucosamine Derivatives in Glycosylation. 2-Picolinyl Ethers as Reactivity-Enhancing Replacement for Benzyl Ethers. J. Am. Chem. Soc. 2001, 123, 6819–6825. 10.1021/ja010086b. [DOI] [PubMed] [Google Scholar]
- Kandasamy J.; Schuhmacher F.; Hahm H. S.; Klein J. C.; Seeberger P. H. Modular Automated Solid Phase Synthesis of Dermatan Sulfate Oligosaccharides. Chem. Commun. 2014, 50, 1875–1877. 10.1039/C3CC48860H. [DOI] [PubMed] [Google Scholar]
- Walvoort M. T. C.; Volbeda A. G.; Reintjens N. R. M.; Van Den Elst H.; Plante O. J.; Overkleeft H. S.; Van Der Marel G. A.; Codée J. D. C. Automated Solid-Phase Synthesis of Hyaluronan Oligosaccharides. Org. Lett. 2012, 14, 3776–3779. 10.1021/ol301666n. [DOI] [PubMed] [Google Scholar]
- Guberman M.; Bräutigam M.; Seeberger P. H.. Automated Glycan Assembly of Lewis Type I and II Oligosaccharide Antigens, submitted. [DOI] [PMC free article] [PubMed]
- Kandasamy J.; Hurevich M.; Seeberger P. H. Automated Solid Phase Synthesis of Oligoarabinofuranosides. Chem. Commun. 2013, 49, 4453–4455. 10.1039/c3cc00042g. [DOI] [PubMed] [Google Scholar]
- Geert Volbeda A.; Reintjens N. R. M.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. The Cyanopivaloyl Ester: A Protecting Group in the Assembly of Oligorhamnans. Eur. J. Org. Chem. 2016, 2016, 5282–5293. 10.1002/ejoc.201600956. [DOI] [Google Scholar]
- Weishaupt M. W.; Matthies S.; Hurevich M.; Pereira C. L.; Hahm H. S.; Seeberger P. H. Automated Glycan Assembly of a S. Pneumoniae Serotype 3 CPS Antigen. Beilstein J. Org. Chem. 2016, 12, 1440–1446. 10.3762/bjoc.12.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walvoort M. T. C.; van den Elst H.; Plante O. J.; Kröck L.; Seeberger P. H.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. Automated Solid-Phase Synthesis of β-Mannuronic Acid Alginates. Angew. Chem., Int. Ed. 2012, 51, 4393–4396. 10.1002/anie.201108744. [DOI] [PubMed] [Google Scholar]
- Kröck L.; Esposito D.; Castagner B.; Wang C.-C.; Bindschädler P.; Seeberger P. H. Streamlined Access to Conjugation-Ready Glycans by Automated Synthesis. Chem. Sci. 2012, 3, 1617. 10.1039/c2sc00940d. [DOI] [Google Scholar]
- Lai C.-H.; Hahm H. S.; Liang C.-F.; Seeberger P. H. Automated Solid-Phase Synthesis of Oligosaccharides Containing Sialic Acids. Beilstein J. Org. Chem. 2015, 11, 617–621. 10.3762/bjoc.11.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fair R. J.; Hahm H.-S. S.; Seeberger P. H. Combination of Automated Solid-Phase and Enzymatic Oligosaccharide Synthesis Provides Access to α(2,3)-Sialylated Glycans. Chem. Commun. 2015, 51, 6183–6185. 10.1039/C5CC01368B. [DOI] [PubMed] [Google Scholar]
- Kamena F.; Tamborrini M.; Liu X.; Kwon Y. U.; Thompson F.; Pluschke G.; Seeberger P. H. Synthetic GPI Array to Study Antitoxic Malaria Response. Nat. Chem. Biol. 2008, 4, 238–240. 10.1038/nchembio.75. [DOI] [PubMed] [Google Scholar]
- Hewitt M. C.; Snyder D. A.; Seeberger P. H. Rapid Synthesis of a Glycosylphosphatidylinositol-Based Malaria Vaccine Using Automated Solid-Phase Oligosaccharide Synthesis. J. Am. Chem. Soc. 2002, 124, 13434–13436. 10.1021/ja027538k. [DOI] [PubMed] [Google Scholar]
- Hurevich M.; Seeberger P. H. Automated Glycopeptide Assembly by Combined Solid-Phase Peptide and Oligosaccharide Synthesis. Chem. Commun. 2014, 50, 1851–1853. 10.1039/C3CC48761J. [DOI] [PubMed] [Google Scholar]
- Oyelaran O.; Gildersleeve J. C. Glycan Arrays: Recent Advances and Future Challenges. Curr. Opin. Chem. Biol. 2009, 13, 406–413. 10.1016/j.cbpa.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissner A.; Seeberger P. H. Glycan Arrays: From Basic Biochemical Research to Bioanalytical and Biomedical Applications. Annu. Rev. Anal. Chem. 2016, 9, 223–247. 10.1146/annurev-anchem-071015-041641. [DOI] [PubMed] [Google Scholar]
- Geissner A.; Anish C.; Seeberger P. H. Glycan Arrays as Tools for Infectious Disease Research. Curr. Opin. Chem. Biol. 2014, 18, 38–45. 10.1016/j.cbpa.2013.11.013. [DOI] [PubMed] [Google Scholar]
- Ruprecht C.; Dallabernardina P.; Smith P. J.; Urbanowicz B. R.; Pfrengle F. Analyzing Xyloglucan Endotransglycosylases by Incorporating Synthetic Oligosaccharides into Plant Cell Walls. ChemBioChem 2018, 19, 793–798. 10.1002/cbic.201700638. [DOI] [PubMed] [Google Scholar]
- Hofmann J.; Hahm H. S.; Seeberger P. H.; Pagel K. Identification of Carbohydrate Anomers Using Ion Mobility-Mass Spectrometry. Nature 2015, 526, 241–244. 10.1038/nature15388. [DOI] [PubMed] [Google Scholar]
- Hokke C. H.; Yazdanbakhsh M. Schistosome Glycans and Innate Immunity. Parasite Immunol. 2005, 27, 257–264. 10.1111/j.1365-3024.2005.00781.x. [DOI] [PubMed] [Google Scholar]
- Schindler B.; Barnes L.; Renois G.; Gray C.; Chambert S.; Fort S.; Flitsch S.; Loison C.; Allouche A.-R.; Compagnon I. Anomeric Memory of the Glycosidic Bond upon Fragmentation and Its Consequences for Carbohydrate Sequencing. Nat. Commun. 2017, 8, 973. 10.1038/s41467-017-01179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]