

Abstract
Adverse neurodevelopmental consequences remain a primary concern when evaluating the effects of thyroid hormone (TH) disrupting chemicals. Though the developing brain is a known target of TH insufficiency, the relationship between THs in the serum and the central nervous system is not well characterized. To address this issue, dose response experiments were performed in pregnant rats using the goitrogen propylthiouracil (PTU) (dose range 0.1–10 ppm). THs were quantified in the serum and brain of offspring at gestational day 20 (GD20) and postnatal day 14 (PN14), two developmental stages included in OECD and EPA regulatory guideline/guidance studies. From the dose response data, the quantitative relationships between THs in the serum and brain were determined. Next, targeted gene expression analyses were performed in the fetal and neonatal cortex to test the hypothesis that TH action in the developing brain is linked to changes in TH concentrations within the tissue. Results show a significant reduction of T4/T3 in the serum and brain of the GD20 fetus in response to low doses of PTU; interestingly, very few genes were significantly different at any dose tested. In the PN14 pup significant reductions of T4/T3 in the serum and brain were also detected; however, twelve transcriptional targets were identified in the neonatal cortex that correlated well with reduced brain THs. These results show that serum T4 is a good predictor of brain THs, and offer several target genes that could serve as pragmatic readouts of T4/T3 dysfunction within the PN14 cortex.
Keywords: hypothyroidism, brain development, gene expression, adverse outcome pathway (AOP), developmental neurotoxicity, thyroid hormone
The debilitating effects of maternal hypothyroidism on the developing child have been well documented, taken mainly from extreme cases such as cretinism and untreated congenital hypothyroidism. These conditions result in significant deficiencies in somatic growth and severe mental retardation (Grosse and Van Vliet, 2011). More recently, a number of epidemiological studies have indicated that more subtle degrees of maternal thyroid hormone (TH) insufficiency during pregnancy are correlated with lower IQ, learning deficits, and structural abnormalities of the cortex and hippocampus in the developing child (Casey et al., 2017; Korevaar et al., 2016; Murphy et al., 2015; Willoughby et al., 2014). Given the relationship between THs and the developing brain, evaluation of environmental exposures to thyroid disrupting chemicals (TDCs) is of paramount importance to children’s health (Brucker-Davis, 1998; Gore et al., 2015; Massart et al., 2006).
Although there is no question that THs are critical for normal brain development, predicting alterations in brain THs, and potential adverse neurodevelopmental consequences, is challenging (Browne et al., 2017; Crofton et al., 2017). There are limited data that address how lowered thyroxine (T4) and triiodothyronine (T3) in the circulation may, or may not, alter hormonal status in the brain during critical phases of development (Bastian et al., 2010, 2012; Escobar del Rey et al., 1987; Morreale de Escobar et al., 1985; Morte et al., 2010). To date, most published rodent studies utilize severe models of hypothyroidism, and rarely are measures of both serum and brain hormones provided within the same report. Additionally, the phenotypes described are often not assessed using regulatory standard histological and behavioral methods, which pose several challenges in systematically evaluating the risk of TDCs (OECD/OCDE, Test 426; OECD/OCDE, Test 421; US EPA Guidance for Thyroid Assays, 2005).
To address these complexities, the U.S. Environmental Protection Agency (U.S. EPA) has endorsed the use of several in vivo testing approaches. This includes guidance for evaluating TH dysregulation; namely by measuring serum T4 concentrations in rat dams and offspring at key developmental stages during pregnancy (gestational day [GD] 20) and lactation (postnatal day [PN] 2 and 21) following a xenobiotic exposure (US EPA Guidance for Thyroid Assays, 2005). In order to contextualize how serum T4 changes affect neurodevelopmental processes, U.S. EPA is also supporting the development of adverse outcome pathways (AOPs) (US EPA Adverse Outcome Pathway Research Brief, 2017). The AOP is a conceptual framework, organizing biological knowledge to aide in interpretation of chemical risk (Villeneuve et al., 2014a,b). The current OECD sponsored AOP for thyroid peroxidase (TPO) inhibition describes the qualitative relationships between serum and brain TH reductions, in addition to disturbances in gene expression of the central nervous system (Crofton et al. 2017). As THs classically signal through ligand activated receptors, it is expected that TH insufficiency may alter gene expression in the brain, which could then lead to an adverse phenotype. Although such relationships are supported experimentally in the aforementioned AOP, the precision between serum T4 measures and brain THs, as well as the brain THs and transcriptional changes, lacks quantitative support (Crofton et al., 2017).
In order to address these knowledge gaps and to aid in the interpretation of regulatory tests recommended by U.S. EPA and OECD, this report utilizes an extensive dose-response analysis with the TPO inhibitor propylthiouracil (PTU).We hypothesized that measures of serum and brain THs, coupled with gene expression analyses, would strengthen the understanding of how a chemical exposure relates to both hormonal and cellular perturbations of the brain in vivo. Specifically, this work (1) provides quantitative information regarding the relationships between serum and brain THs at two developmental stages (GD20 fetus and PN14 pup) assessed in regulatory studies (OECD/OCDE, Test 426; OECD/OCDE, Test 421; US EPA Guidance for Thyroid Assays, 2005), and (2) identifies potential transcriptional bioindicators of TH dysregulation in the developing cortex. We propose that a transcriptional bioindicator could serve as an additional screening tool, strengthening the plausibility of a TH-mediated cellular response in the brain following exposure to TDCs.
MATERIALS AND METHODS
Animals
All animal husbandry was conducted with prior approval from the U.S. EPA’s Institutional Animal Care and Usage Committee, in an AALAAC approved facility. In all studies sperm positive was considered GD0 and pup birth PN0. Timed pregnant Long Evans rats were purchased from Charles River (Morrisville, North Carolina) and delivered on GD2. Pregnant animals were single housed in hanging cages, maintained on a 12:12-light cycle, and offered chow (Purina 5008) and tap water ad libitum.
Treatments to Induce Developmental Hypothyroidism Two dose response experiments were performed, differing in both dose concentration and duration. In both experiments, doses of PTU were chosen to induce graded decreases of circulating THs in the dams.
In Experiment I (gestational study), pregnant dams (n 1/4 6–9 per treatment) were administered PTU (CAS No. 51–52-5, purity >98%) dissolved in deionized drinking water from GD6 to GD20 at the following five concentrations: 0.1 ppm (0.00001%), 0.5 ppm (0.00005%), 1 ppm (0.0001%), 2 ppm (0.0002%), and 3 ppm (0.0003%). Control animals received drinking water only (Figure 1B). Dams were euthanized by decapitation on GD20 and fetuses collected by casarean section. Dam and fetal serum T4 and T3 were previously reported in Hassan et al. (2017).
Figure 1.
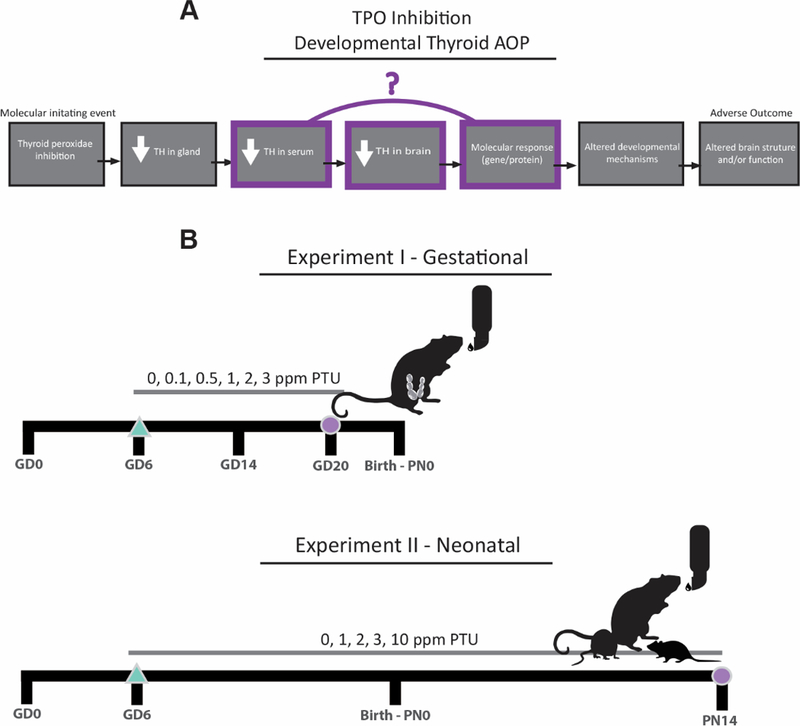
The AOP framework and experimental design. A, An example of a generic neurodevelopmental AOP with TPO inhibition as the molecular initiating event (first box, left). Each subsequent linkage is a key event; quantitative support for key events related to serum TH reductions, brain TH reductions, and gene expression changes are investigated in this study (highlighted). B, In the experimental design, an extensive dose response of PTU was administered to pregnant rats beginning on GD6 (triangle). In experiment I, pregnant rats were euthanized on GD20 and the fetuses analyzed for serum THs, brain THs, and cortical gene expression (circle); in experiment II, pups were euthanized on postnatal day 14 (PN14) for similar comparisons (circle). GD20 and PN14 are developmental stages currently requested and/or recommended for analysis in regulatory studies.
In Experiment II (neonatal study), dams (n 1/4 13–15 per treatment) were exposed to 0, 1, 2, 3, and 10 ppm PTU (CAS No. 51–52-5, purity >98%) beginning in early GD6 and continuing throughout lactation (GD6–PN21) (Figure 1B). All litters were culled to 10 pups on PN4. On PN14 pups were euthanized by decapitation. Dam euthanasia by decapitation occurred on PN21 with the following exception: dams exposed to 10 ppm PTU were not sacrificed until PN27 to permit late weaning of pups. Dam and serum data from this study were previously published in Gilbert et al. (2014). Pups were also assessed for brain morphology, neurochemistry, electrophysiology, and behavior, which is also reported elsewhere (Gilbert et al., 2016, 2017; Johnstone et al., 2013).
Tissue collection
All animals were sacrificed by decapitation between the hours of 08:00–12:00 and counterbalanced by dose over the collection period.
Serum for hormone analysis.
On GD20, trunk blood was pooled across the litter for all fetuses (experiment I). On PN14, blood was collected from one male and one female pup per litter and pooled into a single sample (Experiment II). Trunk blood was collected from dams on GD20 (Experiment I) or when pups were weaned on PN21 or PN27 (Experiment II). Whole blood was allowed to clot on ice for at least 30 min in serum separator tubes before centrifugation at 4_C for 30 min. Serum was then aliquoted and stored at _80_C until analysis.
Brain for hormone analysis.
To determine TH concentrations in brain tissue, whole brain from multiple male and female fetuses from each litter (3–4) were pooled together in a single 2-ml Eppendorf tube and snap frozen in liquid nitrogen. For the PN14 pup, the cortex was isolated from one male pup from each litter and preserved in a similar manner. All samples were stored at −80C until further analysis.
Brain for gene expression analysis.
For experiment I, the brain of one GD20 fetus per litter was removed from the skull, the forebrain dissected, and preserved in RNAlater (Qiagen) according to the manufacturer’s recommendations. For experiment II, the brain was removed from the skull of male pups on PN14. An oblique slice of neocortex from the frontal/parietal lobe was dissected using a razor blade. The tissue was then placed in a sterile 2-ml Eppendorf tube, snap frozen in liquid nitrogen, and stored at _80_C until RNA extraction.
Serum TH analysis.
Fetal and dam serum T4/T3 (Experiment I) were analyzed by mass spectrometry (MS) (T4 limit of quantification [LLOQ] 1/4 0.025 ng/ml and T3 1/4 0.02 ng/ml). Pup samples on PN14 and dams on PN21 (experiment II) were quantified by radioimmunoassay (LLOQ of T4 1/4 4.51 ng/ml and T3 1/4 6.20 ng/ml). Methods for quantification are previously reported by Hassan et al. (2017) and Gilbert and Sui (2006), respectively. Thyroid stimulating hormone (TSH) was also evaluated with methods previously reported in Stoker et al. (2010) and Thibodeaux et al. (2003). TSH was not measured in the GD20 fetus due to limitations in sample volume.
Brain hormone analysis
Brain samples were thawed on ice and homogenized by hand using a pestle mixer. An aliquot of the homogenate was weighed and transferred to a 15 ml polypropylene conical centrifuge tube. Methanol containing 1 mM PTU (methanol-PTU) was added to the brain homogenate (4 ml methanol-PTU per gram tissue) and spiked with a mixed stable isotope solution containing 13C6-T2, 13C6-T3, and 13C6-T4. THs were extracted from the brain using methanol, chloroform and an aqueous solution of 0.05% CaCl2 as described elsewhere in Bastian et al. (2010). The extract was then subjected to nitrogen evaporation to remove remaining methanol and chloroform from the aqueous mixture containing the THs. A 1 ml of water was added and the total volume of the aqueous mixture was determined. An equal volume of 80/20 (v/v) 4% formic acid (aq) and acetonitrile was added and the sample was vortex mixed. The sample was cleaned up through an Evolute 50 mg, 3 ml CX solid phase extraction (SPE) cartridge (Biotage, Charlotte, North Carolina) using a vacuum manifold. The SPE cartridge was conditioned with methanol followed by 2% formic acid. After sample application, the cartridge was washed with 50 mM ammonium acetate (pH 6), 2% formic acid and then methanol. TH were eluted into a 15 ml polypropylene conical centrifuge tube using 5% NH4OH in methanol and evaporated to dryness using nitrogen. Afterwards, the residue was dissolved in 100 ml of 5% acetonitrile: 95% water with 0.1% formic acid and transferred to an amber liquid chromatography (LC) micro-vial. SPE reconstituted extracts were analyzed for THs by stable isotope dilution LC/MS/MS as described in Hassan et al. (2017).
Gene Expression
All candidate genes analyzed in this study were chosen based on previously published work demonstrating their transcriptional sensitivity in the brains of developmentally hypothyroid rodents, or in cell culture models. Preference was given to candidates that were reported to be differentially expressed in the cortex, and/or at similar developmental stages as a GD20 and PN14 rat. All candidate genes analyzed in this study and associated information can be found in Table 4 and Supplementary Table 1.
Table 4. Complete list of genes interrogated and Supplementary material.
All genes were previously shown or speculated to be thyroid hormone targets (see extended version, Supplementary Table 1). Taqman primer codes represent the Assay ID (Thermofisher Scientific).
| Stage Analyzed | Gene name | Gene Symbol | NCBI Gene ID | TaqMan Primer | Differentially expressed |
|---|---|---|---|---|---|
| GD20 | Bone morphogenetic protein 7 | Bmp7 | 85272 | Rn01528889_m1 | No |
| GD20 | Roundabout guidance receptor 1 | Robo1 | 58946 | Rn00573395_m1 | No |
| GD20 | Slit guidance ligand 2 | Slit2 | 360272 | Rn00575268_m1 | No |
| GD20 | Neurofilament medium polypeptide | Nefm | 24588 | Rn00566763_m1 | No |
| GD20 | Sonic hedgehog | Shh | 29499 | Rn00568129_m1 | No |
| GD20 | Thyroid receptor alpha | Thra (TRα) | 81812 | Rn01464140_m1 | No |
| GD20 | Thyroid receptor beta | Thrb (TRβ) | 24831 | Rn00562044_m1 | Yes |
| GD20, PN14 | Calcium/calmodulin-dependent protein kinase type IV | Camk4 | 25050 | Rn00664802_m1 | Yes (GD20) |
| GD20, PN14 | Reelin | Reln | 24718 | Rn00589609_m1 | Yes (PN14) |
| GD20, PN14 | Sernaphorin 7a | Sema7a | 315711 | Rn01417778_m1 | Yes (PN14) |
| GD20, PN14 | Hairless | Hr | 60563 | Rn00577605_m1 | Yes (PN14) |
| GD20, PN14 | Nerve growth factor | Ngf | 310738 | Rn01533872_m1 | Yes (PN14) |
| GD20, PN14 | Inter-Alpha-Trypsin Inhibitor Heavy Chain 3 | Itih3 | 50693 | Rn00569293_m1 | Yes (PN14) |
| GD20, PN14 | Prepronociceptin | Pnoc | 25516 | Rn01637101_m1 | Yes (PN14) |
| GD20, PN14 | Deiodonase 11 | Dio2 | 65162 | Rn00581867_m1 | No |
| GD20, PN14 | Brain derived neurotophic factor total | BDNF-t | 24225 | Rn02531967_s1 | Yes (P1414) |
| GD20, PN14 | ICruppel like factor 9 | Klf9 | 117560 | Rn00589498_m1 | Yes (PN14) |
| GD20, PN14 | SRY-box 2 | Sox2 | 499593 | Rn01286286_m1 | Yes |
| GD20, PN14 | Beta-2-microglobulin | B2M | 24223 | Rn00560865_m1 | Reference gene |
| PN14 | Monocarboxylate transporter 8 | Slc16a2 (Mct8) | 259248 | Rn00596041_m1 | No |
| PN14 | Organic anion transporter polypeptide IC | Slcolc1 (Oatpc1c) | 84511 | Rn00584891_m1 | Yes (PN14) |
| PN14 | Solute Carrier Family 7 Member 3 | Sl7a3 | 29485 | Rn00500256_m1 | Yes (PN14) |
| PN14 | Neurotrophin-3 | Ntf3 | 81737 | Rn00579280_m1 | Yes (PN14) |
| PN14 | Growth Associated Protein 43 | Gap43 | 29423 | Rn01474579_m1 | Yes (PN14) |
| PN14 | Paired box 6 | Pax6 | 25509 | Rn00689608_m1 | Yes (PN14) |
| PN14 | Calcium/cahnodulin-dependent protein kinase type II | Camk2d | 24246 | Rn00560913_m1 | No |
| PN14 | Early growth response protein 1 | Egr1/Cuzd1 | 24330 | Rn00561138_m1 | Yes (PN14) |
| PN14 | Activity regulated cytoskeleton associated protein | Arc | 54323 | Rn00571208_g1 | Yes (PN14) |
| PN14 | Angiotensinogen | Agt | 24179 | Rn00593114_m1 | Yes (PN14) |
| PN14 | Tenascin C | Tnc | 116640 | Rn01454953_m1 | Yes (PN14) |
| PN14 | Gap junction beta 6 | Gjb6 | 84403 | Rn02042582_s1 | Yes (PN14) |
| PN14 | Collagen type XI alpha 2 chain | Col11a2 | 294279 | Rn01428773_g1 | Yes (PN14) |
| PN14 | Hop homeobox | Hopx | 171160 | Rn00592446_m1 | Yes (PN14) |
| PN14 | Nuclear receptor related 1 protein | Nurr1/Nr4a2 | 54278 | Rn00570936_m1 | Yes (PN14) |
| PN14 | Myelin oligodendrocyte glycoprotein | Mog | 24558 | Rn00575354_m1 | Yes (PN14) |
| PN14 | Parvalbumin | Parv/Parvlb | 25269 | Rn00574541_m1 | Yes (PN14) |
| PN14 | Mesenchymal-epithelial transition factor | Met | 24553 | Rn00580462_m1 | Yes (PN14) |
| PN14 | Sortilin Related Receptor 1 | Sorla/Sorl1 | 300652 | Hs00983770_m1 | No |
RNA Extraction
Total RNA was extracted using Trizol (Invitrogen) according to the manufacturer’s protocol; DNase treatment (Promega) was then utilized to remove genomic DNA contamination. RNA quantity and purity was quantified via a Nanodrop 2000c (Thermofisher), and sample integrity measured using a Bioanalyzer 2100 (Agilent). Only samples with a 260/280 > 1.9 and a RNA-integrity number > 8.0 were used in downstream applications. cDNA was synthesized using random primers (Applied Biosystems), and subsequently diluted in nanopure water.
Quantitative PCR.
Quantitative PCR (qPCR) was performed on a 7600HT Fast Real-Time PCR System (Applied Biosystems), using TaqMan Universal PCR Master Mix and commercially purchased TaqMan probes (Applied Biosystems). The thermocycler program was as follows: 95_C for 5 min, followed by 40 cycles of 15 s at 95_C, and 1 min at 60_ C. Each sample was run in duplicate, and the data analyzed using 2_DDct method (Livak and Schmittgen, 2001) with b2-microglobulin (B2M) as the reference gene. Information on all Taqman probes can be found in Table 4.
Statistics.
All TH and gene expression experiments were analyzed by 1-way analysis of variance (ANOVA) using SAS 9.4 (Cary, North Carolina), with the litter serving as the biological unit. In all assays, N 1/4 6–9 animals were analyzed for each endpoint. In the event of a significant main effect, mean contrast tests were performed using Dunnett’s t-tests (a 1/4 0.05). In analysis of gene expression, type I error was controlled by dividing the alpha level by the square root of the number of genes analyzed (a 1/4 0.01 for experiment I, a 1/4 0.009 for experiment II). To investigate the mathematical relationship between serum T4/T3 and brain T4/T3 concentrations, both mean and percent control data were fit using various linear and exponential equations in SigmaPlot 13.0 (Systat). To determine the correlation between TH status and gene expression changes in the cortex, qPCR fold changes were expressed as percent control values and plotted as a function of serum T4, brain T4, and brain T3. These relationships were also fit using various standard and exponential equations. In all cases model fit was measured by both R2 and significance values.
RESULTS
Serum THs Are Altered in a Dose-Dependent Manner Portions of serum hormone data were previously reported but included here for completeness; all serum data are summarized in Tables 1 and 2. In experiment I, dam T4 was significantly reduced on GD20 in response to 2 and 3 ppm PTU (Table 1). No changes were observed in dam serum T3 at any dose tested (Table 1, adapted from Hassan et al., 2017). Serum TSH concentrations in the GD20 dam were significantly elevated at the two highest doses of PTU (F[5, 37] 1/4 27.98 p < .0001). In the fetus, T4 was significantly decreased in response to 0.5, 1, 2, and 3 ppm PTU; reductions in serum T3 were also observed in animals from the 2 and 3 ppm exposures (Table 1).
Table 1. Dam and pup TH measures on GD20.
All PTU doses represent the maternal dose administered. Dam serum T4 was reduced at 2 and 3 ppm. T3 levels were not significantly altered in dam serum. TSH was significantly increased in response to 2 and 3 ppm PTU. Fetal serum T4 was significantly reduced at PTU doses of 0.5, 1, 2, and 3 ppm. Fetal T3 was reduced at 2 and 3 ppm dose groups. All analyses reflect n 1/4 6–9 biological replicates, and were measured by LC/MS/MS. The LLOQ for T4 is 0.025 ng/ml and 2.0 ng/dl for T3. Asterisks represents p < .05, and error terms 6 SEM. All but dam TSH data were originally published in Hassan et al. (2017).
| GD20 Dam | Thyroxine (T4) ng/mL |
Triiodothyronine (T3) ng/dL |
Thyroid stimulating hormone (TSH) |
|---|---|---|---|
| 0 ppm PTU (control) | 17.33 ± 1.96 | 53.66 ± 5.43 | 2.71 ± 0.51 |
| 0.1 ppm PTU | 21.34 ± 1.79 | 55.79 ± 4.43 | 1.92 ± 0.26 |
| 0.5 ppm PTU | 17.88 ± 2.66 | 52.37 ± 4.71 | 3.13 ± 0.38 |
| 1 ppm PTU | 13.98 ± 1.48 | 63.83 ± 7.68 | 3.37 ± 0.42 |
| 2 ppm PTU | 7.71 ± 1.04** | 56.78 ± 4.00 | 8.59 ± 1.05** |
| 3 ppm PTU | 3.28 ± 0.30** | 42.01 ± 3.82 | 12.18 ± 1.40** |
| GD20 Fetus | Thyroxine (T4) ng/mL |
Triiodothyronine (T3) ng/dL |
Thyroid stimulatine hormone (TSH) |
| 0 ppm PTU (control) | 3.33 ± 0.24 | 3.79 t 0.39 | - |
| 0.1 ppm PTU | 3.25 ± 0.15 | 3.33 ± 0.37 | - |
| 0.5 ppm PTU | 2.14 ± 0.25** | 2.99 ±0.38 | - |
| 1 ppm PTU | 1.32 ± 0.08** | 2.77 ± 0.54 | - |
| 2 ppm PTU | 0.54 ± 0.06** | 2.00 ± 0.001** | - |
| 3 ppm PTU | 0.32 ± 0.03** | 2.02 ± 0.02** | - |
Table 2. PN 21 Dam and PN14 pup Serum TH measures.
All PTU doses represent the maternal dose administered. Mean (6 SEM ) dam T4 on PN21 was significantly lower in response to 2 and 3 ppm PTU. Dam T3 was significantly higher in the 3 ppm dose group. TSH was significantly increased in dams of the 2 and 3 ppm PTU treatment groups. Note that dam THs of the 10 ppm PTU dose group were measured on PN28, one week following PTU withdrawal and showed recovery (#, see methods). On PN14 pup serum T4 was significantly reduced following exposure to 1, 2, 3, and 10 ppm PTU. Only PN14 pups from the 10 ppm PTU treatment had a significant reduction in T3. Pup serum TSH was significantly increased in all treatment groups. The limit of quantification is 11.3 ng/mL for T4 and 4.51 ng/ml for T3 as measured by RIA. All analyses reflect biological replicates from 13–15 litters.
| PN21 Dam | Thyroxine (T4) ng/mL |
Triiodothvronine (T3) ng/dL |
Thyroid stimulating honnone (TSH) |
|---|---|---|---|
| 0 ppm PTU (control) | 60.04 ± 2.43 | 92.15 ± 2.42 | 4.70 ± 0.63 |
| 1 ppm PTU | 50.34 ± 3.65 | 91.03 ± 3.67 | 7.62 ± 1.07 |
| 2 ppm PM | 34.16 ± 2.71** | 96.73 ± 5.14 | 9.91 ± 0.65** |
| 3 ppm PTU | 27.88 ± 1.91** | 105.87 ± 2.60** | 14.02 ± 1.26** |
| 10 ppm PTU | 64.24 ± 5.64 # | 91.29 ± 4.35 # | 4.42 ± 1.50 # |
| PN14 Pup | Thyroxine (T4) ng/mL |
Triiodothyronine (T3) ng/dL |
Thyroid stimulating honnone (TSH) |
| 0 ppm PTU (control) | 54.58 ± 1.78 | 75.03 ± 2.74 | 1.71 ± 0.13 |
| 1 ppm PTU | 29.57 ± 2.29** | 83.79 ± 5.00 | 4.00 ± 0.47** |
| 2 ppm PTU | 18.55 ± 2.20** | 81.53 ± 4.95 | 7.66 ± 0.46** |
| 3 ppm PTU | 9.89 ± 1.15** | 61.82 ± 3.92 | 11.39 ± 0.80** |
| 10 ppm PTU | 4.51 ± .0005** | 30.62 ± 5.66** | 6.71 ± 0.49** |
p < 0.05. All serum hormone data were previously published in Johnstone et al., 2013 and Gilbert et al., 2014.
In experiment II, dams exposed to 2 and 3 ppm had decreased T4 concentrations on PN21; T3 was reduced in the 3 ppm PTU dose group only (Table 2, adapted from (Gilbert et al., 2014). Dam TSH concentrations were increased in response to 2 and 3 ppm PTU. Dams from the 10 ppm treatment were assessed one week after termination of exposure due to concerns with litter viability (see “Materials and Methods” section); examination of serum T4, T3, and TSH concentrations on PN28 in the 10 ppm PTU dams showed TH concentrations comparable to that of control dams on PN21. In the PN14 pup, serum T4 was significantly reduced in a dose-dependent manner; significant reductions in pup serum T3 were restricted to the 10 ppm PTU dose group only (Table 2). Additionally, serum TSH was significantly elevated in the PN14 pup in response to 1, 2, 3, and 10 ppmPTU.
Brain TH Concentrations
As with serum, control concentrations of T4 and T3 in the fetal brain were much lower than those observed in the neonatal brain (Figs. 2A and 2B vs 2C and 2D). On GD20, PTU reduced both T4 (Figure 2A) and T3 (Figure 2B) in the fetal brain in a dose dependent manner (F[5, 37] 1/4 27.16 p < .0001 and F[5, 37] 1/4 48.95 p < .0001, respectively). A nominal decrease in brain T4 and T3 was evident in the 0.5 ppm treatment group; however, statistically significant reductions in brain THs were limited to animals exposed to 1, 2, and 3 ppm PTU (Figs. 1A and 1B).
Figure 2.
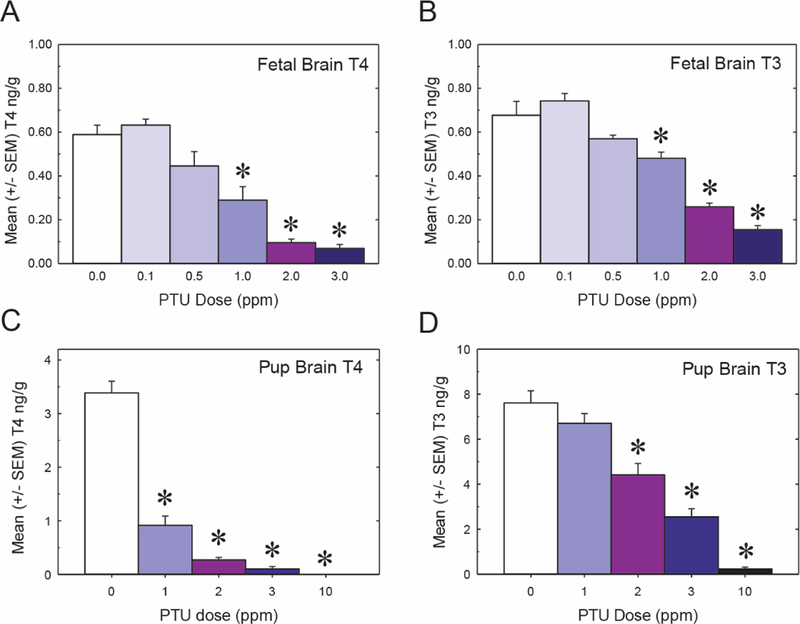
Fetal and pup brain hormones. All PTU doses represent the maternal dose administered. A, Fetal T4 levels on GD20 in whole brain. B, Fetal brain T3 levels. C, T4 levels in the PN14 in cortex. D, Cortical T3 on PN14. In all graphs asterisks represent p < .05 and error bars are 6 SEM (n 1/4 7–9 biological replicates per treatment for fetal measurements, and n 1/4 8 biological replicates per treatment for pups). LLOQ is 0.005 ng/g for both T4 and T3.
In the PN14 pup brain, a significant reduction in T4 was also observed in the 1, 2, 3, and 10 ppm PTU exposure groups (Figure 2C) (F[4, 35) 1/4 122.85, p < .0001); additionally, the magnitude of the observed T4 decrement was greater in the pup than in the fetus (Figs. 2A vs 2C). Pup brain T3 concentrations were also reduced, but statistically significant observations were restricted to the 2, 3, and 10 ppm dose groups (Figure 2D) (F[4, 25] 1/4 51.79 p < .0001).
The Quantitative Relationship Between Serum and Brain THs
In the fetus, a strong linear correlation was observed between T4 concentrations in the serum and T4 levels in the brain (Figure 3A, R2 1/4 0.980 and p < .0001). In the pup, this relationship was best fit as a quadratic function (Figure 3B, R2 1/4 0.998 and p < .0001). In both the GD20 fetus and PN14 pup, mean brain T3 concentrations correlated well to mean serum T4; at both stages an exponential relationship was observed (Figs. 3C and 3D, R2 1/4 0.983 and p < .0001 for GD20; R2 1/4 0.900 and p < .0001 for PN14). Brain T3 expressed as a function of serum T3, rather than serum T4, was also analyzed. However, the observed R2 values were lower and the confidence limits broader, for both the GD20 fetus and the PN14 pup (Supplementary Figs. 1A and 1B, R2 1/4 0.863 and p < .0047 for GD20; R2 1/4 0.807 and p < .0026 for PN14). Interrogation of the relationship between T4 and T3 in the GD20 brain revealed a highly significant linear relationship (Figure 3E R2 1/4 0.969 and p < .0001), whereas this relationship in the neonatal cortex was better described by an exponential function (Figure 3F, R2 1/4 0.989 and p < .0001 for PN14).
Figure 3.
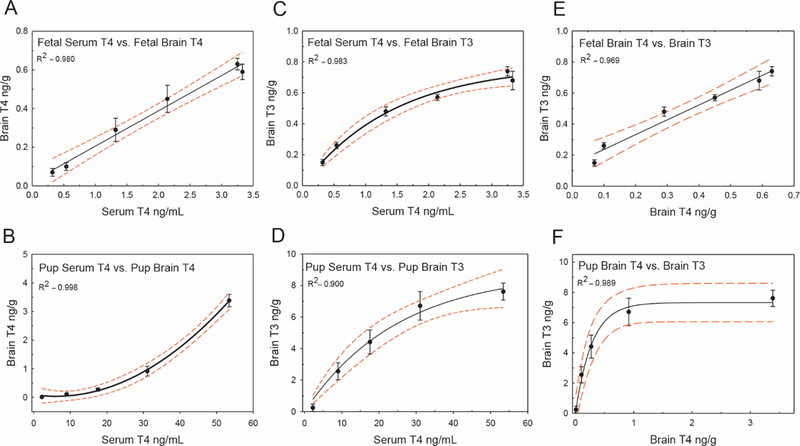
Quantitative relationships between serum and brain THs. All hormone values plotted are means, as determined by LC/MS/MS or RIA (see “Materials and Methods” section). A, Serum T4 versus brain T4 in the GD20 fetus. Observations correlate well to a standard linear equation. B, Pup serum T4 correlates to brain T4 concentrations on PN14 as a quadratic relationship. C, Serum T4 versus brain T3 in the GD20 fetus is fitted by an exponential equation relationship. D, Pup serum T4 and brain T3 are also modeled well as an exponential equation, similar to what is observed in the fetus. E, Fetal brain T3 levels versus brain T4 are well represented by a linear equation. F, Pup brain T3 versus brain T4 is best fit as an exponential relationship. R2 values for each relationship are shown. In all graphs error bars represent 6 SEM, and dashed lines the 95% CIs.
These same data were transformed to percent control values and again analyzed. Expressed in this manner, distinct differences in the relationship between serum and brain T4 of the GD20 fetus and PN14 pup were again apparent. In the fetus, a 40% decrease in serum T4 predicted a 35% decrease in brain T4 (Figure 4A). In the pup an equivalent 40% serum decrease is associated with a dramatic 70% decrement in brain T4 (Figure 4B). Despite these observed stage-specific differences in brain T4 concentrations, the associated T3 reduction in the tissue are comparable between the fetus and pup (approximately 15%) when assuming this serum T4 decrement (Figs. 4C and 4D). However, developmental differences are again observed when predicting brain T3 given a specific brain T4 estimate; a 20% decrease in brain T4 results in a more severe reduction in T3 on GD20 (approximately 20%) as opposed to PN14 (approximately 2%) (Figs. 4E and 4F). For both the absolute and percent control data, the equations describing the TH dynamics are presented in Table 3.
Figure 4.
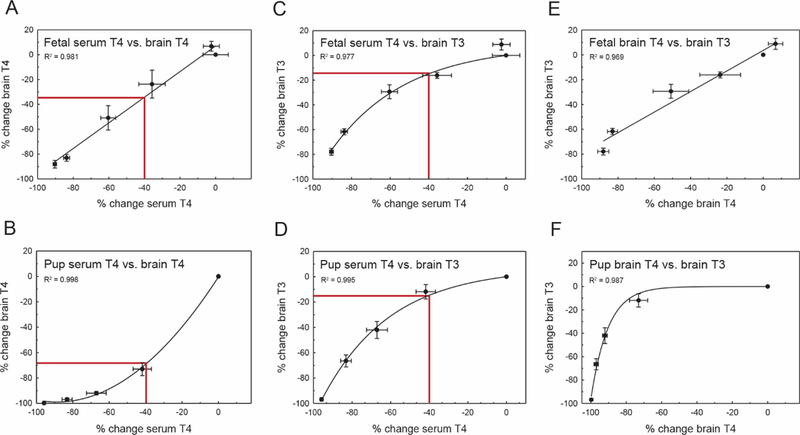
Stage-specific differences in TH economy. All values plotted are percent control. A, Percent change serum T4 versus brain T4 in the GD20 fetus. Prediction estimate demonstrates the relationship between the periphery and brain assuming a 40% decrease in serum T4. B, PN14 pup brain T4 is more adversely affected when assuming a 40% decrease in serum T4 (prediction line in both panels A and B). Given this large decrease in brain T4 on PN14, brain T3 is similarly affected in both the GD20 fetus (C) and the pup (D) E and F, However, when only examining the relationship between THs in the brain, a T4 deficit in the fetal brain produces a more significant decrement in T3 as compared with the pup. R2 values for each relationship are shown on their respective panels, and error bars represent 6 SEM for both the x and y axes.
Table 3. Mathematical relationship describing thyroid hormone dynamics in the GD20 fetus and PN14 pup.
Equations resulting from analyses of serum and brain THs at both stages. Equations were derived from mean or percent change T4 and T3 as indicated. In all equations x 1/4 quantified serum or brain T4. The top equations are for use with mean values, and the bottom percent control. In the percent control equations, solve for y given negative x values (to represent TH detriment).
| Means | Serum T4 vs. brain T4 | Serum T4 vs. brain T3 | Brain T4 vs. brain T3 |
|---|---|---|---|
| GD20 fetus | y = 0.183x + 0.023 | y = 0.780 (1 − e−6.97x) | y = 0.955x + 0.141 |
| PN14 neonate | y = 0.0015x2 − 0.018x + 0.090 | y = 0.126 (1 − e−4.87x) | y = 7.328 (1 − e−3.52x) |
| Percent change | Serum T4 vs. brain T4 | Serum T4 vs. brain T3 | Brain T4 vs. brain T3 |
| GD20 fetus | y = 0.013x2 + 2.219x − 0.395 | y = 7.000 (1 − e−0.028x) | y = 0.003 (1 − e−0.103x) |
| PN14 neonate | y = 1.031x + 7.030 | y = 8.236 (1 − e−0.026x) | y = 0.828x + 3.582 |
Differential Gene Expression as a Function of PTU Dose
Fetal cortex. A total of eighteen genes that were previously reported to be TH-targets in the fetal brain were assessed in the GD20 cortex (Table 4). Only two genes, Camk4 and Thrb, were found to be differentially expressed. Camk4 was significantly downregulated in the 2 and 3 ppm PTU treatment groups (F[5, 41] 1/4 9.07, p < .0001); it is noted that the magnitude of fold changes in these treatments were modest, with an observed 31% and 43% percent decrease respectively (Figure 5A). Camk4 encodes a calcium/calmodulin dependent kinase, and has been implicated as a direct target of thyroid receptor (Krebs et al., 1996; Liu and Brent, 2002; Morte et al., 2010). One of the thyroid receptors (Thrb) was also significantly downregulated (51% compared with control), but this was only observed in the cortex of animals exposed to the highest dose tested, 3 ppm PTU (Figure 5B) (F[5, 39] 1/4 10.10, p < .0001). The general functional classification, magnitude of change, and associated statistics of Camk4 and Thrb are summarized in Table 5. No other candidate genes analyzed were significantly different at any PTU dose tested (Supplementary Figure 2).
Figure 5.
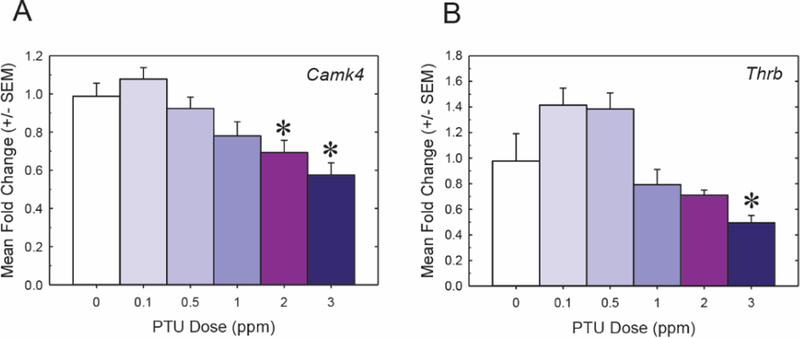
Differential gene expression in the forebrain of the GD20 fetus. All PTU doses represent the maternal dose administered. A, The gene Camk4 was downregulated in response to 2 and 3 ppm PTU. B, Thyroid receptor beta (Thrb) was the only other differentially expressed gene in the GD20 fetus, and a significant change was only observed in the 3 ppm PTU dose group. An n 1/4 6–9 was analyzed per treatment; significant downregulation of both genes was determined by an overall ANOVA, a1/4 0.01. All asterisks represent significant dose effects as determined by mean contrast tests, a1/4 0.05. All error bars represent 6 SEM.
Table 5. Statistical summary of the candidate genes with the greatest potential to serve as a bioindicator of TH insufficiency.
All PTU doses represent the maternal dose administered. Positive values represent upregulation and negative downregulation. Values listed are the percent change in gene expression, 6 SEM. Resulting overall ANOVA F-statistics and p-values for each gene are shown. Mean contrast tests were then conducted using Dunnett’s t-test with a=0.05. Significant dose effects are indicated by asterisks.
| Experiment 1: GD20 forebrain | ||||||||
|---|---|---|---|---|---|---|---|---|
| 0.1 ppm PTU |
0.5 ppm PTU |
1 ppm PTU |
2 ppm PTU |
3 ppm PTU |
F statistic |
p-value |
||
| Calcium Signaling - CREB pathway | Camk4 | −7±8 | 8±6 | −22±5 | −31±5** | −43±5** | F(5,41)=9.07 | <0.0001 |
| Thyroid signaling | Thwb | 34±13 | 38±14 | −21±12 | −38110 | −51±6** | F(5,39)=10.10 | <0.0001 |
| Experiment II: PN14 neocortex | ||||||||
| 1 ppm PTU |
2 ppm PTU |
3 ppm PTU |
10 ppm PTU |
F statistic |
p-value |
|||
| Calcium signaling - GABAergenic neurons | Parv | −30±19** | −60±7** | −81±3** | −97±.6** | F(4,30)=49.49 | <0.0001 | |
| Colla2 | −26±8** | −54±6** | −640** | −86±1** | F(4,30)=37.34 | <0.0001 | ||
| Gjb6 | −25±6** | −44±3** | −48±3** | −58±3** | F(4,30)=33.51 | <0.0001 | ||
| Cell and tissue structure | Itih3 | −28±6** | −48±6** | −65±4** | −82±2** | F(4,30)=38.60 | <0.0001 | |
| Mog | −22±12 | −46±14** | −71±7** | −90±3** | F(4,30)=13.63 | <0.0001 | ||
| Agt | −26±10** | −53±7** | −71±4** | −85±1** | F(4,30)=32.49 | <0.0001 | ||
| Pnoc | −13±2** | −23±3** | −32±4** | −55±2** | F(4,30)=48.62 | <0.0001 | ||
| Neuronal signaling and migration | Ngf | −10±7 | −30±9** | −46±4** | −58±4** | F(4,30)=14.03 | <0.0001 | |
| Sema7a | −30±9 | −60±7** | −81±3** | −97±.6** | F(4,30)=16.41 | <0.0001 | ||
| Hod | −20±5** | −44±5** | −52±3** | −73±1** | F(4,30)=58.70 | <0.0001 | ||
| Cell differentiation and death | Hr | −17±8 | −46±12** | −71±6** | −90±.5** | F(4,30)=27.39 | <0.0001 | |
| Kfl9 | −30±9 | −60±7** | −81±3** | −97±.6** | F(4,30)=45.72 | <0.0001 | ||
Neonatal cortex.
Expression of thirty genes was evaluated in the cortex of the PN14 pup, twenty-five of which were differentially expressed following maternal PTU treatment (summarized in Table 5). In contrast to the fetus, seven of these targets (Parv, Col11a2, Gjb6, Itih3, Agt, Pnoc, and Hod) were significantly reduced at all PTU doses tested (1, 2, 3, and 10 ppm) (Figs. 6A–G). This pattern of significant gene expression changes aligns to the significant decrease of T4 within the brain, suggesting that these genes are highly sensitive to brain T4 status (compare Figs. 2C with 6A–G). These genes encode a calcium-binding albumin protein expressed in inhibitory neurons (Parv), extracellular matrix and cytoskeletal proteins (Col11a2, Gjb6, and Itih3), neuropeptides (Pnoc and Agt), and a homeodomain-only factor (Hod) (see Table 4 for gene information). Among these seven candidates, Parv demonstrated a large magnitude of change, with a highly significant 97% reduction in response to 10 ppm PTU (Figure 6A) (F[4, 30] 1/4 49.49, p < .0001). Five additional genes (Hr, Mog, Ngf, Klf9, and Sema7a) were differentially expressed at 2, 3, and 10 ppm PTU (Figs. 6H–L). Gene expression changes at these PTU doses coincide with significant reductions in brain T3 (compare Figs. 2D with 6H–L). Although a statistically significant difference in gene expression was not detected for these targets at the lowest dose of PTU, every candidate demonstrated a dose response pattern in expression changes. This second class of markers includes genes encoding for a transcriptional corepressor (Hr), a zinc finger transcription factor (Klf9), a myelin specific glycoprotein (Mog), a semaphorin glycoprotein (Sema7a), and a nerve growth factor (Ngf). Similar to what was observed with Parv, the gene Hr exhibited a large magnitude of change in response to PTU exposure, with a 90% decrease in expression in the 10 ppm PTU dose group (Figure 6H) (F[4, 30] 1/4 27.39, p <.0001). A summary of the general functional classification, magnitude of gene expression changes, and associated statistics of these targets are summarized in Table 5.
Figure 6.
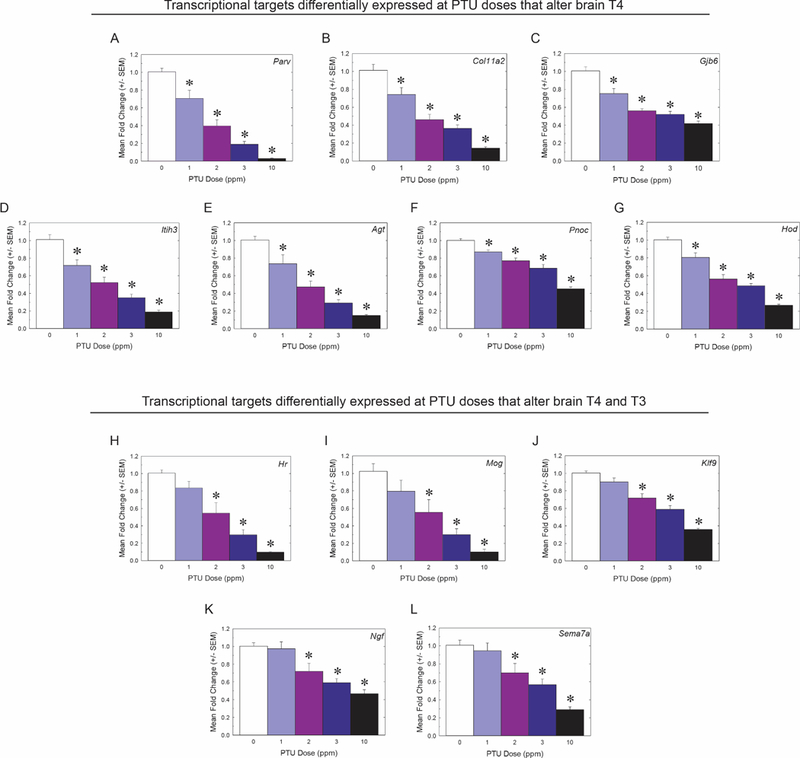
Gene expression changes in the PN14 neocortex that reflect the brain T4/T3 deficit. All PTU doses represent the maternal dose administered. A–G, Transcriptional targets that were differentially expressed at every PTU dose tested at this stage (1, 2, 3, and 10 ppm); these PTU exposures also induced a significant T4 decrease in the PN14 brain. H–K, These candidate genes were differentially expressed in the PTU treatments that induced statistically significant decreases in both T4 and T3 within the brain (2, 3, and 10 ppm PTU). n 1/4 7 were analyzed for each dose group; significant differential expression of genes was determined by an overall ANOVA, a= 0.009. All asterisks represent significant dose effects as determined by mean contrast tests, a= 0.05. All error bars represent 6 SEM.
Gene expression analyses also identified transcripts that were differentially regulated only in the highest PTU doses administered. Five genes (Arc, Met, Egr1, Gap43, and Ntf3) were detected to be significantly different in the 3 and 10 ppm PTU treatments; three of these targets demonstrated upregulation (Gap43, Met, and Ntf3; Supplementary Figure 3). Four additional candidates (Pax6, Sox2, Slco1c1, and Slc7a2) were differentially expressed in the neocortex of animals exposed to 10 ppm PTU (Supplementary Figure 4), but the overall pattern of gene expression changes did not necessarily follow a clear dose response pattern. Genes that were not detected to be differentially regulated on PN14 are shown in Supplementary Figure 5.
The Relationship Between TH Status and Cortical Gene Expression
The relationship between TH concentrations in serum and brain were then compared with transcripts tested in both the GD20 fetus and PN14 pup. The only two differentially expressed genes identified in the fetal forebrain, Camk4 and Thrb, were plotted relative to serum T4 (Figs. 7A and 7B), brain T4 (Figs. 7C and 7D), and brain T3 (Figs. 7E and 7F). Gene expression as a function of serum T4 exhibited lesser fit values (Figure 7A Camk4, R2 1/4 0.946, p 1/4 .001; Figure 7B Thrb, R2 1/4 0.646, p 1/4 .051), whereas gene expression as a function of brain T3 was the best supported relationship for both targets (Figure 7E Camk4, R2 1/4 0.981, p < .001; Figure 7F Thrb, R2 1/4 0.749, p 1/4 .026). Gene expression as a function of brain T4 was also highly supported for Camk4 (Figure 7C Camk4, R2 1/4 0.967, p < .001; Figure 7D Thrb, R2 1/4 0.744, p 1/4 .027).
Figure 7.
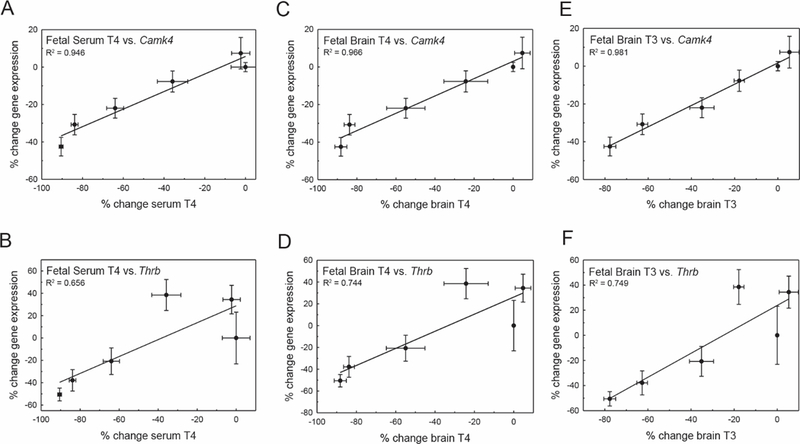
Correlation of gene expression and THs in the GD20 fetus. A, Camk4 expression in the fetal forebrain is well correlated to serum T4. B, Thrb is not as linearly related to graded reductions in serum T4. For both Camk4 and Thrb, percent change in gene expression is also linearly correlated to percent change in fetal brain T4, but with higher support values than those observed for serum. Gene expression is also highly correlated to changes in brain T3 (E, F) in the fetus. Error bars represent 6 SEM for both the x and y values, and R2 values are noted on each panel.
The same relationships between gene expression and THs were then investigated in the PN14 pup. For these analyses two transcriptional targets, Parv and Hr, were examined given their large magnitude of change in response to PTU treatment. Both Parv and Hr expression displayed a strong linear relationship with serum T4 (Figure 8A Parv, R2 1/4 0.981, p 1/4 .001; Figure 8B Hr, R2 1/4 0.924, p 1/4 .009). Gene expression as a function of brain T4 was best fit by an exponential equation (Figure 8C Parv, R2 1/4 0.993, p < .001; Figure 8D Hr, R2 1/4 0.992, p < .001), whereas the relationship to brain T3 was best described by a quadratic relationship (Figure 8E Parv, R2 1/4 0.989, p < .001; Figure 8F Hr, R2 1/4 0.999, p 1/4 .002).
Figure 8.
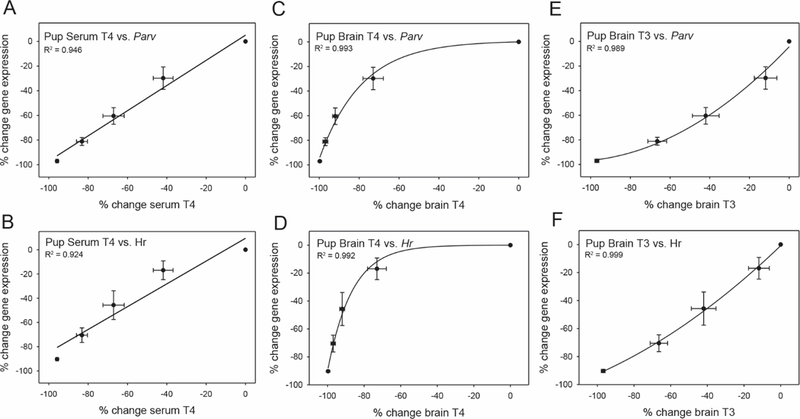
Correlation of gene expression and THs in the PN14 rat. Percent change in serum T4 correlated to percent change in (A) Parv and (B) Hr in the pup neocortex. These candidate genes were also highly correlated to brain T4 (C, D), but in an exponential manner. Percent change in expression of Parv and Hr, interrogated as a function of brain T3, were best represented by quadratic equations. Expectedly, differential gene expression in the cortex was more highly predicted by brain TH status, as opposed to serum T4. Error bars represent 6 SEM for both the x and y data, and R2 values are noted on each panel.
Potential Bioindicators of TH Homeostasis in the Developing Cortex
Despite the limited transcriptional changes in response to PTU treatment within the fetal brain, Camk4 exhibited an overall dose dependent downregulation. However, a significant decrease in gene expression was only observed at the two highest doses of PTU tested in experiment I (2 and 3 ppm), suggesting this gene has limited potential as a sensitive marker of small T4/T3 reductions in the brain.
The most robust changes in gene expression that coincided with significant reductions in brain T4 and/or T3 were deemed sensitive readouts of TH status in the PN14 pup. Of these Parv (Figure 6A), Col11a2 (Figure 6B), Itih3 (Fig, 6D), Agt (Figure 6E), Hr (Figure 6H), and Mog (Figure 6I), exhibited the greatest magnitude of change (_80% at 10 ppm PTU, Table 5), and were statistically significant (a < 0.009) at PTU doses that decreased brain T4 and/or T3. The associated percent change of these aforementioned genes as function of PTU dose can be found in Table 5.
DISCUSSION
A rat model was used to interrogate the quantitative relationship between THs in the serum and brain during two important stages of neurodevelopment. Additionally, potential transcriptional readouts of TH activity in the developing cortex were investigated. Results demonstrate that serum T4 was an excellent predictor of brain T4/T3 at both developmental stages tested. Although very few differentially expressed genes were identified in the fetal forebrain, a number were observed in the PN14 cortex. The quantitative relationship between serum and brain T4/T3, accompanied by correlated transcriptional changes in a vulnerable tissue, suggests that these genes may be downstream targets of TH signaling. These candidate genes may be useful tools to identify perturbations in TH status within the developing cortex, and could assist in the generation of new hypotheses regarding how TH insufficiency may negatively impact the developing brain.
TH Insufficiency in the Developing Rat
Our previous dose response work documented that the hormone suppressive effects of PTU were greater in the GD20 fetus (Hassan et al., 2017) and PN14 pup (Gilbert et al., 2014) than in the dams. The present findings extend these observations to include T4/T3 within the brain tissue of these same individuals, demonstrating that the central nervous system also experiences alterations in TH homeostasis in response to low maternal doses of PTU (1, 2, and 3 ppm on GD20 and 1, 2, 3, and 10 ppm on PN14). Although brain T4 and T3 are best predicted by serum T4 in both the fetus and the pup, the precise relationship between the periphery and the brain is stage specific. Comparing hormone profiles from the GD20 fetus and neonate clearly demonstrates that the brain responds differently to T4/ T3 decreases in the serum at these two distinct life stages. If an equivalent percent reduction in serum T4 (ie, 40%) is assumed for both the fetus and pup, brain T4 in the PN14 pup is drastically reduced (approximately 70%) as compared with the GD20 fetus (approximately 35%). Though pup brain T4 concentrations are more sensitive to serum T4 decrements, brain T3 concentrations are comparably affected at both stages (approximately 15% reduction). It is acknowledged that these stage-specific relationships, which are based solely on the percent change of THs in the serum relative to the brain, may be influenced by the specific toxicokinetics of the PTU exposure. Nonetheless, this may be an important observation regarding the ontogeny of the thyroid system.
Although the biological underpinnings of these stage specific differences between serum and brain THs are not fully understood, it may be due in part to the dissimilar physiological state of the rat on GD20 and PN14. Previous work has shown that, though the fetal thyroid gland is developed and begins functioning on GD17, the thyroid axis in the near term fetus remains immature (Morreale de Escobar et al., 1985, 1990). It is therefore reasonable to consider that T4/T3 regulation within the fetal central nervous system may also be immature. What other scenarios could account of our stage-specific observations of brain T4/T3? First, rodent studies have shown that T3 is not preferentially transported into the fetal brain despite expression of its transporter (Monocarboxylate transporter 8, MCT8), and instead nearly all T3 is derived via local 5’-deiodination (Barez-Lopez et al., 2018; Calvo et al., 1990; Grijota-Martinez et al., 2011). Therefore, one potential compensatory mechanism (direct T3 transport into the brain) is present in the PN14 pup that is absent and/or reduced in the fetus. Secondly, 5’-deiodinase activity in the GD20 fetus may not be as efficient as that in a 2-week-old pup, as suggested by the work of Kaplan and Yaskoski (1981). Together, a more comprehensive understanding of how TH transport and 5’-deiodination changes in developing hypothyroid animals, would aide in deciphering the periods that may be acutely susceptible to hormone perturbation.
Although the drastic decrease in brain T4 within the pup did not necessarily result in a large change in brain T3, this large change in T4 may still be of biological significance. Classically, T4 is considered the prohormone and T3 the active ligand, but this canonical view of thyroid signaling has been recently challenged by the identification of T4-specific signaling mechanisms in the brain (Flamant et al., 2017; Galton, 2017; Gil-Ibanez et al., 2017a; Maher et al., 2016; Oyanagi et al., 2015; Schroeder and Privalsky, 2014; Zamoner et al., 2008). Therefore, the possibility that a brain T4 deficit alone could alter neurodevelopmental processes cannot be discounted. Additionally, in both the GD20 and PN14 brain, T4 and/or T3 reductions were observed that were not necessarily statistically significant (Figure 2). Despite the lack of statistical reliability of brain T3 reductions in PN14 pups exposed to 1 ppm PTU, this modest deficit is correlated with alterations in cortical structure, synaptic physiology, and learning and memory (Gilbert, 2011; Gilbert et al., 2016, 2017).
Predictive Capabilities of Dose Response Data
It is clear that more studies are needed to illuminate how minor T4/T3 reductions alter brain structure and function; however, the data presented here offer a first step to understanding the relationship between THs in the general circulation and the brain at two developmental stages. Although serum T4 in the GD20 fetus and PN14 pup are measured in some regulatory studies (OECD/OCDE, Test 421; US EPA Guidance for Thyroid Assays, 2005), it was unclear how TH insufficiency does, or does not, alter brain hormones. Here, we present the quantitative relationships of these two compartments derived from dose response data utilizing the well-characterized goitrogen PTU. The predictive power of the described associations will need to be tested in the context of other TDCs that reduce circulating levels of THs. Although some chemicals may disrupt brain development through a TH-dependent mechanism without altering serum T4/T3 (DeVito et al., 1999), a reduction in serum T4 remains a convergent key event common to many TDCs. Therefore, we propose that the equations reported here can be used across laboratories to predict T4 and T3 concentrations within the rodent brain on GD20 and PN14. These predictions can be generated given a simple serum T4 or T3 measurement, and could eventually be correlated to adverse neurodevelopmental phenotypes. Thus, these quantitative relationships represent an important initial milestone in the understanding of how thyroid disrupting chemicals may impact neurodevelopment.
As discussed earlier, the physiological differences in TH regulation of the late term fetus and a 2-week-old pup are striking; however here, only dose response data from GD20 and PN14 are reported. Evaluation of serum and brain TH concentrations at other developmental stages will be needed to investigate other potential stage-specific differences. Current guidelines recommend inclusion of a perinatal (PN2–4) collection period for serum TH analysis (US EPA Guidance for Thyroid Assays, 2005; OECD/OCDE Test 421), in addition to the two stages presented here. The perinatal period in the rat corresponds to the second trimester of human pregnancy (Bernal, 2007). This is a crucial neurodevelopmental phase in mammals characterized by rapid neurogenesis, cell differentiation, and cell migration (Berger-Sweeney and Hohmann, 1997; Bernal, 2007; Tramontin et al., 2003), and previous work has shown that TH disruption during this developmental phase is associated with structural and functional deficits in rodents (Blanco et al., 2013; Boyes et al., 2018; Gilbert et al., 2014; Goldey et al., 1995; Li et al., 2004; Negishi et al., 2005; O’Shaughnessy et al., 2018; Slotkin et al., 1984). As these data suggest that the rodent perinatal period may be another window of susceptibility, evaluation of serum and brain hormones during this period would enhance understanding of stage-specific differences in TH homeostasis.
The Identification and Utility of a TH Bioindicator
There are few elucidated adverse neurodevelopmental outcomes in rats following moderate reductions in THs (Gilbert et al., 2014, 2016, 2017; Gilbert and Sui, 2006; Goldey et al., 1995; Sharlin et al., 2008), and these phenotypes may escape identification in standard developmental neurotoxicity testing (OECD/OCDE, Test 426). As the primary action of ligand activated nuclear receptors is the regulation of transcription, we hypothesized that gene expression could serve as a readily obtainable measure that could reflect TH status within the brain tissue. Although a sensitive transcriptional signature of compensatory-like activity may be expected in animals with reduced T4/T3 brain concentrations, this was not experimentally reproducible in these studies. This was despite assaying for TH transporters and deiodinases in both the GD20 and PN14 brain.
Fetal bioindicator. Of the eighteen candidate genes measured in fetal cortex, only two candidates, Camk4, a calcium binding protein, and Thrb, thyroid receptor beta, were differentially regulated in response to PTU. Though Camk4 was downregulated at the highest doses of PTU (2 and 3 ppm, respectively), this was not the case in the 1 ppm exposure, despite a significant decrease in brain THs (50% decrease in T4, and 23% in T3). Thrb was only significantly downregulated in response to 3 ppm PTU. Other groups have identified robust changes of Camk4 in the hypothyroid cortex (Dong et al., 2015; Morte et al., 2010; Navarro et al., 2014; Zhang et al., 2015), and demonstrated that it is likely a direct target of T3 (Liu and Brent, 2002; Morte et al., 2010). Our data extend these observations of Camk4 in the context of a milder level of serum hormone reduction. However, taken together with the dose response profile of brain T4/T3, it is difficult to conclude if Camk4 is an accurate bioindicator of TH status in the GD20 fetal forebrain.
Identification of only two significant transcriptional responses in the cortex during late gestation is somewhat perplexing, but consistent with previous reports (Dong et al., 2015; Morte et al., 2010; Schwartz et al., 1997). There are several scenarios that could account for the observed negative data. First, the cortex is comprised of a heterogeneous cellular population, and may not be the most TH sensitive tissue at this developmental stage. Even if the cerebral cortex is vulnerable to perturbations of THs at GD20, this may not result in differential expression of protein coding genes. A previous genome-wide study demonstrated that despite a lack of transcriptional differences in coding genes within the GD20 brain, there were a plethora of microRNA changes (Dong et al., 2015). This suggests that TH action may be biased towards other transcriptional regulatory processes like noncoding RNA expression. To more adequately address this issue, future work using whole transcriptome sequencing is needed to assay for both protein coding and noncoding RNA in a nonbiased fashion. If and once TH bioindicators are identified in the GD20 forebrain, these targets could be easily interrogated by qPCR. Despite the difficulties in identifying transcriptional readouts in the late gestational brain, a TH decrement during this period may still induce adverse phenotypes, which is supported experimentally (Alvarez-Dolado et al., 1999; Auso et al., 2004; Berbel et al., 2010; Goodman and Gilbert, 2007; Martinez-Galan et al., 2004; Morte et al., 2010; Navarro et al., 2014; Rabie et al., 1980). Additionally, while the data presented here focuses on gene expression, it is understood that regulation of cell activity is not just mediated by transcription. Instead, the hormonal control of processes like posttranslational modification could lead to adverse consequences in the developing brain that cannot be resolved by gene expression alone (Bajpai and Chaudhury, 1999; Barez-Lopez et al., 2018; Tezuka et al., 2015; Zhang and Su, 2014). In the future, utilization of techniques related to protein expression and/or function are needed to address this possibility.
Neonatal bioindicator.
In contrast to the fetal brain, analysis of the PN14 neocortex yielded several potential targets that were differentially expressed following reductions in brain T4 and/or T3. Affected targets fell into three general functional categories: mediators of cell polarity and adhesion (Col11a2, Gjb6, Itih3, and Mog), neural function (Parv, Agt, Pnoc, Ngf, and Sema7a), and cell differentiation and regeneration (Hod, Hr, and Klf9). Although in this report we do not claim to provide functional implications of these differentially expressed genes in the PN14 cortex, phenotypic alterations have been linked to perturbations of these signaling pathway in TH insufficient animals (Gilbert et al., 2016). Interestingly, we also note an apparent bias in gene expression changes of structural proteins (Table 5). Proper cell structure, cell-cell contact, and cellular organization is a key physical property that is under tight control during central nervous system development, and others have demonstrated that these processes are affected in hypothyroid animals (Alvarez-Dolado et al., 2000; Berbel et al., 2001; Gould et al., 1990; Lavado-Autric et al., 2003; Liu et al., 2013; Martinez-Galan et al., 1997, 2004; Pathak et al., 2011; Paul et al., 1996; Rami and Rabie, 1988). Although it is unknown precisely how TH is affecting these processes, it appears these pathways represent convergent downstream targets of TH modulation.
Of those genes altered in the PN14 cortex, it is notable that several were differentially regulated in a manner that reflected a statistically significant change in T4, but not T3, within the brain (1 ppm PTU dose; Gjb6, Col11a2, Hopx, Itih3, Agt, Pnoc, and Parv). Above we discussed that slight changes in brain TH could still mediate adverse neurodevelopmental consequences, in addition to the potential role of T4-specific cell signaling. Given these points, these genes represent the best current candidates to serve as potential TH bioindicators. If these transcriptional targets indeed represent a readout of TH action, it is expected that they would, (1) remain stably expressed unless there is a change in at least brain T4, and (2) show a reproducible direction of change (upregulation or downregulation) given a xenobiotic exposure. These predictions are currently under investigation in the context of other TDC exposures.
Limitations of Gene Expression
Ideally a bioindicator of TH-disruption would be ubiquitously expressed throughout the brain and sensitive to hormonal perturbations across developmental time. However, this is not necessarily a biologically plausible scenario. Neurodevelopment is highly dynamic and morphogenetic processes are constrained by both time (developmental stage) and space (anatomical location) (Naumova et al., 2013). Therefore, the lack of common gene expression changes in the GD20 fetus and PN14 pup is not surprising. Given the complexities of the developing central nervous system, cognizance of developmental constraint and temporal susceptibilities to TH disruption are essential for incorporation of bioindicator data into a regulatory framework.
In addition to stage-specific transcriptional events, gene expression data is often difficult to reproduce. Although all of the proposed fetal and neonatal bioindicators were previously shown to be differentially regulated in the hypothyroid animal by others (Supplementary Table 1), gene expression data can be influenced by tissue collection methods, RNA preparation, and PCR chemistry. Adherence to established Minimum Information for Publication of Quantitative Real-Time PCR Experiments guidelines, which was designed to enhance reproducibility in qPCR studies (Bustin et al., 2009), will be essential in the implementation of a bioindicator approach. Furthermore, the lowest maternal dose of 1 ppm in the neonatal study still decreased serum and brain T4 by a large magnitude in the pups (50% and 70%, respectively). In the future, investigation of the presented biomarkers should be verified under conditions where serum and brain THs are not as detrimentally affected. Taken together, acknowledgement of the limitations of this biomarker framework is critical, though standardization of experimental design, execution, and data reporting can aid in the interpretation of gene expression data in the regulatory arena.
Linking Gene Expression to Apical Endpoints
Transcriptional changes alone do not imply that a gene is directly TH sensitive, nor does it imply adverse development. Of all the differentially regulated genes identified here, only Camk4 (Gil-Ibanez et al., 2017b; Krebs et al., 1996; Liu and Brent, 2002; Morte et al., 2010; Navarro et al., 2014), Pnoc (Gil-Ibanez et al., 2014), Klf9 (Bagamasbad et al., 2015; Denver and Williamson, 2009; Gil-Ibanez et al., 2017b; Zhang et al., 2017), Hr (Dudazy-Gralla et al., 2013; Gil-Ibanez et al., 2017b; Potter et al., 2002; Thompson and Bottcher, 1997), Sema7a (Gil-Ibanez et al., 2017b), and Sox2 (Lemkine et al., 2005; Lopez-Juarez et al., 2012), have been shown to be direct TH targets. This designation was met by (1) TH-dependent gene expression changes in the presence of a translation inhibitor (cyclohexamide), (2) in silico demonstration of thyroid receptor binding elements within regulatory regions of these genes, and/or (3) evidence of thyroid receptor/gene interaction via chromatin immunoprecipitation or gel shift assay. Though this study confirms that several transcripts are likely TH sensitive (direct or otherwise), it cannot be ruled out that these results may be due to hormone independent effects. Careful work is needed in the future to link the gene expression changes observed in this study to potential alterations at the cellular, tissue, and organ level at both GD20 and PN14, in order to determine the precise adverse effects of TH insufficiency.
CONCLUSIONS
This work clarifies the relationship between TH levels in the serum and the brain, at two developmental time points commonly assayed in regulatory studies. As a dose response experiment was performed for both stages analyzed, the resulting quantitative relationships can be utilized to predict brain TH concentrations from serum levels in both the GD20 fetus and the PN14 pup. Additionally, this work provides several candidate genes in the GD20 and PN14 cortex that may serve as bioindicators of TH status. Although it is unknown if some of these candidates are direct transcriptional targets of T4/T3, they nonetheless could be used for inference purposes in lieu of hormone and/or phenotypic measures of TH-induced developmental neurotoxicity.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jermaine Ford for his countless advice and consultation, and Drs. Andrew Johnstone and Masashi Hasegawa for their technical assistance. We also thank Drs. Kevin Crofton and Thomas Zoeller for reviewing previous versions of this article. This document has been subjected to review by the National Health and Environmental Effects Research Laboratory and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
FUNDING
This work was supported by The United States Environmental Protection Agency’s Office of Research and Development.
Footnotes
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
REFERENCES
- Alvarez-Dolado M, Cuadrado A, Navarro-Yubero C, Sonderegger P, Furley AJ, Bernal J, and Munoz A (2000). Regulation of the L1 cell adhesion molecule by thyroid hormone in the developing brain. Mol. Cell. Neurosci. 16, 499–514. [DOI] [PubMed] [Google Scholar]
- Alvarez-Dolado M, Ruiz M, Del Rı_o JA, Alc_antara S, Burgaya F, Sheldon M, Nakajima K, Bernal J, Howell BW, and Curran T. (1999). Thyroid hormone regulates reelin and dab1 expression during brain development. J. Neurosci. 19, 6979–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auso E, Lavado-Autric R, Cuevas E, Del Rey FE, Morreale DeEscobar G, and Berbel P (2004). A moderate and transient deficiency of maternal thyroid function at the beginning of fetal neocorticogenesis alters neuronal migration. Endocrinology 145, 4037–4047. [DOI] [PubMed] [Google Scholar]
- Bagamasbad PD, Bonett RM, Sachs L, Buisine N, Raj S, Knoedler JR, Kyono Y, Ruan Y, Ruan X, and Denver RJ (2015). Deciphering the regulatory logic of an ancient, ultraconserved nuclear receptor enhancer module. Mol. Endocrinol. 29, 856–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai M, and Chaudhury S (1999). Transcriptional and posttranscriptional regulation of Naþ, K(þ)-ATPase alpha isoforms by thyroid hormone in the developing rat brain. Neuroreport 10, 2325–2328. [DOI] [PubMed] [Google Scholar]
- Barez-Lopez S, Obregon MJ, Bernal J, and Guadano-Ferraz A (2018). Thyroid hormone economy in the perinatal mouse brain: Implications for cerebral cortex development. Cereb. Cortex 28(5), 1783–1793. [DOI] [PubMed] [Google Scholar]
- Bastian TW, Anderson JA, Fretham SJ, Prohaska JR, Georgieff MK, and Anderson GW (2012). Fetal and neonatal iron deficiency reduces thyroid hormone-responsive gene mRNA levels in the neonatal rat hippocampus and cerebral cortex. Endocrinology 153, 5668–5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian TW, Prohaska JR, Georgieff MK, and Anderson GW (2010). Perinatal iron and copper deficiencies alter neonatal rat circulating and brain thyroid hormone concentrations. Endocrinology 151, 4055–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbel P, Auso E, Garcia-Velasco JV, Molina ML, and Camacho M (2001). Role of thyroid hormones in the maturation and organisation of rat barrel cortex. Neuroscience 107, 383–394. [DOI] [PubMed] [Google Scholar]
- Berbel P, Navarro D, Auso E, Varea E, Rodriguez AE, Ballesta JJ, Salinas M, Flores E, Faura CC, and deEscobar GM (2010). Role of late maternal thyroid hormones in cerebral cortex development: An experimental model for human prematurity. Cereb. Cortex 20, 1462–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger-Sweeney J, and Hohmann CF (1997). Behavioral consequences of abnormal cortical development: Insights into developmental disabilities. Behav. Brain Res. 86, 121–142. [DOI] [PubMed] [Google Scholar]
- Bernal J (2007). Thyroid hormone receptors in brain development and function. Nat. Clin. Pract. Endocrinol. Metab. 3, 249–259. [DOI] [PubMed] [Google Scholar]
- Blanco J, Mulero M, Heredia L, Pujol A, Domingo JL, and Sanchez DJ (2013). Perinatal exposure to BDE-99 causes learning disorders and decreases serum thyroid hormone levels and BDNF gene expression in hippocampus in rat offspring. Toxicology 308, 122–128. [DOI] [PubMed] [Google Scholar]
- Boyes WK, Degn L, George BJ, and Gilbert ME (2018). Moderate perinatal thyroid hormone insufficiency alters visual system function in adult rats. Neurotoxicology 67, 73–83.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne P, Noyes PD, Casey WM, and Dix DJ (2017). Application of adverse outcome pathways to U.S. EPA’s endocrine disruptor screening program. Environ. Health Perspect. 125, 096001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brucker-Davis F (1998). Effects of environmental synthetic chemicals on thyroid function. Thyroid 8, 827–856. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. (2009). The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. [DOI] [PubMed] [Google Scholar]
- Calvo R, Obregon MJ, Ruiz de Ona C, Escobar del Rey F, and Morreale de Escobar G (1990). Congenital hypothyroidism, as studied in rats. Crucial role of maternal thyroxine but not of 3, 5, 3’-triiodothyronine in the protection of the fetal brain. J. Clin. Invest. 86, 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey BM, Thom EA, Peaceman AM, Varner MW, Sorokin Y, Hirtz DG, Reddy UM, Wapner RJ, Thorp JM Jr., Saade G, et al. (2017). Treatment of Subclinical Hypothyroidism or Hypothyroxinemia in Pregnancy. N. Engl. J. Med. 376, 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofton K, Gilbert ME, Paul Friendman K, Demeneix B,Marty MS, and Zoeller RT (2017). Inhibition of Thyroperoxidase and Subsequent Adverse Neurodevelopmental Outcomes in Mammals AOP 42. AOP Wiki https://aopwiki.org/aops/42, last accessed April 11, 2018. [Google Scholar]
- Denver RJ, and Williamson KE (2009). Identification of a thyroid hormone response element in the mouse Kruppel-like factor 9 gene to explain its postnatal expression in the brain. Endocrinology 150, 3935–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVito M, Biegel L, Brouwer A, Brown S, Brucker-Davis F, Cheek AO, Christensen R, Colborn T, Cooke P, Crissman J, et al. (1999). Screening methods for thyroid hormone disruptors. Environ. Health Perspect. 107, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, You SH, Williams A, Wade MG, Yauk CL, and Thomas Zoeller R (2015). Transient maternal hypothyroxinemia potentiates the transcriptional response to exogenous thyroid hormone in the fetal cerebral cortex before the onset of fetal thyroid function: A messenger and microrna profiling study. Cereb. Cortex 25, 1735–1745. [DOI] [PubMed] [Google Scholar]
- Dudazy-Gralla S, Nordstrom K, Hofmann PJ, Meseh DA, Schomburg L, Vennstrom B, and Mittag J (2013). Identification of thyroid hormone response elements in vivo using mice expressing a tagged thyroid hormone receptor alpha1. Biosci. Rep. 33, e00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar del Rey F, Mallol J, Pastor R, and Morreale de Escobar G (1987). Effects of maternal iodine deficiency on thyroid hormone economy of lactating dams and pups: Maintenance of normal cerebral 3, 5, 3’-triiodo-L-thyronine concentrations in pups during major phases of brain development. Endocrinology 121, 803–811. [DOI] [PubMed] [Google Scholar]
- Flamant F, Gauthier K, and Richard S (2017). Genetic investigation of thyroid hormone receptor function in the developing and adult brain. Curr. Top. Dev. Biol. 125, 303–335. [DOI] [PubMed] [Google Scholar]
- Galton VA (2017). The ups and downs of the thyroxine prohormone hypothesis. Molecular and Cellular Endocrinology 458, 105–111. [DOI] [PubMed] [Google Scholar]
- Gil-Ibanez P, Belinchon MM, Morte B, Obregon MJ, and Bernal J (2017a). Is the intrinsic genomic activity of thyroxine relevant in vivo? Effects on gene expression in primary cerebrocortical and neuroblastoma cells. Thyroid 27, 1092–1098. [DOI] [PubMed] [Google Scholar]
- Gil-Ibanez P, Bernal J, and Morte B (2014). Thyroid hormone regulation of gene expression in primary cerebrocortical cells: Role of thyroid hormone receptor subtypes and interactions with retinoic acid and glucocorticoids. PloS One 9, e91692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Ibanez P, Garcia-Garcia F, Dopazo J, Bernal J, and Morte B (2017b). Global transcriptome analysis of primary cerebrocortical cells: Identification of genes regulated by triiodothyronine in specific cell types. Cereb. Cortex 27, 706–717. [DOI] [PubMed] [Google Scholar]
- Gilbert ME (2011). Impact of low-level thyroid hormone disruption induced by propylthiouracil on brain development and function. Toxicol. Sci. 124, 432–445. [DOI] [PubMed] [Google Scholar]
- Gilbert ME, Goodman JH, Gomez J, Johnstone AF, and Ramos RL (2017). Adult hippocampal neurogenesis is impaired by transient and moderate developmental thyroid hormone disruption. Neurotoxicology 59, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert ME, Ramos RL, McCloskey DP, and Goodman JH (2014). Subcortical band heterotopia in rat offspring following maternal hypothyroxinaemia: Structural and functional characteristics. J. Neuroendocrinol. 26, 528–541. [DOI] [PubMed] [Google Scholar]
- Gilbert ME, Sanchez-Huerta K, and Wood C (2016). Mild thyroid hormone insufficiency during development compromises activity-dependent neuroplasticity in the hippocampus of adult male rats. Endocrinology 157, 774–787. [DOI] [PubMed] [Google Scholar]
- Gilbert ME, and Sui L (2006). Dose-dependent reductions in spatial learning and synaptic function in the dentate gyrus of adult rats following developmental thyroid hormone insufficiency. Brain Res. 1069, 10–22. [DOI] [PubMed] [Google Scholar]
- Goodman JH, and Gilbert ME (2007). Modest thyroid hormone insufficiency during development induces a cellular malformation in the corpus callosum: a model of cortical dysplasia. Endocrinology 148(6), 2593–2597. [DOI] [PubMed] [Google Scholar]
- Goldey ES, Kehn LS, Rehnberg GL, and Crofton KM (1995). Effects of developmental hypothyroidism on auditory and motor function in the rat. Toxicol. Appl. Pharmacol. 135, 67–76. [DOI] [PubMed] [Google Scholar]
- Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS, Toppari J, and Zoeller RT (2015). EDC-2: The endocrine society’s second scientific statement on endocrine-disrupting chemicals. Endocr. Rev. 36, 593–E150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Frankfurt M, Westlind-Danielsson A, and McEwen BS (1990). Developing forebrain astrocytes are sensitive to thyroid hormone. Glia 3, 283–292. [DOI] [PubMed] [Google Scholar]
- Grijota-Martinez C, Diez D, Morreale de Escobar G, Bernal J, and Morte B (2011). Lack of action of exogenously administered T3 on the fetal rat brain despite expression of the monocarboxylate transporter 8. Endocrinology 152, 1713–1721. [DOI] [PubMed] [Google Scholar]
- Grosse SD, and Van Vliet G (2011). Prevention of intellectual disability through screening for congenital hypothyroidism: How much and at what level? Arch. Dis. Childhood 96, 374–379. [DOI] [PubMed] [Google Scholar]
- Hassan I, El-Masri H, Kosian PA, Ford J, Degitz SJ, and Gilbert ME (2017). Neurodevelopment and thyroid hormone synthesis inhibition in the rat: Quantitative understanding within the adverse outcome pathway framework. Toxicol. Sci. 160, 57–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone AF, Gilbert ME, Aydin C, Grace CE, Hasegawa M, and Gordon CJ (2013). Thermoregulatory deficits in adult Long Evans rat exposed perinatally to the antithyroidal drug, propylthiouracil. Neurotoxicol. Teratol. 39, 1–8. [DOI] [PubMed] [Google Scholar]
- Kaplan MM, and Yaskoski KA (1981). Maturational patterns of iodothyronine phenolic and tyrosyl ring deiodinase activities in rat cerebrum, cerebellum, and hypothalamus. J. Clin. Invest. 67, 1208–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korevaar TI, Muetzel R, Medici M, Chaker L, Jaddoe VW, de Rijke YB, Steegers EA, Visser TJ, White T, Tiemeier H, et al. (2016). Association of maternal thyroid function during early pregnancy with offspring IQ and brain morphology in childhood: A population-based prospective cohort study. Lancet Diabetes Endocrinol. 4, 35–43. [DOI] [PubMed] [Google Scholar]
- Krebs J, Means RL, and Honegger P (1996). Induction of calmodulin kinase IV by the thyroid hormone during the development of rat brain. J. Biol. Chem. 271, 11055–11058. [DOI] [PubMed] [Google Scholar]
- Lavado-Autric R, Aus_o E, Garc_ıa-Velasco JV, del Carmen Arufe M, Escobar del Rey F, Berbel P, and Morreale deEscobar G. (2003). Early maternal hypothyroxinemia alters histogenesis and cerebral cortex cytoarchitecture of the progeny. J. Clin. Invest. 111, 1073–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkine GF, Raj A, Alfama G, Turque N, Hassani Z, Alegria-Prevot O, Samarut J, Levi G, and Demeneix BA (2005). Adult neural stem cell cycling in vivo requires thyroid hormone and its alpha receptor. FASEB J. 19, 863–865. [DOI] [PubMed] [Google Scholar]
- Li GH, Post J, Koibuchi N, and Sajdel-Sulkowska EM (2004). Impact of thyroid hormone deficiency on the developing CNS: Cerebellar glial and neuronal protein expression in rat neonates exposed to antithyroid drug propylthiouracil. Cerebellum 3, 100–106. [DOI] [PubMed] [Google Scholar]
- Liu CR, Miao J, Zhang YL, Liu YM, and Yu BG (2013). Effects of hypothyroidism on expression of CRMP2B and ARPC5 during development of the rat frontal cortex. Int. J. Biol. Sci. 9, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YY, and Brent GA (2002). A complex deoxyribonucleic acid response element in the rat Ca(2þ)/calmodulin-dependent protein kinase IV gene 5’-flanking region mediates thyroid hormone induction and chicken ovalbumin upstream promoter transcription factor 1 repression. Mol. Endocrinol. 16, 2439–2451. [DOI] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lopez-Juarez A, Remaud S, Hassani Z, Jolivet P, Pierre Simons J, Sontag T, Yoshikawa K, Price J, Morvan-Dubois G, and Demeneix BA (2012). Thyroid hormone signaling acts as a neurogenic switch by repressing Sox2 in the adult neural stem cell niche. Cell Stem Cell 10, 531–543. [DOI] [PubMed] [Google Scholar]
- Maher SK, Wojnarowicz P, Ichu TA, Veldhoen N, Lu L, Lesperance M, Propper CR, and Helbing CC (2016). Rethinking the biological relationships of the thyroid hormones, l-thyroxine and 3, 5, 3’-triiodothyronine. Comparat. Biochem. Physiol. Part D Genomics Proteomics 18, 44–53. [DOI] [PubMed] [Google Scholar]
- Martinez-Galan JR, Escobar del Rey F, Morreale de Escobar G, Santacana M, and Ruiz-Marcos A (2004). Hypothyroidism alters the development of radial glial cells in the term fetal and postnatal neocortex of the rat. Brain Res. Dev. Brain Res. 153, 109–114. [DOI] [PubMed] [Google Scholar]
- Martinez-Galan JR, Pedraza P, Santacana M, Escobar del Rey F, Morreale de Escobar G, and Ruiz-Marcos A (1997). Myelin basic protein immunoreactivity in the internal capsule of neonates fromrats on a low iodine intake or onmethylmercaptoimidazole (MMI). Brain Res. Dev. Brain Res. 101, 249–256. [DOI] [PubMed] [Google Scholar]
- Massart F, Massai G, Placidi G, and Saggese G (2006). Child thyroid disruption by environmental chemicals. Minerva Pediatr. 58, 47–53. [PubMed] [Google Scholar]
- Morreale de Escobar G, Calvo R, Obregon MJ, and Escobar Del Rey F (1990). Contribution of maternal thyroxine to fetal thyroxine pools in normal rats near term. Endocrinology 126, 2765–2767. [DOI] [PubMed] [Google Scholar]
- Morreale de Escobar G, Pastor R, Obregon MJ, and Escobar del Rey F (1985). Effects of maternal hypothyroidism on the weight and thyroid hormone content of rat embryonic tissues, before and after onset of fetal thyroid function. Endocrinology 117, 1890–1900. [DOI] [PubMed] [Google Scholar]
- Morte B, Diez D, Auso E, Belinchon MM, Gil-Ibanez P, Grijota-Martinez C, Navarro D, de Escobar GM, Berbel P, and Bernal J (2010). Thyroid hormone regulation of gene expression in the developing rat fetal cerebral cortex: Prominent role of the Ca2þ/calmodulin-dependent protein kinase IV pathway. Endocrinology 151, 810–820. [DOI] [PubMed] [Google Scholar]
- Murphy NC, Diviney MM, Donnelly JC, Cooley SM, Kirkham CH, Foran AM, Breathnach FM, Malone FD, and Geary MP (2015). The effect of maternal subclinical hypothyroidism on IQ in 7- to 8-year-old children: A case control review. Aust. N. Z. J. Obstetr. Gynaecol. 55, 459–463. [DOI] [PubMed] [Google Scholar]
- Naumova OY, Lee M, Rychkov SY, Vlasova NV, and Grigorenko EL (2013). Gene expression in the human brain: The current state of the study of specificity and spatiotemporal dynamics. Child Dev 84, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro D, Alvarado M, Morte B, Berbel D, Sesma J, Pacheco P, Morreale de Escobar G, Bernal J, and Berbel P (2014). Late maternal hypothyroidism alters the expression of Camk4 in neocortical subplate neurons: A comparison with Nurr1 labeling. Cerebral Cortex 24, 2694–2706. [DOI] [PubMed] [Google Scholar]
- Negishi T, Kawasaki K, Sekiguchi S, Ishii Y, Kyuwa S, Kuroda Y, and Yoshikawa Y (2005). Attention-deficit and hyperactive neurobehavioural characteristics induced by perinatal hypothyroidism in rats. Behav. Brain Res. 159, 323–331. [DOI] [PubMed] [Google Scholar]
- OECD/OCDE (Test No. 426). Developmental Neurotoxicity Study. OECD Guidelines for the Testing of Chemicals, Section 4. 10.1787/20745788. [DOI] [Google Scholar]
- OECD/OCDE (Test No. 421). Reproduction/Developmental Toxicity Study. OECD Guidelines for the Testing of Chemicals, Section 4. 10.1787/2074578. [DOI] [Google Scholar]
- O’Shaughnessy KL, Kosian PA, Ford JL, Oshiro WM, Degitz SJ, and Gilbert ME (2018). Developmental Thyroid Hormone Insufficiency Induces a Cortical Brain Malformation and Learning Impairments: A Cross-Fostering Study. Toxicological sciences 163(1), 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyanagi K, Negishi T, and Tashiro T (2015). Action of thyroxine on the survival and neurite maintenance of cerebellar granule neurons in culture. J. Neurosci. Res. 93, 592–603. [DOI] [PubMed] [Google Scholar]
- Pathak A, Sinha RA, Mohan V, Mitra K, and Godbole MM (2011). Maternal thyroid hormone before the onset of fetal thyroid function regulates reelin and downstream signaling cascade affecting neocortical neuronal migration. Cereb. Cortex 21, 11–21. [DOI] [PubMed] [Google Scholar]
- Paul S, Das S, Poddar R, and Sarkar PK (1996). Role of thyroid hormone in the morphological differentiation and maturation of astrocytes: Temporal correlation with synthesis and organization of actin. Eur.J. Neurosci. 8, 2361–2370. [DOI] [PubMed] [Google Scholar]
- Potter GB, Zarach JM, Sisk JM, and Thompson CC (2002). The thyroid hormone-regulated corepressor hairless associates with histone deacetylases in neonatal rat brain. Mol. Endocrinol. 16, 2547–2560. [DOI] [PubMed] [Google Scholar]
- Rabie A, Clavel MC, and Legrand J (1980). Analysis of the mechanisms underlying increased histogenetic cell death in developing cerebellum of the hypothyroid rat: Determination of the time required for granule cell death. Brain Res. 190, 409–414. [DOI] [PubMed] [Google Scholar]
- Rami A, and Rabie A (1988). Effect of thyroid deficiency on the development of glia in the hippocampal formation of the rat: An immunocytochemical study. Glia 1, 337–345. [DOI] [PubMed] [Google Scholar]
- Schroeder AC, and Privalsky ML (2014). Thyroid hormones, t3 and t4, in the brain. Front. Endocrinol. 5, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz HL, Ross ME, and Oppenheimer JH (1997). Lack of effect of thyroid hormone on late fetal rat brain development. Endocrinology 138, 3119–3124. [DOI] [PubMed] [Google Scholar]
- Sharlin DS, Tighe D, Gilbert ME, and Zoeller RT (2008). The balance between oligodendrocyte and astrocyte production in major white matter tracts is linearly related to serum total thyroxine. Endocrinology 149, 2527–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA, Johnson A, Whitmore WL, and Slepetis RJ (1984). Ornithine decarboxylase and polyamines in developing rat brain and heart: Effects of perinatal hypothyroidism. Int. J. Dev. Neurosci. 2, 155–161. [DOI] [PubMed] [Google Scholar]
- Stoker TE, Gibson EK, and Zorrilla LM (2010). Triclosan exposure modulates estrogen-dependent responses in the female wistar rat. Toxicol. Sci. 117, 45–53. [DOI] [PubMed] [Google Scholar]
- Tezuka Y, Herai N, Inomata Y, Kagami K, Yamauchi J, Nishigori H, and Sanbe A (2015). Upregulation of inorganic pyrophosphatase 1 as a JNK phosphatase in hypothyroid embryonic chick cerebellum. Life Sci. 128, 94–100. [DOI] [PubMed] [Google Scholar]
- Thibodeaux JR, Hanson RG, Rogers JM, Grey BE, Barbee BD, Richards JH, Butenhoff JL, Stevenson LA, and Lau C (2003). Exposure to perfluorooctane sulfonate during pregnancy in rat and mouse. I: Maternal and prenatal evaluations. Toxicol. Sci. 74, 369–381. [DOI] [PubMed] [Google Scholar]
- Thompson CC, and Bottcher MC (1997). The product of a thyroid hormone-responsive gene interacts with thyroid hormone receptors. Proc. Natl. Acad. Sci. U.S.A. 94, 8527–8532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramontin AD, Garcia-Verdugo JM, Lim DA, and Alvarez-Buylla A (2003). Postnatal development of radial glia and the ventricular zone (VZ): A continuum of the neural stem cell compartment. Cereb. Cortex 13, 580–587. [DOI] [PubMed] [Google Scholar]
- US EPA Adverse Outcome Pathway (AOP) Research Brief. (2017). Research Brief. [Google Scholar]
- US EPA Guidance for Thyroid Assays in Pregnant Animals, Fetuses and Postnatal Animals, and Adult Animals. (2005). Office of Pesticide Programs (OPP) Guidance Document. [Google Scholar]
- Villeneuve DL, Crump D, Garcia-Reyero N, Hecker M, Hutchinson TH, LaLone CA, Landesmann B, Lettieri T, Munn S, Nepelska M, et al. (2014a). Adverse outcome pathway (AOP) development I: Strategies and principles. Toxicol. Sci. 142, 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve DL, Crump D, Garcia-Reyero N, Hecker M, Hutchinson TH, LaLone CA, Landesmann B, Lettieri T, Munn S, Nepelska M, et al. (2014b). Adverse outcome pathway development II: Best practices. Toxicol. Sci. 142, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby KA, McAndrews MP, and Rovet JF (2014). Effects of maternal hypothyroidism on offspring hippocampus and memory. Thyroid: official journal of the American Thyroid Association 24(3), 576–84. [DOI] [PubMed] [Google Scholar]
- Zamoner A, Heimfarth L, Oliveira Loureiro S, Royer C, Mena Barreto Silva FR, and Pessoa-Pureur R (2008). Nongenomic actions of thyroxine modulate intermediate filament phosphorylation in cerebral cortex of rats. Neuroscience 156, 640–652. [DOI] [PubMed] [Google Scholar]
- Zhang HM, and Su Q (2014). PKC in developmental hypothyroid rat brain. Neurol. Sci. 35, 1161–1166. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Fan Y, Yu X, Wang X, Bao S, Li J, Fan C, Shan Z, and Teng W (2015). Maternal subclinical hypothyroidism impairs neurodevelopment in rat offspring by inhibiting the CREB signaling pathway. Mol. Neurobiol. 52, 432–441. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xue Y, Cao C, Huang J, Hong Q, Hai T, Jia Q, Wang X, Qin G, Yao J, et al. (2017). Thyroid hormone regulates hematopoiesis via the TR-KLF9 axis. Blood 130, 2161–2170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.