

Abstract
Identification of novel chemotypes with antimalarial efficacy is imperative to combat the rise of Plasmodium species resistant to current antimalarial drugs. We have used a hybrid target-phenotype approach to identify and evaluate novel chemotypes for malaria. In our search for drug-like aspartic protease inhibitors in publicly available phenotypic antimalarial databases, we identified GNF-Pf-4691, a 4-aryl-N-benzylpyrrolidine-3-carboxamide, as having a structure reminiscent of known inhibitors of aspartic proteases. Extensive profiling of the two terminal aryl rings revealed a structure-activity relationship in which relatively few substituents are tolerated at the benzylic position, but the 3-aryl position tolerates a range of hydrophobic groups and some heterocycles. Out of this effort, we identified (+)−54b (CWHM-1008) as a lead compound. 54b has EC50 values of 46 nM and 21 nM against drug sensitive Plasmodium falciparum 3D7 and drug resistant Dd2 strains, respectively. Furthermore, 54b has a long half-life in mice (4.4 h) and is orally efficacious in a mouse model of malaria (q.d.; ED99 ~ 30 mg/kg/day). Thus, the 4-aryl-N-benzylpyrrolidine-3-carboxamide chemotype is a promising novel chemotype for malaria drug discovery.
Keywords: aspartic protease inhibitors, pyrrolidines, antiplasmodial, antimalarial
Graphical Abstract
Introduction
Efforts at reducing incidents of malaria have stalled in recent years after a decade of progressive reduction in disease burden. The majority of malaria-related deaths occur in Africa and are caused by Plasmodium falciparum, the most lethal to man of the Plasmodium species. The introduction of artemisinin and artemisinin combination therapies (ACT) in 2005 led to a remarkable 60% reduction in mortality rates between 2000–2015.1 However, according to the World Health Organization, there were ~219 million new cases of malaria and 435,000 deaths in 2017, highlighting a leveling out of progress in reducing global malarial disease burden since 2015.2 Furthermore, there are reports of emerging resistance to artemisinin in Southeast Asia.1, 3–4 As the artemisinins are the only fully effective class of antimalarial drugs available today, it is crucial that additional antimalarial drugs be developed with new mechanisms of action as the next line of defense to combat developing resistance to known drugs. These efforts, along with efforts to control malaria transmission will be needed if successful global eradication of malaria is to be achieved.
The Plasmodium parasite has a complex lifecycle including sexual replication in the mosquito stage and asexual replication in the human liver and blood stages. These lifecycle stages involve numerous potential opportunities for intervention. The challenge is to identify unexploited biological targets that will result in effective parasite killing. A broad class of proteases, the aspartic proteases, have been successfully exploited for the treatment of AIDS—more than 10 FDA approved drugs have been developed that inhibit the HIV aspartic protease. Plasmodium has multiple aspartic proteases that play key roles in the survival of the parasite in its human host.5–6 Identification of inhibitors of the Plasmodium aspartic proteases has the potential to provide a novel mechanism for anti-malarial therapies. However, a major challenge to developing protease inhibitors as drugs is identifying compounds with cellular activity commensurate with their enzyme inhibition potency. This is often due to poor physicochemical properties, efflux mechanisms, protein binding or poor target-related biochemical efficiencies.
Rather than focus on a substrate-based inhibitor design approach that may provide potent but non-drug-like peptidomimetics, we elected to identify known, drug-like aspartic protease inhibitors with inherent antimalarial cell activity for optimization. We termed this approach as a “hybrid target-phenotype drug discovery”. Scientists at GlaxoSmithKine (GSK),7–8 Novartis,9 and St. Jude’s Children’s Hospital10 recently screened large chemical libraries for antimalarial activity in a standard Plasmodium falciparum 3D7-infected red blood cell assay. This effort identified ~20,000 compounds with antimalarial activity, and the data were made public. Since a number of aspartic proteases have been shown to play essential roles in the Plasmodium life cycle, we hypothesized that aspartic protease inhibitors with antimalarial activity may be acting through one or more Plasmodium aspartic proteases. Such inhibitors would provide a powerful starting point for drug discovery: phenotypic hits (cell active) with a limited but efficacious set of potential drug targets. By mining these databases for aspartic protease-inhibiting chemotypes, we and others have found multiple inhibitor classes with drug-like properties that may be acting on the parasite through an aspartic protease mechanism due to their structural similarity to known aspartic protease inhibitors.11–13 Indeed, we have shown drug-like compounds from the aminohydantoin series inhibits plasmespins II, IV, V, and X.6, 11 The pyrrolidine class described herein also appears to be particularly drug-like and is ideal for lead optimization to identify novel clinical candidates for treatment of malaria.
In our previous work, we evaluated a set of spiropiperidine hydantoins such as CWHM-505 (1) (Fig. 1).12 While we were able to improve the potency of this series to sub-100 nanomolar, they suffered from particularly poor metabolic stability, presumably due to the cleavage of the benzylic piperidine. A number of pyrrolidine and piperidine-based inhibitors of aspartic proteases BACE, renin and Pf plasmepsin 2 (PM-II) have been reported in the literature (2–5).14–18 These inhibitors have been shown to bind to the aspartic acid residues in the active site via the protonated nitrogen of the pyrrolidine (e.g., PDB ID: 3UFL).14
Figure 1.

Antimalarial spiropiperidine hydantoin and examples of pyrrolidine- and piperidine-based aspartic protease inhibitors. Common basic group is highlighted in blue.
We were attracted to pyrrolidine as an antimalarial pharmacophore as it would avoid the primary metabolic liability in our previous work on benzyl piperidines (e.g., 1) as the basic nitrogen is not substituted in these compounds. Indeed, renin-inhibiting pyrrolidine (4) had been reported to have a two-hour half-life in rats.15 Using the simple unsubstituted pyrrolidine core (Fig. 2), we searched the GSK TCAMS and Novartis malaria phenotypic screening databases and found 45 hits containing this pharmacophore. Many of these hits were aminoquinoline analogs of chloroquine and amodiaquine and were thus eliminated from our consideration for lack of novel mechanism of action. Six compounds, however, were members of the unique antimalarial pharmacophore represented by racemic Novartis compound GNF-Pf-4691 (6; Fig. 2). We resynthesized GNF-Pf-4691 (6) and assayed it in a SYBR Green Pf 3D7 assay19 and found it to have an IC50 of 385 nM, within 3-fold of the reported database value. To our delight, we found this compound to have a 100-fold cytotoxicity selectivity index and an improved metabolic stability profile in mouse liver microsomes. On the basis of this data, we elected to initiate structure-activity relationship (SAR) studies to determine whether this pyrrolidine series should be pursued for full lead optimization. We independently varied the amide benzyl ring and the pyrrolidine aryl ring to develop some preliminary SAR and identified the preferred stereoisomers of the pyrrolidine core. Using this SAR, we combined preferred groups at each position to identify lead compounds suitable for in vivo efficacy studies.
Figure 2.

Strategy to identify phenotypic protease inhibitor-like hits with drug-like properties while eliminating liabilities of earlier series.
Results and Discussion
Synthesis.
It is well established that trans-3,4-disubstituted pyrrolidines can be readily and stereo selectively synthesized through a 3+2 cycloaddition reaction using N-(methoxymethyl)-N-(trimethylsilylmethyl)benzylamine.20–21 Accordingly, racemic trans-α,β-unsaturated esters (8) were prepared by the Wittig reaction and then converted to the corresponding pyrrolidines ((a) through the unstabilized azomethine ylide generated by TFA (Scheme 1). To simplify preparation of the final products, the benzyl group was replaced with Boc (9b). Ester hydrolysis followed by amide coupling and Boc-deprotection yielded the final trans-pyrrolidines (11) as racemates.
Scheme 1. Synthesis of racemic pyrrolidines.

Reagents and conditions: (a) Ph3P=CHCO2Me; (b) TFA, Me3SiCH2N(Bn)CH2OMe; (c) NH4CO2H, Pd/C; (d) Et3N, Boc2O; (e) LiOH; (f) ArCH2NH2, HATU; (g) HCl.
Amide benzyl ring SAR.
Holding the right-hand side constant as 4-trifluoromethylphenyl, we systematically modified the terminal amide aryl ring. Initially new analogs were synthesized as racemates to more efficiently profile the SAR and develop our understanding of the pharmacophore. Representative analogs are shown in Fig. 3. Substitution on the phenyl ring is essential for potency. 4-Methoxy (6) and 4-methyl (12; GNF-Pf-3587) groups give 10-fold increases in potency relative to H (13). The most potent substituent in this position identified to date is dimethylamine (14), approximately two-fold more potent than the lead methoxy analog. Conversion of the dimethyl aniline into an indoline ring (15) was tolerated but extending it as a diethyl aniline (16) reduced potency by 2–3-fold.
Figure 3. Initial amide benzyl ring SAR.

IC50 values are given as average potency values in the Pf 3D7 assay (n≥3). Standards chloroquine and artemesinin have IC50 values of 54 ± 0.4 nM and 33 ± 0.6 nM, respectively.
Pyrrolidine aryl ring SAR.
Holding the amide aryl ring constant as the dimethyl aniline, we systematically explored SAR of the pyrrolidine aryl ring(Fig. 4). This position is also quite sensitive to substitution patterns. 4-CF3 (14) is the preferred substituent with t-butyl (19) being an appropriate replacement, but replacement with methyl (17) or chloro (18) led to 3- to 4-fold reductions in potency. We also explored potential replacements for the trifluoromethyl group such as difluoromethyl (20), CF2CH3 (21), and pentafluorosulfide (22–23). These groups were suitable replacements with IC50 values ranging from 130 to 470 nM.
Figure 4. Initial pyrrolidine aryl ring SAR.

IC50 values are given as average potency values in the Pf 3D7 assay (n≥3).
3,4-Disubstitution is tolerated and may even be favorable for enhancement of potency (24–29). For example, with an IC50 of 83 nM, the 4-CF3-3-Cl analog (26) is one of the most potent racemates we have identified. 2,4-Disubstitution was less well tolerated leading to 6–8-fold reductions in potency (25, 27).
Extension of the aryl ring with phenylether was tolerated in the 4-position (30) but not the 3-position (31) or as a pyridylether (32). Extending the aryl ring itself led to loss of potency (33).
The phenyl ring itself can be replaced with pyridine without loss in potency (34). Incorporation of a second ring nitrogen (e.g., pyrimidine 35) leads to modest erosion of potency. A number of other heterocycles were explored (36–44). Benzothiophenes (36–37, 39), benzofuran (38), and 2-quinoline (40) were all essentially equipotent with 4-trifluoromethylphenyl (14). Thiophene analogs designed to mimic the CF3 and t-butyl phenyl group were tolerated as t-butyl (41) but not so as CF3 (42). Other heterocycles such as thiazole (43) and pyrazole (44) led to >8-fold losses in potency.
Pyrrolidine Stereochemical SAR.
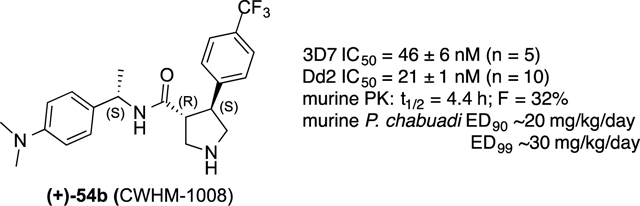
The stereochemistry of the pyrrolidine was also investigated(Fig. 5). The cycloaddition chemistry leads to stereospecific formation of the pyrrolidine ring as a racemic mixture of trans isomers (46a-b). After replacement of the benzyl groups for Boc groups (47a-b) and hydrolysis of the methyl esters to the acids, the (S)-benzyloxazolidinone chiral auxiliary coupled to enable resolution of the resulting diastereomers 48a-b (first eluent) and 49a-b (second eluent) by silica gel chromatography. The isolated diastereomers were then hydrolyzed to furnish the carboxylic acids 50a-b and 51a-b as single enantiomers. These acids were then coupled to benzylic amines and deprotected to give the final products 52–55. The absolute stereochemistry was determined by x-ray crystallography for 54b to be the (3R,4S)-configuration (Fig. 5B).
Figure 5. Synthesis and SAR of pyrrolidine enantiomers 52–55.

(A) IC50 values are given as average potency values in the Pf 3D7 assay (n≥3). Reagents and conditions: (a) Ph3P=CHCO2Me; (b) TFA, Me3SiCH2N(Bn)CH2OMe; (c) NH4CO2H, Pd/C; (d) Et3N, Boc2O; (e) LiOH; (f) (S)-4-benzyl-2-oxazolidinone, pivaloyl chloride, TEA, LiCl; (g) ArCH2NH2, HATU; (h) HCl. (B) ORTEP representation of the crystal structure of 54b as an HCl salt.
There is a modest three-fold difference in potency between (3R,4S)-54a and (3S,4R)-52a enantiomers with the (3R,4S)-configuration being preferred (Fig. 5A). In contrast, stereochemistry at the α-benzyl carbon has a dramatic effect on potency with the (S)-Me group being tolerated (53) and the (R)-Me group being detrimental to potency by >30-fold (55). Comparing pyridine analog 54a versus phenyl analog 54b shows a nearly 3-fold preference for phenyl. A similar relationship holds for pyridine analog 52a versus phenyl analog 52b.
Revisiting the amide aryl ring SAR.
Having identified the (3R,4S) stereochemistry as preferred, we revisited our studies on the amide aryl ring SAR using the 4-trifluoropyridine moiety as the pyrrolidine aryl ring substituent as this would give lower lipophilicity. As described in the synthesis section, we utilized enantiomer 50a as a synthetic intermediate to derive a series of analogs. SAR with the 4-trifluoropyridine moiety is shown in Fig. 6. As with the 4-trifluoromethylphenyl series, the dimethylaniline is preferred (34). Modest changes to pyridine (56), pyrrolidine (57), and N-pyrazole (58) led to modest to dramatic losses in potency. Other substitutions (59–61) and extension of the dimethylamine (62) were not tolerated.
Figure 6. Amide aryl ring SAR with 4-trifluoromethylpyridine.

IC50 values are given as average potency values in the Pf 3D7 assay (n≥3).
Pyrrolidines are potent on the drug-resistant Dd2 strain of P. falciparum.
With an IC50 value of 51 nM, compound 54b was selected as our lead compound for further profiling. 54b was profiled in extensive side-by-side studies with chloroquine (CQ) in the multi-drug resistant Dd2 strain of P. falciparum. 54b has equivalent potency to CQ in the Pf 3D7 strain (IC50 = 46 ± 6 Nm vs. CQ IC50 = 38 ± 2 nM) and 10-fold greater potency than CQ against the multi-drug resistant Dd2 (IC50 = 21 ± 1 nM vs. CQ IC50 = 196 ± 14 nM).
Inhibition of Aspartic Proteases.
Since our original hypothesis was that these pyrrolidines might be aspartic protease inhibitors, we profiled a select set of seven compounds for inhibition of human β-secretase (BACE1), Pf plasmepsin II (PM-II) and Pf plasmepsin IV (PM-IV) enzymes (Table 1; entries 1–7). However, none of the seven compounds inhibited these aspartic proteases. 6 was also tested for inhibition of Pf PM-V, PM-IX and PM-X in a knockdown assay (PM-V) and in western blots looking for PM-IX and X substrate processing in compound-treated parasites but was found to be inactive against these proteases (data not shown).6,11,22–23 Thus it appears that these pyrrolidines are not inhibitors of plasmepsins II, IV, V, IX or X. To date, we have not identified a biomolecular target for these pyrrolidines, and we cannot rule out inhibition of other Plasmodium aspartic proteases.
Table 1.
In vitro Profiling Data
| Entry | Cmpd | Pf 3D7 IC50, nM | BACE1 IC50, nM | Pf PM-II IC50, nM | Pf PM-IV IC50, nM | hERG IC50, nM | hERG/3D7 Ratio | MLM t1/2, min | MLM Clint, μL/min/mg |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 12 | 460 | >10,000 | >10,000 | >10,000 | 8,950 | 19 | 161 | 1.4 |
| 2 | 14 | 200 | >10,000 | >10,000 | >10,000 | 9,410 | 47 | 35 | 6.2 |
| 3 | 16 | 600 | >10,000 | >10,000 | >10,000 | 4,740 | 8 | n.d. | n.d. |
| 4 | 24 | 140 | >10,000 | >10,000 | >10,000 | 8,540 | 61 | 34 | 6.4 |
| 5 | 26 | 83 | >10,000 | >10,000 | >10,000 | 7,560 | 91 | 33 | 6.7 |
| 6 | 30 | 170 | >10,000 | >10,000 | >10,000 | 2,110 | 12 | 31 | 7.2 |
| 7 | 34 | 230 | >10,000 | >10,000 | >10,000 | 21,700 | 94 | 70 | 3.1 |
| 8 | 35 | 450 | n.d. | n.d. | n.d. | >50,000 | >111 | n.d. | n.d. |
| 9 | 54a | 140 | n.d. | n.d. | n.d. | >50,000 | >357 | n.d. | n.d. |
| 10 | 54b | 51 | n.d. | n.d. | n.d. | 32,000 | 627 | 38 | n.d. |
n.d. = not determined.
Affinity for the hERG channel.
A potential concern with this series is its potential for binding the hERG channel given all of these compounds contain a basic amine pyrrolidine core.24–25 To address this, we evaluated ten compounds in this series for hERG binding in a competitive binding assay (Table 1). Compounds tested have binding affinities for hERG ranging from 2 μM to >50 μM. While some of the compounds have modest 8- to 20-fold hERG/3D7 ratios, our best compounds had hERG binding affinities of >20 μM and selectivity ratios of 90- to 600-fold. Compounds with a pyrrolidine phenyl ring (entries 1–6) tended to have stronger affinity for the hERG channel versus pyridine (34 and 54a) and pyrimidine (35) (entries 7–9). The exception to this trend is 54b (entry 10) which had a >600-fold selectivity vs. the hERG channel. It is possible that this is due to this compound being tested as a single enantiomer or the chiral α-methyl group may also reduce hERG potency.
In vitro and in vivo pharmacokinetics.
Six compounds were profiled for metabolic stability in mouse liver microsomes (MLM; Table 1). All six compounds were moderately to very stable with MLM half-lives ranging from 31 to 161 min. 12 and 34 and 54b were selected for mouse PK studies (Table 2). 12 has relatively high clearance and a modest half-life in mice. In contrast, and 34 and 54b have good half-lives and low clearance in mice. 54b was found to have suitable oral bioavailability for in vivo efficacy studies.
Table 2.
Mouse Pharmacokinetic Data
| Compound | Route | Animals | Dose, mg/kg | t1/2, h | CLz, mL/min/kg | Vz, L/kg | F, % |
|---|---|---|---|---|---|---|---|
| 12 | i.v. | 4 | 1 | 1.7 | 52 | 7.3 | - |
| 34 | i.v. | 4 | 2 | 6.6 | 5.9 | 3.2 | - |
| 34 | p.o. | 4 | 5 | - | - | - | 13 |
| 54b | i.v. | 4 | 2 | 4.4 | 3.3 | 1.3 | - |
| 54b | p.o. | 3 | 5 | - | - | - | 32 |
CLz = apparent rate of clearance; Vz = apparent volume of distribution.
54b is orally efficacious in a mouse model of malaria.
Given the oral bioavailability of 54b, we evaluated it as a tool compound to provide in vivo proof-of-concept in the murine Peters 4-day suppressive test using P. chabaudi ASCQ (a CQ-resistant strain).19, 26–27 NIH mice were inoculated with P. chabaudi ASCQ parasitized red blood cells. After 4 h, 54b was dosed orally at 3, 10 and 30 mg/kg/day once daily for four days. Parasitemia levels were determined 24 h after the last treatment (Table 3). At 30 mg/kg/day, 54b inhibits parasitemia at 98.7%. The dose-response data allow us to approximate an ED90 of ~20 mg/kg/day and an ED99 of ~30 mg/kg/day. Compound concentrations in the plasma were determined at 1 h, 6 h and 24 h post dose on the last day of treatment. Compound concentrations in the plasma (110 nM at 24 h) remained above the Pf 3D7 IC50 (46 nM) for the full 24 h period only for the most fully efficacious dose of 30 mg/kg/day.
Table 3.
In vivo efficacy of pyrrolidine 54b in P. chabaudi ASCQ infected mice.
| Oral Dose (qd) | % Inh. of Growth | Plasma Compound Concentration (nM) | ||
|---|---|---|---|---|
| 1h | 6h | 24 h | ||
| 54b, 3 mg/kg/day | 22.4 ± 11.6 | 640 | 140 | 0 |
| 54b, 10 mg/kg/day | 75.2 ± 10.6 | 1,160 | 960 | 10 |
| 54b, 30 mg/kg/day | 98.7 ± 0.2 | 2,150 | 1,630 | 110 |
| CQ, 10 mg/kg/day | 95.0 ± 1.1 | - | - | - |
Inhibition of parasitemia after 4 days of qd oral dosing and plasma compound concentrations on Day 4. n=6 animals/group.
Conclusions
Herein, we describe the profiling of aryl pyrrolidine carboxamides as a novel class of antimalarial agents. We have generated an extensive SAR for the terminal aryl rings. The SAR is rather narrow in that relatively few substituents are permissible without significant losses in potency. On the carboxamide aryl ring, dimethylaniline is vastly superior to nearly every functional group evaluated. On the pyrrolidine aryl ring, para-trifluoromethyl can be replaced by a limited set of small lipophilic moieties, including t-butyl, CHF2, and CF2CH3. Addition of a halogen in the meta-position is beneficial. Replacement of the phenyl ring with heterocycles such as pyridine, pyrimidine, benzofuran and thiophene, is also tolerated. (3R,4S) is the preferred stereochemistry on the pyrrolidine ring and (S)-Me is preferred in the benzyl position. Out of this effort, we identified 54b as a lead compound for further profiling. 54b is more potent on the drug resistant Dd2 strain, has suitable selectivity over the hERG channel, has a long half-life in mice, and is orally efficacious in a mouse model of malaria (q.d.; ED99 ~ 30 mg/kg/day). Thus, compound 54b (CWHM-1008) presents a promising lead for optimization as an antimalarial drug with a low molecular weight, modest lipophilicity, excellent antimalarial potency, and long half-life oral bioavailability in mice.
Experimental Section
General.
Commercially obtained reagents were used without further purification. All reactions were monitored by TLC with silica gel-coated plates. Chemical structures and IUPAC names were generated using CambridgeSoft ChemDraw Ultra 10.0 or CDD Vault (www.collaborativedrug.com). Specific rotation value were recorded on AUTOPOL®μOJ-H, 4.6mm × 250mm 5 IV-T (γ= 589 nm, 50 mm cell, 20°C). MS analyses were performed on the API 2000 electrospray mass spectrometer in positive/negative ion mode. The scan range was 100–1000d. 1H NMR spectra were recorded on a Bruker AV-400 or 500 MHz spectrometer. Chemical shifts (δ) are given in relative to tetramethylsilane (δ 0.00 ppm) in CDCl3. Coupling constants, J, were reported in hertz unit (Hz). All compounds were ≥95% pure by HPLC conducted on an Agilent 1260 system using a reverse phase C18 column with diode array detector and a methanol/water (0.1% NH4OH) gradient unless stated otherwise. HRMS spectra were recorded on an ABSciex 5600+ instrument.
Compounds were filtered for PAINS substructures using the PAINS (Pan-assay interference compounds) filters available at http://fafdrugs4.mti.univ-paris-diderot.fr. 38 of the 47 final compounds described here have the dialkyl aniline substructure PAIN alert. However, this substructure is only considered an interference substructure for AlphaScreen technology as they can be quenchers of singlet oxygen and are not considered general PAINS.28 The assays used herein are cell-based assays and include correlation of compound potency from cell based assays to in vivo efficacy models.
(±)-(3R,4S)-methyl1-benzyl-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-3-carboxylate (46a).
To a stirred solution of methyl (triphenylphosphoranylidene)acetate (20.1 g,60.0 mmol) in dichloromethane (100 mL) was added 6-(trifluoromethyl)nicotinaldehyde (10.0g,57.1 mmol) at 0 °C, and then the resulting mixture was stirred at room temperature for 4 h . The reaction was monitored by TLC (10% ethyl acetate in petroleum ether). The reaction was concentrated in vacuo. Flash chromatography (petroleum ether/ethyl acetate: 95/5) afforded (E)-methyl 3-(6-(trifluoromethyl)pyridin-3-yl)acrylate (13.2 g, 99 % yield) as a white solid.
To a stirred solution of (E)-methyl 3-(6-(trifluoromethyl)pyridin-3-yl)acrylate (13.2 g, 57.1 mmol) in dichloromethane (150 mL) was added N-(methoxymethyl)-N-(trimethylsilymethyl)-benzylamine (17.8 g, 74.3 mmol). The resulting mixture was cooled to 0 °C and a solution of TFA (0.8 mL, 0.1 eq) in dichloromethane (2.0 mL) was added dropwise. The reaction mixture was allowed to warm to room temp and stirred for 16 h. The solvent was removed by evaporation in vacuo and the resulting oil was purified by flash column chromatography (petroleum ether/ethyl acetate: 80/20) to give the title compound (16.8 g, 81% yield). MS: m+1=365.3.
(±)-(3R,4S)-1-tert-butyl3-methyl4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-1,3-dicarboxylate (47a).
To a flask was added 46a (16.8 g, 46.2 mmol), ammonium formate (9.2 g,146 mmol), Pd/C (1.7 g, 10%) and MeOH (200 mL). The reaction flask was flushed with argon three times and then heated at 70°C for 30min. The reaction was cooled to room temp and filtered through Celite®, rinsing with MeOH (50 mL). To this mixture was added triethylamine (23.3 g, 231 mmol), then cooled to 0 °C and di-tert-butyl decarbonate (30.2 g ,139 mmol) was added dropwise. The reaction mixture was allowed to warm to room temp and stirred for 16 h. The solvent was removed by evaporation in vacuo and the resulting oil was purified by flash column chromatography (petroleum ether/ethyl acetate: 80/20) to give the title compound (15.6 g, 90% yield). MS: m+1=375.2.
(+)-(3R,4S)-1-(tert-butoxycarbonyl)-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-3-carboxylic acid (51a) and (−)-(3S,4R)-1-(tert-butoxycarbonyl)-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-3-carboxylic acid (51a).
To a flask was added 47a (15.6 g, 44.4 mmol) and methanol (150 mL). A solution of LiOH (4.7 g, 111 mmol) in water (100 mL) was added dropwise. The reaction mixture was stirred at room temperature for 4 h. The most of methanol was removed by evaporation, diluted with water and basified with hydrochloric acid (2M) to pH 4. The mixture was extracted with EtOAc (3 × 150 mL). The combined organic extracts were washed with water then brine, dried over sodium sulfate, filtered and concentrated to afford (±)-(3R,4S)-1-(tertbutoxycarbonyl)-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-3-carboxylic acid (14.5 g, 91% yield).
To a suspension of (±)-(3R,4S)-1-(tert-butoxycarbonyl)-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-3-carboxylic acid (14.5 g, 40.3 mmol) in anhydrous THF (300 mL) was added triethylamine (10.2 g, 101 mmol). The resulting mixture was cooled to −20 °C and a solution of pivaloyl chloride (5.8g, 48.4 mmol) in THF (20 mL) was added dropwise. The reaction mixture was stirred at −20 °C for 2 h and then added LiCl (1.8 g, 44.3 mmol) was added followed by benzyl-2-oxazolidinone (7.1 g, 40.3 mmol). The reaction mixture was allowed to warm to room temp and stirred for 4 h. The solvent was removed by evaporation in vacuo and the resulting oil was purified by flash column chromatography (petroleum ether/ethyl acetate: 90/10) to separate the mixture of 48a and 49a.
The first eluting compound was (3R,4S)-tert-butyl3-((S)-4-benzyl-2-oxooxazolidine-3-carbonyl)-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-1-carboxylate (48a) obtained as a white solid (6.7 g, 32% yield). TLC Rf = 0.52 (petroleum ether/ethyl acetate: 3/1). To a flask was added 48a (6.7 g, 12.9 mmol) and THF (100 mL). A solution of LiOH (1.1 g, 25.8 mmol) in water (25 mL) was added dropwise. The reaction mixture was stirred at room temperature for 4 h. The most of methanol was removed by evaporation, diluted with water and basified with hydrochloric acid (2M) to pH 4. The mixture was extracted with EtOAc (3×150 mL). The combined organic extracts were washed with water then brine, dried over sodium sulfate, filtered and concentrated to afforded (+)-(3R,4S)-1-(tert-butoxycarbonyl)-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-3-carboxylic acid (50a) (3.8 g, 82% yield). MS: m-1=359.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.74 (s, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 3.81 (t, J = 8.8 Hz, 1H), 3.73 (td, J = 8.8,1.6 Hz, 1H), 3.67 (m, 1H), 3.49–3.36 (m, 2H), 3.29 (t, J = 10.0 Hz, 1H), 1.41 (d, J = 8.4 Hz, 9H). [α]D20 +30.1 (c 0.09, MeOH).
The second eluting compound was diastereomer (3S,4R)-tert-butyl3-((S)-4-benzyl-2-oxooxazolidine-3-carbonyl)-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-1-carboxylate (49a) obtained as a white solid (7.1 g, 34% yield). TLC Rf = 0.41 (petroleum ether/ethyl acetate: 3/1). 49a was hydrolyzed with LiOH as described above to give (−)-(3S,4R)-1-(tert-butoxycarbonyl)-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-3-carboxylic acid (51a) (3.7 g, 75% yield). MS: m-1=359.2. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.75 (s, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 3.81 (t, J = 8.8 Hz, 1H), 3.77–3.66 (m, 2H), 3.44 (br, 2H), 3.30 (t, J = 9.6 Hz, 1H), 1.41 (d, J = 9.6 Hz, 9H). [α]D20 −30.4 (c 0.13, MeOH).
(+)-(3R,4S)-N-(4-(dimethylamino)benzyl)-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-3-carboxamide (53).
To a suspension of 50a (400 mg, 1.11 mmol), 4-(aminomethyl)-N,N-dimethylaniline (200 mg, 1.33 mmol), HATU (630 mg, 1.66 mmol) in dichloromethane (10 mL) was added triethylamine (0.31 ml, 2.22 mmol). The reaction mixture was stirred at room temp for 16 h. The mixture was diluted with dichloromethane (30 mL) and washed with saturated sodium carbonate solution then brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography (petroleum ether/ethyl acetate: 1/1) to give (330 mg, 61% yield) of (3R,4S)-tert-butyl3-(4-(dimethylamino)benzylcarbamoyl)-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-1-carboxylate as a white solid.
To a suspension of (3R,4S)-tert-butyl3-(4-(dimethylamino)benzylcarbamoyl)-4-(6-(trifluoromethyl)pyridin-3-yl)pyrrolidine-1-carboxylate (330 mg, 0.67 mmol), in dichloromethane (6 mL) was added TFA (3.0 ml). The reaction mixture was stirred at room temp for 2 h. The solvent was removed by evaporation in vacuo and the resulting oil was diluted with dichloromethane (30 mL) and washed with saturated sodium carbonate solution then brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography (DCM/NH3 in MeOH: 20/1) to give the title compound 160 mg (61% yield). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.67(s, 1H), 8.25(t, J=5.5, 1H),7.96 (d, J=8.0, 1H), 7.83 (d, J=8.0, 1H), 6.86 (d, J=8.5, 2H), 6.58(d, J=8.5, 2H), 4.18(dd, J=14.5, 6.0, 1H), 4.01(dd, J=14.5, 6.0, 1H),3.52(q, J=10.5, 1H), 3.26 (t, J=10.0, 1H), 2.99 (q, J=8.0, 1H), 2.97(q, J=8.0, 1H),2.85 (t, J=10.0, 1H), 2.83 (s, 6H). [α]D20 +73.2 (c 0.11, MeOH). HRMS (ESI) m/z: [M + H]+ Calcd for C20H24F3N4O 393.1902; found 393.1889.
(−)-(3S,4R)-N-[(1S)-1-[4-(dimethylamino)phenyl]ethyl]-4-[6-(trifluoromethyl)pyridin-3-yl]pyrrolidine-3-carboxamide (52a).
The title compound was synthesized from 51a and (S)-4-(1-aminoethyl)-N,N-dimethylbenzenamine as according to the method described for 53. Yellow solid (110 mg, 54% yield). 1H NMR (400 MHz, DMSOd6) δ ppm 8.66 (s, 1H), 8.15 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.06 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 4.79 (m, 1H), 3.36 (m, 1H), 3.30 (1H, overlapped with the peak of H2O), 3.20 (m, 1H), 2.90–2.84 (m, 9H), 1.23 (s, 1H), 1.19 (t, J = 6.8 Hz, 3H). [α]D20 −122.4 (c 0.12, MeOH). HRMS (ESI) m/z: [M + H]+ Calcd for C21H26F3N4O 407.2058; found 407.2043.
(−)-(3S,4R)-N-[(1S)-1-[4-(dimethylamino)phenyl]ethyl]-4-[4-(trifluoromethyl)phenyl]pyrrolidine-3-carboxamide (52b).
The title compound was synthesized from 51b and (S)-4-(1-aminoethyl)-N,N-dimethylbenzenamine as according to the method described for 53. White solid, 95 mg (56% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.10 (d, J = 8.4Hz, 1H), 7.65 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.06 (d, J = 8.4 Hz, 2H), 6.66 (d, J = 8.4 Hz, 2H), 4.79 (m, 1H), 3.49 (q, J = 16.0 Hz, 1H), 3.30 (1H, overlapped with the peak of H2O), 3.18 (t, J = 8.0 Hz, 1H), 2.90–2.80 (m, 7H), 2.75 (dd, J = 14.4,8.8 Hz, 1H). [α]D20 −156.7 (c 0.10, MeOH). HRMS (ESI) m/z: [M + H]+ Calcd for C22H27F3N3O 406.2106; found 406.2102.
(+)-(3R,4S)-N-[(1S)-1-[4-(dimethylamino)phenyl]ethyl]-4-[6-(trifluoromethyl)pyridin-3-yl]pyrrolidine-3-carboxamide (54a).
The title compound was synthesized from 50a and (S)-4-(1-aminoethyl)-N,N-dimethylbenzenamine as according to the method described for 53. White solid, 78 mg (48% yield). 1H NMR (400 MHz, DMSOd6) δ ppm 8.62 (s, 1H), 8.15 (d,J = 8.0 Hz , 1H), 7.90 (dd, J = 8.0 Hz, 1H), 7.81 (d, J = 8.0 Hz, 1H), 6.78 (d, J = 8.4 Hz, 2H), 6.51 (d, J = 8.8 Hz, 2H), 4.79 (m, 1H), 3.41 (m, 1H), 3.30 (2H, overlapped with the peak of H2O), 3.20 (t, J = 7.2 Hz, 1H), 2.98 (m, 2H), 2.85 (t, J = 6.4 Hz, 1H), 2.83 (s, 6H), 1.24 (d, J = 7.2 Hz, 3H). [α]D20 +38.3 (c 0.12, MeOH). HRMS (ESI) m/z: [M + H]+ Calcd for C21H26F3N4O 407.2058; found 407.2048.
(+)-(3R,4S)-N-[(1S)-1-[4-(dimethylamino)phenyl]ethyl]-4-[4-(trifluoromethyl)phenyl]pyrrolidine-3-carboxamide (54b).
The title compound was synthesized from 50b and (S)-4-(1-aminoethyl)-N,N-dimethylbenzenamine as according to the method described for 53. White solid, 125 mg (63% yield). 1H NMR (500 MHz, DMSO-d6) δ ppm 8.09 (d, J = 8.0 Hz, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H), 6.83 (d, J = 8.5 Hz, 2H), 6.53 (d, J = 8.5 Hz, 2H), 4.81 (m, 1H), 3.39 (q, J = 17.0 Hz, 1H), 3.28 (t, J = 9.5 Hz, 1H), 3.19 (t, J = 8.0 Hz, 1H), 2.97 (t, J = 8.0 Hz, 1H), 3.39 (q, J = 16.0 Hz, 1H), 2.82 (s, 6H), 2.77 (t, J = 10.0 Hz, 1H), 1.25 (d, J = 8.0 Hz, 3H). [α]D20 +39.1 (c 0.14, MeOH). HRMS (ESI) m/z: [M + H]+ Calcd for C22H27F3N3O 406.2106; found 406.2098.
(+)-(3R,4S)-N-[(1R)-1-[4-(dimethylamino)phenyl]ethyl]-4-[6-(trifluoromethyl)pyridin-3-yl]pyrrolidine-3-carboxamide (55).
The title compound was synthesized from 50a and (R)-4-(1-aminoethyl)-N,N-dimethylbenzenamine as according to the method described for 53. White solid, 102 mg (58% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.66 (s, 1H), 8.16 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.05 (d, J = 8.0 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 4.78 (m, 1H), 3.50 (q, J = 15.2 Hz, 1H), 3.30 (2H, overlapped with the peak of H2O), 3.20 (t, J = 8.0 Hz, 1H), 2.87–2.67 (m, 8H), 1.18 (d, J = 6.8 Hz, 3H). [α]D20 +149.6 (c 0.12, MeOH). HRMS (ESI) m/z: [M + H]+ Calcd for C21H26F3N4O 407.2058; found 407.2057.
Biological Assays.
Aspartic protease enzyme assays, mouse liver microsome assays, in vivo PK studies, in vivo efficacy studies were performed as we have previously described.11
In vitro Antimalarial Assays (3D7 and Dd2).
In vitro antimalarial activity was determined by a malaria SYBR Green I-based fluorescence (MSF) method described previously by Smilkstein et al.29 with slight modification.30 Stock solutions of each test drug were prepared in DMSO at a concentration of 20 mM. The drug solutions were serially diluted with culture medium and distributed to asynchronous parasite cultures on 96-well plates in quadruplicate in a total volume of 100 μl to achieve 0.5% parasitemia with a 2% hematocrit in a total volume of 100 μl. The plates were then incubated for 72 h at 37°C. After incubation, 100 μl of lysis buffer with 0.2 μl/ml SYBR Green I was added to each well. The plates were incubated at 37°C for an hour in the dark and then placed in a 96-well fluorescence plate reader (Spectramax Gemini- EM; Molecular Diagnostics) with excitation and emission wavelengths at 497 nm and 520 nm, respectively, for measurement of fluorescence. The 50% inhibitory concentration (IC50) was determined by nonlinear regression analysis of logistic dose-response curves (GraphPad Prism software). Antimalarial potency of compounds was determined by this technique for both Plasmodium falciparum 3D7 (CQ-sensitive) and Dd2 (multi-drug resistant) strains. 3D7 assay results are given as an average of at least three replicates.
In vivo Antimalarial Efficacy Suppressive Assay.
In vivo antimalarial activity was determined for 54b against the rodent CQ-resistant Plasmodium chabaudi ASCQ strain according to the 4-day suppressive test.19, 31 Briefly, 5-week old female NIH mice (n = 6 per group) were inoculated i.p. with 2×107 parasitized (Plasmodium chabaudi ASCQ) red blood cells. Thereafter, the compounds were administered as a suspension in 0.5% carboxymethyl cellulose orally to the animals once daily at 4 h, 24 h, 48 h and 72 h post inoculation. Groups including a vehicle control and chloroquine (CQ) as a reference drug were included. Parasitemia levels were determined on the day following the last treatment (Day 4). Plasma drug concentration was determined by LCMS using 100 |μL orbital blood plasma sample from Day 3, which was collected at 1 h, 6 h, 24 h after dosing.
The Institutional Animal Care and Use Committee at the Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, reviewed and approved the animal use in these studies. The animal care and use program is run entirely according to Association for Assessment and Accreditation of Laboratory Animal Care, International (AAALACi) standards and is assured by the Office of Laboratory Animal Welfare (OLAW identification number A5748-01).
Fluorescence polarization hERG assay.
The inhibition activity of compounds on hERG potassium channel was determined using Predictor® hERG Fluorescence Polarization Assay Kit (Life Technologies, Carlsbad, CA, USA). Experiments were performed according to the manufacturer’s instructions. In brief, the reactions were carried out in 384-well plates including 10 μL of Predictor® hERG Membrane and 10 μL Predictor® hERG Tracer Red with appropriate amount of compound or positive control. Reactions were incubated for 2 h at room temp and then read on an EnVision Multilabel Reader (Perkin Elmer, Inc.) using polarized excitation and emission filters. Data were analyzed using Graphpad Prism 5 (GraphPad Software Inc., San Diego, CA).
Supplementary Material
Acknowledgments.
The authors would like to thank Dr. Bryan Yeung for helpful discussions and Mr. Chris Eickhoff for assaying compounds.
Funding. Research reported in this publication conducted at Saint Louis University, was supported by Saint Louis University and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI106498. Research reported in this publication conducted at the Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, was supported by Bureau of Science and Information Technology of Guangzhou Municipality Grant number 2009Z1-E841 and Natural Science Foundation of China (NSFC) and by the Ministry of Sciences and Technology Key Program (No. 2016YFE0107300). DEG is supported by the National Institutes of Health under Award Number AI047798. The content is solely the responsibility of the authors and does not necessarily represent the official views of Saint Louis University, the Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, the Natural Science Foundation of China, or the National Institutes of Health.
Abbreviations.
- ACT
artemisinin combination therapy
- Pf
Plasmodium falciparum
- PM
plasmepsin
- SI
Selectivity Index
- TCAMS
Tres Cantos Antimalarial Set
- MLM
mouse liver microsomes
- CQ
chloroquine
- mg/kg/day
milligrams per kilogram
Footnotes
Conflict of Interest: The authors declare no competing financial interest.
Supporting Information. Synthetic procedures and characterization data for final products 6, 12–44, and 56–62, 1H and 13C NMR spectra for 54b, and x-ray crystallographic data for 54b. Molecular Formula Strings are provided as a .csv file. This information is available free of charge on the ACS Publications Website at DOI: 10.1021/acs.jmedchem.8b01972.
References
- 1.World Malaria Report 2015; World Health Organization: Geneva, 2015. [Google Scholar]
- 2.World Malaria Report 2018; World Health Organization: Geneva, 2018. [Google Scholar]
- 3.Dondorp AM; Yeung S; White L; Nguon C; Day NP; Socheat D; von Seidlein L, Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol 2010, 8 (4), 272–280. [DOI] [PubMed] [Google Scholar]
- 4.Artemisinin resistance and artemisinin-based combination therapy efficacy Global Malaria Programme Status Report; World Health Organization: Geneva, 2018. [Google Scholar]
- 5.Meyers MJ; Goldberg DE, Recent advances in plasmepsin medicinal chemistry and implications for future antimalarial drug discovery efforts. Curr Top Med Chem 2012, 12 (5), 445–455. [DOI] [PubMed] [Google Scholar]
- 6.Nasamu AS; Glushakova S; Russo I; Vaupel B; Oksman A; Kim AS; Fremont DH; Tolia N; Beck JR; Meyers MJ; Niles JC; Zimmerberg J; Goldberg DE, Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science 2017, 358 (6362), 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gamo F-J; Sanz LM; Vidal J; de Cozar C; Alvarez E; Lavandera J-L; Vanderwall DE; Green DVS; Kumar V; Hasan S; Brown JR; Peishoff CE; Cardon LR; Garcia-Bustos JF, Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465 (7296), 305–310. [DOI] [PubMed] [Google Scholar]
- 8.Calderon F; Barros D; Bueno JM; Coteron JM; Gamo F-J; Lavandera J-L; Leon ML; Macdonald SJF; Mallo A; Manzano P; Porras E; Fiandor JM; Castro J; Fernandez E, An invitation to open innovation in malaria drug discovery: 47 quality starting points from the TCAMS. ACS Med. Chem. Lett 2011, 2 (10), 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meister S; Plouffe DM; Kuhen KL; Bonamy GM; Wu T; Barnes SW; Bopp SE; Borboa R; Bright AT; Che J; Cohen S; Dharia NV; Gagaring K; Gettayacamin M; Gordon P; Groessl T; Kato N; Lee MC; McNamara CW; Fidock DA; Nagle A; Nam TG; Richmond W; Roland J; Rottmann M; Zhou B; Froissard P; Glynne RJ; Mazier D; Sattabongkot J; Schultz PG; Tuntland T; Walker JR; Zhou Y; Chatterjee A; Diagana TT; Winzeler EA, Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 2011, 334 (6061), 1372–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guiguemde WA; Shelat AA; Bouck D; Duffy S; Crowther GJ; Davis PH; Smithson DC; Connelly M; Clark J; Zhu F; Jimenez-Diaz MB; Martinez MS; Wilson EB; Tripathi AK; Gut J; Sharlow ER; Bathurst I; El Mazouni F; Fowble JW; Forquer I; McGinley PL; Castro S; Angulo-Barturen I; Ferrer S; Rosenthal PJ; Derisi JL; Sullivan DJ; Lazo JS; Roos DS; Riscoe MK; Phillips MA; Rathod PK; Van Voorhis WC; Avery VM; Guy RK, Chemical genetics of Plasmodium falciparum. Nature 2010, 465 (7296), 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyers MJ; Tortorella MD; Xu J; Qin L; He Z; Lang X; Zeng W; Xu W; Qin L; Prinsen MJ; Sverdrup FM; Eickhoff CS; Griggs DW; Oliva J; Ruminski PG; Jacobsen EJ; Campbell MA; Wood DC; Goldberg DE; Liu X; Lu Y; Lu X; Tu Z; Lu X; Ding K; Chen X, Evaluation of aminohydantoins as a novel class of antimalarial agents. ACS Medicinal Chemistry Letters 2014, 5, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyers MJ; Anderson EJ; McNitt SA; Krenning TM; Singh M; Xu J; Zeng W; Qin L; Xu W; Zhao S; Qin L; Eickhoff CS; Oliva J; Campbell MA; Arnett SD; Prinsen MJ; Griggs DW; Ruminski PG; Goldberg DE; Ding K; Liu X; Tu Z; Tortorella MD; Sverdrup FM; Chen X, Evaluation of spiropiperidine hydantoins as a novel class of antimalarial agents. Bioorg Med Chem 2015, 23 (16), 5144–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaudzems K; Tars K; Maurops G; Ivdra N; Otikovs M; Leitans J; Kanepe-Lapsa I; Domraceva I; Mutule I; Trapencieris P; Blackman MJ; Jirgensons A, Plasmepsin inhibitory activity and structure-guided optimization of a potent hydroxyethylamine-based antimalarial hit. ACS Med Chem Lett 2014, 5 (4), 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stachel SJ; Steele TG; Petrocchi A; Haugabook SJ; McGaughey G; Katharine Holloway M; Allison T; Munshi S; Zuck P; Colussi D; Tugasheva K; Wolfe A; Graham SL; Vacca JP, Discovery of pyrrolidine-based beta-secretase inhibitors: lead advancement through conformational design for maintenance of ligand binding efficiency. Bioorg Med Chem Lett 2012, 22 (1), 240–244. [DOI] [PubMed] [Google Scholar]
- 15.Lorthiois E; Breitenstein W; Cumin F; Ehrhardt C; Francotte E; Jacoby E; Ostermann N; Sellner H; Kosaka T; Webb RL; Rigel DF; Hassiepen U; Richert P; Wagner T; Maibaum J, The discovery of novel potent trans-3,4-disubstituted pyrrolidine inhibitors of the human aspartic protease renin from in silico three-dimensional (3D) pharmacophore searches. J Med Chem 2013, 56 (6), 2207–2217. [DOI] [PubMed] [Google Scholar]
- 16.Luksch T; Blum A; Klee N; Diederich WE; Sotriffer CA; Klebe G, Pyrrolidine derivatives as plasmepsin inhibitors: binding mode analysis assisted by molecular dynamics simulations of a highly flexible protein. ChemMedChem 2010, 5 (3), 443–454. [DOI] [PubMed] [Google Scholar]
- 17.Efremov IV; Vajdos FF; Borzilleri KA; Capetta S; Chen H; Dorff PH; Dutra JK; Goldstein SW; Mansour M; McColl A; Noell S; Oborski CE; O’Connell TN; O’Sullivan TJ; Pandit J; Wang H; Wei B; Withka JM, Discovery and optimization of a novel spiropyrrolidine inhibitor of beta-secretase (BACE1) through fragment-based drug design. Journal o f Medicinal Chemistry 2012, 55 (21), 9069–9088. [DOI] [PubMed] [Google Scholar]
- 18.Ostermann N; Ruedisser S; Ehrhardt C; Breitenstein W; Marzinzik A; Jacoby E; Vangrevelinghe E; Ottl J; Klumpp M; Hartwieg JC; Cumin F; Hassiepen U; Trappe J; Sedrani R; Geisse S; Gerhartz B; Richert P; Francotte E; Wagner T; Kromer M; Kosaka T; Webb RL; Rigel DF; Maibaum J; Baeschlin DK, A novel class of oral direct renin inhibitors: highly potent 3,5-disubstituted piperidines bearing a tricyclic p3-p1 pharmacophore. J Med Chem 2013, 56 (6), 2196–2206. [DOI] [PubMed] [Google Scholar]
- 19.Li X; He Z; Chen L; Li Y; Li Q; Zhao S; Tao Z; Hu W; Qin L; Chen X, Synergy of the antiretroviral protease inhibitor indinavir and chloroquine against malaria parasites in vitro and in vivo. Parasitol Res 2011, 109 (6), 1519–1524. [DOI] [PubMed] [Google Scholar]
- 20.Hosomi A; Sakata Y; Sakurai H, Chemistry of organosilicon compounds. 195. N-(Trimethylsilylmethyl)aminomethyl ethers as azomethine ylide synthons. A new and convenient access to pyrrolidine derivatives. Chemistry Letters 1984, (7), 1117–1120. [Google Scholar]
- 21.Terao Y; Kotaki H; Imai N; Achiwa K, Trifluoroacetic acid-catalyzed 1,3-cycloaddition of the simplest iminium ylide leading to 3- or 3,4-substituted pyrrolidines and 2,5-dihydropyrroles. Chemical & Pharmaceutical Bulletin 1985, 33 (7), 2762–2766. [Google Scholar]
- 22.Russo I; Babbitt S; Muralidharan V; Butler T; Oksman A; Goldberg DE, Plasmepsin V licenses Plasmodium proteins for export into the host erythrocyte. Nature 2010, 463 (7281), 632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldberg DE; Niles J; Polino AJ; Nasamu AS, Assessment of biological role and insight into druggability of the Plasmodium falciparum protease plasmepsin V. bioRxiv 2018. DOI: 10.1101/426486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pearlstein R; Vaz R; Rampe D, Understanding the structure-activity relationship of the human ether-a-go-go-related gene cardiac K+ channel. A model for bad behavior. J Med Chem 2003, 46 (11), 2017–2022. [DOI] [PubMed] [Google Scholar]
- 25.Jamieson C; Moir EM; Rankovic Z; Wishart G, Medicinal chemistry of hERG optimizations: Highlights and hang-ups. J Med Chem 2006, 49 (17), 5029–5046. [DOI] [PubMed] [Google Scholar]
- 26.He Z; Qin L; Chen L; Peng N; You J; Chen X, Synergy of human immunodeficiency virus protease inhibitors with chloroquine against Plasmodium falciparum in vitro and Plasmodium chabaudi in vivo. Antimicrob Agents Chemother 2008, 52 (7), 2653–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fidock DA; Rosenthal PJ; Croft SL; Brun R; Nwaka S, Antimalarial drug discovery: efficacy models for compound screening. Nature Reviews Drug Discovery 2004, 3, 509–520. [DOI] [PubMed] [Google Scholar]
- 28.Baell JB; Holloway GA, New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 2010, 53 (7), 2719–2740. [DOI] [PubMed] [Google Scholar]
- 29.Smilkstein M; Sriwilaijaroen N; Kelly JX; Wilairat P; Riscoe M, Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrobial Agents and Chemotherapy 2004, 48 (5), 1803–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winter RW; Kelly JX; Smilkstein MJ; Dodean R; Bagby GC; Rathbun RK; Levin JI; Hinrichs D; Riscoe MK, Evaluation and lead optimization of anti-malarial acridones. Exp Parasitol 2006, 114 (1), 47–56. [DOI] [PubMed] [Google Scholar]
- 31.Peters W, The chemotherapy of rodent malaria, XXII. The value of drug-resistant strains of P. berghei in screening for blood schizontocidal activity. Ann Trop Med Parasitol 1975, 69 (2), 155–171. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.