

Abstract
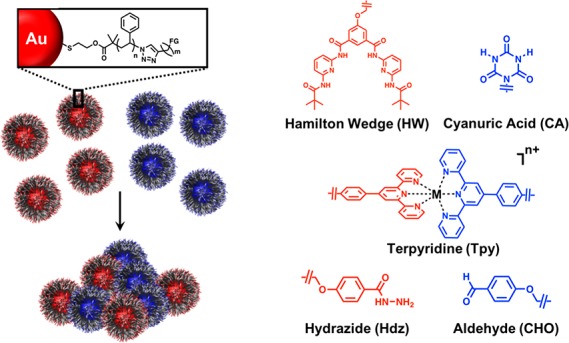
Nanocomposite tectons (NCTs) are a recently developed building block for polymer–nanoparticle composite synthesis, consisting of nanoparticle cores functionalized with dense monolayers of polymer chains that terminate in supramolecular recognition groups capable of linking NCTs into hierarchical structures. In principle, the use of molecular binding to guide particle assembly allows NCTs to be highly modular in design, with independent control over the composition of the particle core and polymer brush. However, a major challenge to realize an array of compositionally and structurally varied NCT-based materials is the development of different supramolecular bonding interactions to control NCT assembly, as well as an understanding of how the organization of multiple supramolecular groups around a nanoparticle scaffold affects their collective binding interactions. Here, we present a suite of rationally designed NCT systems, where multiple types of supramolecular interactions (hydrogen bonding, metal complexation, and dynamic covalent bond formation) are used to tune NCT assembly as a function of multiple external stimuli including temperature, small molecules, pH, and light. Furthermore, the incorporation of multiple orthogonal supramolecular chemistries in a single NCT system makes it possible to dictate the morphologies of the assembled NCTs in a pathway-dependent fashion. Finally, multistimuli responsive NCTs enable the modification of composite properties by postassembly functionalization, where NCTs linked by covalent bonds with significantly enhanced stability are obtained in a fast and efficient manner. The designs presented here therefore provide major advancement for the field of composite synthesis by establishing a framework for synthesizing hierarchically ordered composites capable of complicated assembly behaviors.
Introduction
Nanoparticle–polymer composites are an important class of materials composed of disparate inorganic and polymeric components, combined in a manner that allows the resulting materials to exhibit physical characteristics not observed in either of the single phases.1−6 By mixing nanoparticles and polymers of different sizes,7 shapes, molecular structures,8 and elemental compositions in varying stoichiometric amounts,9 the bulk makeup of the resulting material can be tuned across a diverse range of overall compositions.10−15 In order to fully dictate the properties of these composites, however, it is necessary to control the arrangement of individual polymer and particle building blocks within the composite, as the spatial positioning of different components affects how these different phases interact with one another.16,17 The development of methods to achieve this level of control represents a major challenge for future materials syntheses. It is therefore critical to establish new types of chemical interactions and stimuli that can programmably dictate how the constituent components interact with one another.18 Moreover, significant benefit could be obtained from the ability to dynamically manipulate each of these types of interactions after the composite has been prepared, as this would enable postsynthetic modification of material structure19 or pathway-dependent organization of particles within the polymer matrix.
The “nanocomposite tecton” (NCT) is a recently developed nanoparticle-based construct20 capable of programmed self-assembly that is ideal for synthesizing these types of complex composites. Each NCT is inherently a composite material in itself, and contains multiple independently addressable design elements that can be used to program interactions between NCTs and their surrounding environment. NCTs consist of an inorganic nanoparticle (NP) core functionalized with a dense polymer brush, where each polymer chain that comprises the brush terminates in a supramolecular recognition group; interactions between these groups drive the assembly of NCTs into hierarchically ordered structures. Importantly, the fact that these supramolecular interactions can be tuned separately from the composition of the NPs or polymer brushes potentially enables independent control over the chemical makeup of the particle and polymer components of the composite, the spatial organization of the assembled structure, and the types of interactions governing the assembly process. However, while NCTs have significant potential in the development of unique polymer nanocomposite materials, only a single type of supramolecular interaction (hydrogen bonding) has so far been used to control particle assembly,20 which limits the processing conditions and compositional diversity of NCT assemblies. The development of a wider variety of supramolecular groups21−23 that can be used to direct NCT assembly would therefore be a major step toward addressing the design criteria outline above that are needed to fully manipulate composite structure and properties. In addition, the ability to use multiple orthogonal supramolecular interactions simultaneously would make it possible to develop sophisticated NCT architectures capable of more complex behaviors, such as composites that respond to multiple stimuli, or the formation of different particle arrangements in a processing-path dependent manner. Here, we introduce three unique supramolecular bonding motifs in order to establish new processing and assembly pathways capable of manipulating nanocomposite structure. We subsequently explore how the organization of these supramolecular groups around a nanoscale scaffold affects the thermodynamics of NCT assembly, and demonstrate how different stimuli (e.g., heat, light, pH, small molecules) can be used to regulate their collective binding interactions. Finally, we use the control afforded by the incorporation of many different supramolecular recognition chemistries into the NCT concept to expand the capabilities of this modular materials synthon. Specifically, we develop multistimuli responsive nanoparticle arrays, enhance reaction kinetics for covalent bond formation, and synthesize permanently cross-linked particle lattices that are significantly more robust to heat and solvent changes than prior systems. The designs presented here therefore provide major advancement for the field of composite synthesis by establishing a framework for synthesizing hierarchically ordered composites that are capable of complicated assembly behaviors.
Results and Discussion
System Design
Previously, hydrogen bonding interactions between diaminopyridine (DAP) and thymine (Thy) units have been used to assemble NCT superlattices.20 However, the labile nature of these interactions limits the ability of NCTs to incorporate a diverse range of polymer compositions or different stimuli to control material synthesis. These limitations stem from the fact that the nanoparticle-tethered DAP/Thy complexes are only stable in nonpolar solvents such as toluene, as solvents that contain more polar functional groups potentially disrupt DAP/Thy hydrogen bonding interactions. Therefore, directing NCT-NCT interactions with stronger recognition motifs would be a major step toward expanding the types of polymer and particle compositions that can be used in NCTs.
Hamilton wedge (HW)/cyanuric acid (CA) complexes24 represent a simple alteration to the previously established NCT system (Figure 1b), as they represent a stronger hydrogen-bonding pair than DAP/Thy that should still follow similar assembly behaviors. Compared with DAP/Thy complexes which possess three H-bonds per complex, HW/CA pairs possess six H-bonds per complex, and the Ka of a single HW/CA pair is 2 orders of magnitude higher than a DAP/Thy pair under the same conditions.25 Beyond hydrogen-bonding, metal coordination bonds represent an even stronger type of interaction, and are typically responsive to different stimuli,26 making them another ideal choice for robust NCT assembly. As an example, terpyridine (Tpy) units can form supramolecular complexes in the presence of metal ions, and the strength of these interactions can be controlled as a function of which metal ion species are used.27,28 Finally, dynamic covalent chemistry (DCC) represents an interesting potential means of assembling NCTs, as DCC bonds are only reversible under specific environmental conditions. For instance, hydrazide (Hdz) and aldehyde (CHO) functionalities undergo irreversible covalent bond formation at moderate pH, but these bonds can be made reversible by adding a strong acid.29
Figure 1.
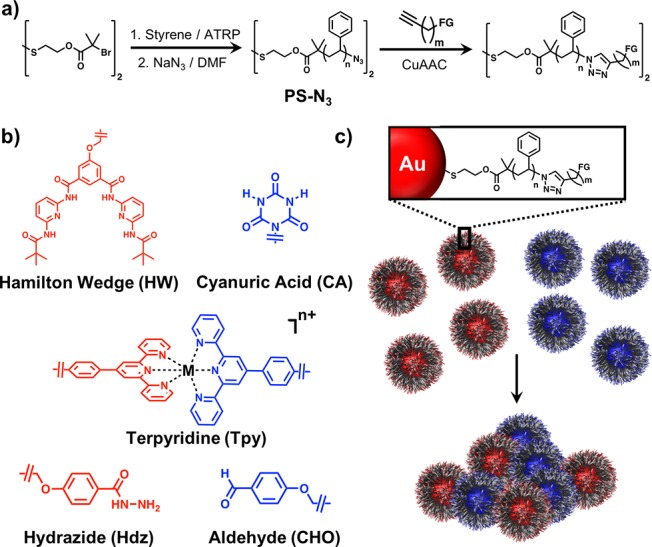
(a) General synthetic route for the preparation of the functionalized polystyrene (PS) used in this study. (b) Chemical structures of the different functional groups used to drive particle assembly. (c) Schematic illustration of the assembly of polymer-functionalized NCTs via supramolecular interactions between end groups.
By synthesizing NCTs with either HW/CA pairs, Tpy complexes, or DCC bonds as their chemical recognition motifs, the stability and stimuli responsiveness of the NCT system should be readily expanded beyond what can be achieved with DAP/Thy driven NCT assembly alone. In order to incorporate each of these functional groups into an NCT construct, individual systems were prepared using a previously established protocol,20 modified to include these new chemistries. Briefly, polystyrene (PS) chains were synthesized using atom transfer radical polymerization (ATRP) from a disulfide-containing initiator, and the binding groups were attached to the ends of these chains through copper catalyzed azide–alkyne cycloaddition (CuAAC, Figure 1a). Once the polymer chains had been functionalized with these binding groups, the disulfide was cleaved and the polymers were attached to gold nanoparticles using gold–thiol chemistry. A complete description of all synthesis protocols and full characterization of all molecular, macromolecular, and nanoparticle materials can be found in the SI.
NCT Assemblies Induced by Different Recognition Motifs
Due to the similar nature of the supramolecular interactions between the previously established DAP/Thy complexes and the HW/CA system, these stronger hydrogen bonding complexes were first investigated to confirm that more robust chemical interactions could still result in NCT assembly without any undesirable side reactions. Preassembled HW coated NCTs were permanently dispersible in nonpolar solvents like toluene, while some CA NCT systems exhibited a small amount of self-aggregation in these same solvents; this aggregation was easily reversed by adding trace amounts of a more polar solvent such as anisole. However, when HW- and CA- NCTs were mixed, the complementary hydrogen bonding interactions resulted in rapid assembly as monitored both visually and via UV–vis spectroscopy, as the assembly of gold nanoparticles (AuNPs) results in a redshift and reduction of the gold nanoparticles’ plasmon resonance, diminishing the solution’s absorption intensity at 520 nm. While previous DAP/Thy linked NCTs of comparable size and polymer length exhibited a melting temperature (Tm, the temperature at which NCT dissociate from one another) of 40 °C,20 HW/CA linked NCTs were irreversibly linked in toluene even at the boiling point of the solvent. This aggregation was also observed to occur in both pure toluene as well as in toluene-anisole mixtures up to 75% anisole (an example with 28 AuNPs and 11 kDa polymers in 50% anisole is shown in Figure 2a). The assembly of the HW/CA-NCTs was also observed in mixtures of toluene and more polar solvents, including CHCl3, CH2Cl2 and o-dichlorobenzene (see SI, Figure S2). The ability of HW/CA pairs to assemble NCTs both at elevated temperatures and in the presence of polar solvents capable of disrupting hydrogen bonding demonstrates that increasing the binding strength of NCT supramolecular bonding groups increases the compatibility of the system with more challenging assembly environments.
Figure 2.
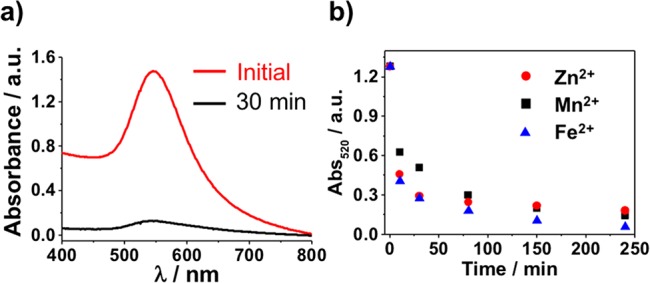
UV–vis spectroscopy investigation of (a) the self-assembly of the complementary 28 nm HW and CA-NCTs (11 kDa PS) in a 1:1 mixture of anisole and toluene at room temperature, and (b) kinetic study of the self-assembly process of the 28 nm Tpy-NCTs (11 kDa PS) induced by different metal ions in toluene.
When investigating the assembly behavior of the Tpy ligand functionalized NCTs, it is important to note that the strength of a Tpy-metal ion complex depends on the identity of the metal ion being coordinated. Therefore, three sets of identical Tpy ligand functionalized NCT solutions were prepared (see SI, Section 3 for experimental details), to which three different metal ions (Mn2+, Zn2+, and Fe2+) were added. Previous work has shown that the association strength of Tpy ligands with these metal ions follows the trend of Mn2+ < Zn2+ < Fe2+.30 In the NCT system, all three metal ions were able to readily induce the assembly of NCTs in toluene at nearly identical rates (Figure 2b). The slow kinetics of this assembly process compared with the hydrogen bond driven assemblies presumably results from the Coulombic repulsion between the positively charged M(Tpy)22+ complexes when forming large NCT aggregates.31 It is noteworthy that in all three cases the metal ion-Tpy coordination-induced NCT assembly remained assembled in toluene up to the boiling point of the solvent.
While the HW/CA and Tpy systems were capable of assembling NCTs in a rapid manner, the initial attempts of using DCC to assemble NCTs were unsuccessful, and no aggregates were observed after mixing the Hdz- and CHO-functionalized NCTs under weak acidic conditions, even after a week at room temperature. This observation indicates that the covalent hydrazone bond formation between Hdz- and CHO-NCTs is negligible under these conditions. The inability of DCC to induce particle assembly is ascribed to the relatively low concentration (<1 μM) of the reacting groups on the surface of NCTs. Unlike the supramolecular interactions in the cases of hydrogen-bonding and Tpy ligands, the chemical reaction-based DCC requires high concentrations in order to facilitate effective collision between the Hdz and CHO groups to drive the formation of the hydrazone bond. These observations are consistent with prior attempts to induce covalent bond formation between nanoparticles, where particle-bound molecules only formed covalent bonds after approximately a month, even when the reacting groups were at a concentration 105 times higher than those on the NCTs in this study.32 Therefore, while the NCTs examined here were unable to form aggregates via dynamic covalent bond formation, in principle it should be theoretically possible to induce covalent bond formation if the local concentration of reactive groups could be increased (vide infra).
External Stimuli-Driven Reversible NCT Assembly/Disassembly
While it is not surprising that the supramolecular interactions above (which are stronger than the previously examined DAP-Thy complexes) are able to induce particle assembly, it is not necessarily obvious that these stronger interactions also allow for particle disassembly, reorganization, or stimuli response. Given that these factors are critical for tackling the design criteria outlined at the beginning of this work, it is important to determine what external stimuli are capable of dynamically regulating the assembly of these HW/CA and Tpy-complex driven particle assemblies.
In order to disassemble the NCTs using external stimuli, both the enthalpic and entropic effects involved in the assembly process need to be taken into account. The enthalpy (ΔH) is determined by both the binding affinity of each individual supramolecular recognition moiety, as well as the number of supramolecular groups that comprise an NCT-NCT bond. Alterations to particle size and polymer length in the NCT system can affect the local concentration of the supramolecular recognition groups, thereby significantly varying the multivalency of the NCT interactions, and thus the collective ΔH of the interparticle bonds.33 The entropy of these interactions (ΔS) is dictated by the reduced conformational freedom of the supramolecular recognition groups and the polymer chains they tether upon assembling.34 While the entropic effects of supramolecular complexation can generally be overlooked in the case of small molecule functionalized NPs, the polymer chains in the NCT system provide unique handles to mediate the thermodynamics of the particle assembly. This added design flexibility arises because the entropy change associated with interparticle bonding is significantly larger for the NCT system compared with the small molecule functionalized NPs, as the number of conformations an NCT polymer chain can adopt is severely restricted upon the formation of NCT-NCT bonds. As a result, the entropic effects have more pronounced influence on the thermodynamics of the NCT systems (larger TΔS), making the free energy change associated with NCT bonding more thermally sensitive. Changes in both particle size and polymer length should therefore shift the strength of each NCT-NCT bond via changes to the chain dynamics of individual polymers comprising the brush. In order to make valid comparisons across the diverse range of design handles that could potentially be used to regulate the thermodynamics of the NCT supramolecular-driven assembly behavior (e.g., particle size, polymer length, supramolecular binding group identity, solvent conditions, etc.), comparisons will primarily be made for NCTs of a standard particle size and polymer length (28 nm particles and 11 kDa polymer chains); additional NCT designs and thermodynamic analyses can be found in the SI.
The HW/CA-NCT assemblies were first treated with heat as a simple means to study the reversibility of the assembly process. Compared with the DAP/Thy NCTs that dissociated at temperatures of ∼25–60 °C when assembled in toluene,20 the stronger HW/CA recognition motif actually prevented particle dissociation in this nonpolar solvent for most of the NCT systems that were assembled, excluding the NCTs with the longest polymer chains (see SI, Section 3). As a result, more polar solvents than toluene needed to be added in order to weaken the hydrogen bonds and enable particle disassembly. By altering the solvent composition to contain different amounts of anisole (a polar solvent that is highly miscible with toluene), the Tm of the HW/CA linked NCTs could be easily tuned by tailoring the ratio of the two solvents. Indeed, it was found that by increasing the polarity of the solvent, the Tm of the NCT assemblies could be gradually decreased (Figure 3a) from a permanent interaction that did not break even at the boiling point of pure toluene, down to 40 °C in a mixture of 1:3 toluene:anisole. In pure anisole, the hydrogen bonding interactions were sufficiently weakened such that no assembly was observed for any of the NCT systems. More interestingly, the change in the Tm of the assemblies exhibited a linear relationship with the anisole/toluene ratio (see SI, Figure S2), which allows the Tm of the system to be precisely tailored by changing the solvent composition. Furthermore, the high stability of the HW/CA pairs enables a wider range of polymer designs to form reversible assemblies than can be achieved when using the DAP/Thy system. HW/CA based NCTs with PS brushes of molecular weight up to 26 kDa were still observed to form stable assemblies in pure toluene at room temperature (Tm = 36 °C, Figure 3b, black trace). In all, the HW/CA recognition motif exhibits the same thermally reversible assembly behavior as the prior DAP-Thy NCT constructs, but its significantly enhanced stability compared with the previous hydrogen-bonded systems allows for much greater flexibility in NCT design, making it a useful expansion of the NCT toolbox.
Figure 3.

Thermal study of complementary 28 nm HW- and CA-NCT mixtures with (a) the same polymer molecular weight (11 kDa) and varying solvent composition, or (b) different polymer weights (11 and 26 kDa), which demonstrates that the stronger HW/CA binding groups allow for the NCTs to reversibly assemble in various solvent compositions with significantly broader range of polymer lengths. (c,d) UV–vis spectroscopy investigation of the responsiveness of the different metal ion-induced 28 nm Tpy-NCT aggregates upon titration of TFA, and the reversible acid–base induced disassembly/assembly of the 28 nm Zn2+-Tpy-NCTs upon adding TFA and TEA in an alternating sequence. The percentage of the disassembled NCTs in (c) is determined by comparing the absorbance of the TFA-treated assemblies with that of the original free NCTs.
While hydrogen-bonded ligands are typically easily modified via changes to solution temperature, coordination bonds are often not as labile. Indeed, the general high binding affinity between the metal ions and the Tpy ligands has been shown to prohibit the disassembly process in response to common external stimuli such as temperature or solvent changes in prior molecular or nanoparticle systems.35,36 Strong chelating reagents such as EDTA that have higher binding enthalpy with the metal ions than Tpy are therefore typically needed to reverse the assembly process.36,37 However, the poor solubility of EDTA in common organic solvents limits its use in disassembling NP aggregates that exist in the organic phase.
As mentioned earlier, though, the polymer chains in the NCT system make it possible in principle to reverse the assembly process using the significant entropy penalty associated with polymer tethering as a driving force. In other words, chemical stimuli that have been shown to be ineffective at dissociating small molecule Tpy-based NP assemblies could potentially be used to force NCT disassembly. For example, while trifluoroacetic acid (TFA) can be used to protonate the pyridine groups on the Tpy complex (thereby reducing the enthalpy of complex formation and shifting equilibrium toward the unbound state),38−40 the addition of acid was not a strong enough driving force to dissociate previous Tpy-based NP assemblies.36,41 In the NCT system, however, addition of 90 mM TFA at 22 °C resulted in complete dissociation of the Mn2+- and Zn2+-Tpy-NCT assemblies (Figure 3c). However, the Fe2+-Tpy assembled NCTs only exhibited ∼15% dissociation even when ∼1 M TFA was added due to the stronger binding affinity between Tpy and Fe2+ (Ka > 1021, compared with 1012 and 109 for Zn2+ and Mn2+ respectively).42 Further increasing the entropic penalty associated with forming Tpy complexes between adjacent particles does facilitate the disassembly process, though, as Fe2+-Tpy-linked NCTs functionalized with 26 kDa PS polymer readily disassembled upon the addition of 260 mM TFA at 22 °C (Figure S4a). The ability to disassemble these Fe2+-Tpy-linked NCTs with longer polymer chains is consistent with the fact that alterations to polymer configuration in the NCT system provide a unique design handle to tune the assembly properties of NPs.
In order to demonstrate complete reversibility of this pH-driven particle assembly process, acid (TFA) and base (triethylamine, (TEA)) were added in an alternating sequence to the Zn2+-Tpy-NCT samples (Figure 3d); the assembly and disassembly processes were easily repeated for multiple cycles without deterioration of the NCT complexes. It is also noteworthy that the more weakly bound Zn2+/Mn2+-Tpy-NCT aggregates can also disassemble upon the addition of free Tpy compound (Figure S4b), which offers an alternative approach to tune the assembly state of the Tpy-NCTs. Together, the use of acid–base and small molecule stimuli to reverse the assembly of Tpy-based NCTs in organic solvents is not only an important step toward broadening the application scope of coordination chemistry in the field of colloidal assembly, but also serves as an example to demonstrate how the introduction of polymer chains in the NCT system leads to unique alterations to the thermodynamics of supramolecular complexation.
Because these HW/CA and Tpy NCTs can be reversibly disassembled using the stimuli of solvent polarity and pH, respectively, this opens up the possibility to use other design handles that indirectly alter these solvent properties to control the NCT assembly process. In principle, a small molecule additive that either changes its dipole or releases H+ in response to an external stimulus could therefore be used to control NCT aggregation. While multiple possible stimuli can be used to alter the structures of small molecules in such a manner, light is a desirable candidate because it can be easily delivered to the system with a high degree of spatial and temporal control. Photoresponsive NP systems have been developed for prior NCT assembly systems, but many of the state-of-the-art NP assembly methods that are responsive to light rely on NPs functionalized directly with monolayers of light-responsive molecular switches.43−45 These NP-bound molecules can often be difficult to synthesize and difficult to stimulate uniformly (e.g., the light has to penetrate the aggregated NPs to trigger the switching process, which may result in low efficiency). The use of solvent-borne photoswitches that alter NP aggregation indirectly therefore enable more facile development of light-responsive particle assemblies.46
On the basis of the results above, it was hypothesized that spiropyran (SP), a molecule known for its rapid and reversible bond-cleavage when stimulated with light,47 could potentially introduce photoresponsive properties to the NCT assemblies through two different mechanisms without the need to directly couple it to the nanoparticles. Ring opening of SP upon exposure to UV light generates the merocyanine (MC) zwitterion (Figure 4a), which is significantly more polar than the starting SP form, a change which would be expected to weaken the hydrogen bond formation between the HW/CA NCTs. Furthermore, the MC species can be protonated yielding MCH+, a photoacid capable of releasing and capturing H+ reversibly in solution47 (Figure 4b) and this process could therefore be coupled to the assembly and disassembly of Tpy-NCTs
Figure 4.
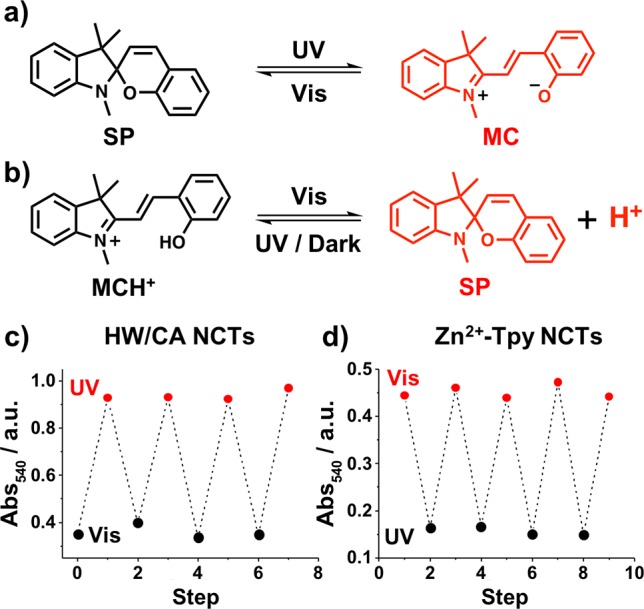
(a,b) Two possible approaches through which spiropyran can induce the self-assembly/disassembly processes of NCTs upon the application of photostimuli, including (a) the generation of the polar zwitterion species MC which can disrupt the hydrogen bonding interactions of HW/CA complexes and (b) the generation of protons which can disrupt the coordination interactions of Zn2+-Tpy complexes. (c,d) Absorption at 540 nm for the 16 nm HW/CA-NCT and Zn2+-Tpy-NCT systems undergoing multiple disassembly assembly cycles upon the application of external photostimuli.
When a solution of 10 μM SP (ca. 50 equiv to the HW/CA groups) was added to NCTs linked via HW/CA pairs, the absorption intensity of these NCT samples increased upon exposure to UV light, an observation consistent with the more polar MC species weakening the hydrogen bonding interactions between NCTs, leading to disassembly. The dispersed NCT sample remained in this state until exposed to visible light, upon which the less polar SP form was regenerated and the hydrogen bonds between NCTs reformed, resulting in particle reassembly (Figure 4c). Upon addition of 4 mM MCH+ (ca. 2 × 104 equiv to the Tpy groups,48 See SI, Section 6 for experimental details) to the Zn2+-Tpy-NCT assemblies, the resulting solution showed a strong absorption band attributed to MCH+ at 460 nm and low absorption intensity at 540 nm attributed to assembled NCTs (Figure S5). When this mixture was irradiated with visible light, MCH+ was converted to SP, releasing a proton which subsequently interacted with the Tpy groups. This change in solution acidity resulted in dissociation of the Zn2+-Tpy complex and disassembly of the NCTs, as confirmed by the decrease in the absorption band at 460 nm and the concomitant increase of the absorption band at 540 nm. Upon removal of the visible light source, the protons bound to the SP molecules (see SI, Section 6), leading to regeneration of MCH+ and NCT reassembly (Figure S7).
These light-reversible assembly processes were subsequently repeated (Figure 4c,d) for multiple cycles for both the HW/CA- and the Tpy-NCTs, demonstrating complete reversibility of the assembly process.
Multistimuli Responsive NCT System
The successful establishment of different stimuli, including heat, pH, small molecules and light to tune the assembly/disassembly state of the NCTs functionalized by different recognition groups makes it possible to construct more complicated NCT architectures with increased functionality. Specifically, the functionalization of multiple binding groups to a single nanoparticle scaffold would introduce the possibility of developing multistimuli responsive NCTs capable of assembling into different states under different conditions, or NCTs capable of pathway-dependent assembly where the resulting morphologies depended on the order in which different stimuli were introduced. In order to explore this possibility, two types of NCT samples were prepared (Figure 5a): 16 nm AuNPs functionalized with CA groups, and 28 nm AuNPs functionalized with HW and Tpy groups using a 9:1 feed ratio (see SI Section 3). Encouragingly, the NCTs readily assembled at room temperature upon mixing in a 3:1 toluene/anisole solution despite the introduction of the Tpy groups (Figure 5b, purple trace), and the assemblies could be melted upon heating the solution to ∼60 °C (Figure 5c, red trace). In contrast, when Zn2+ was present, the NCTs again formed stable assemblies at 22 °C (Figure 5d, purple trace), but the original absorption was only partially recovered when the system was heated (Figure 5d, red trace). The fact that only some of the NCT dissociated upon heating is ascribed to fact that the 16 nm CA-NCTs could melt into solution, while the 28 nm HW-Tpy-NCTs were locked into the assembled form by the Zn2+-Tpy coordination complex. This observation confirms that (i) the formation of the hydrogen bonding interactions is not compromised by the addition of Zn2+ and (ii) both hydrogen bonding and coordination interactions can be used simultaneously to govern the assembly process. Finally, the coordination interactions between the NCTs can be out-competed by the addition of free Tpy compound (3) as demonstrated above, leaving only hydrogen bonding interactions to mediate the assembly process. When excess free Tpy was added to these assemblies, the thermal response of the system matched the previous assembly and disassembly behavior in the absence of Zn2+ (Figure 5e). Taken together, these data allow for the NCTs to exist in four unique states depending on whether the Tpy complexes are formed, the HW/CA complexes are formed, or both or neither are formed (Figure 6a).
Figure 5.

(a) A schematic illustration of the NCTs that were used to examine the multistimuli responsive NCT self-assembly system. (b–e) UV–vis spectroscopic characterization of the NCT samples upon applying different external stimuli.
Figure 6.

(a) A schematic illustration of the various responses of the multistimuli responsive system in Figure 5, demonstrating that the pathway of stimulus addition controls the morphology of the resulting composites. Conditions: (i) cool; (ii) heat; (iii) addition of Zn2+, heat; (iv) addition of Tpy, heat; (v) addition of Zn2+; (vi) addition of Tpy. (b) TEM images of the assembly induced by coordination interactions only (left) and hydrogen bonding interactions only (right). (c) SAXS traces for the HW-Tpy and CA-based multistimuli responsive NCT assemblies under different conditions, where the difference in interparticle spacings demonstrates the pathway-dependent nature of the assembly process.
The multistimuli responsive behavior of the NCTs was also confirmed by transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS), providing information on the structure of the assembled NCTs. Specifically, when the assembly behavior was driven exclusively by coordination interactions, only 16 nm NCTs were observed in electron microscopy images of the supernatant (Figure S8a) and only 28 nm NCTs were observed in the aggregate (Figure 6b, left), and a single peak in low q region in the SAXS data confirms the presence of a single average interparticle distance (d, determined as d = 2π/q0, where q0 is the maximum of the scattering peak),49 consistent with solely 28 nm NCTs being part of the precipitate (Figure 6c, red solid trace). In comparison, when the assembly was driven by hydrogen bonding or both hydrogen bonding and coordination interactions, the aggregate was comprised of a mixture of 28 and 16 nm NCTs (Figure 6b, right, and Figure S8c–e).
Importantly, however, the ability to orthogonally address these two different assembly mechanisms potentially also enables pathway-dependent assembly, allowing the positions of the particles within a macroscopic aggregate to be controlled as a function of the order in which different stimuli are introduced. For example, when the 28 and 16 nm NCTs were assembled (Figure S8d) via steps (i) and (v) as shown in Figure 6a (first hydrogen bonding, then Zn2+ complexation), NCTs formed homogeneous aggregates with both sizes of NCTs interspersed throughout the aggregate. This homogeneity was noted by the presence of a single interparticle distance of 38 nm observed in the SAXS data, as indicated by only a single peak in the low q region. In comparison, when the two sets of NCTs were assembled in the opposite pathway (steps (iii) and (i), Zn2+-Tpy-complex formation followed by hydrogen bonding), the 28 and 16 nm NCTs were segregated into distinct regions within the assemblies—the 28 nm NCTs formed aggregates which were surrounded by 16 nm NCTs (Figure S8e). Although TEM images can potentially make full structure determination challenging, the SAXS data unequivocally show this particle segregation behavior, as two distinct peaks were observed in the low q region, corresponding to the two different interparticle distances that would be expected: a larger distance of 52 nm for 28–28 nm particle bonds linked with Zn2+-Tpy complexes, and a shorter distance of 37 nm for the 28–16 nm particle bonds consisting of HW/CA pairs (Figure 6c, Table S1).
These results show that the differences in interparticle distance are a direct result of the differences in the assembly pathway. In step (i), the 28 and 16 nm NCTs interact with each other via hydrogen bonding and form aggregates consisting of both particle types homogeneously dispersed throughout the sample. The addition of Zn2+ (step (v)) induces additional interactions between adjacent 28 nm NCTs, but the initial assembled structure with interspersed particle types is retained. In comparison, when Zn2+ is added to the NCTs first, the initial aggregates are composed exclusively of 28 nm NCTs associated via coordination interactions (step (iii)). When the solution is subsequently cooled, 16 nm CA-NCTs aggregate (step (i)) around the exterior of the 28 nm HW-Tpy-NCT clusters through hydrogen bonding interactions, giving rise to two distinct interparticle spacings.
Taken together, the UV–vis spectra, TEM images, and SAXS measurements demonstrate that the assembly/disassembly of the multistimuli responsive NCT system can be controlled by (i) hydrogen bonding interactions, (ii) coordination chemistry, or (iii) both interactions in an orthogonal and reversible manner. This pathway dependent assembly process enables the selective self-assembly of a particular NCT type from a mixture of NCTs, as well as the potential for more complex assembly behaviors that can control the organization of particles within a macroscopic composite in a more sophisticated manner.
Accelerating Covalent Bond Formation via Enhanced Local Concentration
In addition to selectively controlling assembly, postassembly functionalization of NP-based materials is another key challenge in the field of nanochemistry and nanocomposite development. For example, while supramolecular interactions are often employed to direct NP aggregation, using covalent bonds to link particles together once assembled would potentially result in significantly more stable NP assemblies. Such enhanced stability would open the possibility to process NCT composites under different conditions than are currently accessible. Despite these potential benefits, forming covalent bonds between NPs is challenging because nanoparticle solutions such as those used in this work are typically several orders of magnitude more dilute than typical organic synthesis schemes used to make covalent bonds between molecules (nM particle solutions vs mM solutions of molecules). While covalently linked NP aggregates have been prepared using a large excess of reactive small molecules as a cross-linking agent, the need of removing the excess reagent after covalent bond formation and the extremely slow (∼month) rates of covalent bond formation limit their use,32 and strategies for directly forming covalent bonds between NPs on more rapid time scales have not yet been established.
In principle, the NCT system offers a distinct advantage in developing methods to form covalent bonds between particles using postassembly functionalization strategy, specifically the fact that NCTs are decorated with a large number of polymer chains that result in a high local concentration of chain ends, as well as conformation flexibility that allows these chain ends to adopt different positions relative to the particle surface. While initial efforts to link Hdz and CHO containing NCTs discussed resulted in no observable particle assembly (Figure 7a), it should in principle be possible to increase the local concentration of the reacting groups in a supramolecular-assisted postassembly functionalization approach. Specifically, by coloading HW and CA group-functionalized NCTs with Hdz and CHO terminated polymers, respectively (Figure 7b, (i)), the use of hydrogen bonding to assemble particles should result in a significant local concentration increase of the reacting Hdz and CHO groups. Moreover, because the polymer chains afford some degree of conformational freedom to these binding groups, the Hdz and CHO terminated polymer chains would be expected to be much more likely to physically contact one another and form covalent bonds than Hdz and CHO groups tethered to a particle surface via small molecule linkers. As a result, the DCC coupling reaction between NCTs should be able to occur in a much shorter time frame than prior DCC-driven nanoparticle assemblies (Figure 7b, (iii)).
Figure 7.
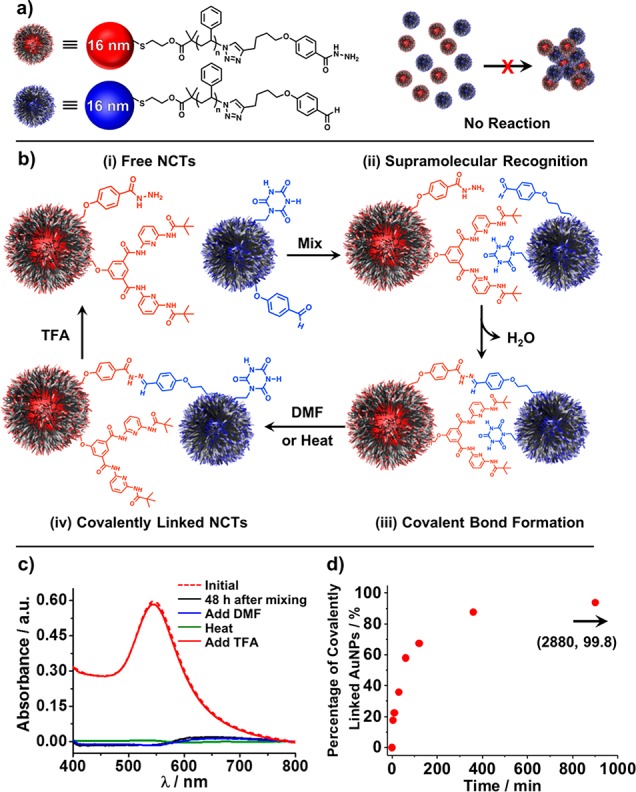
(a) No covalent bond formation is observed upon mixing NCTs functionalized with hydrazide and the aldehyde terminated PS. (b) A schematic illustration of the covalent bond formation process assisted by supramolecular interactions between HW and CA groups, where hydrogen-bond driven assembly results in significant concentration of the reactive hydrazide and aldehyde groups, increasing the rate of covalent bond formation. (c) UV–vis spectra of the NCT samples shown in (b) initial state (red dash), 48 h after mixing (black), followed by the treatment of the NCT samples with DMF (blue), heat (green) and TFA (red), demonstrating that all NCTs are covalently linked within 48 h of mixing. (d) Kinetic study of the covalent bond formation process between the NCTs shown in (b).
To test this hypothesis, HW-Hdz and CA-CHO functionalized 16 nm NCTs were combined (see SI for experimental protocols), and their assembly was monitored using UV–vis spectroscopy. Upon mixing, the NCTs immediately self-assembled due to the hydrogen bonding interactions, indicating that the presence of the Hdz and CHO groups did not significantly affect HW-CA complex formation (Figure S10). After 48 h, aliquots of the NCT aggregates (Figure 7c, black trace) were either treated with DMF or heated to 80 °C; both of these conditions have been shown to disrupt hydrogen bonding interactions between NCTs (vide supra). Notably, the UV–vis spectra (Figure 7c, blue and green traces) show no evidence for the dissolution of the HW-Hdz/CA-CHO NCT aggregates. These observations are consistent with the NCTs in this system being connected by covalent hydrazone bonds formed between the Hdz and CHO groups. The covalent bonding is further confirmed by the addition of TFA, a strong acid known to cleave the hydrazone bond, which causes the aggregates to readily redisperse (Figure 7c, red trace). The kinetics of this covalent bond formation process between the NCTs were also investigated (see SI, Section 3 for experimental details) by UV–vis spectroscopy. It was found (Figure 7d) that the hydrazone bond formation between the NCTs was initiated within 10 min of mixing and that over 90% of the NCTs were covalently linked within 6 h of mixing, suggesting that compared with other reported examples, the strategy described here using supramolecular complexation to form a high local concentration of the reacting Hdz and CHO groups provides an effective approach for covalently linking NPs with significantly shortened reaction time and elevated efficiency.50 More importantly, this strategy of enhancing the local concentration of binding groups to accelerate reaction kinetics should be readily applicable to NP systems in a general manner, meaning that it should be possible to form many different types of covalent bonds between NPs.
Conclusions
In this work, we have demonstrated the ability of a single NCT system to express finely tuned assembly behaviors in response to multiple external stimuli, and the feasibility of using a supramolecular-assisted postassembly functionalization approach to perform chemical reactions between NPs. The unique role of the polymer tethers in the NCT system sheds light on how to take advantage of the entropic effects associated with polymer chain dynamics to tune the assembly process. Moreover, the example of directly linking NPs through covalent bonds provided here has important implications in the field of catalysis,51 as NP scaffolds can potentially be used to enhance the kinetics of different chemical reactions via a local concentration effect. Future work will focus on both the construction of hierarchical self-assembled NCT architectures using the various recognition motifs established here and potentially many others such as host–guest chemistry,52,53 and using covalent bonds to enhance the stability and mechanical properties of long-range ordered NCT superstructures for their applications in the solid phase.
Acknowledgments
This material is based upon work supported in part by the U. S. Army Research Office under grant number W911NF-18-1-0197. This work was supported by an NSF CAREER Grant, award number CHE-1653289 and made use of the MRSEC Shared Experimental Facilities at MIT, supported by the NSF under Award DMR 14-19807. P.J.S. acknowledges support by the NSF Graduate Research Fellowship Program under Grant 1122374. J.M.K. acknowledges support by the Department of Defense (DoD) through the National Defense Science & Engineering Graduate Fellowship (NDSEG) Program.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b06695.
Synthesis and characterization of the new compounds, preparation protocol of the NCT samples for different studies, UV–vis spectroscopic investigations and TEM, SAXS measurements of NCT assemblies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Mark J. E. Some novel polymeric nanocomposites. Acc. Chem. Res. 2006, 39, 881–888. 10.1021/ar040062k. [DOI] [PubMed] [Google Scholar]
- Sambhy V.; MacBride M. M.; Peterson B. R.; Sen A. Silver bromide nanoparticle/polymer composites: Dual action tunable antimicrobial materials. J. Am. Chem. Soc. 2006, 128, 9798–9808. 10.1021/ja061442z. [DOI] [PubMed] [Google Scholar]
- Zou H.; Wu S.; Shen J. Polymer/silica nanocomposites: Preparation, characterization, properties, and applications. Chem. Rev. 2008, 108, 3893–3957. 10.1021/cr068035q. [DOI] [PubMed] [Google Scholar]
- Kao J.; Thorkelsson K.; Bai P.; Rancatore B. J.; Xu T. Toward functional nanocomposites: taking the best of nanoparticles, polymers, and small molecules. Chem. Soc. Rev. 2013, 42, 2654–2678. 10.1039/C2CS35375J. [DOI] [PubMed] [Google Scholar]
- Li K.; Liu B. Polymer-encapsulated organic nanoparticles for fluorescence and photoacoustic imaging. Chem. Soc. Rev. 2014, 43, 6570–6597. 10.1039/C4CS00014E. [DOI] [PubMed] [Google Scholar]
- Ferhan A. R.; Kim D.-H. Nanoparticle polymer composites on solid substrates for plasmonic sensing applications. Nano Today 2016, 11, 415–434. 10.1016/j.nantod.2016.07.001. [DOI] [Google Scholar]
- Chen Y.; Thakar R.; Snee P. T. Imparting nanoparticle function with size-controlled amphiphilic polymers. J. Am. Chem. Soc. 2008, 130, 3744–3745. 10.1021/ja711252n. [DOI] [PubMed] [Google Scholar]
- Nakano T.; Kawaguchi D.; Matsushita Y. Anisotropic self-assembly of gold nanoparticle grafted with polyisoprene and polystyrene having symmetric polymer composition. J. Am. Chem. Soc. 2013, 135, 6798–6801. 10.1021/ja402412q. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Peng W.; Zare Y.; Hui D.; Rhee K. Y. Predicting the electrical conductivity in polymer carbon nanotube nanocomposites based on the volume fractions and resistances of the nanoparticle, interphase, and tunneling regions in conductive networks. RSC Adv. 2018, 8, 19001–19010. 10.1039/C8RA00811F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. Y.; Lo M. K. F.; Yang G. W.; Monbouquette H. G.; Yang Y. Nanoparticle-assisted high photoconductive gain in composites of polymer and fullerene. Nat. Nanotechnol. 2008, 3, 543–547. 10.1038/nnano.2008.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N.; McKeown S. J.; Ma X.; Pochan D. J.; Cloutier S. G. Structure-property correlations in hybrid polymer-nanoparticle electrospun fibers and plasmonic control over their dichroic behavior. ACS Nano 2010, 4, 5551–5558. 10.1021/nn100582f. [DOI] [PubMed] [Google Scholar]
- Jang S. G.; Kramer E. J.; Hawker C. J. Controlled supramolecular assembly of micelle-like gold nanoparticles in ps-b-p2vp diblock copolymers via hydrogen bonding. J. Am. Chem. Soc. 2011, 133, 16986–16996. 10.1021/ja206615c. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Kook K.; Hwang S. K.; Park C.; Cho J. Polymer/perovskite-type nanoparticle multilayers with multielectric properties prepared from ligand addition-induced layer-by-layer assembly. ACS Nano 2014, 8, 2419–2430. 10.1021/nn405988d. [DOI] [PubMed] [Google Scholar]
- Barrow M.; Taylor A.; Murray P.; Rosseinsky M. J.; Adams D. J. Design considerations for the synthesis of polymer coated iron oxide nanoparticles for stem cell labelling and tracking using MRI. Chem. Soc. Rev. 2015, 44, 6733–6748. 10.1039/C5CS00331H. [DOI] [PubMed] [Google Scholar]
- Kim C. R.; Uemura T.; Kitagawa S. Inorganic nanoparticles in porous coordination polymers. Chem. Soc. Rev. 2016, 45, 3828–3845. 10.1039/C5CS00940E. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Liu Y.; Yao D.; Yang B. Hybridization of inorganic nanoparticles and polymers to create regular and reversible self-assembly architectures. Chem. Soc. Rev. 2012, 41, 6066–6088. 10.1039/c2cs35038f. [DOI] [PubMed] [Google Scholar]
- Feng L. H.; Zhu C. L.; Yuan H. X.; Liu L. B.; Lv F. T.; Wang S. Conjugated polymer nanoparticles: preparation, properties, functionalization and biological applications. Chem. Soc. Rev. 2013, 42, 6620–6633. 10.1039/c3cs60036j. [DOI] [PubMed] [Google Scholar]
- Grzelczak M.; Liz-Marzan L. M.; Klajn R. Stimuli-responsive self-assembly of nanoparticles. Chem. Soc. Rev. 2019, 48, 1342–1361. 10.1039/C8CS00787J. [DOI] [PubMed] [Google Scholar]
- Udayabhaskararao T.; Altantzis T.; Houben L.; Coronado-Puchau M.; Langer J.; Popovitz-Biro R.; Liz-Marzan L. M.; Vukovic L.; Kral P.; Bals S.; Klajn R. Tunable porous nanoallotropes prepared by post-assembly etching of binary nanoparticle superlattices. Science 2017, 358, 514–518. 10.1126/science.aan6046. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Santos P. J.; Gabrys P. A.; Lee S.; Liu C.; Macfarlane R. J. Self-assembling nanocomposite tectons. J. Am. Chem. Soc. 2016, 138, 16228–16231. 10.1021/jacs.6b11052. [DOI] [PubMed] [Google Scholar]
- Yang H.; Yuan B.; Zhang X.; Scherman O. A. Supramolecular chemistry at interfaces: host-guest interactions for fabricating multifunctional biointerfaces. Acc. Chem. Res. 2014, 47, 2106–2115. 10.1021/ar500105t. [DOI] [PubMed] [Google Scholar]
- Song N.; Yang Y.-W. Molecular and supramolecular switches on mesoporous silica nanoparticles. Chem. Soc. Rev. 2015, 44, 3474–3504. 10.1039/C5CS00243E. [DOI] [PubMed] [Google Scholar]
- Elacqua E.; Zheng X.; Shillingford C.; Liu M.; Weck M. Molecular Recognition in the Colloidal World. Acc. Chem. Res. 2017, 50, 2756–2766. 10.1021/acs.accounts.7b00370. [DOI] [PubMed] [Google Scholar]
- Chang S.; Hamilton A. D. Molecular Recognition of Biologically Interesting Substrates - Synthesis of an Artificial Receptor for Barbiturates Employing 6 Hydrogen-Bonds. J. Am. Chem. Soc. 1988, 110, 1318–1319. 10.1021/ja00212a065. [DOI] [Google Scholar]
- Chen S. B.; Binder W. H. Dynamic Ordering and Phase Segregation in Hydrogen-Bonded Polymers. Acc. Chem. Res. 2016, 49, 1409–1420. 10.1021/acs.accounts.6b00174. [DOI] [PubMed] [Google Scholar]
- Knight A. S.; Larsson J.; Ren J. M.; Zerdan R. B.; Seguin S.; Vrahas R.; Liu J.; Ren G.; Hawker C. J. Control of amphiphile self-assembly via bioinspired metal ion coordination. J. Am. Chem. Soc. 2018, 140, 1409–1414. 10.1021/jacs.7b11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmeijer B. G. G.; Schubert U. S. Playing LEGO with macromolecules: Design, synthesis, and self-organization with metal complexes. J. Polym. Sci., Part A: Polym. Chem. 2003, 41, 1413–1427. 10.1002/pola.10685. [DOI] [Google Scholar]
- Winter A.; Hager M. D.; Newkome G. R.; Schubert U. S. The marriage of terpyridines and inorganic nanoparticles: synthetic aspects, characterization techniques, and potential applications. Adv. Mater. 2011, 23, 5728–5748. 10.1002/adma.201103612. [DOI] [PubMed] [Google Scholar]
- Deng G.; Tang C.; Li F.; Jiang H.; Chen Y. Covalent cross-linked polymer gels with reversible sol-gel transition and self-healing properties. Macromolecules 2010, 43, 1191–1194. 10.1021/ma9022197. [DOI] [Google Scholar]
- Rossow T.; Habicht A.; Seiffert S. Relaxation and Dynamics in Transient Polymer Model Networks. Macromolecules 2014, 47, 6473–6482. 10.1021/ma5013144. [DOI] [Google Scholar]
- Wu W. J.; Wang J.; Chen M.; Qian D. J.; Liu M. H. Terpyridine-functionalized NanoSiO2 multi-dentate linkers: preparation, characterization and luminescent properties of their metal-organic hybrid materials. J. Phys. Chem. C 2017, 121, 2234–2242. 10.1021/acs.jpcc.6b11510. [DOI] [Google Scholar]
- Kay E. R. Dynamic covalent nanoparticle building blocks. Chem. - Eur. J. 2016, 22, 10706–10716. 10.1002/chem.201601394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard A.; Smith D. K. Self-assembled multivalency: dynamic ligand arrays for high-affinity binding. Angew. Chem., Int. Ed. 2012, 51, 6572–6581. 10.1002/anie.201200076. [DOI] [PubMed] [Google Scholar]
- Martinez-Veracoechea F. J.; Leunissen M. E. The entropic impact of tethering, multivalency and dynamic recruitment in systems with specific binding groups. Soft Matter 2013, 9, 3213–3219. 10.1039/c3sm27766f. [DOI] [Google Scholar]
- Schubert U. S.; Eschbaumer C. Functionalized oligomers and copolymers with metal complexing segments: A simple and high yield entry towards 2,2’:6’,2’’-terpyridine monofunctionalized telechelics. Macromol. Symp. 2001, 163, 177–187. . [DOI] [Google Scholar]
- Norsten T. B.; Frankamp B. L.; Rotello V. M. Metal directed assembly of terpyridine-functionalized gold nanoparticles. Nano Lett. 2002, 2, 1345–1348. 10.1021/nl020217m. [DOI] [Google Scholar]
- Dewi M. R.; Gschneidtner T. A.; Elmas S.; Ranford M.; Moth-Poulsen K.; Nann T. Monofunctionalization and Dimerization of Nanoparticles Using Coordination Chemistry. ACS Nano 2015, 9, 1434–1439. 10.1021/nn5058408. [DOI] [PubMed] [Google Scholar]
- Drew M. G. B.; Hudson M. J.; Iveson P. B.; Russell M. L.; Liljenzin J. O.; Skalberg M.; Spjuth L.; Madic C. Theoretical and experimental studies of the protonated terpyridine cation. Ab initio quantum mechanics calculations, and crystal structures of two different ion pairs formed between protonated terpyridine cations and nitratolanthanate(III) anions. J. Chem. Soc., Dalton Trans. 1998, 2973–2980. 10.1039/a802458h. [DOI] [Google Scholar]
- Dobrawa R.; Ballester P.; Saha-Möller C. R.; Würthner F.. Thermodynamics of 2,2′:6′,2′′-terpyridine-metal ion complexation. In Metal-Containing and Metallosupramolecular Polymers and Materials; ACS Symposium Series; American Chemical Society: Washington, D.C., 2006; Vol. 928, pp 43–62. [Google Scholar]
- Offenhartz P.; George P.; Haight G. P. Ionization constants for ligand 2,2’,2’’-tripyridine. J. Phys. Chem. 1963, 67, 116–118. 10.1021/j100795a027. [DOI] [Google Scholar]
- Zhao Y.; Newton J. N.; Liu J.; Wei A. Dithiocarbamate-coated SERS substrates: sensitivity gain by partial surface passivation. Langmuir 2009, 25, 13833–13839. 10.1021/la902087e. [DOI] [PubMed] [Google Scholar]
- Rossow T.; Seiffert S. Supramolecular polymer gels with potential model-network structure. Polym. Chem. 2014, 5, 3018–3029. 10.1039/c3py01692g. [DOI] [Google Scholar]
- Piech M.; George M. C.; Bell N. S.; Braun P. V. Patterned colloid assembly by grafted photochromic polymer layers. Langmuir 2006, 22, 1379–1382. 10.1021/la051636c. [DOI] [PubMed] [Google Scholar]
- Shiraishi Y.; Shirakawa E.; Tanaka K.; Sakamoto H.; Ichikawa S.; Hirai T. Spiropyran-modified gold nanoparticles: reversible size control of aggregates by uv and visible light irradiations. ACS Appl. Mater. Interfaces 2014, 6, 7554–7562. 10.1021/am5009002. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Sen S.; Udayabhaskararao T.; Sawczyk M.; Kucanda K.; Manna D.; Kundu P. K.; Lee J. W.; Kral P.; Klajn R. Reversible trapping and reaction acceleration within dynamically self-assembling nanoflasks. Nat. Nanotechnol. 2016, 11, 82–88. 10.1038/nnano.2015.256. [DOI] [PubMed] [Google Scholar]
- Kundu P. K.; Samanta D.; Leizrowice R.; Margulis B.; Zhao H.; Borner M.; Udayabhaskararao T.; Manna D.; Klajn R. Light-controlled self-assembly of non-photoresponsive nanoparticles. Nat. Chem. 2015, 7, 646–652. 10.1038/nchem.2303. [DOI] [PubMed] [Google Scholar]
- Klajn R. Spiropyran-based dynamic materials. Chem. Soc. Rev. 2014, 43, 148–184. 10.1039/C3CS60181A. [DOI] [PubMed] [Google Scholar]
- Due to the large association constant of the Zn(Tpy)2 complex (Ka ∼ 1012) and weak basicity of Tpy groups (pKb ∼ 9),40 a relatively high concentration of H+ is needed in order to disassociate the coordination complex.
- Hyland L. L.; Taraban M. B.; Yu Y. Using small-angle scattering techniques to understand mechanical properties of biopolymer-based biomaterials. Soft Matter 2013, 9, 10218–10228. 10.1039/c3sm51209f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The cyanuric acid groups functionalized on the NCTs, which are in close proximity to the covalent bond reaction center, may also serve as a catalyst due to its weak acidic nature, as the HW/CA assisted covalent bond formation process is faster than the Tpy assisted process. See SI, Section 9.
- Daniel M. C.; Astruc D. Gold nanoparticles: Assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem. Rev. 2004, 104, 293–346. 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- Yang Y.-W.; Sun Y.-L.; Song N. Switchable host-guest systems on surfaces. Acc. Chem. Res. 2014, 47, 1950–1960. 10.1021/ar500022f. [DOI] [PubMed] [Google Scholar]
- Wang X.; Liu Z.-J.; Hill E. H.; Zheng Y.; Guo G.; Wang Y.; Weiss P. S.; Yu J.; Yang Y.-W. Organic-inorganic hybrid pillarene-based nanomaterial for label-free sensing and catalysis. Matter 2019, 1, 1–14. 10.1016/j.matt.2019.03.005. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.