

Abstract
Objective
To investigate whether soluble ST2, a prognostic marker in cardiovascular and inflammatory disorders, is associated with neurological injury after aneurysmal subarachnoid hemorrhage (SAH).
Methods
We studied SAH patients from two independent cohorts. Outcome assessments included functional status at 90 days using the modified Rankin Scale (mRS), mortality, and delayed cerebral ischemia (DCI). The relationships between sST2 plasma level and outcome measures were assessed in both cross sectional and longitudinal analysis. Primary blood mononuclear cells from SAH patients and elective aneurysm controls were analyzed by multiparameter flow cytometry.
Results
In the discovery cohort sST2 predicted 90-day mRS 3–6 (C index 0.724, p < 0.001) and mortality in Kaplan-Meier analysis (p < 0.001). The association with functional status was independent of age, sex, World Federation of Neurosurgical Societies (WFNS) score, modified Fisher score, treatment modality and cardiac comorbidities (adjusted OR 2.28, 95% CI 1.04 to 5.00, p = 0.039). Higher sST2 concentration was observed in those patients with DCI (90.8 vs 53.7ng/mL, p = 0.003). These associations were confirmed in a replication cohort. In patients with high sST2, flow cytometry identified decreased expression of CD14 (4.27×105 ± 2950 A.U. vs. 5.64×105 ± 1290 A.U., p < 0.001), and increased expression of CD16 (39,960 ± 272 A.U. vs. 34,869 ± 183 A.U., p < 0.001).
Interpretation
Plasma sST2 predicts DCI, functional outcome and mortality after SAH, independent of clinical and radiographic markers. Elevated sST2 is also associated with changes in CD14+CD16+ monocytes.
Introduction
Early and delayed brain injury after aneurysmal subarachnoid hemorrhage (SAH) contributes to a 30% mortality rate and leads to long-term disability1,2 in those who survive. There is a limited understanding of the molecular pathways involved in early brain injury (EBI)3, delayed cerebral ischemia (DCI)4, and long term functional recovery5. Although the calcium channel blocker nimodipine is approved for SAH6, subsequent trials of cerebral vasodilators have not been shown to alter recovery. This paucity of treatment options highlights the need to identify new biomarkers and candidate pathways that are linked to SAH-associated brain injury.
The immune response is thought to play a key role in the pathophysiology of SAH7, and activation of pro-inflammatory pathways is involved in multiple sequelae including cerebral vasospasm and DCI8–10. One candidate mechanism for regulation of immune response after injury is the ST2/IL-33 pathway. ST2 is a member of the interleukin-1 receptor family that is expressed on multiple immune cell types including T helper cells and macrophages11. IL-33 secreted by injured cells binds to transmembrane ST2, sequestering the signaling adaptor protein MyD88, preventing its interaction with toll-like receptor 4 (TLR4)12. On microglia, monocyte-derived macrophages and helper T cells which express both ST2tm and TLR4,13,14 the binding of IL-33 to ST2tm attenuates the pro-inflammatory TLR4 cascade.12 ST2 also exists as soluble form (sST2), which is generated by differential transcription through a dual promoter system15,16. This alternate transcription has some degree of cell-type specificity, with hematopoietic cells predominantly expressing the transmembrane form and non-hematopoietic cells favoring the soluble form17. Soluble ST2 acts as a decoy receptor, reducing the interaction of IL-33 with transmembrane ST2 and therefore shifting towards a pro-inflammatory response11,18.
ST2 has been implicated in multiple immune-mediated diseases19–22 and also serves as a prognostic biomarker in myocardial infarction23,24 and heart failure25,26. We recently identified sST2 as a marker of secondary brain injury and functional neurologic outcome after ischemic stroke27, suggesting it may also play a role in the immune response to neurovascular injury. In the current study, we evaluated sST2 as a marker of 90-day functional status and mortality in two independent SAH cohorts. To assess whether sST2 was linked to alterations in the immune response, we further explored the relationship between sST2 levels and immune cell subsets using multiparameter flow cytometry.
Methods
Patient Cohorts
Boston Cohort
Consecutive patients aged 18 years or older who presented with subarachnoid hemorrhage (SAH) between 2013 and 2016 were prospectively enrolled in the Massachusetts General Hospital Acute Brain Injury Biobank. All patients or their surrogates provided informed consent following a protocol approved by the Partners Healthcare Institutional Review Board. Those with SAH due to trauma, mycotic aneurysm or arteriovenous malformation were excluded. A total of 190 patients with plasma samples available for analysis were identified. Clinical and demographic data were obtained through patient or surrogate interview and confirmed with medical records. SAH severity was classified using the World Federation of Neurosurgical Societies (WFNS) grading scale28, and hemorrhage burden was classified using the modified Fisher Score29. Plasma samples were available from early, intermediate and late time points, corresponding to median post-bleed time of 3, 8 and 13 days, respectively. In those patients who had external ventricular drains placed for clinical indications, CSF was obtained at the same time points. Sonographic vasospasm was defined as peak systolic velocity > 200 cm/s in the proximal anterior or middle cerebral arteries using transcranial Doppler sonography, per usual clinical practice. Delayed cerebral ischemia (DCI) was defined as a 2-point drop in Glasgow Coma Scale (GCS) or a new focal deficit without another attributable cause for deterioration30. Functional status at 90 days was determined by telephone assessment of mRS with patients and surrogates. Raters were not aware of sST2 levels at the time of assessment. Mortality data through 180 days was obtained from the medical record and confirmed using the Death Master File for the Social Security Administration.
Copenhagen Cohort
A total of 50 consecutive patients with aSAH were prospectively enrolled at Rigshospitalet in Copenhagen, Denmark between November 2014 and May 2015. Patients or their surrogates provided informed consent for inclusion in this study, which was approved by the Danish Scientific Ethics Committee of the Capital Region. Patients were included who were ≥18 years of age, Danish-speaking, and had clearly established symptom onset within 24 hours of presentation. Patients had daily plasma samples collected on post-bleed days 1 through 9. Clinical and demographic data, including DCI designation, were collected in a manner analogous to that as described above for the MGH cohort.
Plasma and CSF sST2 Analysis
Blood was obtained at the timepoints described above and within one hour of collection plasma was isolated and stored at −80°C for later analysis. When available, CSF was similarly collected and stored at −80°C. Soluble ST2 was measured in both sample types using a commercially-available, FDA-approved enzyme-linked immunosorbent assay (ELISA; Presage ST2 Assay Kit, Critical Diagnostics, San Diego, CA). C-reactive protein (CRP) and interleukin-6 (IL-6) were also measured by ELISA (ThermoFisher and R&D Systems, respectively).
Flow Cytometry
Additional whole blood samples from a subset of patients from Massachusetts General Hospital were collected at the early time point for immune cell characterization. Blood was collected into BD Vacutainer CPT tubes and peripheral blood mononuclear cells (PBMCs) were isolated. Samples were stained for the following markers: transmembrane ST2, CD45, CD3, CD4, CD8, CD45RA, CD19, CD56, CD16, CD14, CCR6, CCR4, PD-1, CCR7, CD127, CD28, CXCR3, CD25, HLA-DR, CD80. Samples were analyzed on a Beckman Coulter CytoFLEX LX Flow Cytometer at the Massachusetts General Hospital Pathology Flow Cytometry Core Facility.
Analysis of multiparameter flow cytometry was conducted using the t-distributed Stochastic Neighbor Embedding (tSNE) data reduction package in FlowJo 10 (FlowJo, LLC, Ashland, OR). Each sample was manually gated to identify singlets, and then further to select only viable cells based on staining with iFluor™ 860 maleimide (AAT Bioquest, Inc). Cell populations were down sampled to 10,000 to 30,000 events per subject and tSNE was performed using all parameters listed above. The resulting maps were visually compared and unique populations in each group identified. Changes in expression of specific markers was measured using mean fluorescence intensity. Differences in populations identified by particular marker expression were verified with manual gating.
Statistical Analysis
For baseline characteristics, continuous variables with a normal distribution are described as mean ± standard deviation. Non-normal or ordinal data is described as median and interquartile range. Differences in sST2 between groups at a single time point were determined using Wilcoxon Rank Sum or Kruskal-Wallis tests as appropriate, with Dunn’s test performed for post hoc multiple comparisons. In the MGH cohort, differences in sST2 between groups over time were determined using linear mixed effects models with a random effect for subject. sST2 concentration was log-transformed prior to analysis. In the Copenhagen cohort, which used a balanced design of regular daily sample collection, differences between groups over time were compared using an analysis of response profiles31. Cross sectional analysis in the Copenhagen cohort was conducted using day 3 samples, which was pre-specified to correspond to the early time point in the Boston cohort. The predictive value of sST2 on mRS was determined using receiver operating curve (ROC) analysis with ideal cut points identified using Youden’s Index. Multivariable models of mRS and mortality prediction were calculated using log-transformed sST2 values alongside other clinical variables in logistic regression models. Association of sST2 with mortality was determined using Kaplan-Meier log-rank analysis. All analysis was carried out in Stata version 15 (StataCorp LLC, College Station, TX, USA).
Results
Patient Characteristics
Characteristics of the Boston and Copenhagen cohorts are presented in Table 1. The 190 patients in the Boston cohort had a mean age of 57 ± 11 years, median WFNS grade of 1 [IQR 1, 4] and modified Fisher grade of 3 [IQR 3, 4]. Compared to the Boston patients, the Copenhagen cohort was more predominantly female (90% vs. 62% in Boston) and had a higher WFNS grade (median 3 [1, 4]; p = 0.016), but outcomes were better with a median 90-day mRS of 1 [0, 3] in Copenhagen vs. 2 [1, 3] in Boston (p = 0.045). Plasma sST2 measured at the early time point (median 3 days after ictus) in Boston was similar to day 3 sST2 in Copenhagen (75.1ng/mL vs. 82.4ng/mL, p = 0.73).
Table 1.
Characteristics of the Boston and Copenhagen cohorts
| Boston | Copenhagen | ||
|---|---|---|---|
| n = 190 | n = 50 | p | |
| Age (years) | 57 ± 12 | 61 ± 11 | 0.07 |
| Sex (F) | 117 (62%) | 45 (90%) | < 0.001 |
| WFNS | 1 [1, 4] | 3 [1, 4] | 0.016 |
| Modified Fisher score | 3 [3, 4] | 3 [3, 3] | 0.25 |
| mRS at 90 days | 2 [1, 3] | 1 [0, 3] | 0.045 |
| Mortality at 90 days | 25 (13%) | 5 (10%) | 0.55 |
| DCI* | 61 (43%) | 25 (50%) | 0.39 |
| Plasma sST2 (ng/mL), Day 3 | 75.1 [44.0, 133] | 82.4 [47.8, 167] | 0.73 |
| CSF sST2 (ng/mL), Day 3** | 17.1 [4.1, 44.1] | -- | -- |
DCI designations were available for 142 patients in the MGH cohort
CSF samples were available from 46 patients in the MGH cohort
Plasma sST2 is elevated in SAH and is associated with outcome
Plasma sST2 levels were measured in healthy volunteers (n = 12), patients undergoing elective clipping of unruptured aneurysms (n = 13), those with arteriovenous malformation (n = 6), and those with spontaneous SAH. Spontaneous SAH patients were further subdivided into those with a perimesencephalic pattern (n = 18) and a more diffuse, classical aneurysmal SAH pattern (n = 93). Patients with aneurysmal SAH had significantly higher level of sST2 measured a median of 3 days after ictus compared to those with AVM, perimesencephalic blood, following elective clipping, or in healthy controls (median 80.1ng/ml [IQR 45.7, 139.4] vs. 17.8 [9.24, 50.1], 35.3 [27.6, 40.2], 24.9 [16.6, 30.0] and 25.6 [18.8, 32.1] respectively; adjusted p < 0.0001 for aneurysmal SAH vs. all other conditions; Figure 1A).
Figure 1.

Plasma and CSF sST2 in the Boston Cohort. A) Plasma sST2 is elevated in those patients with aneurysmal SAH compared to those with arteriovenous malformation, perimesencephalic pattern SAH, patients undergoing elective clipping of an unruptured aneurysm, or healthy volunteers (p<0.0001). B) Plasma sST2 is associated with outcome, with significantly higher sST2 in those with poor (mRS 3–6, red squares) compared to good outcome (mRS 0–2, blue circles) using a linear mixed effects model (β = 0.54 [95% CI 0.25 to 0.83], p < 0.001). Markers indicate mean with error bars showing standard deviation. C) A similar pattern was seen in CSF sST2 levels, but there was no significant difference based on outcome (β = 0.61 [95% CI −0.43 to 1.64], p = 0.25). D) Plasma and CSF sST2 are positively correlated (Spearman’s r = 0.37, p = 0.012).
In the Boston cohort, 305 plasma samples were collected from the 190 patients across all three time points. Plasma sST2 levels were highest at the early time point and decreased at the middle and late time points (Figure 1B). Those patients with poor (mRS 3–6) outcome had significantly higher sST2 as compared to those with good (mRS 0–2) outcome (β = 0.54 [95% CI 0.25 to 0.83], p < 0.001). A total of 144 CSF samples were obtained from 46 patients in the Boston cohort. A similar pattern was observed for CSF sST2 levels, but there was no significant difference based on outcome (Figure 1C, β = 0.61 [95% CI −0.43 to 1.64], p = 0.25). However, plasma and CSF sST2 concentrations at the early time point were moderately correlated (Spearman r = 0.37, p = 0.012; Figure 1D).
Early plasma sST2 is an independent predictor of outcome and survival in SAH patients
To further explore the association between sST2 and clinical outcome, we compared early time point sST2 concentration to functional neurologic status and mortality. Elevated plasma sST2 was associated with poor outcome (median 112.6ng/mL [IQR 74.0, 176.4] for mRS 3–6 vs. 55.5ng/mL [39.1, 100.4] for mRS 0–2, p = 0.0002). Similar elevations were seen in plasma IL-6 (median 29.6 pg/ml [IQR 13.7, 51.4] for mRS 3–6 vs. 14.2 pg/ml [6.4, 30.0] for mRS 0–2, p = 0.011) and CRP (29.5μg/mL [9.38, 95.9] for mRS 3–6 vs. 2.24μg/mL [1.40, 6.60] for mRS 0–2, p = 0.021). sST2 remained an independent predictor of poor outcome in multivariable logistic regression models including either IL-6 (adjusted OR for sST2 is 2.06 [1.03, 4.15], p = 0.042; adjusted OR for IL-6 is 1.47 [0.92, 2.33], p = 0.103) or CRP (adjusted OR for sST2 is 2.49 [1.28, 4.85], p = 0.007; adjusted OR for CRP is 1.37 [1.03, 1.81], p = 0.029).
ROC analysis found sST2 to be a significant predictor of poor outcome, with an optimal sST2 cutpoint of 80.8ng/ml (Figure 2A; C index = 0.724, 95% CI 0.620–0.827, p < 0.001). Plasma sST2 was similarly predictive of mortality (Figure 2B; C index = 0.788, 95% CI 0.681–0.896, Youden cut point: 91.3ng/ml, p = 0.002). Kaplan Meier analysis of patients with high versus low sST2 (dichotomized based on the median level at the first timepoint) found those with high sST2 to have a significantly higher risk of death (Figure 2C, p < 0.001).
Figure 2.

Plasma sST2 is associated with outcome and mortality in the Boston cohort. A) ROC analysis finds early sST2 to be a predictor of poor outcome (mRS 3–6) at 90 days (C statistic = 0.724, 95% CI 0.620–0.827, Youden’s cutpoint 80.8ng/mL, p < 0.001). B) Similar results are observed for mortality, where early sST2 is again a predictor of outcome (C statistic = 0.788, 95% CI 0.681–0.896, Youden cut point: 91.3ng/ml, p = 0.002). C) Kaplan Meier analysis of patients with high versus low sST2 (dichotomized based on the median level at the first timepoint) found those with high sST2 to have a significantly higher rate of death (Figure 2D, p < 0.001).
To determine the independence of sST2 as a predictor of functional status and mortality, we constructed multivariable logistic regression models (Table 2). In univariate analysis, log-transformed sST2 at the early timepoint was associated with poor outcome (OR 3.02 [95% CI 1.58–5.76], p < 0.001) and mortality (OR 4.62, 95% CI 1.92–11.1, p < 0.001). sST2 remained an independent predictor of mRS and mortality after adjusting for age, sex and WFNS score (mRS 3–6: adjusted OR 2.31 [1.13, 4.72], p = 0.022; mortality adjusted OR 4.26 [1.43, 12.7], p = 0.009). When including radiographic markers (modified Fisher score and perimesencephalic pattern) and treatment type (aneurysm clipping vs. coiling), sST2 remained an independent predictor (adjusted OR 2.29 [95% CI 1.04–5.01], p = 0.039; mortality adjusted OR 3.54 [95% CI 1.13–11.1], p = 0.03). Given the known role of sST2 as a cardiac biomarker32,33, we further adjusted for a history of atrial fibrillation or congestive heart failure, and sST2 again remained an independent predictor of outcome (adjusted OR 2.28 [95% CI 1.04–5.00], p = 0.039) and mortality (adjusted OR 3.19 [95% CI 1.00–10.2], p = 0.05).
Table 2.
Univariate and multivariable models for prediction of functional outcome and mortality
| Poor outcome (mRS 4–6) | Mortality | ||||||
|---|---|---|---|---|---|---|---|
| sST2 OR | 95% CI | p | sST2 OR | 95% CI | p | ||
| Model 1 | 3.02 | 1.58 – 5.76 | <0.001 | 4.62 | 1.92 – 11.1 | <0.001 | |
| Model 2 | 2.31 | 1.13 – 4.72 | 0.022 | 4.26 | 1.43 – 12.7 | 0.009 | |
| Model 3 | 2.29 | 1.04 – 5.01 | 0.039 | 3.54 | 1.13 – 11.1 | 0.03 | |
| Model 4 | 2.28 | 1.05 – 5.00 | 0.039 | 3.19 | 1.00 – 10.2 | 0.05 | |
Model 1: sST2 univariate
Model 2: Model 1 + age, sex, WFNS
Model 3: Model 2 + modified Fisher, perimesencephalic appearance, clip vs. coil
Model 4: Model 3 + atrial fibrillation, congestive heart failure
sST2 levels are associated with outcome in a replication cohort
To replicate these findings in an independent SAH cohort, we measured sST2 in plasma samples from SAH patients enrolled in an observational study from Rigshospitalet, Copenhagen. Samples were collected daily, up to post-bleed day 9, for a total of 374 samples from the 50 patients. Similar to the discovery cohort (as shown in Figure 1B), patients with poor outcome had a higher sST2 over time (Figure 3A, p = 0.002). sST2 was similarly higher over time in those who died (Figure 3B, p < 0.0001). To parallel our analysis of sST2 at the early timepoint versus outcome in the Boston cohort (Figure 2A), we compared sST2 at day 3 between those with good and poor outcome in the Copenhagen cohort, and again found higher sST2 concentration in those with poor 90-day mRS (Figure 3C, p = 0.012).
Figure 3.
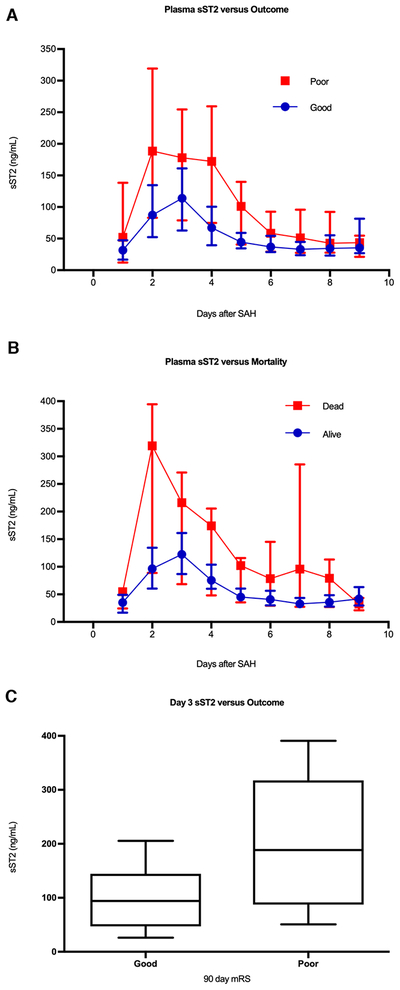
Plasma sST2 and outcome in the Copenhagen cohort. A) sST2 over time was also significantly higher in those with poor outcome in the Copenhagen cohort (analysis of response profiles, p = 0.002). B) Similar results were seen for mortality, with sST2 again higher over time in those who died, but this different did not meet the threshold for significance (p < 0.0001). C) In an effort to replicate the Boston cohort results, sST2 concentration at day 3 after ictus, corresponding to the median day of the early timepoint in the Boston cohort, was compared between those with good and poor outcome. Median day 3 sST2 concentration was higher in those with poor outcome (188.5 [IQR 87.4 – 317.6] ng/mL versus 94.3 [IQR 47.2 – 114.5] ng/mL, p = 0.012).
Early sST2 level predicts delayed cerebral ischemia
To explore the link between sST2 and secondary neurologic injury, we examined the relationship between early timepoint sST2 and sonographic vasospasm, and between sST2 and DCI in the Boston cohort. While the presence of sonographic vasospasm was associated with risk for DCI in the Boston cohort (risk ratio 2.4 [95% CI 1.26–4.43], p =0.003), there was no difference in early timepoint sST2 between those with and without vasospasm (79.0 vs 74.4ng/mL, p = 0.476; Figure 4A). There was however, significantly higher early sST2 in those with DCI (90.8 vs 53.7ng/mL, p = 0.003; Figure 4B). Similar results were seen in the Copenhagen cohort, where sST2 over time was higher in those with DCI (Figure 4C, p = 0.049). Elevated sST2 at the early timepoint predicted DCI in the MGH cohort with an optimal cut point of 76.7ng/mL by Youden’s Index (C index = 0.668, 95% CI 0.548 – 0.789, p = 0.034).
Figure 4.

sST2 levels are associated with DCI. A) No association was seen between presence of sonographic vasospasm (defined as peak middle cerebral artery velocity > 200cm/sec) and median plasma sST2 at the early time point (79.0 vs 74.4ng/mL, p = 0.476). B) There was, however, higher day 3 sST2 in those patients with DCI, defined as a clinical worsening of at least 2 points on the GCS scale over 24 hours (90.8 vs 53.7ng/mL, p = 0.003). C) The relationship between sST2 and DCI was also present in the Copenhagen cohort, where those with DCI had significantly higher sST2 concentration over time (analysis of response profiles, p = 0.049). Markers indicate mean sST2, error bars show standard deviation. D) Elevated early timepoint sST2 predicted DCI in the Boston cohort with an optimal cut point of 76.7ng/mL by Youden’s Index (C index = 0.668 [0.548, 0.789], p = 0.034).
High sST2 is associated with changes in immune cell populations
We next sought to explore the differences in immune cell populations associated with SAH. We first compared PBMCs from aneurysmal SAH and control patients at the time of elective aneurysm clipping (n = 6 for each group; 30,000 cells per patient). The tSNE maps (Figure 5A) identified a cell population selectively increased in aneurysmal SAH which expressed transmembrane ST2, CD14 and in subpopulations, CD16 or CD56. Manual gating found the proportion of cells expressing transmembrane ST2 and CD16 to be increased in SAH patients compared to elective clipping cases (38.0±6.7% in SAH [n=6] vs. 23.0±3.15% in controls [n=13], p = 0.035). There were also higher proportions of ST2+CD14+ (37.7±14.7% vs. 17.0±3.5%, p = 0.095) and ST2+CD56+ populations (25.6±3.3% vs. 22.1±3.1%, p = 0.494), but these differences were not significant.
Figure 5.

Flow cytometric analysis of subarachnoid hemorrhage patients. A) t-distribution Stochastic Neighbor Embedding (tSNE) analysis of subjects with aneurysmal SAH (n = 6) and control patients undergoing elective clipping (n = 6). Patients with aneurysmal rupture appear to have an increase in cells expressing transmembrane ST2 (tmST2) as well as the monocyte marker CD14, with subpopulations of CD16 and CD56 positive cells. B) tSNE analysis of aneurysmal rupture subjects with low-sST2 versus high-ST2 at the early time point (defined as below or above the median value of 75ng/mL, n = 3 in each group). There is a shift in expression of tmST2 (circled population in 3 left panels), with lesser intensity in the high ST2 group. This population also appears to have lower CD14 and variably increased CD16 expression. Unique cell clusters expressing NK cell marker CD56 were identified in low-sST2 and high-sST2 populations (circled clusters in right-most panels) variably expressing CD14 and CD16. C) Analysis of mean fluorescence intensity within the subpopulation of cells identified by tSNE analysis in the aneurysmal rupture patients finds that high-sST2 is associated with lower tmST2 (43,027 ± 860 vs. 86,500 ± 368, p < 0.001), lower CD14 (4.27×105 ± 2950 vs. 5.64×105 ± 1290, p < 0.001) and higher CD16 expression (39,960 ± 272 vs. 34,869 ± 183, p < 0.001). Markers indicate mean, error bars show standard error. D) Manual gating of CD56+ cell subpopulations in aneurysmal rupture patients finds lower percentage of CD14+CD56+ (19.8 ± 0.33% in low sST2 vs. 2.4 ± 1.3% in high ST2, p = 0.0002) and higher percentage of CD16+CD56+ cells in the high-sST2 patients (8.2 ± 4.21% vs. 18.1 ± 2.4%, p = 0.11).
To further examine whether these changes were also related to sST2 expression, the aneurysmal rupture SAH patients were divided into low- and high-sST2 groups and PBMCs from patients in each group were again compared using tSNE. The resulting maps (Figure 5B) highlighted a shift in expression of transmembrane ST2, CD14 and CD16 (circled in Figure 5B left-most panels). This was confirmed by comparison of mean fluorescence intensity (Figure 5C), where high sST2 was associated with lower tmST2 (43,027 ± 860 vs. 86,500 ± 368, p < 0.001), lower CD14 (4.27×105 ± 2950 vs. 5.64×105 ± 1290, p < 0.001), and higher CD16 (39,960 ± 272 vs. 34,869 ± 183, p < 0.001). No changes were seen in expression of T cell markers, which has been associated with sST2 in other conditions34,35, a finding that was confirmed by manual gating for Th1 (16.7±0.75% in low sST2 vs. 17.1±2.1% in high, p = 0.845) and Th2 (7.0±0.46% in low vs. 5.8±0.69% in high, p = 0.191) subsets. In addition, tSNE identifies two populations of CD56+ cells that were differentially present in low and high sST2 patients (circled in rightmost panels of Figure 5). The population present in low sST2 was CD56+CD14+ but CD16-, while that present in high sST2 was CD56+CD16+ but CD14-.
Potential changes in CD56+ NK cell subsets were verified by manually gating the entire PBMC population in each aneurysmal SAH sample to identify the proportion of PBMCs expressing each subgroup of markers (Figure 5D). High sST2 was associated with a higher proportion of CD16+CD56+ cells, although this difference was not significant (8.2 ± 4.21% vs. 18.1 ± 2.4%, p = 0.11). There was a significantly smaller proportion of CD14+CD56+ cells in those patients with high sST2 (19.8 ± 0.33% in low sST2 vs. 2.4 ± 1.3% in high ST2, p = 0.0002).
Discussion
In this study, we demonstrate that plasma level of sST2 is predictive of functional outcome and mortality after subarachnoid hemorrhage. The predictive ability of sST2 was independent of clinical and radiographic factors including age, WFNS, modified Fisher scale, cardiac co-morbidities and treatment modality. These findings were replicated in a second independent cohort of SAH patients. These associations were present in longitudinal as well as cross-sectional analysis early in the hospital course. This latter point demonstrates the potential of sST2 to serve as a clinically relevant biomarker with early levels predicting subsequent clinical events. This is highlighted by the association between day 3 sST2 and subsequent development of DCI. Interestingly, sST2 was not associated with sonographic vasospasm, adding further support to the concept that sonographic changes are poorly correlated with clinical findings36,37 and that other pathophysiologic mechanisms may contribute to clinical deterioration after SAH.
One proposed contributor to delayed deterioration and poor outcome after SAH is activation of the immune response38–42. Consistent with this, we found that aneurysm rupture, and furthermore high levels of sST2 after rupture, were associated with shifts in immune cell populations. Our comparison between SAH patients and control patients undergoing elective aneurysm clipping found an apparent shift in a CD14+ monocyte population, which is consistent with animal studies of SAH43,44. Human studies have similarly show increases in monocyte-associated genes after aneurysm rupture45, elevations in monocyte chemoattractant protein 1 in serum46 and CSF47 after SAH and increase in activated monocytes identified by CSF and peripheral blood flow cytometry7. We further found that in those SAH patients with high sST2, there was a reduction in CD14 expression intensity and increase in CD16 expression as well as a shift in a CD56+ cell population from CD56+CD14+ to CD56+CD16+. CD56+ is a marker of natural killer (NK) cells, and CD16+ is an activating receptor on those cells48. CD14+CD56+ cells, which also express HLA-DR (data not shown), are thought to be a precursor of dendritic cells, and thus are involved in initiation of the adaptive immune response49,50. Increased peripheral blood NK cells have been reported in patients undergoing clipping for ruptured aneurysm, particularly in those with poor outcome51. NK cell subset analysis has similarly shown increased CD16+ NK cell populations in the CSF of SAH patients with vasospasm52. While sST2 has been associated with shifts in T helper cell subsets in autoimmune diseases11,19,22, we did not observe any association between sST2 level and T cell populations in this study. Taken together, our data support alterations in the innate immune response, including monocytes and NK cells, in relation to SAH and increased sST2 level.
This study has several limitations. It was a retrospective analysis, although we used two independent, prospectively-collected cohorts that allowed for replication. Both cohorts consisted of consecutively enrolled patients, but not all patients consented which has potential to introduce bias. Moreover, the two cohorts did differ, where the Copenhagen cohort was predominantly female, and the Boston cohort was more evenly distributed. The Copenhagen cohort had a higher initial WFNS grade, but better median 90 day outcome. Despite these differences, we found similar relationships between sST2, DCI and outcome in the two cohorts. Our flow cytometry analysis was based on a panel design that surveyed a broad array of markers to phenotype as many immune cell types as possible. While this has the advantage of broadly phenotyping immune cells, it limited our ability to further specify the monocyte subtypes that were altered with high sST2. Future study examining multiple timepoints and using additional monocyte/macrophage and NK cell markers with techniques such as mass cytometry will be needed to further characterize these changes.
In summary, we find that plasma sST2 measured early after aneurysm rupture is able to predict DCI, functional neurologic outcome and mortality. Furthermore, elevated sST2 is associated with shifts towards a more pro-inflammatory population of monocytes and NK cells in patients with SAH. These findings have important implications not only for guiding risk-stratification of patients with SAH, but also provide a pathway for further investigation into mechanisms of neurological deterioration after aneurysm rupture that may inform the design of novel therapeutic targets.
Acknowledgements
MBB receives funding from the Andrew David Heitman Neurovascular Foundation and American Academy of Neurology. WTK receives funding from the Andrew David Heitman Neurovascular Foundation, American Heart Association, and NINDS.
Footnotes
Potential Conflicts of Interest
The authors report no relevant conflicts of interest.
References
- 1.Vlak MHM, Algra A, Brandenburg R, Rinkel GJE. Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: A systematic review and meta-analysis. Lancet Neurol. 2011;10(7):626–636. [DOI] [PubMed] [Google Scholar]
- 2.Connolly ES, Rabinstein AA, Carhuapoma JR, et al. Guidelines for the Management of Aneurysmal Subarachnoid Hemorrhage: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2012;43(6):1711–1737. [DOI] [PubMed] [Google Scholar]
- 3.Fujii M, Yan J, Rolland WB, et al. Early Brain Injury, an Evolving Frontier in Subarachnoid Hemorrhage Research. Transl. Stroke Res. 2013;4(4):432–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol 2014;10(1):44–58. [DOI] [PubMed] [Google Scholar]
- 5.Vergouwen MDI, de Haan RJ, Vermeulen M, Roos YBWEM. Effect of statin treatment on vasospasm, delayed cerebral ischemia, and functional outcome in patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis update. Stroke 2010;41(1):e47–52. [DOI] [PubMed] [Google Scholar]
- 6.Allen GS, Ahn HS, Preziosi TJ, et al. Cerebral arterial spasm--a controlled trial of nimodipine in patients with subarachnoid hemorrhage. [Internet]. N. Engl. J. Med 1983;308(11):619–24.Available from: http://www.ncbi.nlm.nih.gov/pubmed/6338383 [DOI] [PubMed] [Google Scholar]
- 7.Moraes L, Grille S, Morelli P, et al. Immune cells subpopulations in cerebrospinal fluid and peripheral blood of patients with Aneurysmal Subarachnoid Hemorrhage. Springerplus 2015;4(1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghaemi A, Alizadeh L, Babaei S, et al. Astrocyte-mediated inflammation in cortical spreading depression. Cephalalgia 2017;033310241770213. [DOI] [PubMed] [Google Scholar]
- 9.Osuka K, Suzuki Y, Tanazawa T, et al. Interleukin-6 and development of vasospasm after subarachnoid haemorrhage. Acta Neurochir. (Wien). 1998;140(9):943–51. [DOI] [PubMed] [Google Scholar]
- 10.Chamling B, Gross S, Stoffel-Wagner B, et al. Early Diagnosis of Delayed Cerebral Ischemia: Possible Relevance for Inflammatory Biomarkers in Routine Clinical Practice? World Neurosurg. 2017;104:152–157. [DOI] [PubMed] [Google Scholar]
- 11.Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. [Internet]. Nat. Rev. Drug Discov. 2008;7(10):827–40.Available from: http://www.ncbi.nlm.nih.gov/pubmed/18827826%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4277436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drieu A, Martinez de Lizarrondo S, Rubio M. Stopping Inflammation in Stroke: Role of ST2/IL-33 Signaling. J. Neurosci 2017;37(40):9614–9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brint EK, Xu D, Liu H, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. [Internet]. Nat. Immunol 2004;5(4):373–9.Available from: http://www.ncbi.nlm.nih.gov/pubmed/15004556 [DOI] [PubMed] [Google Scholar]
- 14.Xu D, Chan WL, Leung BP, et al. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. [Internet]. J. Exp. Med 1998;187(5):787–94.Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2212173&tool=pmcentrez&rendertype=abstract%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/9480988%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2212173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergers G, Reikerstorfer A, Braselmann S, et al. Alternative promoter usage of the Fos-responsive gene Fit-1 generates mRNA isoforms coding for either secreted or membrane-bound proteins related to the IL-1 receptor. EMBO J. 1994;13(5):1176–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwahana H, Yanagisawa K, Ito-Kosaka A, et al. Different promoter usage and multiple transcription initiation sites of the interleukin-1 receptor-related human ST2 gene in UT-7 and TM12 cells. Eur. J. Biochem 1999;264(2):397–406. [DOI] [PubMed] [Google Scholar]
- 17.Thomassen E, Kothny G, Haas S, et al. Role of Cell Type-specific pomoters in developmental regulation of T1, an interleukin 1 receptor homologue. Cell Growth Differ. 1995;6(February):179–184. [PubMed] [Google Scholar]
- 18.Hayakawa H, Hayakawa M, Kume A, Tominaga SI. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J. Biol. Chem 2007;282(36):26369–26380. [DOI] [PubMed] [Google Scholar]
- 19.Xu D, Jiang H-R, Kewin P, et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc. Natl. Acad. Sci. U. S. A 2008;105(31):10913–10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larché M, Robinson DS, Kay AB. The role of T lymphocytes in the pathogenesis of asthma. J. Allergy Clin. Immunol 2003;111(3):450–63. [DOI] [PubMed] [Google Scholar]
- 21.Fletcher JM, Lalor SJ, Sweeney CM, et al. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis [Internet]. Clin. Exp. Immunol 2010;162(1):1–11.Available from: http://doi.wiley.com/10.1111/j.1365-2249.2010.04143.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ali M, Zhang G, Thomas WR, et al. Investigations into the role of ST2 in acute asthma in children. Tissue Antigens 2009;73(3):206–212. [DOI] [PubMed] [Google Scholar]
- 23.Shimpo M, Morrow DA, Weinberg EO, et al. Serum Levels of the Interleukin-1 Receptor Family Member ST2 Predict Mortality and Clinical Outcome in Acute Myocardial Infarction. Circulation 2004;109(18):2186–2190. [DOI] [PubMed] [Google Scholar]
- 24.Sabatine MS, Morrow DA, Higgins LJ, et al. Complementary roles for biomarkers of biomechanical strain ST2 and N-terminal prohormone B-type natriuretic peptide in patients with ST-elevation myocardial infarction. Circulation 2008;117(15):1936–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ky B, French B, McCloskey K, et al. High-sensitivity ST2 for prediction of adverse outcomes in chronic heart failure. Circ. Hear. Fail 2011;4(2):180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dieplinger B, Januzzi JL, Steinmair M, et al. Analytical and clinical evaluation of a novel high-sensitivity assay for measurement of soluble ST2 in human plasma - The Presage™ ST2 assay [Internet]. Clin. Chim. Acta 2009;409(1–2):33–40.Available from: 10.1016/j.cca.2009.08.010 [DOI] [PubMed] [Google Scholar]
- 27.Wolcott Z, Batra A, Bevers MB, et al. Soluble ST2 predicts outcome and hemorrhagic transformation after acute stroke [Internet]. Ann. Clin. Transl. Neurol 2017;4(8):553–563.Available from: http://doi.wiley.com/10.1002/acn3.435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charles G Report of World Federation of Neurological Surgeons Committee on a Universal Subarachnoid Hemorrhage Grading Scale. [Internet]. J. Neurosurg 1988;68(6):985–6.Available from: http://www.ncbi.nlm.nih.gov/pubmed/3131498 [DOI] [PubMed] [Google Scholar]
- 29.Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified fisher scale. [Internet]. Neurosurgery 2006;59(1):21–7.Available from: http://www.ncbi.nlm.nih.gov/pubmed/16823296 [DOI] [PubMed] [Google Scholar]
- 30.Vergouwen MDI, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. [Internet]. Stroke 2010;41(10):2391–5.Available from: http://www.ncbi.nlm.nih.gov/pubmed/20798370 [DOI] [PubMed] [Google Scholar]
- 31.Fitzmaurice GM, Ravichandran C. A primer in longitudinal data analysis. Circulation 2008;118(19):2005–2010. [DOI] [PubMed] [Google Scholar]
- 32.Weinberg EO, Shimpo M, Hurwitz S, et al. Identification of serum soluble ST2 receptor as a novel heart failure biomarker. Circulation 2003;107(5):721–726. [DOI] [PubMed] [Google Scholar]
- 33.Shimpo M, Morrow DA, Weinberg EO, et al. Serum levels of the interleukin-1 receptor family member ST2 predict mortality and clinical outcome in acute myocardial infarction. [Internet]. Circulation 2004;109(18):2186–90.Available from: http://www.ncbi.nlm.nih.gov/pubmed/15117853 [DOI] [PubMed] [Google Scholar]
- 34.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. [Internet]. Immunity 2005;23(5):479–90.Available from: http://www.ncbi.nlm.nih.gov/pubmed/16286016 [DOI] [PubMed] [Google Scholar]
- 35.Kakkar AK, Cimminiello C, Goldhaber SZ, et al. Low-molecular-weight heparin and mortality in acutely ill medical patients [Internet]. N Engl J Med 2011;365(26):2463–2472.Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22204723%5Cnhttp://www.nejm.org/doi/pdf/10.1056/NEJMoa1111288 [DOI] [PubMed] [Google Scholar]
- 36.Dankbaar JW, Rijsdijk M, Van Der Schaaf IC, et al. Relationship between vasospasm, cerebral perfusion, and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Neuroradiology 2009;51(12):813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Etminan N, Vergouwen MDI, Ilodigwe D, MacDonald RL. Effect of pharmaceutical treatment on vasospasm, delayed cerebral ischemia, and clinical outcome in patients with aneurysmal subarachnoid hemorrhage: A systematic review and meta-analysis. J. Cereb. Blood Flow Metab. 2011;31(6):1443–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Provencio JJ. Inflammation in subarachnoid hemorrhage and delayed deterioration associated with vasospasm: a review. Acta Neurochir. Suppl 2013;115:233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGirt MJ, Mavropoulos JC, McGirt LY, et al. Leukocytosis as an independent risk factor for cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J. Neurosurg 2003;98(6):1222–1226. [DOI] [PubMed] [Google Scholar]
- 40.Yoshimoto Y, Tanaka Y, Hoya K. Acute systemic inflammatory response syndrome in subarachnoid hemorrhage. Stroke 2001;32(9):1989–93. [DOI] [PubMed] [Google Scholar]
- 41.Miller BA, Turan N, Chau M, Pradilla G. Inflammation, Vasospasm, and Brain Injury after Subarachnoid Hemorrhage. Biomed Res. Int 2014;2014:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lucke-Wold BP, Logsdon AF, Manoranjan B, et al. Aneurysmal Subarachnoid Hemorrhage and Neuroinflammation: A Comprehensive Review. Int. J. Mol. Sci 2016;17(4):497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu Z, Shi WH, Xu LB, et al. Resident Microglia Activate before Peripheral Monocyte Infiltration and p75 NTR Blockade Reduces Microglial Activation and Early Brain Injury after Subarachnoid Hemorrhage. ACS Chem. Neurosci 2018;Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 44.Schneider UC, Schiffler J, Hakiy N, et al. Functional analysis of Pro-inflammatory properties within the cerebrospinal fluid after subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflammation 2012;9:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pera J, Korostynski M, Golda S, et al. Gene expression profiling of blood in ruptured intracranial aneurysms: In search of biomarkers. J. Cereb. Blood Flow Metab. 2013;33(7):1025–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim GH, Kellner CP, Hahn DK, et al. Monocyte chemoattractant protein–1 predicts outcome and vasospasm following aneurysmal subarachnoid hemorrhage. J. Neurosurg 2008;109(1):38–43. [DOI] [PubMed] [Google Scholar]
- 47.Niwa A, Osuka K, Nakura T, et al. Interleukin-6, MCP-1, IP-10, and MIG are sequentially expressed in cerebrospinal fluid after subarachnoid hemorrhage. J. Neuroinflammation 2016;13(1):217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Montaldo E, Del Zotto G, Della Chiesa M, et al. Human NK cell receptors/markers: A tool to analyze NK cell development, subsets and function. Cytom. Part A 2013;83(8):702–713. [DOI] [PubMed] [Google Scholar]
- 49.Gruenbacher G, Gander H, Rahm A, et al. CD56+ human blood dendritic cells effectively promote TH1-type gammadelta T-cell responses. Blood 2009; [DOI] [PubMed] [Google Scholar]
- 50.Van Acker HH, Capsomidis A, Smits EL, Van Tendeloo VF. CD56 in the Immune System: More Than a Marker for Cytotoxicity? Front. Immunol 2017;8(July):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang Y, Zhou Y, Peng Y, Zhang M. The Quantitative and Functional Changes of Postoperative Peripheral Blood Immune Cell Subsets Relate to Prognosis of Patients with Subarachnoid Hemorrhage: A Preliminary Study [Internet]. World Neurosurg. 2017;108:206–215.Available from: 10.1016/j.wneu.2017.08.091 [DOI] [PubMed] [Google Scholar]
- 52.Spitzer D, Spitzer NJ, Deininger M, et al. Activation of Cytotoxic Natural Killer Cells After Aneurysmal Subarachnoid Hemorrhage. [Internet]. World Neurosurg. 2017;101:666–676.e1.Available from: http://www.ncbi.nlm.nih.gov/pubmed/28323187 [DOI] [PubMed] [Google Scholar]