

Abstract
Previous studies demonstrated that cells inhibit protein synthesis as a compensatory mechanism for mitochondrial dysfunction. Protein synthesis can be attenuated by 1) the inhibition of mTOR kinase, which results in a decrease in the phosphorylation of S6K1 and 4E-BP1 proteins, and 2) an increase in the phosphorylation of eIF2α protein. The present study investigated both of these pathways under conditions of short-term acute and long-term mitochondrial stress. Short-term responses were triggered in mammalian cells by treatment with menadione, antimycin A, or CCCP. Long-term mitochondrial stress was induced by prolonged treatment with menadione or rotenone and expression of genetic alterations, such as knocking down the MIA40 oxidoreductase or knocking out NDUFA11 protein. Short-term menadione, antimycin A, or CCCP cell treatment led to the inhibition of protein synthesis, accompanied by a decrease in mTOR kinase activity, an increase in the phosphorylation of eIF2α (Ser51), and an increase in the level of ATF4 transcription factor. Conversely, long-term stress led to a decrease in eIF2α (Ser51) phosphorylation and ATF4 expression and to an increase in S6K1 (Thr389) phosphorylation. Thus, under long-term mitochondrial stress, cells trigger long-lasting adaptive responses for protection against excessive inhibition of protein synthesis.
INTRODUCTION
Mitochondria are often recognized as organelles that are mainly responsible for energy conversion, but they also play an important role in cellular signaling, such as apoptosis, proliferation, and differentiation. Moreover, under conditions of stress, they are able to signal their state to other organelles of the cell (Nunnari and Suomalainen, 2012; Chandel, 2014). Stress conditions often lead to a reduction of anabolic activity to avoid cellular damage and unnecessary energy expenditures. Indeed, the inhibition of cytosolic protein synthesis reduced mitochondrial degeneration (Wang et al., 2008). However, the mechanisms by which dysfunctional mitochondria potentially induce a reduction of protein synthesis remain unclear.
Several types of initial events that are related to dysfunctional mitochondria that can cause the inhibition of protein synthesis have been discovered. In yeast, mitochondrial defects were shown to trigger response pathways, in which cap-dependent translation is attenuated. Mitochondrial precursor overaccumulation stress (mPOS) down-regulated ribosomal proteins, the target-of-rapamycin (TOR) pathway, and mRNA turnover components (Wang and Chen, 2015). Similar effects on cytosolic ribosomal proteins were observed in the case of the unfolded protein response activated by mistargeted proteins (UPRam; Wrobel et al., 2015). This response was induced by a mutation of yeast Mia40 oxidoreductase, which was defective for protein import in the mitochondrial intermembrane space (Wrobel et al., 2015). Proteins that are involved in translation were down-regulated in cells with the knockout of mitochondrial Complex I accessory subunits (Stroud et al., 2016).
However, mitochondria are also the main source of cellular reactive oxygen species (ROS) because of their central role in energy metabolism. An increase in the production of mitochondrial ROS (mROS) can cause oxidative damage in the cell, but mROS can also serve as secondary messengers (Reczek and Chandel, 2015). Mitochondrial dysfunction that is caused by various pathological conditions and chemicals (e.g., menadione and antimycin A) increases ROS production (Osenbroch et al., 2009; Ma et al., 2011; Monteiro et al., 2013). In yeast, treatment with H202 caused the global and reversible inhibition of protein synthesis (Shenton and Grant, 2003; Grant, 2011), suggesting the involvement of ROS in protein synthesis regulation during mitochondrial dysfunction.
The inhibition of protein synthesis can be executed via several different mechanisms. Two of these mechanisms are well documented: inhibition of the mTOR pathway and phosphorylation of eukaryotic initiation factor 2α (eIF2α). The inhibition of mTOR signaling can lead to a reduction of protein synthesis through a decrease in the phosphorylation of proteins that are responsible for the regulation of protein synthesis, such as ribosomal protein S6 kinase 1 (S6K1) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1). Apart from inhibition of the mTOR pathway, the phosphorylation of eIF2α was shown to be another mechanism for the attenuation of protein synthesis. eIF2α is a core component of the integrated stress response (ISR) and can be phosphorylated under various conditions of stress, such as amino acid deprivation (GCN2 kinase), viral infection (PKR kinase), heme deprivation (HRI kinase), and endoplasmic reticulum (ER) stress (PERK kinase; Harding et al., 2003; Pakos-Zebrucka et al., 2016). Reactive oxygen species that are generated by stressed or dysfunctional mitochondria also induced the phosphorylation of eIF2α in Caenorhabditis elegans via GCN2 kinase. Additionally, GCN2 was required for extension of the lifespan of C. elegans in the presence of mitochondrial stress, suggesting its protective role (Baker et al., 2012). This study suggested that PERK kinase rather than GCN2 in mammalian cells is responsible for the phosphorylation of eIF2α under conditions of severe short-term mitochondrial stress. The phosphorylation of eIF2α has been reported to increase the expression of ATF4 transcription factor, which is considered a main player in the mitochondrial stress response in mammalian cells (Quiros et al., 2017). Moreover, the expression of ATF4 at the transcript level was also activated in vivo in mice and humans with mitochondrial diseases (Quiros et al., 2017) and in mouse hearts with severe mitochondrial dysfunction (Kuhl et al., 2017). Recently, a novel mechanism of inhibition of protein synthesis under conditions of oxidative stress was discovered (Topf et al., 2018). Mitochondrially born ROS can affect the function of cytosolic ribosomes through the oxidation of cysteine residues in several ribosomal proteins that coordinate zinc ions. During mitochondrial dysfunction, these redox switches can reduce global protein synthesis rapidly and reversibly (Topf et al., 2018).
The present study used mammalian cells to investigate the involvement of different pathways that are responsible for the regulation of protein synthesis during short-term acute and long-term mitochondrial stress. We observed decreased phosphorylation of S6K1 and 4E-BP1 proteins upon the induction of short-term acute mitochondrial-born oxidative stress in HEK 293 cells that were treated with menadione, antimycin A, or CCCP. In contrast, in studies that utilized mouse models of mitochondrial diseases (i.e., chronic mitochondrial stress), such as Leigh syndrome (Johnson et al., 2013; Zheng et al., 2016) and mitochondrial myopathy (Khan et al., 2017), an increase in the activity of the mTOR pathway was suggested based on increases in the phosphorylation of S6K1 and S6 proteins, indicating an increase in protein synthesis. However, we also observed an increase in the phosphorylation of S6K1 protein in cells that lacked NDUFA11, in which protein synthesis was reduced and mTOR kinase activity was not significantly changed. Surprisingly, in our research, the expression of ATF4 protein increased only under conditions of short-term acute mitochondrial dysfunction. Under conditions of long-term mitochondrial stress, represented by HEK293 cells treated for 48 h with menadione or rotenone and by cells with the down-regulation of MIA40 oxidoreductase or cells that lacked NDUFA11 protein, eIF2α phosphorylation and ATF4 expression were reduced. Thus, under conditions of long-term mitochondrial stress, the response is different from that under conditions of short-term acute mitochondrial stress. We propose that responses to short-term acute mitochondrial stress, which lead to protein synthesis inhibition, allow cells to restore protein homeostasis to avoid cellular death. In case of long-term responses to mitochondrial stress, cells to survive need to trigger adaptive responses that protect from too high protein synthesis inhibition.
RESULTS
Chemical induction of mitochondrial ROS production inhibited protein synthesis
Our previous work found that oxidative stress in HEK293 cells that were treated with H2O2 decreased protein synthesis (Topf et al., 2018). To determine whether mitochondrial ROS produce a similar effect, we treated HEK293 and HepG2 cells with menadione, antimycin A, or CCCP and analyzed protein synthesis. Menadione generates mROS at the level of Complex I of the mitochondrial respiratory chain. Menadione undergoes reduction that produces a semiquinone radical, which is then autoxidized to the initial quinone form (Osenbroch et al., 2009; Monteiro et al., 2013). This redox cycling increases the production of intracellular ROS. Antimycin A inhibits cytochrome c reductase in mitochondrial Complex III, leading to an increase in ROS production (Ma et al., 2011). CCCP, which is an oxidative phosphorylation uncoupler, also can induce production of ROS by mitochondria (Olsson et al., 2008).
Cells that were treated with menadione for 2 h, antimycin A for 1.5 h, or CCCP for 1.25 h induced production of ROS (Figure 1D; Supplemental Figures 2D and 4C) and exhibited a concentration-dependent reduction of protein synthesis (Figure 1A; Supplemental Figures 1, A and C, 2A, and 4A). This phenotype of protein synthesis inhibition was not related to massive cellular death, in which the survival rate of HEK293 and HepG2 cells was very high (Figure 1B; Supplemental Figures 1, B and D, and 2B). We quantified adenosine triphosphate (ATP) levels under these treatment conditions. Menadione treatment did not lead to a large decrease in ATP levels (Figure 1C), whereas antimycin A and CCCP resulted in a significant decrease in ATP production (Supplemental Figures 2E and 4B). Translation is an energy-consuming process, and a decrease in ATP levels could account for the inhibition of protein synthesis and mask the effect of higher ROS levels. Therefore, in the subsequent experiments, only menadione was used.
FIGURE 1:

Treatment of HEK293 cells with menadione inhibited global protein synthesis. (A, E, F) Incorporation of [35S]-labeled amino acids in HEK293 cells. Total cell extracts were separated by SDS–PAGE and analyzed by autoradiography or immunodecorated with specific antibodies. (A) HEK293 cells were treated for 2 h with menadione as indicated. (B) Survival of HEK293 cells treated for 2 h with menadione as indicated. Mean ± SEM. n = 3. (C) Fold change in ATP concentration in HEK293 cells treated for 2 h with menadione as indicated. Mean ± SEM. n = 3. (D) ROS production in HEK293 cells treated for 2 h with menadione as indicated. Mean ± SEM. n = 6. (E) HEK293 cells were treated for 2 h with menadione in the presence of NAC as indicated. (F) HEK293 cells were treated for 2 h with NAC as indicated. Men, menadione; NAC, N-acetyl-l-cysteine. *p < 0.05; **p < 0.01; ***p < 0.001 by Student’s t test.
To confirm that ROS are specifically responsible for the observed inhibition of protein synthesis, we cotreated cells with menadione and the well-known ROS scavenger N-acetyl-l-cysteine (NAC). The anti-ROS activity of NAC results from its direct scavenging properties and the induction of higher glutathione levels (Halasi et al., 2013). The scavenging of ROS by NAC led to the recovery of protein synthesis, suggesting that the observed phenotype was specific to higher ROS levels (Figure 1E). Additionally, we verified that NAC treatment alone did not have any effect on protein synthesis (Figure 1F). Collectively, these data indicated that the increase in mitochondrion-derived ROS levels led to protein synthesis inhibition in mammalian cells.
Short-term stimulation of mitochondrial ROS production inhibited mTORC1 signaling
mTOR is a Ser/Thr kinase that is a key regulator of many cellular pathways. It is a component of two distinct protein complexes: mTORC1 and mTORC2. The regulation of mTORC1, the core components of which are also Raptor and mLST8 proteins, is far better understood than that of mTORC2 (Saxton and Sabatini, 2017). One of the anabolic processes that can be regulated by mTORC1 is global protein synthesis by inducing ribosome biogenesis and mRNA translation via phosphorylation of S6K1 and 4E-BP1 proteins (Shimobayashi and Hall, 2014).
mTORC1 signaling was previously shown to be attenuated under conditions of stress (Laplante and Sabatini, 2012). Indeed, short-term treatment of HEK293 cells with 10 µM menadione decreased the phosphorylation of S6K1 (Thr389) and 4E-BP1 (Ser65) proteins, indicating that the mTORC1 pathway is involved in the inhibition of protein synthesis under these conditions (Figure 2, A and B, lane 2). However, cells that were cotreated with menadione and NAC exhibited a reversal of these effects, indicating that the decrease in mTORC1 substrate phosphorylation upon menadione treatment was specific to higher ROS levels (Figure 2, A and B, lanes 3 and 4).
FIGURE 2:

Menadione-induced inhibition of mTORC1 activity is responsible for the reduction of global protein synthesis. (A, B) Phosphorylation of 4E-BP1 (Ser65) and S6K1 (Thr389) proteins upon the treatment of HEK293 cells for 2 h with menadione in the presence of NAC as indicated. Mean ± SEM, n = 3. (C) mTOR kinase activity in vitro in HEK293 cells treated for 2 h with menadione as indicated. Mean ± SEM. n = 3. (D) Phosphorylation of AMPK (Thr172) in HEK293 cells treated for 2 h with menadione as indicated. Mean ± SEM. n = 6. (E) Incorporation of [35S]-labeled amino acids in HEK293 cells. Total cell extracts were separated by SDS–PAGE and analyzed by autoradiography or immunodecorated with specific antibodies. HEK293 cells that were transduced only with pLKO.1 vector (Control) and HEK293 cells with shRNA-mediated knockdown of TSC2 protein were treated for 2 h with menadione as indicated. (F) Phosphorylation of S6K1 (Thr389) and 4E-BP1 (Ser65) proteins in HEK293 cells that were transduced only with pLKO vector (Control) and HEK293 cells with shRNA-mediated knockdown of TSC2 protein upon 2 h treatment with menadione as indicated. Men, menadione; NAC, N-acetyl-l-cysteine. **p < 0.01; ***p < 0.001; ns, not significant (p > 0.05) by Student’s t test.
We then analyzed the kinase activity of mTOR after its immunoprecipitation and found that mTOR activity was significantly reduced in cells that were treated with menadione (Figure 2C) and antimycin A (Supplemental Figure 2F). One way of regulating mTORC1 activity is changing stability of the complex (Kim et al., 2002). The stabilization of mTORC1, which can be observed as an increase in the coimmunoprecipitation of Raptor with mTOR protein, decreases its activity (Kim et al., 2002). However, upon the treatment of HEK293 cells with menadione, only a slight stabilization effect was observed (Supplemental Figure 5A). The mTORC1 pathway can also be regulated by adenosine monophosphate (AMP)-activated protein kinase (AMPK). AMPK is activated by phosphorylation in response to various conditions of stress, especially when cellular energy levels are low (i.e., there is a high AMP/adenosine triphosphate [ATP] ratio). It then phosphorylates TSC2 protein, which inhibits the mTORC1 activator RHEB, resulting in a reduction of mTORC1 activity (Ma and Blenis, 2009). mROS was recently shown to stimulate AMPK (Rabinovitch et al., 2017), but menadione treatment did not lead to significant activation (Figure 2D). The mild triggering of known mechanisms of mTORC1 inhibition by menadione treatment suggested the possible existence of another mechanism for the regulation of mTORC1 activity. TSC2 protein is a part of the complex that inhibits RHEB protein activity, which is an mTORC1 activator. We found that the overactivation of mTORC1 by silencing TSC2 protein rescued protein synthesis upon treatment of the cells with menadione (Figure 2E), which was related to increases in the phosphorylation of S6K1 (Thr389) and 4E-BP1 (Ser65) proteins (Figure 2F). These observations confirmed the involvement of mTOR inhibition as an important factor that is responsible for the inhibition of protein synthesis that is caused by the greater production of ROS by mitochondria.
Short-term stimulation of mitochondrial ROS production activated the integrated stress response
We then sought to investigate the involvement of activation of the integrated stress response (ISR) in protein synthesis inhibition under conditions of short-term acute mitochondrial stress. mTOR inhibition with its ATP-competitive inhibitor INK128 led to the complete dephosphorylation of S6K1 (Thr389) and 4E-BP1 (Ser65) proteins. However, the reduction of protein synthesis was lower compared with menadione treatment (Supplemental Figure 3A, lanes 2 and 4). Cells that were treated with both INK128 and menadione exhibited a stronger attenuation of protein synthesis, suggesting the involvement of another mechanism in addition to the mTOR pathway (Supplemental Figure 3A, lanes 4 and 5). The phosphorylation of eIF2α is another well-documented mechanism for the attenuation of protein synthesis. eIF2α is a key player in the ISR and can be phosphorylated under various stress conditions, such as amino acid deprivation (GCN2 kinase), viral infection (PKR kinase), heme deprivation (HRI kinase), and ER stress (PERK kinase; Harding et al., 2003; Pakos-Zebrucka et al., 2016). An increase in the phosphorylation of eIF2α causes the global attenuation of cap-dependent protein synthesis but induces the cap-independent translation of selected mRNAs, such as ATF4 transcription factor, which is a central effector of the ISR (Harding et al., 2003; Pakos-Zebrucka et al., 2016). Indeed, the treatment of HEK293 cells with menadione stimulated the phosphorylation of eIF2α (Ser51) in an mTOR-independent manner (Figure 3A, lane 2; Supplemental Figure 3A, lanes 2 and 5). Additionally, cells that were cotreated with NAC exhibited a reversal of this observed effect, indicating that activation of the ISR by menadione also depended on higher ROS levels (Figure 3A, lanes 3–6). Interestingly, the inhibition of PERK kinase with GSK2606414 decreased the menadione-induced phosphorylation of eIF2α (Ser51), but protein synthesis was not rescued (Figure 4B; Supplemental Figure 3B). This suggested that mTOR inhibition is sufficient and critical for protein synthesis reduction under acute mitochondrial-born oxidative stress. Cell treatment with A92, which is a GCN2 kinase inhibitor, did not reverse phosphorylation of eIF2α induced by menadione (Figure 4A). This suggested that in mammalian cells PERK kinase is responsible for eIF2α phosphorylation under acute short-term mitochondrial stress, in opposition to C. elegans, in which GCN2 kinase played a major role in eIF2α phosphorylation under mitochondrial stress conditions (Baker et al., 2012). Additionally, cells that were treated with menadione exhibited a significant increase in ATF4 expression (Figure 3B). The increased presence of transcription factor ATF4 in the nucleus probably increases the expression of genes critical for the recovery from acute mitochondrial stress (Figure 3, C and D). This effect also depended on ROS, in which cells that were cotreated with NAC exhibited a reversal of increased ATF4 presence in the nucleus (Figure 3, C and D). The enhanced presence of ATF4 in the nucleus was also visible after the cells were treated with CCCP; however, the decrease in ATP levels was very large (Supplemental Figure 4, B, D, and E). Recent studies reported that ATF4 is a key player in the mitochondrial stress response (Khan et al., 2017; Kuhl et al., 2017; Quiros et al., 2017). These results suggest that activation of the ISR by the short-term treatment of cells with menadione was not necessary for the inhibition of protein synthesis but was responsible for the induction of specific gene expression, thus possibly mitigating the consequences of mitochondrial stress.
FIGURE 3:

Increase in mROS production upon treatment of cells with menadione induced the integrated stress response. (A) Phosphorylation of eIF2α (Ser51) protein upon treatment of HEK293 cells for 2 h with menadione in the presence of NAC as indicated. Mean ± SEM. n = 3. (B) Fold change in ATF4 expression in HEK293 cells treated for 2 h with menadione as indicated. Mean ± SEM. n = 3. (C) Localization of ATF4 in HeLa cells. The cells were treated with vehicle (Control), menadione for 2 h, and NAC for 2 h in the presence of menadione as indicated. Nuclei were stained with DAPI. Scale bar = 20 μM. n = 2. (D) ATF4 in cell nuclei. Quantification of fold change in mean fluorescence in cell nuclei in experiment C. Mean ± SEM. n = 9. Men, menadione; NAC, N-acetyl-l-cysteine. *p < 0.05; **p < 0.01 by Student’s t test.
FIGURE 4:
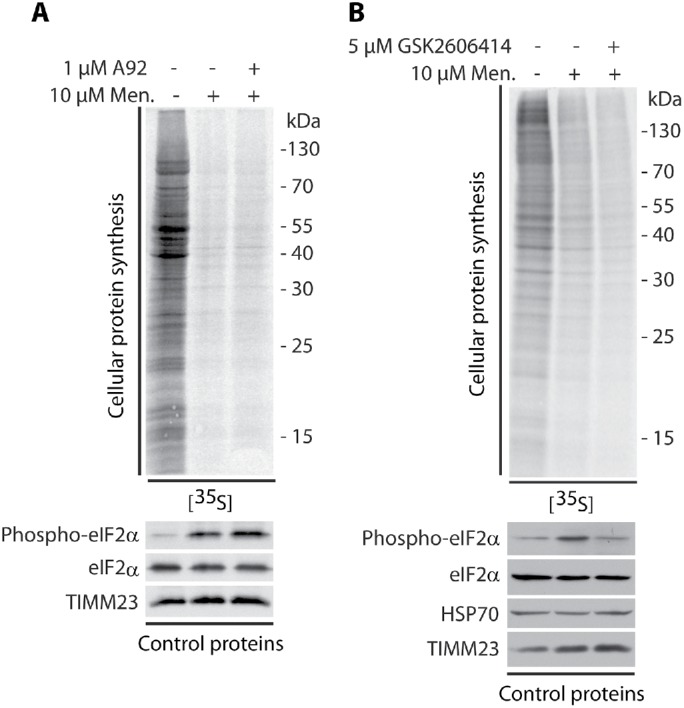
Reversion of eIF2α phosphorylation induced by menadione did not rescue protein synthesis. Incorporation of [35S]-labeled amino acids in HEK293 cells. Total cell extracts were separated by SDS–PAGE and analyzed by autoradiography or immunodecorated with specific antibodies. (A) HEK293 cells were treated with menadione for 2 and 3 h with the GCN2 kinase inhibitor A92 in the presence of menadione (2 h) as indicated. (B) HEK293 cells were treated with menadione for 2 and 3 h with the PERK kinase inhibitor GSK2606414 in the presence of menadione (2 h) as indicated. Men, menadione.
Cytosolic translation stress responses are influenced by long-term mitochondrial alterations
We observed that short-term acute mitochondrial stress led to the inhibition of protein synthesis. Next, we asked about the impact of long-term mitochondrial stress on cytosolic translational responses. To check these responses, we treated HEK 239 cells for 48 h with low concentrations of menadione or rotenone, which is an inhibitor of mitochondrial complex I. Surprisingly, long-term mitochondrial stress had an effect on cytosolic translational responses opposite to that observed under conditions of short-term acute mitochondrial stress. Although protein synthesis was not significantly decreased, the phosphorylation of S6K1 (Thr389) protein and its substrate S6 (Ser235/236) ribosomal protein was increased, indicating an increase in mTOR pathway activity (Figure 5, A, B, D, and E). Moreover, the phosphorylation of eIF2α (Ser51) decreased, indicating that the ISR was inhibited (Figure 5, C and F). Cell treatment with rotenone caused additionally increased expression of a regulatory subunit of PP1 phosphatase, GADD34 (PPP1R15A), which is responsible for eIF2α dephosphorylation, and we noticed slight induction of the expression of CHOP protein, which mediates the activation of GADD34 (Figure 5F; Novoa et al., 2001).
FIGURE 5:

Long-term cell treatment with menadione or rotenone increased S6K1 phosphorylation and decreased eIF2α phosphorylation. (A) Incorporation of [35S]-labeled amino acids in HEK293 cells. Cells were treated with menadione for 48 h as indicated and total cell extracts were separated by SDS–PAGE and analyzed by autoradiography. (B, C) HEK293 cells were treated with menadione for 48 h as indicated. Mean ± SEM. n = 3. (D) Incorporation of [35S]-labeled amino acids in HEK293 cells. Cells were treated with rotenone for 48 h as indicated and total cell extracts were separated by SDS–PAGE and analyzed by autoradiography. (E, F) HEK293 cells were treated with rotenone for 48 h as indicated. Mean ± SEM. n = 3. Men, menadione; Rot, rotenone. *p < 0.05; **p < 0.01; ns, not significant (p > 0.05) by Student’s t test.
For further investigation of cytosolic translational responses under conditions of long-term mitochondrial stress, we silenced MIA40 for 72 h. The oxidoreductase MIA40 is responsible for the import of proteins into the intermembrane space of mitochondria (Chacinska et al., 2004). MIA40 also plays an important role in the biogenesis of respiratory chain complex enzymes in mitochondria (Longen et al., 2009; Mohanraj et al., 2019). The knockdown of MIA40 had an effect similar to that of treating the cells with menadione or rotenone for 48 h, but in HeLa cells, protein synthesis and expression of ATF4 were decreased (Figure 6, C and D; Supplemental Figure 6, A and B); however, ROS production and ATP levels were not significantly changed (Figure 6, A and B). Tunicamycin, which is an ISR activator, significantly induced phosphorylation of eIF2α (Ser51) in control cells but not in HeLa and HEK293 cells after silencing of MIA40, which confirmed that long-term mitochondrial stress caused ISR inhibition (Supplemental Figure 6B).
FIGURE 6:

Knockdown of MIA40 oxidoreductase decreased protein synthesis in the cytosol. (A) ROS production in HeLa cells upon 72 h siRNA-mediated knockdown of MIA40 as indicated. Mean ± SEM. n = 3. (B) Fold change in ATP concentration in HeLa cells upon 72 h siRNA-mediated knockdown of MIA40 as indicated. Mean ± SEM. n = 3. (C) Incorporation of [35S]-labeled amino acids in HeLa cells. Total cell extracts were separated by SDS–PAGE and analyzed by autoradiography or immunodecorated with specific antibodies. The siRNA-mediated knockdown of MIA40 was performed in HeLa cells for 72 h. (D) Phosphorylation of S6K1 (Thr389) and eIF2α (Ser51) proteins and ATF4 expression upon 72 h siRNA-mediated knockdown of MIA40 in HeLa cells. The graphs show the quantification of three (S6K1 [Thr389] and eIF2α [Ser51]) or six biological replicates (ATF4). Mean ± SEM. **p < 0.01; ns, not significant (p > 0.05) by Student’s t test.
To confirm that cytosolic responses under conditions of long-term mitochondrial defects are different from those that are observed under conditions of short-term acute mitochondrial stress, we analyzed protein synthesis efficiency in the HEK293T NDUFA11 knockout cell line (Stroud et al., 2016). NDUFA11 is an accessory subunit of mitochondrial complex I (NADH:ubiquinone oxidoreductase), and it is located at the connection between its hydrophilic matrix arm and a hydrophobic membrane arm (Berger et al., 2008; Stroud et al., 2016). Mutations of complex I subunit genes frequently lead to complex neurodegenerative diseases, such as Leigh’s syndrome (Berger et al., 2008; Rodenburg, 2016). We found that ROS levels in the NDUFA11 knockout cell line were much higher than in wild type (WT) cells, but this was presumably not a reason for the inhibition of protein synthesis in these cells, because even 24 h of incubation with the ROS scavenger NAC did not reverse the effect of protein synthesis attenuation (Figure 7, A and C). Additionally, ATP levels in cells that lacked NDUFA11 were significantly lower (Figure 7B). Although protein synthesis in NDUFA11-deficient cells was inhibited, the phosphorylation of eIF2α (Ser51) and expression of ATF4 both decreased (Figure 8A), in contrast to cells under conditions of short-term acute mitochondrial stress, in which the ISR was activated. Interestingly, previous studies reported higher mRNA levels of the ATF4 gene, independent of the duration of mitochondrial stress (Khan et al., 2017; Kuhl et al., 2017; Quiros et al., 2017). In the present study, however, we observed a decrease in ATF4 transcript levels in the NDUFA11 knockout cell line (Figure 8B). Surprisingly, the phosphorylation of S6K1 (Thr389) protein was enhanced, despite the nonsignificant change in mTOR kinase activity and increase in the phosphorylation (activation) of AMPK (Thr172; Figures 8A and 9, A and B). Interestingly, an increase in S6K1 (Thr389) protein phosphorylation was previously observed in neurons that were treated for 6 h with oligomycin and rotenone/antimycin-A, despite the activation of AMPK, which is often linked to mTOR attenuation (Zheng et al., 2016). Additionally, mTORC1 stability was not significantly altered, indicating that this was not the mode of regulating its activity in cells that lacked NDUFA11 (Supplemental Figure 5B). The observed inhibition of protein synthesis in NDUFA11 knockout cells could be partially achieved by decreasing the phosphorylation of 4E-BP1 (Ser65) protein, but the overactivation of mTORC1 by silencing TSC2 protein rescued protein synthesis nonsignificantly (Figures 8A and 9C). This suggests the involvement of another mechanism of reduction of protein synthesis in cells under conditions of long-term mitochondrial stress. NDUFA11-deficient cells were unable to grow on galactose-containing media, indicating mitochondrial respiration defects (Stroud et al., 2016). We observed that NDUFA11 knockout cells cultured on galactose up to 4 h exhibited stronger protein synthesis inhibition and S6K1 (Thr389) phosphorylation than these cells cultured on glucose (Supplemental Figure 7). This suggested that increased long-term mitochondrial stress induced stronger adaptive responses. Phosphorylation of eIF2α (Ser51), after 4 h in galactose medium, grew in NDUFA11-deficient cells, suggesting that these cells started to reprogram responses characteristic for acute mitochondrial stress (Supplemental Figure 7). Phosphorylation of S6K1 (Thr389) is controlled by mTOR kinase, which phosphorylates this protein and PP2A phosphatase, which is responsible for its dephosphorylation (Peterson et al., 1999). Because, in NDUFA11-deficient cells, mTOR kinase activity was not significantly changed, we checked whether mRNA levels of PP2A phosphatase subunits were altered. Indeed, we observed that while mRNA levels of scaffold subunit (A) were not changed, mRNA levels for regulatory (B) and catalytic (C) subunits were significantly decreased (Supplemental Figure 8), indicating that lower activity of PP2A can be responsible for increased phosphorylation of S6K1 (Thr389) under long-term mitochondrial stress. Although the mechanism of protein synthesis inhibition under long-term mitochondrial stress caused either chemically or by genetic alterations is not completely clear, all these results demonstrated that cytosolic translational responses depend on time and severity of mitochondrial stress.
FIGURE 7:
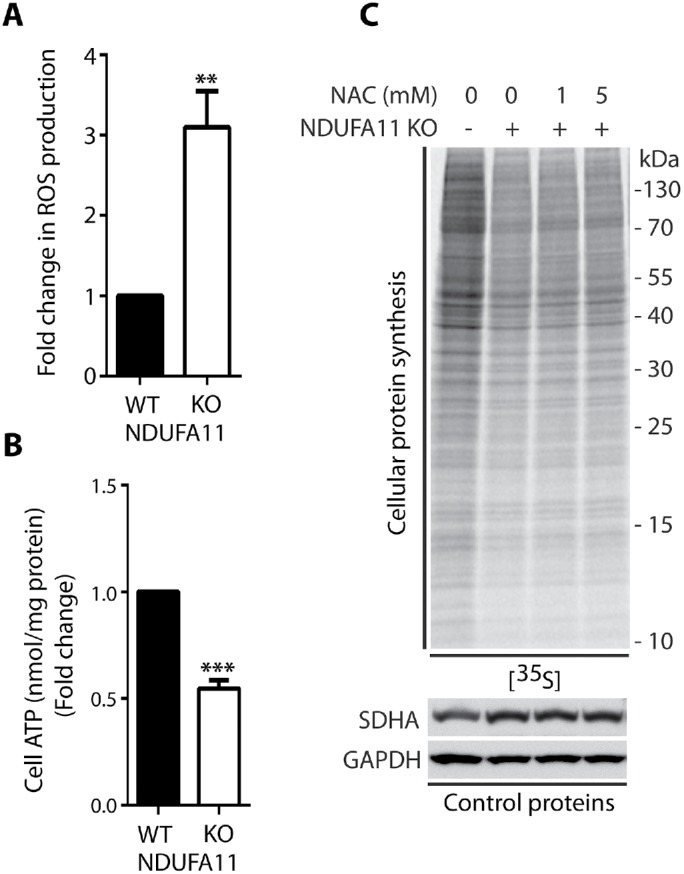
Knockout of NDUFA11 decreased global protein synthesis in a reactive oxygen species–independent manner. (A) ROS production in HEK293T and HEK293T NDUFA11 knockout cells. Mean ± SEM. n = 3. (B) Fold change in ATP concentration in HEK293T and HEK293T NDUFA11 knockout cells. Mean ± SEM. n = 3. (C) Incorporation of [35S]-labeled amino acids in HEK293T and HEK293T NDUFA11 knockout cells in the 24 h presence of NAC as indicated. Total cell extracts were separated by SDS–PAGE and analyzed by autoradiography or immunodecorated with specific antibodies. NAC, N-acetyl-l-cysteine. **p < 0.01; ***p < 0.001 by Student’s t test.
FIGURE 8:

The integrated stress response is inhibited in NDUFA11-deficient HEK293T cells. (A) Incorporation of [35S]-labeled amino acids in HEK293T and HEK293T NDUFA11 knockout cells. Total cell extracts were separated by SDS–PAGE and analyzed by autoradiography or immunodecorated with specific antibodies to assess the phosphorylation of S6K1 (Thr389), 4E-BP1 (Ser65), and eIF2α (Ser51) proteins and ATF4 expression. The graphs show the quantification of three biological replicates of eIF2α phosphorylation and ATF4 expression. Mean ± SEM. (B) ATF4 mRNA levels in HEK293T and HEK293T NDUFA11 knockout cells. Mean ± SEM. n = 3. ***p < 0.001 by Student’s t test.
FIGURE 9:
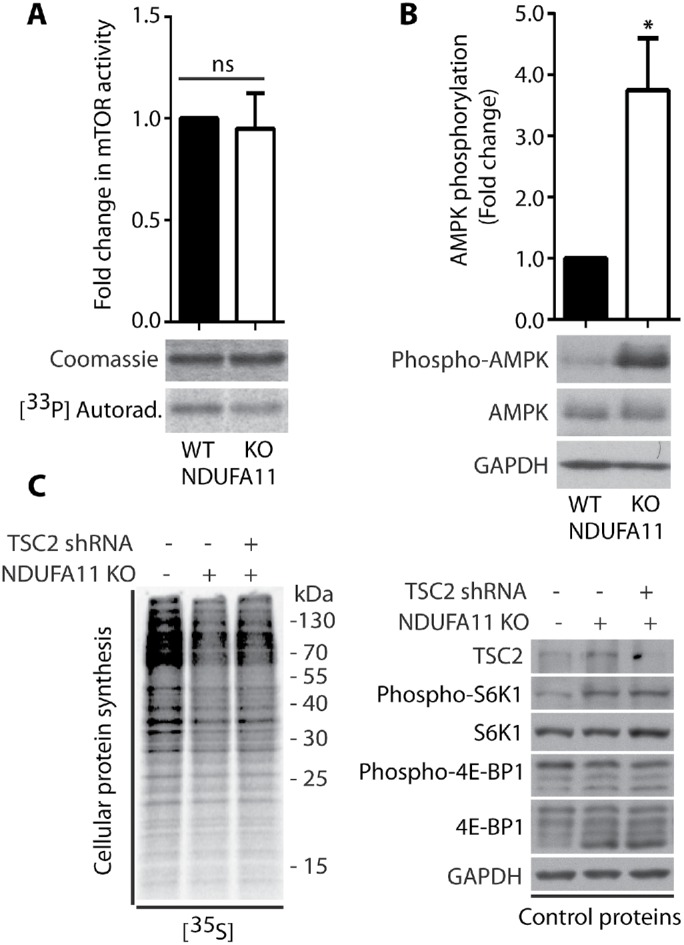
Lower mTORC1 activity is not responsible for global protein synthesis inhibition in NDUFA11-deficient HEK293T cells. (A) mTOR kinase activity in vitro in HEK293T and HEK293T NDUFA11 knockout cells. Mean ± SEM. n = 3. (B) Phosphorylation of AMPK (Thr172) in HEK293T and HEK293T NDUFA11 knockout cells. Mean ± SEM. n = 3. (C) Incorporation of [35S]-labeled amino acids in HEK293T and HEK293T NDUFA11 knockout cells. Total cell extracts were separated by SDS–PAGE and analyzed by autoradiography or immunodecorated with specific antibodies to assess the phosphorylation of S6K1 (Thr389) and 4E-BP1 (Ser65) proteins in HEK293T and HEK293T NDUFA11 knockout cells, which were transduced with pLKO.1 vector (–), and in HEK293T NDUFA11 knockout cells, which were transduced with pLKO.1-shTSC2 vector for the knockdown of TSC2 protein. *p < 0.05; ns, not significant (p > 0.05) by Student’s t test.
DISCUSSION
Previous studies proposed that the reduction of cytosolic protein synthesis can mitigate the harmful consequences of mitochondrial stress (Wang et al., 2008; Liu and Lu, 2010). In the present study, we compared cytosolic translational responses comprehensively to short-term acute and long-term mitochondrial stress. We found that the type of response that was triggered depended on the duration of signaling from defective mitochondria and the severity of mitochondrial stress. Under conditions of short-term acute mitochondrial stress that was induced by menadione, protein synthesis was inhibited in response to higher mROS levels, and the scavenging of ROS by NAC restored protein synthesis. Our previous study (Topf et al., 2018) and the present results suggest that at least two mechanisms can contribute to the reduction of protein synthesis during acute mitochondrial stress: the oxidation of ribosomal proteins and the inhibition of mTORC1 signaling. We proposed that the oxidation of redox switches that are located on ribosomal proteins may act as a rapid and reversible response to oxidative stress (Topf et al., 2018). The reduction of mTORC1 activity was confirmed by the decrease in the phosphorylation of S6K1 and 4E-BP1 proteins, which are responsible for the regulation of protein synthesis and depend on mTORC1. Moreover, the decrease in TOR signaling was also observed in a response caused by mitochondrial precursor accumulation (Wang and Chen, 2015).
Surprisingly, we found that the reversal of menadione-induced eIF2α phosphorylation did not restore protein synthesis, indicating that activation of the ISR was not responsible for the reduction of protein synthesis under conditions of short-term acute mitochondrial stress. In fact, protein synthesis was shown to be inhibited under stress conditions, independent of eIF2α phosphorylation (Knutsen et al., 2015). We showed that activation of the ISR under conditions of short-term mitochondrial stress was responsible for specific gene expression, in which increases in transcription factor ATF4 expression and its presence in the nucleus were observed. These observations are generally consistent with recent findings that ATF4 is a major player in the mitochondrial stress response in mammalian cells (Khan et al., 2017; Kuhl et al., 2017; Quiros et al., 2017).
In contrast, under conditions of long-term mitochondrial stress, represented by long-term chemical stress, knockdown of MIA40 oxidoreductase, and knockout of an accessory subunit of mitochondrial complex I (NDUFA11), cytosolic translational responses were generally opposite to those that were observed during short-term acute mitochondrial stress. Although ROS levels increased significantly in the NDUFA11 knockout cell line, the inhibition of protein synthesis was likely not ROS-dependent, because NAC treatment did not restore protein synthesis or became irreversible. Surprisingly, despite the inhibition of protein synthesis, the ISR was attenuated. eIF2α phosphorylation and the protein and mRNA expression of ATF4 decreased. ISR inhibition was confirmed by the fact that tunicamycin, which is ISR activator, could not induce significantly eIF2α (Ser51) phosphorylation in cells after silencing of MIA40. Interestingly, eIF2α phosphorylation that was induced by mitochondrial dysfunction caused selective dendritic loss in Drosophila melanogaster class IV dendritic arborization neurons (Tsuyama et al., 2017). Moreover, a decrease in ATF4 expression was reported to be an important mechanism of cancer cell survival under conditions of prolonged amino acid deprivation (Mesclon et al., 2017). Interestingly, other studies have reported an increase in the expression of ATF4 at the transcript level, independent of the duration of mitochondrial stress (Khan et al., 2017; Kuhl et al., 2017; Quiros et al., 2017). Thus, activation depends not only on the duration but also on the type and severity of mitochondrial stress.
Strikingly, in cells that lacked NDUFA11, the phosphorylation of S6K1 protein increased, despite the strong activation of AMPK, which usually inhibits mTOR activity and decreases S6K1 phosphorylation. S6K1 phosphorylation was also increased in cells that were treated for 48 h with menadione or rotenone and cells with 72-h knockdown of MIA40 protein. This interesting phenomenon was previously observed in neurons that were treated for 6 h with oligomycin and rotenone/antimycin-A (Zheng et al., 2016). Strikingly, elevation of S6K1 activity was also detected in the brains of Alzheimer’s disease patients (An et al., 2003). Indeed, in our experiments on the NDUFA11 knockout cell line, mTOR kinase activity was not significantly changed in vitro. These results suggest that S6K1 protein “escaped” from its canonical regulation by the mTOR pathway, which may be a prosurvival mechanism, because the strong inhibition of protein synthesis would likely cause cellular death in the long term. Surprisingly, in NDUFA11-deficient cells, transcript levels of regulatory and catalytic subunits of PP2A phosphatase, which dephosphorylates S6K1 protein, were decreased, suggesting its lower activity. This could be a reason for enhanced phosphorylation of S6K1 (Thr389) under long-term mitochondrial stress. Despite the decrease in phosphorylation of the 4E-BP1 protein, the mTOR pathway did not appear to be involved in the inhibition of protein synthesis in NDUFA11 knockout cells under steady-state conditions. We found that the overactivation of mTOR in the NDUFA11 knockout cell line did not significantly affect protein synthesis.
In summary, the present results indicate that long-term mitochondrial stress responses are different from short-term acute mitochondrial stress responses. One possible explanation for this difference is that acute mitochondrial stress induces a rapid reaction, in which the mTOR pathway and cap-dependent translation are inhibited. With prolonged mitochondrial stress, in contrast, this beneficial response is lost (D’Amico et al., 2017; Samluk et al., 2018). Although the mechanism of protein synthesis inhibition under long-term mitochondrial stress is not completely clear, we proposed a model in which long-term mitochondrial stress triggers adaptive responses for protection against excessive inhibition of protein synthesis. In contrast, acute mitochondrial stress induces specific reduction of protein synthesis, probably to achieve protein homeostasis (Figure 10). Under long-term mild mitochondrial stress, cells keep protein synthesis at a high level, despite the pressure for its reduction, thanks to increased S6K1 phosphorylation and ISR inhibition. Enhanced phosphorylation of S6K1 could cause increased phosphorylation of S6 ribosomal protein to stimulate ribosomes for protein synthesis. Inhibited ISR (reduced eIF2α phosphorylation) could be a mechanism of stimulation of translation initiation. These adaptive responses probably enable cell survival under long-term mitochondrial stress.
FIGURE 10:

Schematic illustration of translational responses under mild and acute mitochondrial stress.
Interestingly, not only are cytosolic translational responses influenced by the duration and severity of stress. For example, in the short-term response to mitochondrial dysfunction in yeast (UPRam), proteasome activity increased (Wrobel et al., 2015), whereas prolonged acute oxidative stress that was associated with defective mitochondria led to disassembly and a decrease in proteasome activity (Livnat-Levanon et al., 2014; Segref et al., 2014).
Adaptive responses that we observed under long-term mitochondrial stress may have pathological consequences. It was demonstrated that ISR is involved in resolving Aβ proteotoxic stress in mammalian cells (Sorrentino et al., 2017), so its inhibition may be involved in Alzheimer’s disease pathogenesis. Moreover, increased phosphorylation of S6 and S6K proteins was observed in mitochondrial myopathy, and rapamycin, which is a mTORC1 inhibitor, reverted progression of this mitochondrial disease in mice (Khan et al., 2017). In the context of mitochondrial diseases, both reversal of harmful long-term stress responses and manipulation of short-term responses may be very beneficial for patients. Therefore, a better understanding of adaptive responses that are triggered by mitochondrial stress may have important clinical implications.
MATERIALS AND METHODS
Cell culture conditions
HEK293, HepG2, HeLa, HEK293T, and HEK293T NDUFA11 knockout cell lines were cultured in high-glucose (4.5 g/l) 90% DMEM (Sigma, catalogue no. D5671) supplemented with heat-inactivated (55°C for 30 min) 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin at 37°C in a 5% CO2 humidified atmosphere. The medium was additionally supplemented with 1 mM sodium pyruvate for HepG2 cells and 50 μg/ml uridine for HEK293T WT and HEK293T NDUFA11 knockout cells. The culture medium was changed every other day. For the last 24 h before the experiments, HEK293, HepG2, and HeLa cells were cultured in medium with galactose and 90% DMEM (Sigma, catalogue no. D5030) supplemented with 44 mM NaHCO3, 10 mM galactose, heat-inactivated (55°C for 30 min) 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. HepG2 cells were additionally supplemented with 1 mM sodium pyruvate. The cells were treated with various concentrations of menadione (Sigma, catalogue no. M5625), rotenone (Sigma, catalogue no. R8875), antimycin A (Sigma, catalogue no. A8674), NAC (Sigma, catalogue no. A9165), A-92 (Axon Medchem, catalogue no. Axon 2720), GSK2606414 (Tocris, catalogue no. 5107), INK128 (APExBIO, catalogue no. MLN0128), Tunicamycin (Sigma, catalogue no. T7765), and CCCP (Sigma, catalogue no. C2759) where indicated. HEK293, HepG2, and HeLa cells were obtained from the American Type Culture Collection. HEK293T and HEK293T NDUFA11 knockout cells were a kind gift from David Stroud and Michael Ryan, Monash University, Melbourne, Australia (Stroud et al., 2016).
Translation assay
HEK293, HepG2, HEK293T, and HEK293T NDUFA11 knockout cells were cultivated until 70–90% confluency was reached. Before the experiments, the cells were washed twice with phosphate-buffered saline (PBS) and incubated at 37°C for 60 min in RPMI medium without methionine or serum (Sigma, catalogue no. R7513), and at this step indicated chemicals were added. Newly synthesized proteins were then radiolabeled by adding [35S]methionine and [35S]cysteine (EasyTag EXPRESS35S Protein Labeling Mix, Perkin Elmer, catalogue no. NEG772002MC) to a final concentration of 6 μCi/1 ml and incubated for 0.5 or 1 h at 37°C in the presence of the indicated chemicals. After radiolabeling, the dishes were placed on ice, and the cells were scratched and washed three times with ice-cold PBS. The cells were lysed by 30 min of incubation with ice-cold RIPA buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA) that contained 2 mM phenylmethylsulfonyl fluoride (PMSF; Sigma, catalogue no. P7626) and phosphatase inhibitor cocktail (PhosSTOP, Roche catalogue no. 04 906 837 001). The samples were separated in 15% or 10% polyacrylamide gels. Radioactivity in dried gels was detected using a Typhoon TRIO+ Variable Mode Imager (GE Healthcare). Gel loading was controlled by Western blot.
Cell viability assay
Appropriately diluted HEK293 or HepG2 cell suspensions were mixed with an equal volume of 0.4% Trypan Blue solution (Sigma, catalogue no. T8154) and incubated for 2 min at room temperature. Viable cells (unstained) were counted immediately after cells treatment with indicated chemicals using a hemocytometer and a light microscope in three independent experiments. The values are expressed as mean ± SEM.
Adenosine triphosphate measurements
For the last 24 h before the experiment, HEK293 cells were cultured in DMEM with 10 mM galactose. They were then treated with 10 μM menadione for 2 h, 10 μM antimycin A for 1.5 h, or 10 μM CCCP for 1.25 h. HEK293T and HEK293T NDUFA11 knockout cells were cultured in DMEM with 4.5 g/l glucose supplemented with uridine (50 μg/ml). Knockdown of MIA40 was performed in HeLa cells for 72 h. Cellular ATP levels were assessed using the ATP Assay Kit (Abcam, catalogue no. ab83355) according to the manufacturer’s instructions. Fluorescence was measured at an excitation wavelength of 535 nm and emission wavelength of 587 nm using a plate fluorimeter (Infinite M1000, Tecan). The values were normalized to protein concentrations that were obtained from Bradford assays.
In vitro mTOR kinase assay
HEK293, HEK293T, and HEK293T NDUFA11 knockout cells were harvested in cold TBS (100 mM NaCl and 25 mM Tris-HCl, pH 7.5). After centrifugation, the cell pellets were suspended in lysis buffer (40 mM HEPES-KOH, pH 7.5, 120 mM NaCl, 1 mM EDTA, 0.3% CHAPS, and protease and phosphatase inhibitors) and lysed for 20 min on ice. Cell lysates were cleared by centrifugation, and 5 μg of anti-mTOR antibody (Bethyl, catalogue no. A300-504A) was added. After 2 h of incubation with rotation at 4°C, 20 μl of Dynabeads Protein G (Invitrogen, catalogue no. 10004D) was added to the lysate and incubated for 1 h. The beads were then washed three times with lysis buffer, twice with high-salt buffer (40 mM HEPES-KOH, pH 7.5, 500 mM NaCl, 1 mM EDTA, and 0.3% CHAPS), once with low-salt buffer (40 mM HEPES-KOH, pH 7.5, 150 mM NaCl, and 1 mM EDTA), and once with kinase reaction buffer (25 mM HEPES-KOH, pH 7.5, 50 mM KCl, and 10 mM MgCl2). The kinase reaction was performed by incubating the beads in kinase reaction buffer with the addition of ATP gamma-P33 (Hartmann Analytic, catalogue no. SCF-301) and 1 μg mTOR substrate (Millipore, catalogue no. 12-645) for 20 min at 37°C. The mTOR substrate was stained with Coomassie Brilliant Blue as a loading control. The reaction was stopped by the addition of 4× Laemmli sample buffer.
Stability of mTORC1
For the coimmunoprecipitation experiments, cells were harvested in lysis buffer (40 mm HEPES, pH 7.5, 120 mm NaCl, 1 mm EDTA, 0.3% CHAPS, and protease and phosphatase inhibitors). The lysed cells were incubated on ice for 20 min and centrifuged for 10 min at 14,000 × g at 4°C to remove cellular debris. Supernatants that contained 1.5 mg total protein were then incubated with rabbit anti-mTOR antibody (Cell Signaling Technology, catalogue no. 2972) at 4°C overnight, and then Protein A Sepharose (Thermo Fisher Scientific, catalogue no. 101041) was added. As a negative control, normal rabbit immunoglobulin G (IgG; Sigma, catalogue no. 12-370) was used. After incubation for 2 h at 4°C, the resins were washed four times with lysis wash buffer and boiled in SDS–PAGE sample buffer. The samples were analyzed by Western blot.
shRNA-mediated knockdown of TSC2 protein
The pLKO.1-TRC cloning vector (Addgene, catalogue no. 10878) (Moffat et al., 2006) was the backbone for cloning the shRNA sequence. shTSC2 (382-404) was designed with the siRNA Selection Program (Yuan et al., 2004) against human TSC2 mRNA (GI|4071057). The sequences were the following: shTSC2 4 382-404, GGATTACCCTTCCAACGAA; shTSC2 4 human pLKO, forward, CCGGGGATTACCCTTCCAACGAACTGCAGTTCGTTGGAAGGGTAATCCTTTTTG; reverse, AATTCAAAAAGGATTACCCTTCCAACGAACTGCAGTTCGTTGGAAGGGTAATCC. Lentiviral particles were generated by the transient transfection of HEK293T cells with pLKO.1-TRC control plasmid (Addgene, catalogue no. 10879; Moffat et al., 2006) or pLKO.1-shTSC2 plasmid together with psPAX2-D64V plasmid (Addgene, catalogue no. 163586; Certo et al., 2011) and pMD.RVG.CVS24-B2c plasmid (Addgene, catalogue no. 19713; Mentis et al., 2006). The medium that contained lentiviral particles was collected 48 h after transfection and used for transduction. Transduced cells were selected with puromycin.
Immunofluorescence
HeLa cells were washed twice with PBS and fixed with 3.7% paraformaldehyde for 10 min at 4°C. The cells were then washed three times with PBS and permeabilized for 5 min by treatment with 0.1% Triton X-100 in PBS. Nonspecific binding sites were blocked for 0.5 h with 10% fetal bovine serum in PBS. The cells were then incubated for 2 h with anti-ATF4 antibody (1:100; Cell Signaling, catalogue no. D4B8, 11815). After three washes with PBS, the cells were incubated for 2 h with Alexa Fluor 488 anti-rabbit (1:100; Thermo Fisher Scientific, catalogue no. A-11034). They were then rinsed three times with PBS and once with water, and the samples were mounted in ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific, catalogue no. P36962). The analysis was performed using a Zeiss LSM800 confocal microscope.
siRNA-mediated knockdown of MIA40 protein
HeLa and HEK293 cells (0.1 × 106) were seeded in each well of a six-well plate and grown for 24 h in low-glucose (1 g/l) DMEM. After 24 h, the cells were transfected with the respective oligonucleotides using Oligofectamine (Invitrogen, catalogue no. 12252011) in Opti-MEM I Reduced Serum Medium (Life Technologies, catalogue no. 31-985-070) according to the manufacturer’s instructions. Two solutions were prepared: 1) 124 nM (HeLa cells) or 248 nM (HEK293 cells) of specific small interfering RNA (siRNA) diluted in 175 µl of OptiMEM medium and mixed gently; 2) 4 µl (HeLa cells) or 8 µl (HEK293 cells) of Oligofectamine diluted in 25 µl of Opti-MEM medium and mixed gently. After 5 min of incubation at room temperature, the two solutions were gently mixed and incubated for 15 min at room temperature. The cells were washed once with Opti-MEM, and 800 µl of Opti-MEM was added to each well. After incubation, the mixture was transferred to the cells and incubated at 37°C in 5% CO2 for 4 h. The medium was replaced with low-glucose DMEM with 30% FBS. After 24 h, the cells were shifted to DMEM that contained 10 mM galactose until the end of the experiment and then harvested for analysis. As a control, the cells were treated with Mission siRNA universal negative control (Sigma, catalogue no. SIC001). The following sequences that targeted MIA40 mRNA were used for the knockdown experiments: MIA40_1, 5′-ATAGCACGGAGGAGATCAA-3′; MIA40_2, 5′-GGAATGCATGCAGAAATAC-3′).
Reactive oxygen species measurements
To analyze the levels of ROS production, HEK293T and HEK293T NDUFA11 knockout cells were cultured in DMEM that contained 4.5 g/l glucose supplemented with 50 μg/ml uridine until 70–90% confluency was reached. For the last 24 h before the experiment, HEK293 cells were cultured in DMEM with 10 mM galactose. They were then treated with 10 μM menadione for 2 h, 10 μM antimycin A for 1.5 h, or 10 μM CCCP for 1.25 h. Knockdown of MIA40 was performed in HeLa cells for 72 h. The cells were collected and incubated for 20 min at room temperature in culture medium that contained 2.5 μM CM-H2DCFDA dye (Thermo Fisher Scientific, catalogue no. C6827). Fluorescent signals were corrected for autofluorescence. Fluorescence was measured at an excitation wavelength of 495 nm and emission wavelength of 520 nm using a flow cytometer (FACSCanto II, BD).
Quantitative real-time PCR
Total RNA from cells was extracted using Trizol (Invitrogen, catalogue no. 10296010) and purified using the RNeasy Plus Mini Kit (Qiagen, catalogue no. 74134). cDNA was synthesized from 500 ng of total RNA using the Cloned AMV First-Strand cDNA Synthesis Kit (Invitrogen, catalogue no. 12328-032) according to the manufacturer’s instructions. Endogenous mRNA was measured by quantitative real-time PCR (RT-PCR) with the LightCycler 480 Instrument II (Roche) and SensiFAST SYBR Hi-ROX Kit (Bioline, catalogue no. BIO-92005). The following primer sequences were used for quantitative RT-PCR: ATF4, forward, 5′-CAGCAAGGAGGATGCCTTCT-3′; reverse, 5′-CCAACAGGGCATCCAAGTC-3′; PP2A.Aα/β, forward, 5′-GCTTCAATGTGGCCAAGTCT-3′; reverse, 5′-GGTCCTGGGTCAGCTTCTCT-3′; PP2A.B’γ, forward, 5′-ACAGTGAAGGACGAGGCTCA-3′; reverse, 5′-CTTCCAAGGCTTTCTTGGTG-3′; PP2A.C, forward, 5′-TCGTTGTGGTAACCAAGCTG-3′; reverse, 5′-AACATGTGGCTCGCCTCTAC-3′; ACTB, forward, 5′-GCCGGGACCTGACTGACTAC-3′; reverse, 5′-TTCTCCTTAATGTCACGCACGAT-3′. ACTB was used as the internal control.
Miscellaneous
Total protein extracts of mammalian cells were prepared in RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, and 2 mM PMSF). Laemmli sample buffer that contained 50 mM dithiothreitol (DTT) was added, and proteins were denatured at 65°C for 15 min. Total protein extracts were separated by SDS–PAGE on 15%, 10%, or 6% gels. The following commercially available antibodies against mammalian proteins were used: TIMM23 (BD Biosciences, catalogue no. 611222), SDHA (D-4; Santa Cruz Biotechnology, catalogue no. sc-1669IA47), HSP70 (Enzo Life Sciences, catalogue no. ADI-SPA-812), RPL7 (Bethyl, catalogue no. A300-741A), 4E-BP1 (Cell Signaling Technology, catalogue no. 9644), Phopho-4E-BP1 (Ser65) (Cell Signaling Technology, catalogue no. 9451), eIF2α (Cell Signaling Technology, catalogue no. 9722), Phospho-eIF2α (Ser51) (Cell Signaling Technology, catalogue no. 9721), GAPDH (Santa Cruz Biotechnology, catalogue no. sc-47724), ATP5a (Abcam, catalogue no. ab14748), S6K1 (Cell Signaling Technology, catalogue no. 9202), Phopho-S6K1 (Thr389) (Cell Signaling Technology, catalogue no. 9205), mTOR antibody (Bethyl, catalogue no. A300-504A), mTOR (Cell Signaling Technology, catalogue no. 2972), mTOR (Cell Signaling Technology, catalogue no. 4517), Raptor (Cell Signaling Technology, catalogue no. 2280), AMPK (Cell Signaling Technology, catalogue no. 2532), Phospho-AMPK (40H9) (Thr172) (Cell Signaling Technology, catalogue no. 2535), TSC2 (Cell Signaling Technology, catalogue no. 3612), ATF4 (Cell Signaling Technology, catalogue no. 11815), GADD34 (Proteintech, catalogue no. 10449-1-AP), CHOP (Cell Signaling Technology, catalogue no. 2895), Phospho-S6 (Ser235/236) (Cell Signaling Technology, catalogue no. 2211), and S6 (Cell Signaling Technology, catalogue no. 2217). Protein bands were visualized using secondary anti-rabbit or anti-mouse antibodies conjugated with horseradish peroxidase and chemiluminescence. Chemiluminescence signals were detected using ImageQuant LAS4010 (GE Healthcare) or x-ray films. The images were digitally processed using Adobe Photoshop CS4 software. ImageJ software was used to quantify the Western blots and confocal microscope images. The represented fold changes are the means of fold changes that were obtained from independent biological replicates ± standard error of the mean (SEM). pLKO.1-TRC cloning vector (Addgene, catalogue no. 10878) and pLKO.1-TRC control plasmid (Addgene, catalogue no. 10879) were a gift from David Root, Broad Institute of MIT and Harvard, Cambridge, MA (Moffat et al., 2006). psPAX2-D64V plasmid was a gift from David Rawlings and Andrew Scharenberg, Seattle Children’s Research Institute and University of Washington, Seattle, WA (Addgene, catalogue no. 63586; Certo et al., 2011). pMD.RVG.CVS24-B2c plasmid was a gift from Manfred Schubert, National Institute of Neurological Disorders and Stroke, Bethesda, MD (Addgene, catalogue no. 19713; Mentis et al., 2006). The experiments were replicated at least three times, with the exception of immunofluorescence (n = 2).
Statistical analysis
Statistical significance was tested using Student’s t test. Statistical tests were performed using GraphPad Prism. p scores greater than 0.05 were considered not significant. t test results are indicated consistently in all figures as *p < 0.05, **p < 0.01, ***p < 0.001, and ns for not significant (p > 0.05).
Supplementary Material
Acknowledgments
We thank David Stroud and Michael Ryan for providing the HEK293T NDUFA11 knockout cell line and Ulrike Topf for valuable comments and discussion. This research was funded by the National Science Centre, Poland (NCN; Grants 2013/11/B/NZ3/00974, 2015/18/A/NZ1/00025, and 2012/07/E/NZ3/00503), by Ministerial funds for science within Ideas Plus program 000263 in 2014–2017, and by the “Regenerative Mechanisms for Health” (MAB/2017/2) project, carried out within the International Research Agendas program of the Foundation for Polish Science cofinanced by the European Union under the European Regional Development Fund.
Abbreviations used:
- 4E-BP1
eukaryotic translation initiation factor 4E-binding protein 1
- AMPK
adenosine monophosphate-activated protein kinase
- ATF4
activating transcription factor 4
- CCCP
carbonyl cyanide 3-chlorophenylhydrazone
- eIF2α
eukaryotic initiation factor 2α
- eIF4E
eukaryotic translation initiation factor 4E
- ER
endoplasmic reticulum
- GCN2
general control nondepressible 2
- HRI
heme-regulated inhibitor
- ISR
integrated stress response
- MIA40
mitochondrial intermembrane space import and assembly protein 40
- mLST8
mammalian lethal with SEC thirteen 8
- mPOS
mitochondrial precursor overaccumulation stress
- mROS
mitochondrial ROS
- mTOR
mechanistic target of rapamycin
- mTORC1
mTOR complex 1
- mTORC2
mTOR complex 2
- NAC
N-acetyl- l-cysteine
- NDUFA11
NADH:ubiquinone oxidoreductase subunit A11
- PERK
PKR-like endoplasmic reticulum kinase
- PKR
protein kinase R
- Raptor
regulatory-associated protein of mTOR
- ROS
reactive oxygen species
- RT-PCR
real-time PCR
- S6K1
ribosomal protein S6 kinase 1
- TOR
target of rapamycin
- TSC2
tuberous sclerosis complex 2
- UPRam
unfolded protein response activated by mistargeted proteins.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-10-0628) on May 22, 2019.
REFERENCES
- An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal IG, Winblad B, Pei JJ. (2003). Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am J Pathol , 591–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM, Nargund AM, Sun T, Haynes CM. (2012). Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet , e1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger I, Hershkovitz E, Shaag A, Edvardson S, Saada A, Elpeleg O. (2008). Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann Neurol , 405–408. [DOI] [PubMed] [Google Scholar]
- Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, Scharenberg AM. (2011). Tracking genome engineering outcome at individual DNA breakpoints. Nat Methods , 671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacinska A, Pfannschmidt S, Wiedemann N, Kozjak V, Sanjuan Szklarz LK, Schulze-Specking A, Truscott KN, Guiard B, Meisinger C, Pfanner N. (2004). Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J , 3735–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS. (2014). Mitochondria as signaling organelles. BMC Biol , 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amico D, Sorrentino V, Auwerx J. (2017). Cytosolic proteostasis networks of the mitochondrial stress response. Trends Biochem Sci , 712–725. [DOI] [PubMed] [Google Scholar]
- Grant CM. (2011). Regulation of translation by hydrogen peroxide. Antioxid Redox Signal , 191–203. [DOI] [PubMed] [Google Scholar]
- Halasi M, Wang M, Chavan TS, Gaponenko V, Hay N, Gartel AL. (2013). ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem J , 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell , 619–633. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, et al (2013). mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science , 1524–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NA, Nikkanen J, Yatsuga S, Jackson C, Wang L, Pradhan S, Kivela R, Pessia A, Velagapudi V, Suomalainen A. (2017). mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab , 419–428.e415. [DOI] [PubMed] [Google Scholar]
- Kim D-H, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell , 163–175. [DOI] [PubMed] [Google Scholar]
- Knutsen JH, Rodland GE, Boe CA, Haland TW, Sunnerhagen P, Grallert B, Boye E. (2015). Stress-induced inhibition of translation independently of eIF2alpha phosphorylation. J Cell Sci , 4420–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl I, Miranda M, Atanassov I, Kuznetsova I, Hinze Y, Mourier A, Filipovska A, Larsson NG. (2017). Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife , e30952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. (2012). mTOR signaling in growth control and disease. Cell , 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Lu B. (2010). Reduction of protein translation and activation of autophagy protect against PINK1 pathogenesis in Drosophila melanogaster. PLoS Genet , e1001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livnat-Levanon N, Kevei E, Kleifeld O, Krutauz D, Segref A, Rinaldi T, Erpapazoglou Z, Cohen M, Reis N, Hoppe T, et al (2014). Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep , 1371–1380. [DOI] [PubMed] [Google Scholar]
- Longen S, Bien M, Bihlmaier K, Kloeppel C, Kauff F, Hammermeister M, Westermann B, Herrmann JM, Riemer J. (2009). Systematic analysis of the twin cx(9)c protein family. J Mol Biol , 356–368. [DOI] [PubMed] [Google Scholar]
- Ma X, Jin M, Cai Y, Xia H, Long K, Liu J, Yu Q, Yuan J. (2011). Mitochondrial electron transport chain complex III is required for antimycin A to inhibit autophagy. Chem Biol , 1474–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J. (2009). Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol , 307–318. [DOI] [PubMed] [Google Scholar]
- Mentis GZ, Gravell M, Hamilton R, Shneider NA, O’Donovan MJ, Schubert M. (2006). Transduction of motor neurons and muscle fibers by intramuscular injection of HIV-1-based vectors pseudotyped with select rabies virus glycoproteins. J Neurosci Methods , 208–217. [DOI] [PubMed] [Google Scholar]
- Mesclon F, Lambert-Langlais S, Carraro V, Parry L, Hainault I, Jousse C, Maurin AC, Bruhat A, Fafournoux P, Averous J. (2017). Decreased ATF4 expression as a mechanism of acquired resistance to long-term amino acid limitation in cancer cells. Oncotarget , 27440–27453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell , 1283–1298. [DOI] [PubMed] [Google Scholar]
- Mohanraj K, Wasilewski M, Beninca C, Cysewski D, Poznanski J, Sakowska P, Bugajska Z, Deckers M, Dennerlein S, Fernandez-Vizarra E, et al (2019). Inhibition of proteasome rescues a pathogenic variant of respiratory chain assembly factor COA7. EMBO Mol Med , e9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro JP, Martins AF, Nunes C, Morais CM, Lucio M, Reis S, Pinheiro TJ, Geraldes CF, Oliveira PJ, Jurado AS. (2013). A biophysical approach to menadione membrane interactions: relevance for menadione-induced mitochondria dysfunction and related deleterious/therapeutic effects. Biochim Biophys Acta , 1899–1908. [DOI] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. (2001). Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol , 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. (2012). Mitochondria: in sickness and in health. Cell , 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M, Wilson M, Uller T, Mott B, Isaksson C, Healey M, Wanger T. (2008). Free radicals run in lizard families. Biol Lett , 186–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osenbroch PO, Auk-Emblem P, Halsne R, Strand J, Forstrom RJ, van der Pluijm I, Eide L. (2009). Accumulation of mitochondrial DNA damage and bioenergetic dysfunction in CSB defective cells. FEBS J , 2811–2821. [DOI] [PubMed] [Google Scholar]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. (2016). The integrated stress response. EMBO Rep , 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson RT, Desai BN, Hardwick JS, Schreiber SL. (1999). Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc Natl Acad Sci USA , 4438–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, Auwerx J. (2017). Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol , 2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St-Pierre J, Jones RG. (2017). AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep , 1–9. [DOI] [PubMed] [Google Scholar]
- Reczek CR, Chandel NS. (2015). ROS-dependent signal transduction. Curr Opin Cell Biol , 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenburg RJ. (2016). Mitochondrial complex I-linked disease. Biochim Biophys Acta , 938–945. [DOI] [PubMed] [Google Scholar]
- Samluk L, Chroscicki P, Chacinska A. (2018). Mitochondrial protein import stress and signaling. Curr Opin Physiol , 41–48. [Google Scholar]
- Saxton RA, Sabatini DM. (2017). mTOR signaling in growth, metabolism, and disease. Cell , 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segref A, Kevei E, Pokrzywa W, Schmeisser K, Mansfeld J, Livnat-Levanon N, Ensenauer R, Glickman MH, Ristow M, Hoppe T. (2014). Pathogenesis of human mitochondrial diseases is modulated by reduced activity of the ubiquitin/proteasome system. Cell Metab , 642–652. [DOI] [PubMed] [Google Scholar]
- Shenton D, Grant CM. (2003). Protein S-thiolation targets glycolysis and protein synthesis in response to oxidative stress in the yeast Saccharomyces cerevisiae. Biochem J , 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M, Hall MN. (2014). Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol , 155–162. [DOI] [PubMed] [Google Scholar]
- Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D’Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S, et al (2017). Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature , 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud DA, Surgenor EE, Formosa LE, Reljic B, Frazier AE, Dibley MG, Osellame LD, Stait T, Beilharz TH, Thorburn DR, et al (2016). Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature , 123–126. [DOI] [PubMed] [Google Scholar]
- Topf U, Suppanz I, Samluk L, Wrobel L, Boser A, Sakowska P, Knapp B, Pietrzyk MK, Chacinska A, Warscheid B. (2018). Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat Commun , 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuyama T, Tsubouchi A, Usui T, Imamura H, Uemura T. (2017). Mitochondrial dysfunction induces dendritic loss via eIF2alpha phosphorylation. J Cell Biol , 815–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen XJ. (2015). A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature , 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zuo X, Kucejova B, Chen XJ. (2008). Reduced cytosolic protein synthesis suppresses mitochondrial degeneration. Nat Cell Biol , 1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel L, Topf U, Bragoszewski P, Wiese S, Sztolsztener ME, Oeljeklaus S, Varabyova A, Lirski M, Chroscicki P, Mroczek S, et al (2015). Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature , 485–488. [DOI] [PubMed] [Google Scholar]
- Yuan B, Latek R, Hossbach M, Tuschl T, Lewitter F. (2004). siRNA selection server: an automated siRNA oligonucleotide prediction server. Nucleic Acids Res , W130–W134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Boyer L, Jin M, Kim Y, Fan W, Bardy C, Berggren T, Evans RM, Gage FH, Hunter T. (2016). Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. Elife , e13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.