

Abstract
Tissue regeneration at an injured site depends on proliferation, migration, and differentiation of resident stem or progenitor cells, but solid tissues are often sufficiently dense and constricting that nuclei are highly stressed by migration. In this study, constricted migration of myoblastic cell types and mesenchymal stem cells (MSCs) increases nuclear rupture, increases DNA damage, and modulates differentiation. Fewer myoblasts fuse into regenerating muscle in vivo after constricted migration in vitro, and myodifferentiation in vitro is likewise suppressed. Myosin II inhibition rescues rupture and DNA damage, implicating nuclear forces, while mitosis and the cell cycle are suppressed by constricted migration, consistent with a checkpoint. Although perturbed proliferation fails to explain defective differentiation, nuclear rupture mislocalizes differentiation-relevant MyoD and KU80 (a DNA repair factor), with nuclear entry of the DNA-binding factor cGAS. Human MSCs exhibit similar damage, but osteogenesis increases—which is relevant to bone and to calcified fibrotic tissues, including diseased muscle. Tissue repair can thus be modulated up or down by the curvature of pores through which stem cells squeeze.
INTRODUCTION
For many solid tissues, a stem or progenitor cell must physically travel to a site of injury for regeneration of the tissue. As a cell migrates through either a dense tissue or a diseased fibrotic matrix, cell contortions invariably distort the nucleus, but for a stem cell nucleus, the consequences of constricted migration are largely unknown. Skeletal muscle is representative: a sparse population of muscle stem cells (MuSC), or myoblasts, are squeezed within an interstitial matrix between adjacent myofibers, and the MuSC are activated by nearby injury to proliferate, migrate, and fuse into damaged myofibers (Figure 1A; Yin et al., 2013). Osteogenesis and adipogenesis are additional possible fates of myoblastic cells (Moseychuk et al., 2013; Qi et al., 2016; Jung et al., 2018), and bone and fat are also well-known fates of mesenchymal stem cells (MSCs). MSCs reside in most tissues, including muscle (Sohn et al., 2015) and marrow (Swift et al., 2013). Motility processes conceivably affect all such lineage decisions—either positively or negatively. Intravital imaging has at least established in vivo migration of mouse MuSC after injury (Webster et al., 2016), and within a dorsal skin-fold chamber that squeezes and splays myofibers, tracks between myofibers are <3–4 µm wide, so that as cells migrate, the nucleus deforms (Weigelin et al., 2012). Skeletal muscle is indeed relatively stiff with abundant collagen, as is typical of other solid tissues such as bone (Engler et al., 2004; Swift et al., 2013).
FIGURE 1:

Myoblasts can migrate through small pores but are then less able to regenerate muscle. (A) (i) Myoblasts migrate through extracellular matrix (ECM) barriers to sites of injury to contribute to regeneration. Fibrotic ECM with small pore sizes may support calcification by resident mesenchymal stem cells. (ii) MSCs that contribute to osteogenesis navigate small pores through fibrous ECM and rigid bone. (B) Top: myoblasts and their nuclei (blue) squeeze through small constricting pores in the ECM (green). Bottom: migration through larger, more permissive matrix pores. (C) Live-cell imaging of tdTomato-expressing C2C12s that have migrated through either 3- or 8-μm pores over the course of 4–20 h. Images show migrated cells on the bottoms of nontransparent transwell pore filters. Inset scalebar = 10 μm. (D) (Left) Tibialis anterior (TA) muscles of NSG mice are injected with cardiotoxin to induce muscle damage and subsequent regeneration. After 2 d, tdTomato-expressing C2C12 myoblasts are migrated through 3- or 8-μm transwell pores for 24 h, collected postmigration, and then injected along the TA muscle (3 μm pore–migrated cells in one limb, and 8 μm pore–migrated cells in the contralateral limb). Seven days after cellular injection, muscles are dissected and fixed for histological analysis. Right images: Low- and high-magnification images of thick muscle sections showing tdTomato-expressing C2C12 cells in regenerating myofibers (white arrow) and in interstitial spaces (orange arrow). tdTomato signal is higher in muscle injected with 8 μm than 3 μm pore–migrated cells. Top scalebar = 100 μm; bottom scalebar = 20 μm. (E) The two TA muscles injected with 3 μm pore–migrated myoblasts (red points) exhibit overall fewer regenerating myofibers, as marked by centrally nucleated fibers (CNF), than the two TA muscles injected with 8 μm pore–migrated myoblasts (blue points). Each set of points, connected by a line, represents a single mouse. N = 709–1051 fibers per muscle. (F) TA muscles injected with 3 μm pore–migrated myoblasts exhibit fewer regenerating myofibers that have incorporated tdTomato-positive cells, suggesting that injected myoblasts contribute less to regeneration if they have undergone constricted migration. * indicates significant (p < 0.05) difference from 8-μm migration by a chi-squared test.
After injury and in many diseases, fibrosis develops and tissue stiffens with more collagen cross-linking and decreased porosity – as illustrated in muscular dystrophy (Stedman et al., 1991; Lieber and Ward, 2013; Smith and Barton, 2014a, 2018; Smith et al., 2016). Such factors tend to restrict and constrict migration. MuSCs can in principle crawl and squeeze through extracellular matrix (ECM) (Lund et al., 2014), but fibrosis somehow impairs regeneration: dystrophy-derived MuSCs indeed differentiate robustly in healthy tissue (Smith and Barton, 2018) that normally displays an unrestricted ability to regenerate (Boldrin et al., 2015). On the other hand, in various fibrotic diseases ectopic calcification can occur, and in muscular dystrophy MSCs have a role in this osteogenic process (Sohn et al., 2015). Excess DNA damage is also evident in muscle progenitors from fibrotic and dystrophic patients and animal models (Schmidt et al., 2011), and this excess DNA damage could underlie development of the myoblastic cancer rhabdomyosarcoma (Fanzani et al., 2013). Our two-part hypothesis for a progressive decline in regeneration of fibrotic tissue is that 1) constricted migration damages the nuclei and DNA of key reparative stem cells to thereby impair regeneration, and 2) at the same time, other stem cell types (particularly MSCs) are induced toward misrepair phenotypes.
Migration of multiple cancer lines through 3-µm pores has been seen to cause DNA damage, evident as nucleoplasmic foci of Phospho-Histone γH2AX (Irianto et al., 2017), and also to repress cell cycle progression (Pfeifer et al., 2018). These effects result in part from nuclear rupture during constricted migration, as observed in vivo using GFP-NLS constructs (Denais et al., 2016; Raab et al., 2016). Here, we first show that myoblast migration through small pores—but not large pores—impairs myodifferentiation in vivo and in vitro, and then we describe nuclear rupture, DNA damage, and mislocalization of factors implicated in myogenesis. We finish with studies of MSCs that are relevant not only to muscular dystrophy (Sohn et al., 2015), but also to ongoing osteodifferentiation in bone tissue. In contrast to myogenesis of MuSCs, we find that osteogenesis of human MSCs increases significantly after migration through high-curvature pores, suggesting some generality to a modulating effect of constricted migration on differentiation.
RESULTS
Constricted migration impairs myoblast differentiation in vivo
Absent any serum gradient, mouse myoblastic C2C12 cells seeded at high density on top of a transwell filter migrate over several hours to the bottom (a low-density environment), where they remain adherent and spread. As with other cell types, myoblast migration rate is impeded more by small pores (diameter 3 μm) than by large pores (8 μm; Figure 1, B and C, and Supplemental Figure S1, A and B; Supplemental Movies). Nucleus area increases equally following migration through either size of pore (Supplemental Figure S1, B and C), suggesting similarly adequate adhesion and viable spreading.
C2C12s expressing tdTomato were collected after transwell migration in order to test in vivo the effects of pore migration on regeneration. Muscle regeneration after cardiotoxin injection proceeds along a well-orchestrated time course (Yan et al., 2003), and cell injections were done 3 d after induction of injury in the immunocompromised NSG mouse (Figure 1D). Muscles were harvested 7 d later, at which point fiber regeneration was largely complete and centralized nuclei marked recently regenerated fibers (Ferrari et al., 1998). Although longer studies would allow further maturing of the newly regenerated myofibers (Meng et al., 2015), 10 d following injury and 7 d after transplantation were chosen because myoblast fusion is largely completed within that time frame (Garry et al., 2016), which is consistent with the suggested time frame for transplantation experiments (Motohashi et al., 2014), and because this time frame also corresponds to in vitro differentiation times.
Sectioning and imaging of leg muscle show robust regeneration from resident MuSCs: centrally nucleated fibers are evident at 10 d following injury, and many tdTomato-positive cells locate to an interstitial niche (Figure 1E). Limbs that received C2C12s that had undergone constricted migration (3-μm pores) showed very few tdTomato+ myofibers and therefore minimal fusion into regenerating myofibers. In contrast, contralateral limbs that received C2C12s that had undergone 8-μm pore migration showed many tdTomato+ myofibers and thus abundant fusion (Figure 1F). Many more mouse studies could of course be done, including tests of muscle contraction and physiology over the long term, but reductionist studies in vitro were instead performed on similar approximately week-long timescales in order to assess any differentiation defect and address some of the molecular mechanisms that underlie the cell’s response to constricted migration.
Differentiation defect is independent of cell number and is reversible
Myoblasts were again migrated through small and large pores for 24 h and then counted to replate similar numbers from the tops or bottoms in differentiation media (Figure 2A). Cells from the tops of transwells of either pore size and cells from the bottoms of 8-μm transwells form multinucleated myotubes robustly by day 2–3, but constricted migration delays such differentiation until day 5–6 (Figure 2Bi). 53BP1 is one nuclear marker of DNA damage, and the latter delay in differentiation corresponded well to a 2-d period of elevated anti-53BP1 foci (Supplemental Figure S2A). Cell counts after constricted migration remained low throughout differentiation (Figure 2Bii), and fusion of C2C12s is sensitive to cell number (Supplemental Figure S2, B and C), as others have shown (Messina et al., 2005). To ensure constant cell numbers across conditions, we repeated the experiments with fewer cells from the bottoms of 8-μm transwells; importantly, constricted migration still caused a differentiation defect (Figure 2C). Such experiments done at the same time certainly allow comparisons between conditions, although the full extent of myodifferentiation can differ between separate experiments (e.g., Figure 2, B and C). Plotting the differentiation index at day 3 versus cell density at day 0 (or day 1) for various 2D cultures studied in parallel with 8- and 3-μm pore-migrated cells shows that 8-μm pores allow differentiation that is equal to or greater than that in 2D cultures (Figure 2Di). In contrast, differentiation after 3-μm pore migration consistently falls below the trend lines relating 2D differentiation to cell density, but the effect decays with time (Figure 2Dii) and thus anticipates a reversible defect in differentiation.
FIGURE 2:

Constricted migration of myoblasts delays differentiation. (A) (Left) C2C12 cells are allowed to migrate through 3- or 8-μm transwell pores over the course of 24 h in growth media. Migrated cells (on the bottom [Bot.] of the transwell pore filter) and nonmigrated cells (on the top [Top]) are then collected separately and plated at high density in growth media. After 3 h of adhesion, cells are switched to differentiation media and cultured for 0–6 d. (Right) Representative images of C2C12 myoblasts 0–6 d after migration through 3- or 8-μm pores. Differentiation is indicated by immunofluorescent staining for skeletal myosin heavy chain (Skel.MyoII). Scale bar = 20 μm. (B) (i) Compared with either nonmigrated or 8 μm pore–migrated cells, cells undergoing 3-μm pore migration show significantly reduced differentiation over days 2–3, resulting in an ∼3-d delay in differentiation. N = 3–15 samples. (ii) However, 3 μm pore–migrated cells also show significantly reduced cell density throughout the differentiation time-course. (C) (i) Even when 8 μm pore–migrated cells are plated at low initial density to match the reduced cell density of 3 μm pore–migrated cells, the day-3 differentiation defect persists. N = 3 samples. (ii) When 8 μm pore–migrated cells are plated at low initial density, the 3- and 8-μm populations maintain similar densities throughout the differentiation time course. (D) (i) Differentiation index at day 3 is a linear function of cell density at either day 0 or day 1 (data from Supplemental Figure S2, B and C). The differentiation index for 3 μm pore–migrated cells falls below the line, indicating less differentiation than expected based on cell density. xi denotes the density expected for differentiation, and di denotes the excess density. (ii) The excess density for the expected population-level differentiation decays over days as cells recover from migrating through 3-μm pores. (E) (Top) After 24 h of migration, cells are collected from the bottom of the transwell membrane, plated in growth media for 4–5 d to allow proliferation, and then reseeded on top of a new transwell membrane. This process is repeated twice. Following the third migration, cells are allowed to proliferate to confluence and then differentiated for 4 d. (Bottom) Representative images of cells either cultured in 2D or subjected to three times–repeated migration through 3- or 8-μm pores. Myoblasts exhibit very little differentiation, as indicated by Skel.MyoII, after repeated migration through 3-μm pores. Scalebar = 20 μm. (F) Quantification of differentiation index normalized to 2D: cells undergoing constricted migration have a differentiation defect that persists following 4–5 d of recovery growth and 4 d of differentiation. N = 6 samples. * indicates significant (p < 0.05) difference from 8-μm migration. (G) Cells were grown for 8 rather than 4–5 d before 4-d differentiation. This extended recovery time between serial migration assays eliminates the differentiation defect in 3-μm pore-migrated cells. N = 3 samples.
A single round of constricted migration clearly has important consequences, but a cell and its progeny in vivo might squeeze through multiple barriers. Multiple rounds of constricted migration in cancer cells can lead to genomic variation and altered phenotype (Irianto et al., 2017). C2C12s were migrated up to three times (TW1 to TW3) and grown to high density over 4–5 d before each round and before induction of differentiation, with six replicates of each condition for a total of 54 myotube samples including 2D passage control cultures (Figure 2D). The period of postmigration growth was selected because it showed some rescue of differentiation even after two rounds of migration through large and small pores. However, unless the 4–5 d of growth was extended to 8 d, a third round of constricted migration greatly suppressed differentiation (Figure 2, E and F). Repetitive constricted migration can thus reversibly impair differentiation.
Myoblast nuclei are damaged by constricted migration
Squeezing of nuclei occurs in cell migration through large pores as well as small pores (Figure 3A). Nonetheless, a smaller fraction of cells migrate through small pores, and a larger percentage of those cells exhibit nuclear blebs, characterized by high lamin-A/C and low lamin-B (Figure 3, B–D). Such blebs are indicative of nuclear rupture in other cell types (Denais et al., 2016; Raab et al., 2016; Irianto et al., 2017) and are rare among cells on the top and among cells that migrate through large pores (<5%).
FIGURE 3:

Constricted migration of myoblasts causes nuclear damage. (A) Confocal 3D sections of C2C12 myoblast nuclei exiting large 8 μm– and constrictive 3 μm–diameter pores. Yellow lines indicate positions of orthogonal sections, and dashed pink lines indicate pore diameters and edges of the membranes. Scale bar = 10 μm. (B) Number of migrated C2C12 myoblasts on the bottom of a transwell membrane, normalized to number of nonmigrated cells on top of the membrane. Significantly fewer cells migrate through 3-μm than 8-μm pores. N = 3 samples. (C) Representative images of nuclear blebs following constricted migration through 3-μm pores. Blebs (white arrows) are lamina protrusions deficient in lamin-B and enriched in lamin-A/C. Scale bar = 10 μm. (D) The frequency of nuclear blebbing is low before migration and after migration through large 8-µm pores, but it increases significantly after migration through 3-µm pores. N = 219–598 cells. (E) Nonmigrated and migrated C2C12 myoblast nuclei are shown on the top and bottom, respectively, of transwell membranes. Nuclei exhibit excess DNA damage, as indicated by foci of γH2AX, after migration. Scale bar (main) = 50 μm. Scale bar (expanded) = 10 μm. (F) Number of γH2AX foci per cell, normalized to number of foci per nonmigrated (Top) cell. Migration causes excess DNA damage, with greater damage accrued during migration through constricting vs. large pores. N = 257–358 cells. (G) Superresolution imaging of a C2C12 myoblast after constricted migration shows γH2AX foci are not enriched at sites of nuclear blebs. Multiple blebs are sometimes visible and tend to occur at the high curvature end(s) of a nucleus. Scale bar = 10 μm. (H) C2C12 myoblasts show nuclear bleb formation and excess DNA damage foci after three rounds of constricted migration. DNA damage is more severe on the bottom but is also evident on the top after successive migrations. Nuclear blebs are observed on the bottom only. Scale bar = 10 μm. (I) Numbers of γH2AX foci per C2C12 cell before (Top) and after (Bot.) each of three rounds of migration (TW1 = round 1, etc.). A single round of migration—especially through constricting 3-μm pores—leads to excess DNA damage; additional rounds of migration do not further increase the damage level on transwell Bot. However, the level of DNA damage on transwell Top increases with serial migration, indicating that damage is not fully repaired during the recovery period between migrations. N = 126–203 cells. * indicates significant (p < 0.05) difference from nonmigrated or † from 8 μm.
DNA damage measured by foci of Phospho-Histone H2AX (γH2AX; Figure 3E) suggests a nonzero basal level in cells on the top that increases by ∼50% after migration through large pores and by ∼100% for small pores (Figure 3F). An electrophoretic single-cell “comet” assay provides an orthogonal measure of DNA damage and confirms the significant excess in DNA damage after constricted migration (Supplemental Figure S3, A and B). Such DNA damage has also been observed after constricted migration of cancer cell lines (Irianto et al., 2017; Pfeifer et al., 2018).
In light of the repression of myodifferentiation after repeated rounds of constricted migration (Figure 2G), nuclear rupture and DNA damage were also measured in the same set of experiments. C2C12 cells showed a constant and high incidence of nuclear rupture and DNA damage after migration through 3-µm pores, but DNA damage accumulated progressively in cells as they were put back on top, even though nuclear blebs in those cells remained low (Figure 3, G and H; Supplemental Figure S3C). Larger pores produced modestly elevated DNA damage (Figure 3F) that again did not fully recover after 4–5 d of growth between migration experiments. Sustained DNA damage after constricted migration might thus contribute to the differentiation defect (Figures 1 and 2).
Myosin II inhibition rescues constriction-induced damage
Nonmuscle myosin II inhibition of cancer cell lines limits the rate of constricted migration (Harada et al., 2014), and similar application of blebbistatin to C2C12 myoblasts also slowed migration through large pores and especially through small 3-μm pores (Figure 4A; Supplemental Figure S4A). Nuclear blebbing was almost eliminated, as was the excess DNA damage for the small pores, with no effect for the large pores (Figure 4, B and C). Because very few cells migrate through constrictive pores with blebbistatin, differentiation experiments were not feasible. The results nonetheless point to cytoskeletal forces as being a critical component of the nuclear rupture mechanism by which constricted migration causes DNA damage, consistent with past studies in 2D cultures implicating actomyosin in nuclear rupture (Thomas et al., 2015; Hatch and Hetzer, 2016; Xia et al., 2018, 2019).
FIGURE 4:
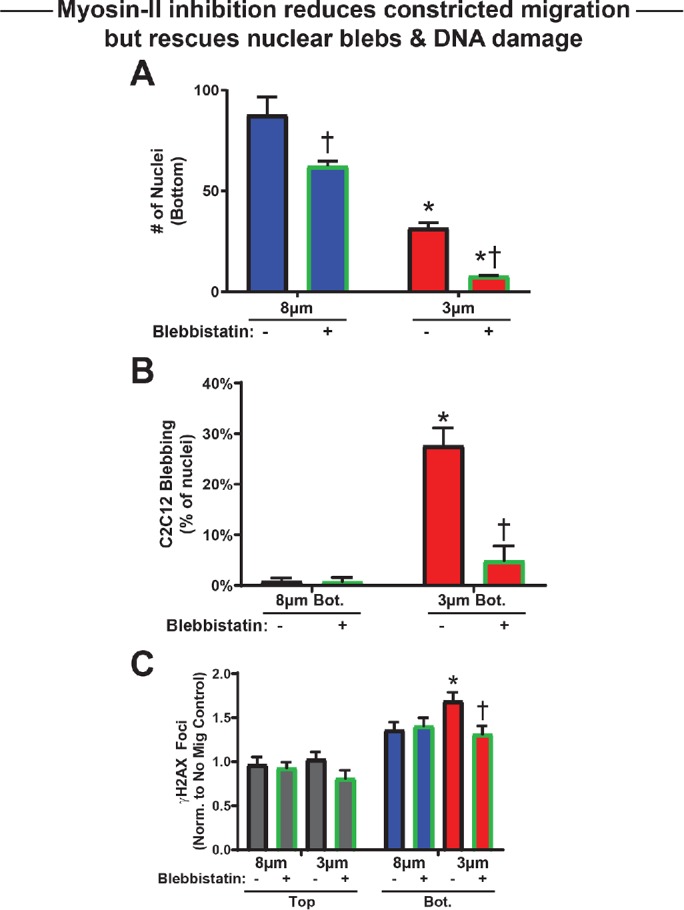
Blebbistatin reduces migration but rescues nuclear damage. (A) Number of migrated C2C12 myoblasts—treated or not with blebbistatin—on the bottom of a 3- or 8-μm pore transwell membrane per 0.12-mm2 field. Blebbistatin reduces migration rate regardless of pore size. N = 5–10 samples. (B) Nuclear bleb formation after 3-μm pore migration is significantly reduced by blebbistatin treatment. N = 80–351 cells. (C) Number of γH2AX foci per cell, normalized to top of membranes without blebbistatin. Blebbistatin treatment has no effect on DNA damage level without migration. DNA damage increases with constricted migration through 3-μm pores, but blebbistatin treatment significantly reduces this excess damage. N = 80–216 cells. * indicates significant (p < 0.05) difference from 8-μm migration or † from migration with blebbistatin.
Cell cycle entry of myoblasts is suppressed by constricted migration
In imaging the transwells, it was noticed that mitotic cells were significantly suppressed in number by constricted migration as compared with migration through large pores (Figure 5A). DNA damage has been demonstrated to induce exit from cell cycle as well as differentiation of myoblasts (Larsen et al., 2010), consistent with DNA damage checkpoints in the cell cycle (Hartwell and Weinert, 1989) but also with a dependence of differentiation on cell density (Messina et al., 2005). Cell cycle analysis was performed by fluorescence imaging of both DNA content and integration of the nucleotide analogue EdU into newly synthesized DNA; EdU was added to the top and bottom of the transwell membrane for the final hour of each 24-h migration experiment (Figure 5B; Supplemental Figure S5, A and B). Constricted migration caused an ∼50% increase in G1 cells, relative to both cells migrating through 8-μm pores and nonmigrating cells, with corresponding decreases in both S and G2 phases (Figure 5C). The delay in cell cycle entry is consistent with the mitotic suppression, and might contribute to the defective differentiation. On the other hand, differentiation conditions (i.e., low serum) also suppress the cell cycle, with very few cells actively synthesizing DNA by day 2 (Supplemental Figure S5, C and D). Mitotic cells after migration and replating under differentiation conditions show a small spike only at day 1 across all conditions (Supplemental Figure S5E). Suppression at day 0 potentially results from replating after detachment from transwells, because similarly dense cells exposed to DMSO but without replating show a higher level of mitosis that decreases within 2 d (Supplemental Figure S5F). Regardless, the DNA-damaging agent etoposide suppresses mitotic cell counts, including the spike upon induction of differentiation.
FIGURE 5:

Constricted migration blocks cell cycle in myoblasts. (A) Percentage mitotic C2C12 myoblasts on the top and bottom of 3- and 8-μm transwell membranes. Migration through 3-μm pores suppresses the mitotic population (N = 337–969 cells). (B, C) After 24-h transwell migration, the thymidine analog EdU was added to the top and bottom of each pore membrane for 1 h before fixation and staining. The EdU and DNA intensities of individual cells—measured by immunofluorescence microscopy—were used to classify the cells as G1, S, G2, or mitotic (M). Cells show less EdU incorporation after migration through 3-μm than 8-μm pores, indicating fewer cells in the S-phase of the cell cycle. Scalebar = 50 μm. Cell cycle phase distributions for nonmigrated (Top) cells, cells that have migrated through 8-μm pores, and cells that have migrated through 3-μm pores. Migration through 3-µm pores leads to enrichment of G1 and corresponding decreases in S and G2 phases, suggesting a cell cycle delay, whereas 8 μm–pore migration does not affect cell cycle. N = 199–201 cells. (D) (Left) Proliferation of C2C12 cells was blocked with CDK4/6 inhibitor (1 µM; CDK4/6i) before and during transwell migration; CDK4/6i was washed out when cells were placed in differentiation media. (E) (i) Even with cell cycle inhibition, differentiation of cells after 5 d is significantly reduced among 3 μm pore–migrated cells. Differentiation is also reduced—to negligible levels—among nonmigrated cells treated for 24 h with the DNA-damaging agent etoposide. N = 3 samples. (ii) Cell density is consistent throughout the differentiation time course, indicating absence of proliferation with CDK4/6i. Cell density is also consistent between conditions, with the exception of the etoposide-treated population, which has low density. The persistence of the migration-induced differentiation defect, even with cell cycle inhibition, indicates that the delay is not purely due to a cell cycle defect. (iii) Differentiation index at day 5 correlates negatively with pore curvature (i.e., inverse of pore diameter); cells show less differentiation after migrating through pores that impose higher Gaussian curvature on the cell membrane. * indicates significant (p < 0.05) difference from no migration or † from 8-μm migration.
To assess the hypothesis that cell cycle repression causes the differentiation defect, the cell cycle inhibitor (CDK4/6i) was applied to cells 3 d before and throughout migration and differentiation of C2C12s (Figure 5D). Cancer cell lines treated with this same inhibitor during constricted migration show the same cell cycle distribution on transwell bottom as untreated cells, and so such migration does not select for a particular phase of the cell cycle (Pfeifer et al., 2018). Differentiation levels at day 5 were modest but significant for nonmigrating cells blocked from proliferating by CDK4/6i with similar results for cells that migrated through 8μm pores (Figure 5E, i and ii), consistent with effects of low cell densities (Figure 2D). Importantly, however, differentiation was greatly suppressed after migration through the high-curvature 3-μm pores with the same CDK4/6i treatment, and the extent of myodifferentiation proved to be linear in pore curvature (Figure 5Eiii), consistent with a physical effect. Cell density was also maintained across migration conditions and the differentiation time course (Figure 5Eii), which demonstrates the efficacy of CDK4/6i. Moreover, treating with etoposide for 24 h and replating with CDK4/6i abolished differentiation, although it also led to lower cell density. Interestingly, equal numbers of suspended cells were replated, and so some form of impaired adhesion could result from the combination of cell cycle inhibition (from CDK4/6i) and DNA damage (from etoposide, an anticancer agent), which raises the possibility of apoptosis.
Apoptosis after constricted migration has been observed for cancer cells migrating from low serum on transwell top to high serum on transwell bottom (Harada et al., 2014). If myoblasts apoptose after constricted migration (even with no serum gradient), then 1) the decreased cell density could limit myodifferentiation distinctly from any cell cycle effects. In addition, 2) early phases of apoptosis in which phosphatidylserine is exposed on the outer leaflet of the plasma membrane can be reversible but can limit cell adhesion, which is also important to cell motility and fusion in myodifferentiation (Iguchi et al., 2001). C2C12s that were migrated through transwells were live-labeled for both early (Apopxin Green) and late (7-AAD) apoptosis markers (Supplemental Figure S5G). Apoptosis is rarely imaged among nonmigrating cells, but early apoptosis is evident in half of the cells treated with staurosporine; migration through large pores leads to ∼2–3% apoptosis, and this percentage roughly doubles after constricted migration (Supplemental Figure S5H).
Flow cytometry analyses of cells detached from transwells and then labeled with apoptotic markers confirmed the trends (Supplemental Figure S5, I–K). However, a high baseline for nonmigrated cells of ∼10% early apoptotic indicates the sensitivity of these cells to a loss of cell adhesion. Such processes as cell death could have a role in the differentiation defect after constricted migration, but the observation that the defect persists even with the same cell density under cell cycle inhibition (Figure 5E) also suggests that other factors contribute.
DNA damage and differentiation: a reciprocal relationship in some ways
Some forms of DNA damage impede myoblast differentiation (Puri et al., 2002; Sinha et al., 2014), but DNA damage also reportedly increases with differentiation of myoblasts (Farzaneh et al., 1982; Coulton et al., 1992). Differentiation of control C2C12s shows robust myotube formation by ∼4 d (Supplemental Figure S6, A and B) with similar kinetics for increased 53BP1, a DNA damage marker with some potential complexity relative to other damage markers (Supplemental Figure S6C). Cell density remains constant beyond day 2 (Supplemental Figure S6D). Nuclei with high levels of 53BP1, although diffuse in immunostaining, also show an accumulation of γH2AX foci (Supplemental Figure S6E).
To assess the effects of DNA damage on differentiation in the absence of nuclear rupture, DNA damage was induced chemically with etoposide and also with radiation. Use of etoposide for 2 h was sufficient to cause an increase in 53BP1 foci as a marker of DNA damage that persisted slightly above control levels for the duration of the 3 d under differentiation conditions (Supplemental Figure S6, F and G). Extending the use of etoposide for the first 24 h of differentiation conditions produced significantly more DNA damage throughout the differentiation time course. The short-term etoposide produced a marginal decrease in the differentiation of myoblasts, while the long-term etoposide eliminated differentiation (Supplemental Figure S6H). However, these levels of DNA damage also limited cell cycle progression, and while the initial cell density was equivalent, 24 h of etoposide blocked proliferation that normally occurs during the first 24–48 h under differentiation conditions (Supplemental Figure S6I). The observation that high initial cell density among 24-h etoposide-treated myoblasts is insufficient for differentiation (Supplemental Figure S2, G and H) suggests that migration-induced DNA damage could cause the delay in differentiation (Figures 1 and 2). On the other hand, acute γ-irradiation of C2C12s also increases 53BP1 foci, but the damage is repaired in 1 d, and neither differentiation nor cell numbers are significantly affected (Supplemental Figure S6, J–L). Duration of DNA damage and the cell cycle delay could ultimately combine with other factors in being key to the repressed differentiation.
Human myoblasts and myogenic factor mislocalization after constricted migration
DNA damage-mediated phosphorylation of the key myogenic transcription factor MyoD has been found to define a DNA damage checkpoint in differentiation of myoblasts (Puri et al., 2002). Loss of MyoD from the ruptured nucleus is also conceivable, given the rupture-induced leakage of other DNA binding factors from the nucleus during constricted migration (Irianto et al., 2017; Pfeifer et al., 2018). For such assessments and for generality of the above results, we switched from mouse to human myoblastic cells, which have the advantage that antibodies such as anti-γH2AX and anti-MyoD are typically more reliable in probing human epitopes. In addition, the immortalized mouse C2C12 cells show unique responses to DNA damage (Martinez et al., 2017).
Primary human muscle stem cells were first examined for nuclear damage. Fewer human myoblasts were used than with the C2C12s, and the transwell migration assays were carried out for twice as long, 48 rather than 24 h (Figure 6A). These myogenic cells again showed nuclear blebs in ∼1/3 of cells after constricted migration, with few blebs in nonmigrated cells on the top (Figure 6B). Curiously, nonmigrated primary human myoblasts showed a near-zero basal level of DNA damage, but γH2AX foci were observed in large excess after constricted migration (Figure 6C).
FIGURE 6:

Constricted migration of human cells induces nuclear damage and MyoD loss. (A) Representative images of nonmigrated and migrated human MuSCs (hMuSCs) on the top and bottom, respectively, of a 3 μm–pore transwell membrane. White arrow indicates a nuclear bleb formed after 3 μm–pore migration. Scale bar = 20 μm; inset scale bar = 5 μm. (B) Migration of hMuSCs through 3-μm pores causes a significant increase in nuclear blebbing. N = 22–37 cells per condition from three experiments. (C) Constricted migration of hMuSCs also leads to a significant increase in DNA damage, as measured by γH2AX foci. (D) (i) Nuclear bleb frequency is elevated among Rh30 cells that migrate through 3-μm pores vs. cells that migrate through 8-μm pores or cells that remain unmigrated on top of the transwell membrane. (ii) Images show a representative Rh30 nucleus with a migration-induced bleb. N = 88–142 cells. Scale bar = 10 μm. (E) Migration of Rh30 cells through 3-μm pores causes excess DNA damage. N = 88–142 cells. (F) Representative images of Rh30 cells after migration through 3- or 8-μm pores. Line scan through two nuclei (a, b) after migration through 3-μm pores. Scale bar = 10 μm. (G) Normalized intensity based on the line scan in F. Cell (a) shows high coincident DNA and MyoD signal and an absence of cGAS. Cell (b) shows cGAS signal overlapping with DNA and low nuclear MyoD intensity. Cell (b) appears to have ruptured, causing nuclear entry of cGAS and mislocalization of MyoD from the nucleus into the cytoplasm. (H) The frequency of MyoD-negative Rh30 nuclei—like cell b—increases significantly following 3 μm–pore migration. N = 314–370 cells. (I) (i) Addition of blebbistatin to both sides of a 3-μm transwell greatly reduces migration. (ii) Bar graph: Rh30 cells expressing DNA repair protein GFP-KU80 and DNA-binding protein mCherry-cGAS migrated through 3-µm pores; DNA repair factor KU80 mislocalizes to cytoplasm at bottom, except with blebbistatin. Inset: cells showing GFP-KU80 mislocalized to cytoplasm in Ctl bottom also exhibit mCherry-cGAS accumulation in the nuclear bleb. (J) After migration through 3-μm transwell pores, cells show mislocalization of GFP-tagged and immunostained Ku80, accompanied by focal mCherry-cGas indicative of nuclear rupture. Blebbistatin prevents such nucleocytoplasmic exchange (17–93 cells on the bottom; >300 cells on the top). * indicates significant (p < 0.05) difference from no migration.
The human rhabdomyosarcoma cancer cell line Rh30 expresses early (MYOD) and late (MYOG) myoblastic transcription factors but does not undergo fusion or terminal differentiation (Tapscott et al., 1993) because of a dominating fusion protein (Hinson et al., 2013). As a stable cell line stuck in a myoblastic state, results might prove more reproducible with this line than with primary human myoblasts from the same or different patients, and results could also prove cancer-relevant. After 48 h of Rh30 migration through transwells, nuclear bleb formation in ∼15% of migrated cells was accompanied by an ∼60% excess of γH2AX foci relative to both nonmigrated cells on the top and cells from the bottoms of large pore transwells (Figure 6, D and E). Results with human myoblastic cells thus support a potentially general conclusion that muscle-related cell types pay a price for constricted migration.
MyoD mislocalization from the nucleus was studied by immunostaining together with cyclic GMP-AMP synthase (cGAS), which is a protein that binds to DNA as part of inflammatory activation. After constricted migration, anti-cGAS marks the site of nuclear rupture in other cancer lines (Harding et al., 2017), and the same finding applies to Rh30 cells (Figure 6, F and G). Such cells also often lose nuclear localization of MyoD: ∼30% of cells undergoing constricted migration lost nucleus-specific MyoD, while low nuclear MyoD was associated with <5% of nonmigrating cells and cells migrating through large pores (Figure 6H). Loss of MyoD was also observed among the mouse C2C12 cells (Supplemental Figure S7), but because specificity of antibody against mouse protein could not be verified, we examined mislocalization of other myogenic-relevant factors after nuclear rupture in human cells.
KU80 is a major DNA repair factor, and mice that are even heterozygous for KU80 deletion exhibit increased DNA damage and show major defects in differentiation of myoblasts (Didier et al., 2012). After constricted migration, ∼30% of Rh30 cells show cytoplasmic mislocalization of KU80, based on both immunofluorescence and transient transfections (Figure 6, I and J). Moreover, ∼100% of cells with substantial cytoplasmic KU80 also showed nuclear cGAS (Figure 6I, inset). Blebbistatin not only inhibited migration rate, consistent with C2C12 results (Figure 4A), but also suppressed KU80 and cGAS mislocalization to levels equivalent to those for nonmigrating cells. These data thus show that nuclear rupture by cytoskeletal forces during constricted migration can lead to partial mislocalization of nuclear or cytoplasmic factors that are known to be important to the ability of myoblasts to terminally differentiate into muscle.
Osteogenesis of low-density human MSCs increases after constricted migration
Mesenchymal stem or stromal cells (MSC) are among the most widely studied adherent human stem or progenitor cells and exhibit multipotency, including osteogenic differentiation (Pittenger et al., 1999) as well as mechanosensitive differentiation (Engler et al., 2006). Cortical bone is also rigid and exhibits a range of pore sizes (Milovanovic et al., 2017), which adds physiological relevance to studies of MSCs in constricted migration. Moreover, in the fibrotic muscle that results from muscular dystrophy, nonmyogenic MSCs have a role in an osteogenic process of ectopic calcification (Sohn et al., 2015).
In studies of MSC migration through rigid transwell pores, these cells migrate more slowly than the myoblastic cell types, and so MSCs were allowed to migrate for 48 h to the transwell bottoms. Despite the longer duration, nuclear blebs after constricted migration were evident in a fraction of MSCs (∼20%) similar to that for the myogenic cell types (e.g., ∼10–20% in Rh30 cells; Figure 6D), with blebs remaining rare in nonmigrating cells (Figure 7, A and B). DNA damage foci labeled by γH2AX also increased in MSCs after constricted migration (Figure 7C).
FIGURE 7:

Constricted migration of human mesenchymal stem cells (hMSC) enhances osteogenic differentiation. (A) Representative images of nonmigrated and migrated hMSCs on the top and bottom, respectively, of a 3 µm–pore transwell membrane. White arrow indicates a nuclear bleb formed after 3 µm–pore migration. Scale bar = 10 µm. (B) Migration of hMSCs through 3-µm pores causes a significant increase in nuclear blebbing. (C) Constricted migration of hMSCs also leads to a significant increase in DNA damage, as measured by γH2AX foci. N = 46–61 cells. * indicates significant (p < 0.05) difference from the no-migration condition. (D) Bone marrow–derived hMSCs are either cultured in monolayer (No mig.) or subjected to 3- and 8-µm transwell migration (Mig.); the nonmigrated and migrated cells are then subjected to either growth media or osteogenic media to induce osteogenic differentiation. (E) Level of osteogenic differentiation was measured by alkaline phosphatase (ALP) staining with Fast Blue RR salt (Sigma). Increased ALP activity is a well-known marker for osteogenic differentiation, as shown by the blue staining. Scale bar = 1 mm. (F) Increase in ALP activity is highest for cells that migrated through 3-µm constrictions and were then cultured at low seeding density (low = 10,000 cells/cm2; high = 20,000 cells/cm2). This is a combination of suppressed spontaneous osteogenic differentiation and enhanced induced differentiation. High seeding density seems to suppress the migration-induced enhancement of osteogenic differentiation. N = 9057–16,814 cells. * indicates significant difference between the slope coefficients of low and high seeding density (p < 0.05).
To test for subsequent effects on osteogenesis and again control for cell density, migrating and nonmigrating cells were plated at either high or low density and then placed in either growth media (control) or osteogenic media to induce differentiation for 7 d (Figure 7D). Alkaline phosphatase (ALP) staining (Swift et al., 2013) was used to quantify the fold increase in osteogenic induction relative to growth conditions (Figure 7E). At low MSC densities, importantly, osteogenesis increases linearly with the degree of constriction as measured by pore curvature (Figure 7F, with 2D culture having 0 curvature). Differentiation was robust at high cell densities regardless of migration. Thus, in contrast to the suppression of myoblast differentiation that increases with cell density, 1) constricted migration of MSCs increases differentiation, and 2) the effect is evident at low cell density.
DISCUSSION
Many types of stem cells are migratory, including myoblasts (Siegel et al., 2009; Webster et al., 2016) and MSCs (Raab et al., 2012), and we find that both cell types will not only squeeze their nucleus through a pore but will even cause self-inflicted damage to the nucleus. Similar increases in nuclear lamina blebs and DNA damage are observed for the mouse cells (with smaller genomes) and for the human normal, cancerous (larger genomes), and primary cells studied here. Actomyosin seems to pull the nucleus through deformable and adhesive collagenous matrix (Petrie and Yamada, 2012), and with the rigid pores here, blebbistatin shows myosin II has the same effect, but myosin II inhibition greatly slows migration and also minimizes nuclear damage. The results implicate stress level or duration of stress on the nuclear lamina—likely related to nuclear curvature (Xia et al., 2018)—in nuclear rupture and its downstream consequences. The results for MSCs further suggest that osteodifferentiation is favored by the same myosin II generated stress that is already known to be important to matrix elasticity–directed differentiation (Engler et al., 2006). Interestingly, the antimyogenic effects and the proosteogenic effects of relevant cell types in constricted migration mesh reasonably well with processes in muscular dystrophy: loss of muscle mass, increased fibrosis, and ectopic calcification (Sohn et al., 2015).
DNA damage can impair myoblast differentiation (Puri et al., 2002; Sinha et al., 2014), but differentiation also associates with increased DNA damage (Farzaneh et al., 1982; Coulton et al., 1992), and so the findings here were not obvious a priori. DNA damage has been speculated to instigate differentiation of muscle stem cells as a means of preserving the stem cells’ genomic integrity (Larsen et al., 2010). Our results indicate that nuclear damage that results from constricted migration leads to an ∼2-d delay in the differentiation of myoblasts. Such a delay is consistent with the time scale for which excess DNA damage is observed in differentiating myoblasts. Repeated migration through 3-μm constrictive pores can also increase the duration of impaired differentiation and prolong the period over which myoblasts exhibit excess DNA damage. However, the myoblasts’ differentiation capacity recovers over the course of a week. If transwells selected a subpopulation of more migratory cells, then we would expect migration rate to increase in the serial transwell experiments—but this was not observed. The delay in differentiation reflects the entire population of cells. Thus, it is conceivable that an individual cell does not have delayed differentiation requiring DNA repair, but that the damaged cells are overtaken by the undamaged cells during the recovery period to restore differentiation capacity over longer periods. However, in single migration experiments, the replacement of substantial damaged cells following constricted migration with undamaged cells would require an increase in proliferation. Our data do not show any increase in proliferation following the damage signal of constricted migration, and indeed show cell cycle delay at early time points. Our data thus suggest that individual myoblasts eventually recover their ability to regenerate rather than being replaced.
In considering potential mechanisms for delayed differentiation of myoblasts, we considered three non–mutually exclusive hypotheses. 1) Delayed differentiation is secondary to reduced effective cell density, which can result from either well-known DNA damage checkpoints in the cell cycle (Hartwell and Weinert, 1989) or through induction of apoptosis (Wang, 2001). 2) Delayed differentiation is due to a direct DNA damage check point in differentiation (Puri et al., 2002). Alternatively, 3) delayed differentiation is caused by mislocalization of nuclear myogenic factors (Xia et al., 2018). As demonstrated for cancer cells (Pfeifer et al., 2018; Xia et al., 2019) constricted migration causes a delay in the cell cycle that limits proliferation and thus cell density following constricted migration. However, the CDK4/6 inhibitor stops proliferation under all conditions and does not affect the differentiation defect (Figures 2Ci and 5Ei both show ∼60% less differentiation). Constricted migration from low to high serum can induce apoptosis in cancer lines (Harada et al., 2014), and we observed only a small fraction of cells in early apoptosis immediately following constricted migration (Supplemental Figure S5), although overt signs of apoptosis (e.g., floating cells) were lacking in subsequent differentiation. Replating for differentiation could have sorted out some of the less viable cells, because phosphatidylserine exposure can limit cell adhesion (Iguchi et al., 2001), but cells were counted after replating (e.g., Figure 2C). Furthermore, extensive apoptosis would be expected to result in decreased cell density, especially when proliferation was blocked, but cell losses are both minimal and the same for 3- and 8-μm pores (Figure 5Eii). Ultimately, the myogenic differentiation defect here after constricted migration exceeds expectations from cell density (Figure 2D). DNA damage has been postulated to be a differentiation checkpoint (Puri et al., 2002), and while etoposide virtually eliminates differentiation (Figure 5E; Supplemental Figure S6, G–I), irradiation caused significant DNA damage but did not impact differentiation (Supplemental Figure S6, J–L).
Mislocalization of crucial DNA repair factors from the nucleus into the cytoplasm reportedly delays repair and favors accumulation of ongoing DNA damage in cancer lines after constricted migration (Irianto et al., 2017; Xia et al., 2018). Although some of the same antibodies were found not to work with the mouse C2C12 cells, we examined KU80—which contributes to myogenesis (Didier et al., 2012)—as well as the myogenic factor MyoD. Both of these nuclear factors mislocalize to cytoplasm in the ∼20–30% of cells that exhibit nuclear rupture with entry of the cytoplasmic DNA-binding factor cGAS (Figure 6, F–J). Differentiation-competent cells are likely to be reduced by this ∼20–30% of rupture with mislocalized factors, and the quantitative analysis of differentiation versus cell density (Figure 2Dii) indeed reveals at day 0 an ∼30–40% excess of cells relative to the expected differentiation. Frequent nuclear rupture has also been documented in standard 2D culture of cells derived from laminopathy patients (which often exhibit muscular dystrophy as well as bone and fat deficits), and observations included 1) mislocalization of multiple transcription factors for extended periods and 2) dysregulation of some downstream genes (five of nine analyzed; De Vos et al., 2011). Neither lamins nor other components were perturbed in the studies here of normal, wild-type cells.
Physiologically, muscle regeneration is a regulated process with a specific time-course (Yan et al., 2003). Evidence here suggests that pore-migrated myoblasts contribute less to new myofibers 7 d after myoblast implantation in injured, immunodeficient NSG mice. It is possible that constricted migration within the tissue further impacts engraftment; and the same mechanisms that compromise engraftment (e.g., apoptosis) would be expected to limit the myoblasts’ ability to contribute to muscle regeneration. Thus the shorter term, 2- to 3-d, delay in differentiation may have a long-lasting impact on muscle regeneration in vivo. Furthermore, the DNA damage that results from constricted migration has been shown to portend genomic variation (Irianto et al., 2017), and there is an intriguing link between dystrophic muscles with prominent fibrosis and the development of muscle progenitor tumorigenesis as rhabdomyosarcoma (Fanzani et al., 2013; Boscolo Sesillo et al., 2019).
Differentiation programs beyond myogenesis can also be impacted by constricted migration. MSCs can differentiate into osteoblasts and potentially migrate through rigid porous bone as well as scaffolds intended for bone regeneration (Bružauskaitė et al., 2016). Surprisingly, constricted migration of MSCs enhanced their differentiation (at low cell density) rather than delaying it. Migration through 8-μm pores caused only a slight perturbation relative to “flat” 2D cultures for both myogenesis and osteogenesis, whereas a linear dependence on curvature is about equal but opposite: 3-μm pores suppress myogenesis to ∼1/3 (Figure 5Eiii) while enhancing osteogenesis by ∼3-fold (Figure 7F). The latter rules out a generic role for apoptosis after constricted migration and instead suggests, for example, that loss of some key nuclear factors that undermine efficient myogenesis have the opposite magnitude effect in osteogenesis. One mechanorepressor that is lost from stressed nuclei of MSCs is NKX-2.5, which leads to up-regulation of α-smooth muscle actin and higher contractility that should favor osteogenesis (Dingal et al., 2015). Regardless of mechanism, it is interesting to speculate that all of this somehow relates to observations that healthy bone has a dense rigid matrix with smaller pore sizes than diseased bone (Osterhoff et al., 2016); this is in contrast to skeletal muscle, where disease is often associated with dense fibrotic tissue (Mahdy, 2019) where pore size should be small. Another distinction between osteogenesis and myogenesis is the genetic stability of the differentiated cell type. The average lifespan of a human myonucleus is estimated at ∼15 years (Gundersen, 2016), while bone forming osteoblasts have a much faster turnover of ∼3 months (Manolagas, 2000), so that compromised DNA would not impact transcription in bone as much as in muscle. The impact of constricted migration thus needs to be studied in the context of a specific differentiation program.
When the muscle environment becomes fibrotic, the ability of muscle stem cells to contribute to regeneration is compromised (Smith and Barton, 2018). The fibrotic process in muscle is common to aging and many diseases, with muscular dystrophies being the most studied. In mdx mice, the most common animal model of Duchenne muscular dystrophy, the diseased muscle is able to regenerate effectively—that is, until the muscle becomes fibrotic, at which point regeneration is steadily reduced as fibrosis progresses (Cholok et al., 2017). The fibrotic process is known to stiffen muscle (Stedman et al., 1991; Smith et al., 2016), making it more challenging for motile cells to squeeze between fibers. Fibrosis also increases matrix density, thus presenting cells with additional barriers to migration. Ectopic calcification by MSC type cells (Sohn et al., 2015) adds to such barriers and seemingly reinforces an antimyogenic microenvironment that is simultaneously proosteogenic—which fits the pathophysiology. While further studies are required to confirm that constricted migration is increasingly necessary in states of fibrosis, there is evidence that cells in such an environment do indeed have enhanced DNA damage (Schmidt et al., 2011). Constricted migration in fibrotic tissue might, along with other factors, help explain the ability of quiescent cells from fibrotic tissue to perform regenerative functions in healthy tissue, while healthy stem cells are not as effective when transplanted into diseased tissue (Boldrin et al., 2012, 2015). Further understanding of how constricted migration of stem cells can alter their contribution to regeneration may open up new therapeutic options in regenerative medicine that could apply broadly to fibrotic tissues.
MATERIALS AND METHODS
Cell culture
C2C12 mouse myoblast cell line (American Type Culture Collection [ATCC]) and Rh30 human rhabdomyosarcoma cell line (Children’s Oncology Group) were cultured in high-glucose DMEM (Life Technologies), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (MilliporeSigma). Human primary skeletal muscle stem cells (PromoCell) were cultured using skeletal muscle cell growth medium (PromoCell). Cells were incubated at 37°C and 5% CO2. Human mesenchymal stem cells (hMSCs) were isolated from bone marrow aspirates and cultured in low-glucose DMEM (Life Technologies) supplemented with 1% FBS and 1% penicillin/streptomycin. C2C12 cells for in vivo studies were transduced with tdTomato using a Lentivirus construct (Harada et al., 2014). C2C12 cells were induced into differentiation with differentiation media consisting of high-glucose DMEM (Life Technologies), supplemented with 1% FBS and 1% penicillin/streptomycin. Generally, cells were plated at high density, 5 × 104 cells per well in a 96-well plate, in growth media and switched to differentiation media after 3 h. Differentiation media were refreshed every 2 d or until fixation. Osteogenic differentiation of hMSCs was induced with osteogenic induction media (R&D Systems).
Transwell migrations
Migration assays were conducted using either 24-well or six-well inserts with 3- and 8-µm pore diameter inserts (Corning) with 2 × 106 and 1 × 105 pores per cm2, respectively. FluoroBlok (Corning) 24-well inserts were used for live imaging of live cells using wide-field microscopy. The tops of 24- and six-well inserts were seeded with 5 × 104 and 5 × 105 cells, respectively. Cell culture medium was added to the top and bottom of the insert so that no nutrient or chemical gradient existed across the membrane. The transwell inserts were incubated for 24 h for C2C12 and Rh30 cells and 48 h for primary muscle stem cells and hMSCs. Following the migration period, membranes were fixed, imaged live on the membrane, or cells were harvested for further cell culture. To harvest cells from membranes, 0.25% Trypsin EDTA solution (Life Technologies) was applied to both sides of the membranes for 5 min and then membranes were agitated manually to suspend cells without touching or scraping the membranes.
Immunostaining
Cells were fixed in 4% formaldehyde (MilliporeSigma) for 15 min. Permeabilization was conducted with 0.5% Triton-X (MilliporeSigma) for 15 min for cells or 1 h for vibratome tissue sections. Samples were blocked for 30 min using 5% BSA (MilliporeSigma) and incubated overnight in primary antibody at 4°C. Antibodies used include lamin-A/C (1:500, sc-7292, mouse, Santa Cruz), laminB (1:500, goat, sc-6217, Santa Cruz and 1:500, rabbit, ab16048, Abcam), γH2AX (1:500, mouse, 05-636-I, MilliporeSigma), 53BP1 (1:300, rabbit, NB100-304, Novus), skeletal-muscle myosin heavy chain (1:500, mouse, MF-20, Novus Biologicals), cGas (1:500, rabbit, D1D3G, Cell Signaling), TdTomato (1:300, rabbit, Rockland, 600-401-379), and MyoD (1:20, mouse, D7F2, Developmental Studies Hybridoma Bank). Following a 90-min incubation in secondary antibodies (1:500, donkey anti-mouse, goat, or rabbit, Thermo Fisher), the nuclei were labeled with 8 µM Hoechst 33342 (Thermo Fisher) for 15 min. When designated, 1 µg/ml phalloidin-TRITC (MilliporeSigma) or 5.0 µg/ml wheatgerm agglutinin conjugated to AlexaFlour488 (Thermo Fisher) was added with secondary antibodies.
Imaging
Widefield epifluorescence images were taken using an Olympus IX71 microscope with a 40×/0.6 NA objective and a digital EMCCD camera (Cascade 512B, Photometrics). Confocal imaging was done on a Leica TCS SP8 system with a 63×/1.4 NA oil-immersion objective. Live-cell imaging was conducted using an EVOS FL autoimaging system with an environmental chamber using a 20× objective. Live images were taken every 15 min using Fluroblok 24-well inserts.
Transfection of cell lines
Cells were plated at 1 × 104 cell/cm2. TdTomato containing lentiviral vector (Addgene #62733) was added at a multiplicity of infection ratios from 10 to 2000, which are the ratios of the number of lentivectors to the number of cells. Lentivector number was determined by a functional titer assay of HEK 293T cells. C2C12 cells (ATCC) and lentivector were coincubated for 1 h, unless otherwise specified. At 72 h posttransduction, cells were assayed for reporter gene expression by fluorescence microscopy. Total cell number was determined by DNA stain (Hoechst 33342 Invitrogen). Rh30 Cells were passaged 24 h before transfection. A complex of GFPs (0.2–0.5 ng/ml) and 1 µg/ml Lipofectamine 2000 (Invitrogen, Life Technologies) was prepared according to manufacturer’s instructions and then added for 24 h to cells in corresponding media supplemented with 10% FBS. GFP-KU80 was a gift from Stuart L. Rulten of the University of Sussex in Brighton, UK (Grundy et al., 2013), and mCherry-cGAS was a gift from Roger Greenburg of the University of Pennsylvania, Philadelphia, PA (Harding et al., 2017).
Alkaline comet assay
The comet assay was performed in accordance with manufacturer’s instructions (Cell Biolabs). Cells were detached from membranes or plates, mixed with liquefied agarose at 37°C, and deposited on a specially treated glass slide to gel for 15 min at 4°C. The gel containing cells was then incubated in lysis buffer for 45 min, followed by alkaline solution for 30 min. Electrophoresis was performed at 300 mA for 30 min and then the slide was washed with 70% ethanol and air-dried overnight. DNA dye was applied for 15 min before widefield epifluorescence imaging, as described above.
EdU labeling and staining
Edu (10 µM, Abcam) was added to each side of the transwell membrane or to a multiwell plate 1 h before fixation. Following permeabilization, samples were stained with 100 mM Tris (pH 8.5; MilliporeSigma), 1 mM CuSO4 (MilliporeSigma), 100 µM Cy5 azide dye (Cyandye), and 100 mM ascorbic acid (MilliporeSigma) for 30 min at room temperature. Samples were thoroughly washed with PBS and immunostaining proceeded as described above.
Apoptosis assay
Prior to application of the apoptosis assay, C2C12 cells in a 24-well plate were incubated under standard conditions with 1 µM staurosporine for 4 h to induce apoptosis. C2C12 cells were detached from membranes or multiwell plates and then suspended in Apoptosis Assay kit (Abcam) buffer with Apopxin Green indicator, 7-AAD, and CytoCalcein 450 for 60 min. Samples were then analyzed using flow cytometry on a BD LSRII as indicated by the kit protocol. For fluorescence microscopy analysis of apoptosis, the assay buffer with labeling components was added to Fluroblok transwell inserts or well plates for 45 min. Cells were washed with PBS and imaged using wide-field epifluorescence imaging as described above. Prior to application of the apoptosis assay, C2C12 cells in a 24-well plate were incubated under standard conditions with 1 µM staurosporine for 4 h to induce apoptosis.
Alkaline phosphatase assay
Cells were fixed in 4% formaldehyde (MilliporeSigma) and washed with 10 mM Tris buffer at pH 7.2. Fast Blue RR Salt (MilliporeSigma) was supplemented with 1:25 naphthol AS-MX phosphate solution (0.25% wt/vol, pH 8.6, MilliporeSigma) and used to stain alkaline phosphatase at room temperature for 30 min. Cells were then rinsed with distilled water and imaged to determine alkaline phosphatase activity per cell.
Cell treatments
DNA damage was induced chemically by adding 10 µM etoposide (MilliporeSigma) to growth media for 24 h before replating for differentiation or for the first 2 or 24 h within differentiation media. Irradiation-based DNA damage was performed under ambient oxygen using a Cs-137 Gammacell irradiator (Nordion) at a dose rate of ∼0.8 Gy/min while in growth media, 1 h before application of differentiation media. To arrest cells in G1 phase, CDK4/6 inhibitor (1 µM, PD332991, Cayman Chemicals) was applied to growth media 3 d prior and throughout cell migration and the differentiation protocol. For myosin II inhibition, 20 µM blebbistatin (EMD Millipore) was added to cell culture media on both sides of the membrane during the 24 h of migration of C2C12 and Rh30 cells.
In vivo differentiation of migrated cells
NSG mice were purchased from the Stem Cell and Xenograft Core at the University of Pennsylvania and experiments were planned and performed according to Institute of Animal Care and Use Committee protocols. Bilateral intramuscular injection into the tibialis anterior of 50 µl of cardiotoxin (MilliporeSigma) at 10 µM was used to induce active regeneration based on differentiation of resident muscle stem cells. Following 24 h of migration through 3- or 8-µm pore membranes, 4 × 105 TdTom+ C2C12 cells were suspended in 30 µl of DMEM and injected into the tibialis anterior muscle at three locations 3 d after the injection of cardiotoxin. Seven days after the cellular injection, mice were killed and tibialis muscles were harvested and fixed overnight in 4% paraformaldehyde at 4°C. Muscles were embedded in 4% agarose and 100-µm sections were cut using a vibratome (Leica) and underwent immunostaining as indicated above.
Quantification and statistical analysis
Image analysis was primarily performed using Fiji (Schindelin et al., 2012) to define stained areas or count foci. The differentiation index was calculated as the ratio of nuclei within a cell expressing skeletal muscle myosin to the total number of nuclei. Quantification of the Olive Moment for comet assay was conducted using OpenComet (Gyori et al., 2014). Quantification of centrally nucleated fiber and TdTom+ fibers was conducted using SMASH (Smith and Barton, 2014b).
The figure legends specify the sample size for each condition. Samples were considered individual membranes or individual wells for differentiation; typically ≥3 samples per experiment were independently analyzed. When a cell is reported as a sample the total number of experiments is typically two to four, with the exception of flow cytometry and human myoblasts. All statistical analyses were performed using GraphPad Prism 7. Analysis includes two-way analysis of variance (ANOVA) with post hoc Holm–Šídák tests with migration and differentiation time as independent variables. Analysis was conducted using one-way ANOVA for samples measured at one timepoint with a Holm–Šídák post hoc test. Comparison of cell or fiber proportions within categories between conditions was conducted using a chi-squared test. All statistical tests were considered significant if p < 0.05. Unless otherwise stated, all plots show mean ± SEM or mean ± SD for cell or fiber proportions.
Supplementary Material
Acknowledgments
We thank the Children’s Oncology Group for providing the myoblast cancer cell line Rh30 and Jake Hsu for transducing tdTomato into the C2C12 cell line. We thank Sarah Brashear for help in manuscript preparation. We are grateful for support from National Institutes of Health/National Cancer Institute PSOC Award U54 CA193417, National Heart Lung and Blood Institute Awards R01 HL124106 and R21 HL128187, National Institute of Arthritis and Musculoskeletal and Skin Diseases Award K99/R00 AR067867, a National Science Foundation Materials Science and Engineering Center grant, and a CEMB grant to the University of Pennsylvania and the US–Israel Binational Science Foundation. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other granting agencies. Finally, we thank Andrea Stout and Jasmine Zhao for training and access to Penn’s Cell and Developmental Biology Microscopy Core as well as the excellent staff of the Stem Cell & Xenograft Core.
Abbreviations used:
- CDKi
cyclin-dependent kinase inhibitor
- MSC
mesenchymal stem cell or mesenchymal stromal cell
- MuSC
muscle stem cell
- TW
transwell.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-02-0090) on June 12, 2019.
REFERENCES
- Boldrin L, Neal A, Zammit PS, Muntoni F, Morgan JE. (2012). Donor satellite cell engraftment is significantly augmented when the host niche is preserved and endogenous satellite cells are incapacitated. Stem Cells , 1971–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldrin L, Zammit PS, Morgan JE. (2015). Satellite cells from dystrophic muscle retain regenerative capacity. Stem Cell Res , 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscolo Sesillo F, Fox D, Sacco A. (2019). Muscle stem cells give rise to rhabdomyosarcomas in a severe mouse model of Duchenne muscular dystrophy. Cell Rep , 689-701.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bružauskaite˙ I, Bironaite˙ D, Bagdonas E, Bernotiene˙ E. (2016). Scaffolds and cells for tissue regeneration: different scaffold pore sizes—different cell effects. Cytotechnology , 355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholok D, Lee E, Lisiecki J, Agarwal S, Loder S, Ranganathan K, Qureshi AT, Davis TA, Levi B. (2017). Traumatic muscle fibrosis: from pathway to prevention. J Trauma Acute Care Surg , 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulton GR, Rogers B, Strutt P, Skynner MJ, Watt DJ. (1992). In situ localisation of single-stranded DNA breaks in nuclei of a subpopulation of cells within regenerating skeletal muscle of the dystrophic mdx mouse. J Cell Sci (Pt 3), 653–662. [DOI] [PubMed] [Google Scholar]
- Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, Lammerding J. (2016). Nuclear envelope rupture and repair during cancer cell migration. Science , 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos WH, Houben F, Kamps M, Malhas A, Verheyen F, Cox J, Manders EMM, Verstraeten VLRM, van Steensel MAM, Marcelis CLM, et al (2011). Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum Mol Genet , 4175–4186. [DOI] [PubMed] [Google Scholar]
- Didier N, Hourdé C, Amthor H, Marazzi G, Sassoon D. (2012). Loss of a single allele for Ku80 leads to progenitor dysfunction and accelerated aging in skeletal muscle. EMBO Mol Med , 910–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingal PCDP, Bradshaw AM, Cho S, Raab M, Buxboim A, Swift J, Discher DE. (2015). Fractal heterogeneity in minimal matrix models of scars modulates stiff-niche stem-cell responses via nuclear exit of a mechanorepressor. Nat Mater , 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler AJ, Griffin MA, Sen S, Bönnemann CG, Sweeney HL, Discher DE. (2004). Myotubes differentiate optimally on substrates with tissue-like stiffness: pathological implications for soft or stiff microenvironments. J Cell Biol , 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, Discher DE. (2006). Matrix elasticity directs stem cell lineage specification. Cell , 677–689. [DOI] [PubMed] [Google Scholar]
- Fanzani A, Monti E, Donato R, Sorci G. (2013). Muscular dystrophies share pathogenetic mechanisms with muscle sarcomas. Trends Mol Med , 546–554. [DOI] [PubMed] [Google Scholar]
- Farzaneh F, Zalin R, Brill D, Shall S. (1982). DNA strand breaks and ADP-ribosyl transferase activation during cell differentiation. Nature , 362–366. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Cusella-De Angelis G, Coletta M, Paolucci E, Stornaiuolo A, Cossu G, Mavilio F. (1998). Muscle regeneration by bone marrow-derived myogenic progenitors. Science , 1528–1530. [DOI] [PubMed] [Google Scholar]
- Garry GA, Antony ML, Garry DJ. (2016). Cardiotoxin induced injury and skeletal muscle regeneration. Methods Mol Biol , 61–71. [DOI] [PubMed] [Google Scholar]
- Grundy GJ, Rulten SL, Zeng Z, Arribas-Bosacoma R, Iles N, Manley K, Oliver A, Caldecott KW. (2013). APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J , 112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen K. (2016). Muscle memory and a new cellular model for muscle atrophy and hypertrophy. J Exp Biol , 235–242. [DOI] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, Clement M-V. (2014). OpenComet: an automated tool for comet assay image analysis. Redox Biol , 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada T, Swift J, Irianto J, Shin J-W, Spinler KR, Athirasala A, Diegmiller R, Dingal PCDP, Ivanovska IL, Discher DE. (2014). Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J Cell Biol , 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature , 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. (1989). Checkpoints: controls that ensure the order of cell cycle events. Science , 629–634. [DOI] [PubMed] [Google Scholar]
- Hatch EM, Hetzer MW. (2016). Nuclear envelope rupture is induced by actin-based nucleus confinement. J Cell Biol , 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson ARP, Jones R, Crose LES, Belyea BC, Barr FG, Linardic CM. (2013). Human rhabdomyosarcoma cell lines for rhabdomyosarcoma research: utility and pitfalls. Front Oncol , 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi K, Hirano K, Hamatake M, Ishida R. (2001). Phosphatidylserine induces apoptosis in adherent cells. Apoptosis , 263–268. [DOI] [PubMed] [Google Scholar]
- Irianto J, et al (2017). DNA damage follows repair factor depletion and portends genome variation in cancer cells after pore migration. Curr Biol , 210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J-I, Park K-Y, Lee Y, Park M, Kim J. (2018). Vitamin C-linker-conjugated tripeptide AHK stimulates BMP-2-induced osteogenic differentiation of mouse myoblast C2C12 cells. Differentiation , 1–7. [DOI] [PubMed] [Google Scholar]
- Larsen BD, Rampalli S, Burns LE, Brunette S, Dilworth FJ, Megeney LA. (2010). Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proc Natl Acad Sci USA , 4230–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber RL, Ward SR. (2013). Cellular mechanisms of tissue fibrosis. 4. Structural and functional consequences of skeletal muscle fibrosis. Am J Physiol Physiol , C241–C252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund DK, Mouly V, Cornelison DDW. (2014). MMP-14 is necessary but not sufficient for invasion of three-dimensional collagen by human muscle satellite cells. Am J Physiol Cell Physiol , C140-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdy MAA. (2019). Skeletal muscle fibrosis: an overview. Cell Tissue Res , 575–588. [DOI] [PubMed] [Google Scholar]
- Manolagas SC. (2000). Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev , 115–137. [DOI] [PubMed] [Google Scholar]
- Martinez AR, Kaul Z, Parvin JD, Groden J. (2017). Differential requirements for DNA repair proteins in immortalized cell lines using alternative lengthening of telomere mechanisms. Genes Chromosomes Cancer , 617–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J, Bencze M, Asfahani R, Muntoni F, Morgan JE. (2015). The effect of the muscle environment on the regenerative capacity of human skeletal muscle stem cells. Skelet Muscle , 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina G, Blasi C, La Rocca SA, Pompili M, Calconi A, Grossi M. (2005). p27Kip1 acts downstream of N-cadherin-mediated cell adhesion to promote myogenesis beyond cell cycle regulation. Mol Biol Cell , 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milovanovic P, Vukovic Z, Antonijevic D, Djonic D, Zivkovic V, Nikolic S, Djuric M. (2017). Porotic paradox: distribution of cortical bone pore sizes at nano- and micro-levels in healthy vs. fragile human bone. J Mater Sci Mater Med , 71. [DOI] [PubMed] [Google Scholar]
- Moseychuk O, Akkiraju H, Dutta J, D’Angelo A, Bragdon B, Duncan RL, Nohe A. (2013). Inhibition of CK2 binding to BMPRIa induces C2C12 differentiation into osteoblasts and adipocytes. J Cell Commun Signal , 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi N, Asakura Y, Asakura A. (2014). Isolation, culture, and transplantation of muscle satellite cells. J Vis Exp , 50846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterhoff G, Morgan EF, Shefelbine SJ, Karim L, McNamara LM, Augat P. (2016). Bone mechanical properties and changes with osteoporosis. Injury , S11–S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Yamada KM. (2012). At the leading edge of three-dimensional cell migration. J Cell Sci , 5917–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer CR, Xia Y, Zhu K, Liu D, Irianto J, García VMM, Millán LMS, Niese B, Harding S, Deviri D, et al (2018). Constricted migration increases DNA damage and independently represses cell cycle. Mol Biol Cell , 1948–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. (1999). Multilineage potential of adult human mesenchymal stem cells. Science , 143–147. [DOI] [PubMed] [Google Scholar]
- Puri PL, Bhakta K, Wood LD, Costanzo A, Zhu J, Wang JYJ. (2002). A myogenic differentiation checkpoint activated by genotoxic stress. Nat Genet , 585–593. [DOI] [PubMed] [Google Scholar]
- Qi R, Long D, Wang J, Wang Q, Huang X, Cao C, Gao G, Huang J. (2016). MicroRNA-199a targets the fatty acid transport protein 1 gene and inhibits the adipogenic trans-differentiation of C2C12 myoblasts. Cell Physiol Biochem , 1087–1097. [DOI] [PubMed] [Google Scholar]
- Raab M, Gentili M, De Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon-Duménil AM, Manel N, et al (2016). ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science , 359–362. [DOI] [PubMed] [Google Scholar]
- Raab M, Swift J, Dingal PCDP, Shah P, Shin J-W, Discher DE. (2012). Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J Cell Biol , 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods , 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt WM, Uddin MH, Dysek S, Moser-Thier K, Pirker C, Höger H, Ambros IM, Ambros PF, Berger W, Bittner RE. (2011). DNA damage, somatic aneuploidy, and malignant sarcoma susceptibility in muscular dystrophies. PLoS Genet , e1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel AL, Atchison K, Fisher KE, Davis GE, Cornelison DDW. (2009). 3D timelapse analysis of muscle satellite cell motility. Stem Cells , 2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, Miller C, Regalado SG, Loffredo FS, Pancoast JR, et al (2014). Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science , 649–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LR, Barton ER. (2014a). Collagen content does not alter the passive mechanical properties of fibrotic skeletal muscle in mdx mice. Am J Physiol Cell Physiol , C889–C898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LR, Barton ER. (2014b). SMASH—semi-automatic muscle analysis using segmentation of histology: a MATLAB application. Skelet Muscle , 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LR, Barton ER. (2018). Regulation of fibrosis in muscular dystrophy. Matrix Biol , 602–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LR, Hammers DW, Sweeney HL, Barton ER. (2016). Increased collagen cross-linking is a signature of dystrophin-deficient muscle. Muscle Nerve , 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn J, Lu A, Tang Y, Wang B, Huard J. (2015). Activation of non-myogenic mesenchymal stem cells during the disease progression in dystrophic dystrophin/utrophin knockout mice. Hum Mol Genet , 3814–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, Narusawa M, Leferovich JM, Sladky JT, Kelly AM. (1991). The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature , 536–539. [DOI] [PubMed] [Google Scholar]
- Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PCDP, Pinter J, Pajerowski JD, Spinler KR, Shin J-W, Tewari M, et al (2013). Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science , 1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ, Thayer MJ, Weintraub H. (1993). Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science , 1450–1453. [DOI] [PubMed] [Google Scholar]
- Thomas DG, Yenepalli A, Denais CM, Rape A, Beach JR, Wang Y, Schiemann WP, Baskaran H, Lammerding J, Egelhoff TT. (2015). Non-muscle myosin IIB is critical for nuclear translocation during 3D invasion. J Cell Biol , 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY. (2001). DNA damage and apoptosis. Cell Death Differ , 1047–1048. [DOI] [PubMed] [Google Scholar]
- Webster MT, Manor U, Lippincott-Schwartz J, Fan C-M. (2016). Intravital imaging reveals ghost fibers as architectural units guiding myogenic progenitors during regeneration. Cell Stem Cell , 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelin B, Bakker G-J, Friedl P. (2012). Intravital third harmonic generation microscopy of collective melanoma cell invasion: principles of interface guidance and microvesicle dynamics. Intravital , 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Ivanovska IL, Zhu K, Smith L, Irianto J, Pfeifer CR, Alvey CM, Ji J, Liu D, Cho S, et al (2018). Nuclear rupture at sites of high curvature compromises retention of DNA repair factors. J Cell Biol , 3796–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Pfeifer CR, Zhu K, Irianto J, Liu D, Pannell K, Chen EJ, Dooling LJ, Tobin MP, Wang M, et al (2019). Rescue of DNA damage after constricted migration reveals a mechano-regulated threshold for cell cycle. J Cell Biol, jcb.201811100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Choi S, Liu X, Zhang M, Schageman JJ, Lee SY, Hart R, Lin L, Thurmond FA, Williams RS. (2003). Highly coordinated gene regulation in mouse skeletal muscle regeneration. J Biol Chem , 8826–8836. [DOI] [PubMed] [Google Scholar]
- Yin H, Price F, Rudnicki MA. (2013). Satellite cells and the muscle stem cell niche. Physiol Rev , 23–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.