

Abstract
Purpose of review:
Von Willebrand disease (VWD) is a common bleeding disorder, but diagnosis of VWD is challenging, particularly with type 1 VWD. While most clinicians use specific tests of von Willebrand factor (VWF) activity to classify patients with VWD, genetic testing for VWF defects is another potential method of diagnosis.
Recent findings:
Studies of patients with type 1 VWD report consistently that many, but not all, subjects have VWF gene defects. Certain populations, including those with VWF levels < 30 IU/dL and those with clearance defects, are more likely to have a VWF sequence variant. In addition, a number of loci outside the VWF gene have been shown to affect VWF levels, including ABO, CLEC4M, STXBP5 and STAB2.
Summary:
Genetic defects in VWF are common, but not all defects lead to disease. Type 1 VWD in particular does not always have an associated VWF sequence variant. New data stemming from genome wide association studies on modifier genes suggests that the etiology of type 1 VWD is multifactorial.
Keywords: von Willebrand disease, von Willebrand factor, genetic
Introduction
Type 1 von Willebrand disease (VWD) is the most common form of VWD which is in turn the most common inherited bleeding disorder (1). Unlike other coagulation factor deficiencies, however, VWD is notoriously hard to diagnose, and type 1 VWD is the most challenging group. Part of this difficulty stems from the need to perform multiple assays of von Willebrand factor (VWF) function, and part stems from the controversy around the appropriate cut-off VWF level to use for diagnosis of type 1 VWD (2). Some studies argue for use of a cut-off VWF level of 30 IU/dL (3,4) while many groups classify patients as type 1 if their VWF levels are lower than the lower limit of the normal range (<50 IU/dL) . It should be noted that regardless of the cutoff used, low VWF does appear to be a risk factor for bleeding, as recent studies from Ireland demonstrate increased bleeding and sustained decreased VWF levels in a cohort of subjects with VWF levels between 30–50 IU/dL (5).
Current VWF testing includes both testing for total protein (VWF antigen, or VWF:Ag) and platelet binding function (either by ristocetin cofactor activity assay [VWF:RCo] or GPIb binding assays [VWF:GPIbM or VWF:GPIbR]) (3). While the VWF:Ag assay is relatively straightforward, the VWF:RCo assay is prone to error due to high inter- and intra- laboratory variability, and the effect of sequence variants in the VWF gene on VWF binding to ristocetin (6–8). The high variability in the VWF:RCo has led to some institutions replacing it with a GPIb binding assay (9–11). The current assays most commonly use a gain-of-function GPIb to induce VWF binding to GPIb independent of ristocetin (11). The International Society on Thrombosis and Haemostasis has recommended using the nomenclature VWF:GPIbM for such assays (12).
The limitations of laboratory testing call for improved methods of diagnosis. Given the ready availability and decreasing cost of gene sequencing, it would seem that genetic diagnosis of type 1 VWD would solve this problem. Several issues, however, preclude the clinical utility of genetic diagnosis, at least in type 1 patients. New findings in the area of type 1 VWD genetics, however, continue to improve our understanding of VWF function and type 1 VWD.
Genetic defects in VWF
In type 2 VWD, defects in VWF function map to specific regions of the VWF protein, and therefore the genetic defects tend to be found in the DNA region corresponding to the affected functional domain (13). In types 1 and 3 VWD, however, defects have been reported across the coding region of the gene (13). These range from point mutations that cause missense or nonsense mutations to deletions and insertions of varying size (13). In some cases, the latter may not be detected on conventional sequencing (14). Copy number variant assays, such as comparative genomic hybridization, can detect how many alleles are present for specific regions of the VWF gene (15,16). In some cases, patients receive whole exome or whole genome sequencing while in other cases, if a specific coagulation defect is under evaluation, the VWF gene may be analyzed specifically.
The most common deletion appears to be one affecting exons 4 and 5 (14). Several other deletions have been described, many of which appear to be unique to the affected family (17). Interestingly, while patients with type 3 VWD likely have two genetic defects causing their VWD, their obligate carrier parents may or may not exhibit findings consistent with type 1 VWD (18). This means that a deletion, in and of itself, does not necessarily confer low VWF levels.
Another problem with sequence variants found in type 1 VWD stems from the highly polymorphic nature of VWF. In healthy individuals, relatively high frequencies of genetic variants have been reported (19). Of note, several variants were seen at high frequency, particularly in the African American population, and some of these variants had been previously considered as causative of disease. This highlights the need for caution when interpreting novel variants. The advent of larger, more diverse cohorts of genetic data has improved our ability to understand ethnic differences, but unique novel variants still may not necessarily cause disease.
One cautionary tale is that of the VWF p.D1472H variant. This variant has been associated with decreased VWF:RCo, and decreased VWF:RCo/VWF:Ag ratios across several studies, yielding levels that could result in a diagnosis of type 2M VWD (8,20). However, when VWF-platelet interactions are examined using other assays (either the VWF antibody assay or assays such as the VWF:GPIbM that do not require ristocetin), normal results are obtained (8,10,21). This “mutation” is actually a common variant, found at high frequencies in the African American population (up to 60% of subjects) but also present at not insubstantial frequencies in the Caucasian population (8). Another variant in the same region has been reported, p.P1467S, but so far described in only one family (22).
Recent VWF genetic findings
A recent study from Spain by Borras and colleagues demonstrated genetic defects in 91% of type 1 subjects (23). Interestingly in their study of 556 subjects a total of 155 novel variants were found, highlighting the high rate of variability in VWF (23). The high rate of genetic variants observed in type 1 VWD in their study can be attributed in part to their stringent entry criteria (VWF levels < 30 IU/dL on two occasions) in contrast to previous reports including patients with levels < 50 IU/dL where rates of genetic variants ranged between 55 and 70% (24–28).
A new comprehensive study in patients with “Low VWF” (VWF levels between 30–50 IU/dL) by Lavin and colleagues showed only 40% of patients had genetic variants in the VWF gene predicted to be damaging, although 60% of the cohort had at least one sequence variant observed (5). This cohort is remarkable for having reproducible low VWF levels (measured on at least 2 occasions) and comprehensive bleeding score data available on their subjects to confirm that a bleeding phenotype was present (5).
While many variants reduce VWF expression, some variants have been shown to increase VWF expression. Several VWF D’D3 variants, VWF c.2880G>A and VWF c.2365A>G(;)c.2385T>C, have been linked with increased VWF levels and therefore a decreased risk of diagnosis of type 1 VWD (29). These results are important to demonstrate that not all changes in VWF gene sequence result in VWD.
Genetic modifiers outside the VWF locus
While variants in VWF logically can cause VWD, a number of genes outside the VWF locus have also been implicated in altering VWF levels (table 1). Perhaps the most well characterized is ABO blood group. Gill and colleagues reported in 1987 that individuals with blood type O had lower VWF levels than those individuals with other blood types (30). The normal range for healthy individuals with blood group O was actually reported as 36–157 IU/dL (30). This is important for two reasons. First, patients with type O are over-represented in studies of type 1 VWD. Second, the diagnosis of VWD typically utilizes levels of <50 IU/dL, and some healthy individuals with type O will fall into this category. For the physician faced with a patient who has bleeding symptoms and low VWF levels, this may be an academic distinction, as low levels of VWF could certainly cause bleeding regardless of the mechanism. It has been pointed out, however, that VWF exists on a continuum, and perhaps low VWF is better considered a risk factor for bleeding as opposed to a definite cause of bleeding (31).
Table:
Modifier genes with defined pathology implicated in VWD
| Modifier gene | Putative function |
| ABO | Clearance |
| CLEC4M | Clearance |
| STXBP5 | Endothelial cell exocytosis |
| STAB2 | Clearance, immunoregulation |
Over the last few years, a number of other genes have been reported to affect VWF gene expression (32,33). Some of these have now been characterized and their mechanism of action described, while others are simply the result of genome-wide association studies with exact pathology to be determined. Further details on those best characterized in terms of mechanism and association with true VWD are discussed below.
CLEC4M in particular has been shown to associate with low VWF levels (32,34). CLECM is a lectin receptor that has been shown to both bind and internalize VWF, thus functioning to clear VWF from plasma (35). A recent study by Manderstedt and colleagues showed enrichment of two CLEC4M genotypes in the Swedish type 1 VWD population (36). Recent work shows that CLEC4M also clears FVIII, both with and without associated VWF (37). Such findings may have implications beyond just diagnosis of VWD, with potential utility in improving treatment of both VWD and hemophilia.
STXBP5 is another gene associated with low VWF levels (32,34). This gene encodes for syntaxin binding protein 5, and is known to inhibit endothelial cell exocytosis (38). Work from the Netherlands demonstrated an association of variants in STX2 and STXBP5 in patients with type 1 VWD and bleeding symptoms (39). Data from Sweden now show that the p.N436S variant in the STXBP5 gene is associated with type 1 VWD (40). No other potentially causative variants were found in this group, but it is possible that other ethnicities may have different variants in STXBP5 affecting VWF regulated secretion from endothelial cells.
STAB2 was also identified by GWAS as a potential influencer of VWF levels (32). STAB2 encodes a protein known as stabilin-2, which is a scavenger receptor. Swystun and colleagues have now demonstrated the mechanism for the effect of stabilin-2, which appears to work as both a clearance receptor and as an immunoregulatory receptor (41). The clinical implications for stabilin-2 are exciting, given that it appears that absence of stabilin-2 prolongs the half-life of VWF, and by association, FVIII (41).
Genome wide association studies continue to implicate additional factors (32). Recent data by Sabater-Lleal show 11 new genes that may affect VWF levels, including PDHB/PXK/KCTD6, SLC39A8, FCHO2/TMEM171/TNPO1, HLA, GIMAP7/GIMAP4, OR13C5/NIPSNAP, DAB2IP, C2CD4B, RAB5C-KAT2A, TAB1/SYNGR1, and ARSA (42). Functional characterization of these new candidate genes will be required to better understand their role in VWD.
Utility of genetics in VWD diagnosis
Given the issues above, use of genetic diagnosis in type 1 VWD is difficult. Other genes beyond VWF may affect VWF levels, so lack of a pathogenic mutation in VWF does not exclude the diagnosis of VWD. On the other hand, the frequency of variants in the VWF gene is such that discovery of a variant does not necessarily equate to a diagnosis of VWD. The one potential caveat is discovery of a clearance defect.
Those variants that cause type 1C VWD are of potential interest from a therapy standpoint. Patients with type 1C VWD clear their VWF more rapidly than normal (43). Desmopressin, while causing a temporary increase in VWF level, will not result in a sustained rise in these patients (43), so treatment for type 1C VWD typically requires administration of VWF concentrate. If a variant previously reported to cause type 1C VWD is found on sequencing, desmopressin may not the best therapy for that patient. However, other assays such as the VWF propeptide (VWFpp) or results of a desmopressin trial including late (2–4 hour) timepoints may be sufficient to make this diagnosis. A VWFpp/VWF:Ag ratio greater than 3 supports the diagnosis of type 1C VWD without an extended desmopressin trial (44).
Otherwise, utility of sequencing for type 1 VWD is debatable. In general, diagnosis is made based on symptoms and VWF level, and the addition of VWF gene sequencing will not usually provide any additional actionable information. Sequencing VWF remains of use from a research standpoint, as we seek to understand more about the myriad functions of the VWF protein, but is less helpful from a clinical standpoint.
Sequencing in type 2 VWD, on the other hand, may be very useful. Differentiation of type 2 VWD subtypes in the current classification system requires plasma based assays, including VWF multimers, FVIII binding, and platelet binding, which may not be readily available at some centers. DNA sequencing technology is now widespread, and may provide quicker answers. If a type 2 variant of VWD is part of the differential diagnosis, VWD sequencing could yield a therapeutically meaningful result, as most type 2 variants would also require treatment with von Willebrand concentrate rather than desmopressin.
Conclusion
In summary, sequence variants are common in type 1 VWD, but their location throughout the VWF gene and the difficulties surrounding classification of pathogenicity of novel variants limit utility of genetic testing for type 1 VWD. When other types of VWD are part of the differential however, VWF gene sequencing may be the fastest method of excluding a potential type 1C or type 2 variant (figure 1). It is important to remember that not all type 1 VWD patients will have a genetic defect in the VWF gene, and further work needs to be done to understand the importance of modifier genes in the pathogenesis of type 1 VWD. However, new studies demonstrating the novel effects of modifier genes continue to expand our knowledge of VWF secretion and clearance and may have promise for future therapeutic improvements for patients with VWD.
Figure 1. Utility of VWF gene sequencing.
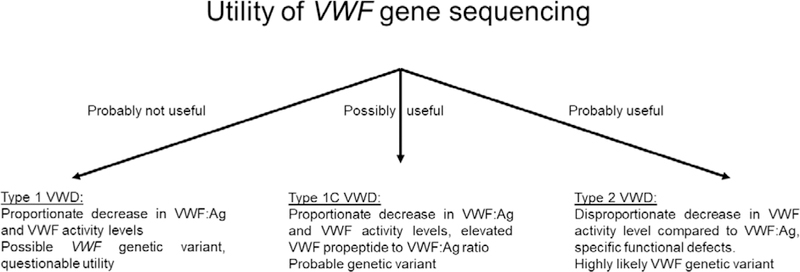
The figure shows typical laboratory results for several types of VWD and the likely utility of VWF gene sequencing for these diagnoses.
Key Points:
The VWF gene is highly polymorphic.
Not all patients with type 1 VWD have genetic variants found in the VWF gene.
Genetic variants outside the VWF locus have also been associated with VWD.
Acknowledgements
The authors would like to acknowledge many collaborators and discussions at the Blood Research Institute, including Robert Montgomery, Kenneth Friedman, and Joan Cox Gill.
Funding: National Institutes of Health (NHLBI) and the MACC Fund Center for Cancer and Blood Disorders
Financial support and sponsorship
This work was supported in part by funding from the National Institutes of Health (HL102260, HL081588, HL126810 and HL136430) and the MACC Fund Center for Cancer and Blood Disorders.
Footnotes
Conflicts of Interest
None of the authors have conflicts of interest to report.
References
- 1.Bowman M, Hopman WM, Rapson D, Lillicrap D, James P. The prevalence of symptomatic von Willebrand disease in primary care practice. JThrombHaemost. 2010;8(1):213–6. [DOI] [PubMed] [Google Scholar]
- 2.Sadler JE. Von Willebrand disease type 1: a diagnosis in search of a disease. Blood. 2003;101(6):2089–93. [DOI] [PubMed] [Google Scholar]
- 3.Nichols WL, Hultin MB, James AH, Manco-Johnson MJ, Montgomery RR, Ortel TL, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008;14(2):171–232. [DOI] [PubMed] [Google Scholar]
- 4.Laffan MA, Lester W, O’Donnell JS, Will A, Tait RC, Goodeve A, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. BrJHaematol. 2014;167(4):453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.**Lavin M, Aguila S, Schneppenheim S, Dalton N, Jones KL, O’Sullivan JM, et al. Novel insights into the clinical phenotype and pathophysiology underlying low VWF levels. Blood. 2017. November 23;130(21):2344–53.This study describes the phenotype seen in patients with low VWF and demonstrated that the majority had bleeding symptoms. This is important in terms of our understanding of what constitutes true VWD.
- 6.Kitchen S, Jennings I, Woods TA, Kitchen DP, Walker ID, Preston FE. Laboratory tests for measurement of von Willebrand factor show poor agreement among different centers: results from the United Kingdom National External Quality Assessment Scheme for Blood Coagulation. SeminThrombHemost. 2006;32(5):492–8. [DOI] [PubMed] [Google Scholar]
- 7.Meijer P, Haverkate F. An external quality assessment program for von Willebrand factor laboratory analysis: an overview from the European concerted action on thrombosis and disabilities foundation. SeminThrombHemost. 2006;32(5):485–91. [DOI] [PubMed] [Google Scholar]
- 8.Flood VH, Gill JC, Morateck PA, Christopherson PA, Friedman KD, Haberichter SL, et al. Common VWF exon 28 polymorphisms in African Americans affecting the VWF activity assay by ristocetin cofactor. Blood. 2010;116(2):280–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanhoorelbeke K, Cauwenberghs N, Vauterin S, Schlammadinger A, Mazurier C, Deckmyn H. A reliable and reproducible ELISA method to measure ristocetin cofactor activity of von Willebrand factor. ThrombHaemost. 2000;83(1):107–13. [PubMed] [Google Scholar]
- 10.Flood VH, Gill JC, Morateck PA, Christopherson PA, Friedman KD, Haberichter SL, et al. Gain-of-function GPIb ELISA assay for VWF activity in the Zimmerman Program for the Molecular and Clinical Biology of VWD. Blood. 2011;117(6):e67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patzke J, Budde U, Huber A, Mendez A, Muth H, Obser T, et al. Performance evaluation and multicentre study of a von Willebrand factor activity assay based on GPIb binding in the absence of ristocetin. Blood CoagulFibrinolysis. 2014;25(8):860–70. [DOI] [PubMed] [Google Scholar]
- 12.Bodo I, Eikenboom J, Montgomery R, Patzke J, Schneppenheim R, Di Paola J, et al. Platelet-dependent von Willebrand factor activity. Nomenclature and methodology: communication from the SSC of the ISTH. JThrombHaemost. 2015;13(7):1345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev. 2010;24(3):123–34. [DOI] [PubMed] [Google Scholar]
- 14.Sutherland MS, Cumming AM, Bowman M, Bolton-Maggs PH, Bowen DJ, Collins PW, et al. A novel deletion mutation is recurrent in von Willebrand disease types 1 and 3. Blood. 2009;114(5):1091–8. [DOI] [PubMed] [Google Scholar]
- 15.Askree SH, Chin EL, Bean LH, Coffee B, Tanner A, Hegde M. Detection limit of intragenic deletions with targeted array comparative genomic hybridization. BMC Genet. 2013;14(Journal Article):116–2156-14–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue Y, Ankala A, Wilcox WR, Hegde MR. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. GenetMed. 2015;17(6):444–51. [DOI] [PubMed] [Google Scholar]
- 17.Yadegari H, Driesen J, Hass M, Budde U, Pavlova A, Oldenburg J. Large deletions identified in patients with von Willebrand disease using multiple ligation-dependent probe amplification. J Thromb Haemost. 2011. May;9(5):1083–6. [DOI] [PubMed] [Google Scholar]
- 18.Bowman M, Tuttle A, Notley C, Brown C, Tinlin S, Deforest M, et al. The genetics of Canadian type 3 von Willebrand disease: further evidence for co-dominant inheritance of mutant alleles. JThrombHaemost. 2013;11(3):512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bellissimo DB, Christopherson PA, Flood VH, Gill JC, Friedman KD, Haberichter SL, et al. VWF mutations and new sequence variations identified in healthy controls are more frequent in the African-American population. Blood. 2012;119(9):2135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnsen JM, Auer PL, Morrison AC, Jiao S, Wei P, Haessler J, et al. Common and rare von Willebrand factor (VWF) coding variants, VWF levels, and factor VIII levels in African Americans: the NHLBI Exome Sequencing Project. Blood. 2013;122(4):590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.**Boender J, Eikenboom J, van der Bom JG, Meijer K, de Meris J, Fijnvandraat K, et al. Clinically relevant differences between assays for von Willebrand factor activity. J Thromb Haemost. 2018. December;16(12):2413–24. This study documents differences in VWF levels using the different commercially available VWF activity assays. This work is important in showing that the different assays can in some cases give different results.
- 22.Flood VH, Friedman KD, Gill JC, Morateck PA, Wren JS, Scott JP, et al. Limitations of the ristocetin cofactor assay in measurement of von Willebrand factor function. JThrombHaemost. 2009;7(11):1832–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.*Borras N, Batlle J, Perez-Rodriguez A, Lopez-Fernandez MF, Rodriguez-Trillo A, Loures E, et al. Molecular and clinical profile of von Willebrand disease in Spain (PCM-EVW-ES): comprehensive genetic analysis by next-generation sequencing of 480 patients. Haematologica. 2017. December;102(12):2005–14.This study shows a high level of sequence variants, including a number of novel variants, in a type 1 VWD population from Spain.
- 24.Cumming A, Grundy P, Keeney S, Lester W, Enayat S, Guilliatt A, et al. An investigation of the von Willebrand factor genotype in UK patients diagnosed to have type 1 von Willebrand disease. ThrombHaemost. 2006;96(5):630–41. [PubMed] [Google Scholar]
- 25.Goodeve A, Eikenboom J, Castaman G, Rodeghiero F, Federici AB, Batlle J, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM-1VWD). Blood. 2007;109(1):112–21. [DOI] [PubMed] [Google Scholar]
- 26.James PD, Notley C, Hegadorn C, Leggo J, Tuttle A, Tinlin S, et al. The mutational spectrum of type 1 von Willebrand disease: Results from a Canadian cohort study. Blood. 2007;109(1):145–54. [DOI] [PubMed] [Google Scholar]
- 27.Yadegari H, Driesen J, Pavlova A, Biswas A, Hertfelder HJ, Oldenburg J. Mutation distribution in the von Willebrand factor gene related to the different von Willebrand disease (VWD) types in a cohort of VWD patients. ThrombHaemost. 2012;108(4):662–71. [DOI] [PubMed] [Google Scholar]
- 28.Flood VH, Christopherson PA, Gill JC, Friedman KD, Haberichter SL, Bellissimo DB, et al. Clinical and laboratory variability in a cohort of patients diagnosed with type 1 VWD in the United States. Blood. 2016;127(20):2481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.*Flood VH, Johnsen JM, Kochelek C, Slobodianuk TL, Christopherson PA, Haberichter SL, et al. Common VWF sequence variants associated with higher VWF and FVIII are less frequent in subjects diagnosed with type 1 VWD. Res Pract Thromb Haemost. 2018. April;2(2):390–8.This study shows several common sequence variants associated with elevated VWF levels appeared to protect against a diagnosis of VWD.
- 30.Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ Jr, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood. 1987;69(6):1691–5. [PubMed] [Google Scholar]
- 31.Sadler JE. von Willebrand factor: two sides of a coin. JThrombHaemost. 2005;3(8):1702–9. [DOI] [PubMed] [Google Scholar]
- 32.Smith NL, Chen MH, Dehghan A, Strachan DP, Basu S, Soranzo N, et al. Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand factor: The CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) Consortium. Circulation. 2010;121(12):1382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Loon J, Dehghan A, Weihong T, Trompet S, McArdle WL, Asselbergs FF, et al. Genome-wide association studies identify genetic loci for low von Willebrand factor levels. EurJHumGenet. 2016;24(7):1035–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.*Sanders YV, van der Bom JG, Isaacs A, Cnossen MH, de Maat MP, Laros-van Gorkom BA, et al. CLEC4M and STXBP5 gene variations contribute to von Willebrand factor level variation in von Willebrand disease. JThrombHaemost. 2015;13(6):956–66.This study shows the association of two sequence variants with type 1 VWD.
- 35.**Rydz N, Swystun LL, Notley C, Paterson AD, Riches JJ, Sponagle K, et al. The C-type lectin receptor CLEC4M binds, internalizes, and clears von Willebrand factor and contributes to the variation in plasma von Willebrand factor levels. Blood. 2013;121(26):5228–37.This study demonstrates the mechanism of action of CLEC4M in VWD, and shows the importance of variants in the CLEC4M gene in VWD.
- 36.*Manderstedt E, Lind-Hallden C, Lethagen S, Hallden C. Genetic variation in the C-type lectin receptor CLEC4M in type 1 von Willebrand Disease patients. PLoS One. 2018;13(2):e0192024.This study also shows the connection between CLEC4M variants and VWD.
- 37.*Swystun LL, Notley C, Georgescu I, Lai JD, Nesbitt K, James PD, et al. The endothelial lectin clearance receptor CLEC4M binds and internalizes Factor VIII in a VWF-dependent and -independent manner. J Thromb Haemost. 2019. February 11;This study adds to our understanding of how CLEC4M works by demonstrating its effect on FVIII as well as VWF.
- 38.*Zhu Q, Yamakuchi M, Ture S, de la Luz Garcia-Hernandez M, Ko KA, Modjeski KL, et al. Syntaxin-binding protein STXBP5 inhibits endothelial exocytosis and promotes platelet secretion. J Clin Invest. 2014. October;124(10):4503–16.This paper demonstrates the mechanism of action of STXBP5.
- 39.van Loon JE, Sanders YV, de Wee EM, Kruip MJHA, de Maat MPM, Leebeek FWG. Effect of genetic variation in STXBP5 and STX2 on von Willebrand factor and bleeding phenotype in type 1 von Willebrand disease patients. PLoS One. 2012;7(7):e40624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.*Lind-Hallden C, Manderstedt E, Carlberg D, Lethagen S, Hallden C. Genetic Variation in the Syntaxin-Binding Protein STXBP5 in Type 1 von Willebrand Disease Patients. Thromb Haemost. 2018. August;118(8):1382–9.This study links variants in the STXBP5 gene directly to the presence of type 1 VWD.
- 41.**Swystun LL, Lai JD, Notley C, Georgescu I, Paine AS, Mewburn J, et al. The endothelial cell receptor stabilin-2 regulates VWF-FVIII complex half-life and immunogenicity. J Clin Invest. 2018. August 31;128(9):4057–73.This critical paper demonstrates not only how stabilin-2 affects VWF, but also provides new evidence on VWF and FVIII immunogenicity, which is important for the development of inhibitors.
- 42.*Sabater-Lleal M, Huffman JE, de Vries PS, Marten J, Mastrangelo MA, Song C, et al. Genome-Wide Association Transethnic Meta-Analyses Identifies Novel Associations Regulating Coagulation Factor VIII and von Willebrand Factor Plasma Levels. Circulation. 2019. January 29;139(5):620–35.This association study provides several new potential candidate genes that may affect VWF secretion and function.
- 43.Haberichter SL, Balistreri M, Christopherson P, Morateck P, Gavazova S, Bellissimo DB, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood. 2006;108(10):3344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haberichter SL. VWF propeptide in defining VWD subtypes. Blood. 2015. May 7;125(19):2882–3. [DOI] [PubMed] [Google Scholar]