

Abstract
Recombineering inserts PCR products into DNA using homologous recombination. A pair of short homology arms (50 base pairs) on the ends of a PCR cassette target the cassette to its intended location. These homology arms can be easily introduced as 5′ primer overhangs during the PCR reaction. The flexibility to choose almost any pair of homology arms enables the precise modification of virtually any DNA for the purposes of sequence deletion, replacement, insertion, or point mutation. Recombineering often offers significant advantages relative to previous homologous recombination methods that require the construction of cassettes with large homology arms and relative to traditional cloning methods that become intractable for large plasmids or DNA sequences. However, the tremendous number of variables, options, and pitfalls that can be encountered when designing and performing a recombineering protocol for the first time introduce barriers that can make recombineering a challenging technique for new users to adopt. This article focuses on three recombineering protocols we have found to be particularly robust, providing a detailed guide for choosing the simplest recombineering method for a given application, and for performing and troubleshooting experiments.
Keywords: recombineering, homologous recombination, genetic engineering, Escherichia coli, viral genome engineering
INTRODUCTION
Recombineering is a versatile genetic engineering method that can be used to introduce deletions, insertions, gene replacements or point mutations virtually anywhere in a DNA sequence and is particularly useful for editing large Bacterial Artificial Chromosomes (BAC), DNA virus genomes, or bacterial genomes. The method utilizes the temporary expression of lambda phage genes (red αβγ) that facilitate recombination between the target BAC or target genome and a DNA targeting cassette that is introduced into the cell through electroporation. Because only about 50 base pairs (bp) of homology are required to facilitate recombination, the homology arms of a recombineering targeting cassette can be conveniently introduced as primer overhangs in a single PCR step (Murphy, 2016). Other homologous recombination-based methods generally require 500–1000 bp homology arms that need to be appended to a targeting cassette through time-consuming restriction cloning or difficult overlap extension PCR (Hamilton, Aldea, Washburn, Babitzke, & Kushner, 1989; Kong, Yang, & Geller, 1999; Uil et al., 2011; Winans, Elledge, Krueger, & Walker, 1985). Another major advantage of recombineering is the ability to modify plasmids of virtually any size. Other popular in vitro DNA editing methods, such as endonuclease cloning or in vitro assembly-based techniques rely on transferring the recombinant DNA plasmid into E. coli after the modifications have been made in a process known as “transformation”. The efficiency of transformation and the ability to modify DNA plasmids in vitro decreases dramatically for plasmids greater than 10 kb (Inoue, Nojima, & Okayama, 1990; Siguret, Ribba, Chérel, Meyer, & Piétu, 1994), rendering most in vitro methods impractical for modifying large plasmids. Because recombineering modifies plasmids that already reside in the E. coli cell, it circumvents the transformation bottleneck. Additionally, recombineering creates modifications without the need to cut or ligate DNA, thereby avoiding other challenges associated with in vitro methods when modifying very large plasmids, such as the lack of unique restriction sites or the difficulty of amplifying large DNA fragments by PCR (Cheng, Fockler, Barnes, & Higuchi, 1994).
Typical applications of recombineering include modifying the E. coli genome itself or the genomes of large human or animal viruses as BACs in E. coli that are later reintroduced into the host organism (Berman et al., 2018; Moore et al., 2018; Narayanan & Chen, 2011). The modified genomes can then produce genetically-engineered virions in a process known as “viral rescue”. Recombineering is also ideal for editing BACs with large fragments of human or animal genomes, such that these modified fragments can then be used as targeting cassettes to modify human or animal cells via non-recombineering homologous recombination (Murphy, 2016). Note that, in human and animal cells, long homology arms are required and thus targeting cassettes must necessarily be large (Baker et al., 2017; Hasty, Rivera-Pérez, & Bradley, 1991). Importantly, recombineering is not limited to modifying the E. coli genome, viral genomes, or large targeting cassettes. Rather, it can be used whenever a large DNA construct needs to be modified in E. coli.
There are many decisions to make when choosing an application-appropriate recombineering strategy. In particular, the multitude of available recombineering-based methods and their many variations can make it daunting for a new user to choose the best option for a specific purpose. This article aims to distill the many possible iterations down to just three simple protocols that, based on our extensive experience engineering adenovirus vectors (Berman et al., 2018; Wong et al., 2018), E. coli genomes (Moore et al., 2018), and other DNA sequences, function reliably and efficiently. Further, although recombineering can be incredibly simple and efficient to apply, it is also sensitive to many critical experimental parameters. Thus, each protocol provides, as a companion resource, detailed annotations for avoiding common pitfalls. In order of increasing difficulty and required time, the protocols are: Basic Protocol 1—One-Step Recombineering (Datsenko & Wanner, 2000; Yu et al., 2000), Basic Protocol 2—Direct and Inverted Repeat stimulated Excision (DIRex) Recombineering (Näsvall, 2017), and Basic Protocol 3—Standard Two-Step Recombineering (H. Wang et al., 2014). As explained below, the ideal choice of protocol depends on factors such as the type of modification to be made, the size of the modification to be made, and the number of modifications to be made on a given BAC or genome. Mastery of the protocols and concepts described here should provide a strong foundation for understanding, utilizing, and adapting these and other recombineering-based methods.
STRATEGIC PLANNING
Under standard double-stranded DNA recombineering conditions, only 1 in 10,000–100,000 cells yields successful recombinants that incorporate the targeting cassette at the desired location (Datta, Costantino, Zhou, & Court, 2008). This frequency is far too low to reasonably screen colonies one-by-one for recombinants, so one must employ selection-based methods where only the rare recombinants survive after selection. Unfortunately, most modifications do not significantly impact the survival of the E. coli cell and therefore cannot be selected for directly. Thus, either an antibiotic resistance gene must be included in the modification to yield a “marked” modification (Basic Protocol 1), or a two-step selection-counterselection method must be employed to yield a “seamless” modification in which the final recombinant does not contain an antibiotic resistance gene (Basic Protocol 2 or 3). To choose the simplest protocol for a desired modification, refer to the decision tree in Figure 1A. Detailed potential uses and time requirements for each of the protocols are provided in Figure 1B.
Figure 1.
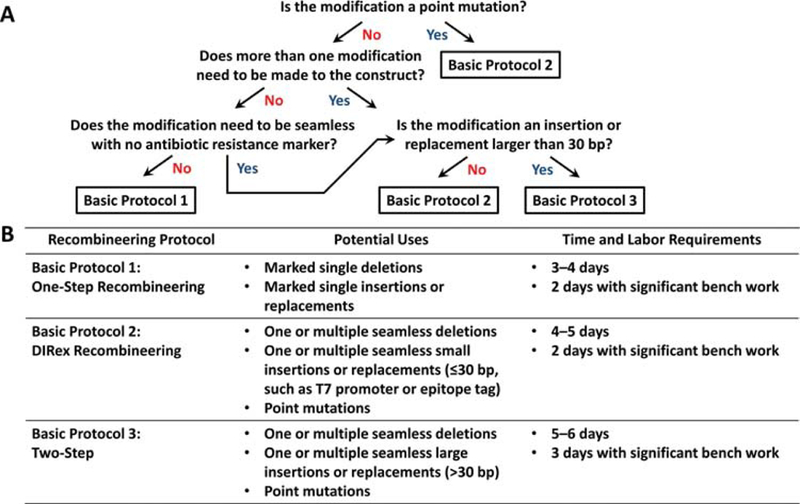
Protocol selection guide. (A) Decision tree for choosing the quickest, simplest protocol for making a desired type of modification. (B) The potential uses and time requirements of each protocol.
For all three Basic Protocols described in this article, it is essential to obtain the target DNA in a BAC unless the E. coli genome itself is to be modified. BACs have low-copy origins (1–2 copies per cell), which is necessary ensure that any plasmids that are modified by recombineering will be isolated in a given cell without any copies of the parent plasmid. If a high-copy plasmid were used as the target, successful recombinants would be mixed with unmodified plasmid within a single cell, even after selection pressure is applied (Thomason, Costantino, Shaw, & Court, 2007). For Basic Protocol 1, the mixture would need to be re-transformed to purify the successful recombinant. For Basic Protocols 2 and 3, the counterselection step would be impossible. Fortunately, it is straightforward to replace any plasmid origin with the BAC origin using one-step recombineering (via Basic Protocol 1). We note that a significant drawback of BACs is that their low copy-number makes it very difficult to purify large quantities of DNA, which is often necessary for downstream applications such as DNA transfection into mammalian cells. Therefore, once all of the desired modifications have been made, it is often useful to replace the low-copy BAC origin with a high-copy pUC origin using one-step recombineering (via Basic Protocol 1). We further note that, while there are some BACs that can switch to high-copy numbers upon arabinose induction by utilizing a second origin, oriV (Westenberg, Bamps, Soedling, Hope, & Dolphin, 2010; Wild, Hradecna, & Szybalski, 2002), all the protocols described here use arabinose induction for other purposes. If oriV-containing BACS were used, the consequence would be undesired high-copy replication of the BACs. Thus, it is essential to ensure that any target BAC employed does not contain oriV.
BASIC PROTOCOL 1
ONE-STEP RECOMBINEERING FOR MARKED INSERTIONS, REPLACEMENTS OR DELETIONS
Introducing a marked modification is the most straightforward way to create DNA insertions, deletions or replacements—large or small—and can be accomplished quickly in a single step (Datsenko & Wanner, 2000). To introduce a marked modification, an antibiotic resistance gene needs to either replace the region that is to be deleted or be included with any regions that are to be inserted, such that recombinants can be selected for by streaking cells on an antibiotic-containing plate (Fig. 2). Marked modifications can be useful for when only one or a few deletions, insertions, or replacements need to be introduced in a given piece of DNA. Nonetheless, the number of sequential modifications that can be introduced is necessarily limited by the requirement for a new antibiotic resistance gene at each step (Thomason, Sawitzke, Li, Costantino, & Court, 2014). Finally, we note that although various scales can be used, we strongly recommend (and describe below) performing the protocol on a 1 mL scale for ease of sample processing and using heat blocks, as described in previous work (H. Wang et al., 2014).
Figure 2.
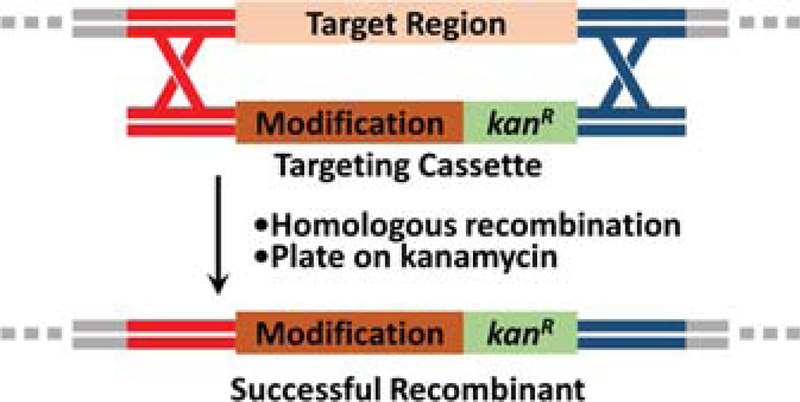
Schematic of one-step recombineering in which a kanamycin resistance gene (kanR) is used to select for recombinants and thereafter becomes part of the modification.
Materials
Target BAC (supplied by user)
psc101-gbaA plasmid (see Table 1)
Table 1.
Plasmids used in these protocols
| Plasmid Name | Purpose | Source |
|---|---|---|
| psc101-gbaA | Induction of recombineering genes and/or the ccdA antitoxin in Basic Protocols 1–3 | Stewart Laboratory, Biotechnology Center, TU Dresden (psc101-gbaA = pSC101-BAD-ccdA-RhaB-gbaA-tet http://www.biotec.tu-dresden.de/research/stewart/group-page.html) (H. Wang et al., 2014) |
| R6K-kan-ccdB | PCR template for the generation of kanR or kanR+ccdB targeting cassettes in Basic Protocol 1 and 3 | Shoulders Laboratory, Department of Chemistry, Massachusetts Institute of Technology (Genbank Accession MH325106.1) (Berman et al., 2018) |
| pBeloBAC11loxP2272 | PCR template for the generation of BAC origin-containing targeting cassettes in Basic Protocol 1, which are used to convert other types of plasmids to BACs | Addgene #60342 (https://www.addgene.org/60342/) (Coren, 2017) |
| pcDNA-DEST40 | PCR template for the generation of pUC origin-containing targeting cassettes in Basic Protocol 1, which are used to convert low-copy BAC plasmids to high-copy pUC plasmids | Invitrogen Cat #12274015 (https://www.thermofisher.com/order/catalog/product/12274015) |
| R6K-KCA | PCR template for the generation of DIRex kanR+ccdB targeting half-cassettes in Basic Protocol 2 | Shoulders Laboratory, Department of Chemistry, Massachussetts Institute of Technology (Genbank Accession MK787297) (Moore, Papa, & Shoulders, 2018) |
| R6K-AKC | PCR template for the generation of DIRex kanR+ccdB targeting half-cassettes in Basic Protocol 2 | Shoulders Laboratory, Department of Chemistry, Massachussetts Institute of Technology (Genbank Accession MK787296) (Moore et al., 2018) |
Targeting primers (designed and ordered by user)
Sequencing primers (designed and ordered by user)
R6K-kan-ccdB plasmid (see Table 1)
Template plasmid (used for insertions; supplied by user)
pBeloBAC11loxP2272 (see Table 1)
pcDNA-DEST40 (see Table 1)
1.7 mL microcentrifuge tubes (e.g., VWR 87003–294)
Bunsen burner and gas line
18-gauge needles (e.g., Becton Dickinson 305195)
Sterile 10 μL pipette tips (e.g., VWR 89079–466)
Sterile 200 μL pipette tips (e.g., VWR 89079–460)
Sterile 1000 μL pipette tips (e.g., VWR 89079–472)
Lysogeny broth (LB) media (see recipe)
1000X streptomycin stock solution (see recipe)
DH10B E. coli cells (Invitrogen 18297010) or other target E. coli strain
50 mL conical tubes (e.g., Cellstar 227261)
0.1 cm electroporation cuvettes (e.g., Bio-Rad 1652083)
Kimwipes (Kimberly-Clark 34155)
Super optimal broth with catabolic repression (SOC) media (see recipe)
LB agar plates with 1X chloramphenicol and 1X tetracycline (see recipe)
9” soda-lime glass Pasteur pipettes (e.g., VWR 14672–380)
1000X tetracycline stock solution (see recipe)
1000X chloramphenicol stock solution (see recipe)
Sterile 50% glycerol (see recipe)
10% rhamnose stock solution (see recipe)
LB agar plates with 1X kanamycin (see recipe)
14 mL sterile culture tubes (e.g., VWR 60818–703)
10 mL serological pipette (e.g., Cellstar 607180)
1000X kanamycin stock solution (see recipe)
Miniprep DNA isolation kit (e.g., Omega Bio-tek Plasmid Mini Kit D6942–01)
Isopropanol (e.g., Macron 3032–16)
70% ethanol (e.g., Koptec V1401)
Restriction endonucleases (selected by user; e.g., New England Biolabs)
Agarose (e.g., Lonza 50004)
0.5X TBE buffer (see recipe)
Paper towels
GelGreen Nucleic Acid Gel Stain (Biotium 41005)
Quick-Load Purple 1 kb DNA Ladder (New England Biolabs N0552S)
6X Gel Loading Dye (New England Biolabs B7024S)
DNA Editing Software (e.g., Snapgene, ApE, Serial Cloner)
P10 pipettor (e.g., VWR 89079–962)
P200 pipettor (e.g., VWR 89079–970)
P1000 pipettor (e.g., VWR 89079–974)
Shaking heat block for 1.5 mL tubes (e.g., Eppendorf Thermomixer F1.5 5384000020)
UV-Vis spectrometer (e.g., Thermo Fisher Nanodrop 2000c)
Ice maker and ice buckets
Refrigerated microcentrifuge ≥16,000 × g (e.g., Thermo Fisher Heraeus Fresco 21 75002426)
Electroporator (Bio-Rad Micropulser 1652100)
30 °C incubator (e.g., VWR Gravity Convection Incubator 89511–422)
Pipette gun (e.g., Drummond Portable Pipet-Aid XP 4–000-101)
30 °C shaking incubator (e.g., Thermo Fisher MaxQ 4000 SHKE4000)
Precision balance (e.g., Mettler Toledo ME1002TE 30216559)
250 mL glass Erlenmeyer flask (e.g., VWR 10536–914)
Agarose gel electrophoresis system (e.g., Bio-Rad Mini-Sub Cell GT System 1664401)
Microwave oven
Mini-gel caster (e.g., Bio-Rad 1704422)
Blue LED transilluminator (e.g., Maestrogen LED Transilluminator SLB-01W)
Design targeting primers and create the targeting cassette
First, choose appropriate homology arms to make the desired modification. Immediately to the left of the region to be deleted or replaced, highlight 50 bp, not including the region to be deleted or replaced, and copy the sequence from the top strand (in the 5′ to 3′ direction) for the left homology arm (Figs. 3A–B).
Immediately to the right of the region to be deleted or replaced, highlight 50 bp, not including the region to be deleted or replaced, and copy or mark the sequence from the BOTTOM strand for the right homology arm. Ensure that the sequence copied and designated as the right homology arm is the bottom strand in the 5′ to 3′ direction (i.e., the reverse complement of the TOP strand). In the case of an insertion, the 3′ ends of the left homology arm and the right homology arm will be adjacent to each other if the homology arms are properly designed.
- To create targeting primers and targeting cassettes for:
- A deletion: Append the left homology arm sequence to the Kan.For primer sequence and append the right homology arm to the Kan.Rev primer sequence (see Table 2 for primer sequences). Use these primers to generate the targeting cassette using R6K-kan-ccdB as a PCR template (see Support Protocol 1). Bear in mind, this targeting cassette will have the kanR resistance gene facing from left to right. To switch the direction of the kanR gene, append the left homology arm to Kan.Rev and the right homology arm to Kan.For.The protocols in this article use kanamycin resistance for selection of recombinants because it works reliably. Other antibiotic resistance genes can also be used for selection. However, ampicillin is not as reliable as kanamycin for killing susceptible bacteria because the β-lactamase enzyme that confers ampicillin resistance is secreted and destroys extracellular antibiotic, allowing some nearby susceptible cells to survive and grow (Medaney, Dimitriu, Ellis, & Raymond, 2016). Therefore, we recommend against using ampicillin resistance for routine recombineering.
- An insertion or replacement: Create or obtain a plasmid that has the genes or region to be inserted into the target. The region to be inserted must include an antibiotic resistance gene for selection. Design primers that have melting temperatures (Tm) of ~60 °C—note that most DNA editing softwares display the Tm as DNA is highlighted—and that amplify the entire region to be inserted (Fig. 3C) along with the antibiotic resistance gene (Sawitzke et al., 2013). In the primer design, append the left homology arm to one primer and the right homology arm to the other primer (Fig. 3D). Note that the choice of which homology arm to append to which primer will determine the ultimate orientation of the insertion. Once the primers are obtained, use them in conjunction with the template plasmid to create the targeting cassette (see Support Protocol 1).
- Converting a plasmid into a BAC: Design homology arms that flank, and thus can be used to replace, the origin and antibiotic resistance gene of the plasmid to be converted to a BAC. Append these homology arms onto the BAC.For and BAC.Rev primer sequences in Table 2. Once the primers are obtained, use them in conjunction with pBeloBac11loxP2272 to create the targeting cassette (see Support Protocol 1). Note that this targeting cassette will confer chloramphenicol resistance, so it is important for selection purposes that the origin that it replaces does not also confer chloramphenicol.Replacing the origin of a high-copy plasmid with the BAC origin nearly always results in the formation of a heteromultimer in which multiple high-copy plasmids and BACs fuse together into one large plasmid with mixed origins. Any successful recombinant cells also likely contain unmodified parent plasmids. Heteromultimers and/or mixtures are unsuitable targets for further recombination. Therefore, it is generally necessary to “remonomerize” and isolate the recombinant BAC via restriction digestion as described in Support Protocol 4. Alternatively, target DNA can be ligated to the origin of pBeloBac11loxP2272 using standard restriction endonuclease cloning.
- Converting a BAC into a high-copy pUC plasmid: Obtain and use primers pUC.For and pUC.Rev (Table 2) in conjunction with pcDNA-DEST40 to create the targeting cassette (see Support Protocol 1). The pUC.For and pUC.Rev primers already have the necessary homology arms, assuming the BAC is derived from pBeloBac11loxP2272. If in doubt, double-check the vector sequence to ensure that the homology arms in pUC.For and pUC.Rev exist in the target BAC and flank the BAC origin. Note that this targeting cassette confers ampicillin resistance. The pcDNA-DEST40 plasmid is particularly useful not only because it provides a template for the pUC origin, but also because it encodes the toxic ccdB gene. If any template plasmid enters the cell during the recombineering electroporation step, the ccdB gene will kill that cell and prevent false positives resulting from template plasmid replication. Templates other than pcDNA-DEST40 can be used to generate a targeting cassette with the pUC origin, but then precautions should be taken to inactivate the template (see Support Protocol 1) and the user may need to redesign the pUC.For and pUC.Rev primers, depending on the alternate template chosen.
Replacing the origin of a BAC with the pUC origin almost always results in the formation of a multimers in which multiple pUCs and/or BACs fuse together into one large plasmid. Multimers may be unsuitable for later applications. Therefore, it is wise to “remonomerize” the recombinant pUC via restriction digestion as described in Support Protocol 4. - For every modification to be made, order “sequencing primers” that amplify the target region and can be used to sequence the recombination junctions and confirm successful recombination.In general, sequencing primers should be ~200 bp away from the homology arms so that any PCR products generated of the region will be at least 400 bp in length (Murphy, 2016; Thomason et al., 2014). PCR products shorter than 400 bp can be difficult to visualize on and purify from a standard agarose gel. In addition to PCR amplifying the region of interest, the sequencing primers will also be used in the subsequent Sanger sequencing reaction. The first 50–100 bp of a Sanger sequencing trace tends to be noisy and unreliable for interpreting the sequence. Placing the sequencing primer 200 bp upstream of the homology arm ensures that the trace is reliable in the region of the recombination junction, thus allowing confirmation of successful recombination.
Figure 3.
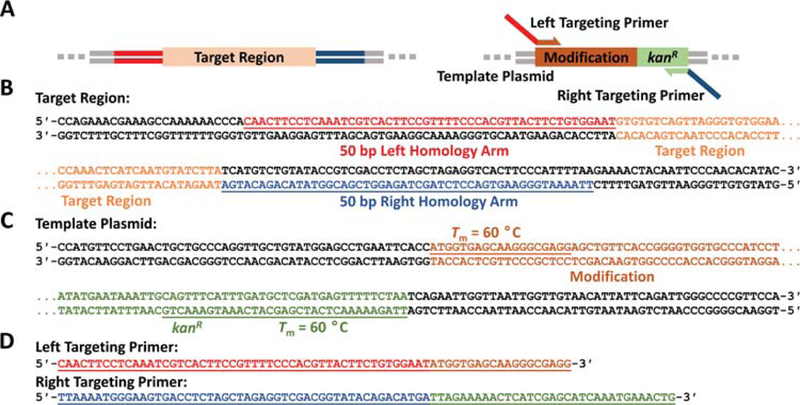
Primer design example for a marked insertion via one-step recombineering. (A) The left and right targeting primers amplify the region to be inserted, which contains a kanamycin resistance marker (kanR). The left and right targeting primers have overhangs that are homologous to the regions flanking the target region to be replaced. (B) The left homology arm (red underlined) is copied from the top strand while the right homology arm (blue underlined) is copied from the bottom strand. (C) The left primer that amplifies the modification from the template plasmid (brown underlined) is copied from the top strand, and the right primer that amplifies the modification from the template plasmid (green underlined) is copied form the bottom strand. (D) The homology arms are appended to the primers to form the full targeting primers. Note that any sequences copied from the bottom strand are reversed here to be represented in the 5′ to 3′ direction.
Table 2.
Primer sequences
| Primer Name | Primer Sequence | Template | Purpose |
|---|---|---|---|
| Kan.For | CCCTCATCAGTGCCAACATAGTAAG | R6K-kan-ccdB | Introducing marked modifications (Basic Protocol 1) |
| Kan.Rev | GTCGAGGCTGATCAGCGAG | R6K-kan-ccdB | Introducing marked modifications (Basic Protocol 1) |
| BAC.For | CGTTTAAGGGCACCAATAACTGCC | pBeloBac11loxP2272 | Converting other plasmids to BACs |
| BAC.Rev | GCCCGGTTAACGTGCCG | pBeloBac11loxP2272 | Converting other plasmids to BACs |
| pUC.For | CGTTTAAGGGCACCAATAACTGCCTTAAAAAAATTACGCCCCGCCCTGCCGAACCGTAAAAAGGCCGC | pcDNA-DEST40 | Converting BACs into high-copy pUC plasmids |
| pUC.Rev | GCCCGGTTAACGTGCCGGCACGGCCTGGGTAACCAGGTATTTTGTCCACACTGGCCCGTACATCGCGAAG | pcDNA-DEST40 | Converting BACs into high-copy pUC plasmids |
| DIRexPrimer | GGCGAACCAACCGGTTTAGG | R6K-AKC or R6K-KCA | Introducing seamless modifications (Basic Protocol 2) |
| DIRexKan.For | GCTCGACGTTGTCACTGAAGC | R6K-KCA | Introducing seamless modifications (Basic Protocol 2) |
| DIRexKan.Rev | CGCCGTCGGGCATGC | R6K-AKC | Introducing seamless modifications (Basic Protocol 2) |
| KanccdB.Rev | CCGCTCATTAGGCGGGC | R6K-kan-ccdB | Introducing seamless modifications (Basic Protocol 3) |
Transforming the target BAC and psc101-gbaA recombineering plasmid into DH10B E. coli
-
5.Heat an 18-gauge needle over a Bunsen burner until red-hot. Poke a needle-sized hole in the lid of a closed, sterile 1.7 mL microcentrifuge tube with the needle while it is still hot. The needle can be re-used after burning off excess melted plastic. Cultures throughout these protocols should be grown in 1.7 mL microcentrifuge tubes with holes.Heating the needle sterilizes it and helps melt a needle-sized hole into the plastic. This hole is critical for proper aeration and growth of the bacterial culture. Never hold the microcentrifuge tube in your hand while poking a hole in the lid. Instead, secure the tube in a rack.
-
6.
If the target BAC and the psc101-gbaA plasmid are already both present in the same E. coli strain, proceed to step 23. If the intent is to engineer the E. coli genome instead of a target BAC, proceed to step 7 but omit the addition of chloramphenicol to all agar plates and media in subsequent steps.
-
7.Add 1 mL of LB media to the tube, followed by 1 μL of 1000X streptomycin stock solution.When preparing or manipulating bacterial cultures in general, use sterile technique to prevent contamination. Keep tubes, bottles, and tips boxes closed and only open momentarily as needed. Everything that comes in contact with the culture must be sterile. Perform any manipulations that require sterility near the base of a Bunsen burner or inside a biosafety cabinet to minimize the chance of airborne contamination.
-
8.
Inoculate the media with DH10B E. coli using either a stab from a glycerol stock or a colony from a plate streaked with DH10B E. coli. Place the tube in a shaking heat block set to 30 °C and 1000 r.p.m. and incubate overnight for ~16 hours.
-
9.Use 40 μL of the DH10B overnight culture, which should now appear opaque, to inoculate a fresh 1.7 mL microcentrifuge tube with 1 mL of LB media containing 1X streptomycin and a hole in the lid (as described in Basic Protocol 1 steps 5–6). Incubate this fresh culture at 30 °C for exactly 2.5 hours. While the culture is growing, chill 50 mL of molecular biology-grade water in a 50 mL conical tube on ice.After 2.5 hours, the culture should appear slightly cloudy with an OD600 of about 0.6, but it is not necessary to measure the OD every time. The slightly cloudy culture is in “mid-log” phase, meaning the cells are growing rapidly and are displaying logarithmic growth. Mid-log phase is when the cells are most “competent,” meaning that the physiology of the cells is most amenable to DNA uptake through electroporation (Calvin & Hanawalt, 1988). Do not try to electroporate cells with an OD of 1 or greater (a culture that appears opaque in a 1.7 mL microcentrifuge tube), as they will not efficiently uptake DNA. It is also not sufficient to dilute a “stationary” culture (i.e., one that has stopped growing) such as an overnight to an OD of 0.6 directly. It is not the actual concentration of cells that matters, but rather their state, which is dependent on their rate of growth at the time they are harvested.
-
10.
Place the culture on ice to arrest growth. Next, pellet the cells at 10,000 × g in a refrigerated microcentrifuge at 4 °C for 1 minute. Carefully remove the media without disturbing the pellet.
-
11.
Gently resuspend the pellet in 1 mL of ice-cold molecular biology-grade water by pipetting several times. Pellet again at 10,000 × g in a refrigerated microcentrifuge at 4 °C for 1 minute.
-
12.Repeat step 11.The purpose of these washing steps is to remove the salt and other electrolytes surrounding the cells, which is critical. During the later electroporation step, the cells will be placed into an electroporation cuvette, which essentially acts as a capacitor. In the cuvette, the cells will be briefly exposed to a strong electric field, which will induce the formation of temporary pores in the cell membrane through which DNA can enter. If there is too much salt or other electrolytes surrounding the cells, the sample will conduct electricity between the positive and negative plates of the cuvette during the electroporation, which will kill the cells.
-
13.Resuspend the pellet in a final volume of ~20–30 μL of molecular biology-grade water. Keep the tube on ice. These cells are now “electrocompetent”.Often there is ~20 μL of residual water around the pellet, in which case it is unnecessary to add additional water to the tube.
-
14.Add ~100 ng of the target BAC and ~100 ng of the psc101-gbaA plasmid to the electrocompetent cells and mix gently by pipetting a few times. Keep the tube on ice.It is critical that the target BAC and psc101-gbaA plasmid were eluted from their purification columns in molecular biology-grade water or 0.2X elution buffer (see recipe). The salt concentration of the elution buffer in most plasmid purification kits will cause the sample to conduct electricity and kill the cells. If the DNA sample was eluted in 1X elution buffer or the salt concentration is unknown, the sample can easily be desalted using a PCR clean-up kit, such as the E.Z.N.A. Omega Bio-tek Cycle Pure Kit. Follow manufacturer instructions, but be sure to elute in molecular biology-grade water or 0.2X elution buffer for the final step. Also, do not add more than 5 μL of DNA sample to the cells, as doing so often causes the samples to conduct electricity even if the DNA samples were eluted in molecular biology-grade water or 0.2X elution buffer.
-
15.Before electroporating, set up all of the necessary electroporation materials. Plug in the Bio-Rad micropulser and turn it on. The screen should display the default setting “Ec1”, which is the proper setting to use. Press the measurement button twice until a light appears next to the “ms” label and the screen reads “0.00”. Set a bottle of SOC media close to the electroporator with the cap completely loose for quick access. Ensure that a box of Kimwipes is nearby.It is important to have everything set up as described before electroporating, because all the subsequent steps need to be completed very quickly.
-
16.Add the electrocompetent cells that have been mixed with DNA into the 0.1 cm gap between the metal plates of a pre-chilled electrocuvette. Next, quickly place the cap back onto the electrocuvette.Do not add more than 40 μL of cells to the cuvette. Adding large volumes decreases the resistance of the sample and can cause the sample to conduct electricity.
-
17.Wipe any condensation or ice off the outside of the electrocuvette, then place it into the sample holder with the notch facing forward. Gently slide the electrocuvette into the electroporation chamber until the electrocuvette “clicks” between the flexible metal electrodes in the electroporation chamber.While it is important to move quickly, do not slam the electrocuvette into the electroporation chamber. Doing so often launches the sample out from between the plates of the electrocuvette, resulting in a failed electroporation because the cells were not exposed to the strong electric field between the plates.
-
18.Press the “Pulse” button. After about a second, a buzzer should indicate that the electroporation is complete and a number representing the pulse length will appear on the screen. Quickly remove the cuvette from the sample holder and resuspend the cells in 1 mL of SOC media.The time between electroporation and resuspension should ideally not exceed 10 seconds. Fewer and fewer cells will survive the longer it takes to resuspend the cells (Dower, Miller, & Ragsdale, 1988). While SOC media helps maximize the number of cells that survive, it is not strictly necessary and can be substituted with LB media.If the electroporation was successful, the pulse length displayed on the screen should be between 4.80 and 6.00 ms. If the pulse length is less than 4.80 ms or if the screen says “Arc”, it means that the voltage applied to the electrocuvette decayed too quickly because significant current passed through the cells. In this case, repeat the electroporation with new electrocompetent cells and add a smaller quantity of DNA. It may be necessary to desalt the DNA again or wash the cells an extra time. It is often useful to prepare a few batches of electrocompetent cells in parallel in case the electroporation needs to be repeated a few times before it is successful.
-
19.
Return the resuspended cells to the 1.7 mL microcentrifuge they came from and incubate them in the heat block at 30 °C for 1 hour at 1,000 r.p.m.
-
20.After recovery, pellet the cells at 10,000 × g for 1 minute. Resuspend the pellet in 50 μL of LB media.It is essential to provide the cells enough time (~1 hour) to recover and express any antibiotic resistance genes before spreading them on an antibiotic containing plate.
-
21.The psc101-gbaA plasmid confers tetracycline resistance and, in this protocol, it is assumed that the target BAC confers chloramphenicol resistance. Therefore, to select for cells that contain both the psc101-gbaA plasmid and the target BAC, streak the resuspended cells on an LB agar plate with 1X chloramphenicol and 1X tetracycline. If using a BAC or plasmid with an antibiotic resistance gene other than chloramphenicol, substitute chloramphenicol with the appropriate antibiotic.Melt the thin end of a 9” soda-lime Pasteur pipette into a hockey stick shape using the Bunsen burner. Do not use borosilicate Pasteur pipettes, as they do not melt easily. Give the hockey stick 10 seconds to cool, then use it to spread the cells evenly on the plate. Allow the plate to dry with the lid off near the base of the Bunsen burner. The updraft created by the Bunsen burner prevents dust or other contaminants from settling onto the plate. Cells can also be spread on the plate with sterile glass beads or a sterile 1000 μL plastic pipette tip, but best results are usually achieved with a glass hockey stick.
-
22.
After the plate has dried, meaning there is no more running liquid on top of the agar, incubate it at 30 °C for ~18 hours or until colonies appear.
If colonies do not appear after 2 days, return to step 5. If the DNA samples are of sufficient quality (>20 ng/μL with an A260/280 of ~1.8–1.9), it should be possible to obtain cells that simultaneously take up the psc101-gbaA plasmid and the target BAC. However, if electroporating both plasmids at once is not successful, instead electroporate the target BAC alone into DH10B cells first, then repeat the process to electroporate the psc101-gbaA cells into DH10B cells that contain the target BAC. An alternative is to maintain a strain of DH10B cells that already contain the psc101-gbaA recombineering plasmid, in which case only the target BAC needs to be electroporated into the DH10B strain that already contains psc101-gbaA. When troubleshooting, electroporating 100 ng of any high-copy positive control plasmid less than 6000 base pairs in length (which should yield thousands of colonies) can help to evaluate whether the electroporation apparatus or electrocompetent cells are the source of any problems.
Recombineering with the targeting cassette
-
23.Start a new 1 mL culture of LB media with 1 μL of 1000X chloramphenicol stock solution and 1 μL of 1000X tetracycline stock solution in a 1.7 mL microcentrifuge tube with a hole in the lid. Inoculate the culture with a colony from step 22 or with a stab from a glycerol stock of a DH10B strain containing the target BAC and the psc101-gbaA plasmid. Place the tube in a shaking heat block set to 30 °C and 1000 r.p.m. and incubate overnight for ~16 hours.The replication of the psc101-gbaA plasmid is temperature-sensitive. Thus, it is important not to exceed 30 °C for any extended period of time to prevent loss of the psc101-gbaA plasmid.
-
24.
Use 40 μL of the overnight culture, which should now appear opaque, to inoculate a new 1 mL LB culture with 1X chloramphenicol and 1X tetracycline. Incubate the new culture at 30 °C for exactly 2.0 hours. While the culture is growing, chill 50 mL of molecular biology-grade water in a 50 mL conical tube on ice. Optionally, thoroughly mix 500 μL of the overnight culture with 500 μL of sterile 50% glycerol in a sterile 1.7 mL microcentrifuge tube (without a hole in the lid) and store immediately at –80 °C to recombineer the target BAC with other targeting cassettes in the future.
-
25.After 2.0 hours, add 20 μL of 10% rhamnose stock solution to the culture and increase the temperature of the heat block to 37 °C. Incubate the culture for exactly 40 more minutes.Rhamnose induces the expression of the lambda phage proteins that carry out recombineering (Red αβγ) from the psc101-gbaA plasmid. The short period of growth at 37 °C assists with the expression of these proteins. After 40 minutes, the culture should be in mid-log phase and slightly cloudy.
-
26.Carry out steps 10–20 to electroporate ~200 ng of the targeting cassette instead of the target BAC or recombineering plasmid and to recover the cells.Once the targeting cassette is introduced into the cells, a small number of the cells will replace the region between the homology arms with the targeting cassette. These cells are the rare recombinants and occur at a rate of 1 in 10,000–100,000 cells (Datta et al., 2008).
-
27.Assuming the targeting cassette confers kanamycin resistance, streak the resuspended cells on an LB agar plate with 1X kanamycin to select for the rare recombinants. If using a targeting cassette with an antibiotic resistance gene other than kanamycin, substitute kanamycin with the appropriate antibiotic.Do not use a tetracycline resistance gene to select for recombinants, as psc101-gbaA already confers tetracycline resistance. If editing a BAC that already confers chloramphenicol resistance, do not use a chloramphenicol resistance gene to select for recombinants. In general, verify what antibiotics the target strains are already resistant to, because these antibiotics cannot be used to select for recombinants.
-
28.After the plate has dried, meaning there is no more running liquid on top of the agar, incubate it at 30 °C for ~18 hours or until colonies appear.There should be a few dozen to a few hundred colonies on the plate. If there is a lawn or thousands of colonies, the targeting cassette was likely contaminated with template plasmid from the PCR. Any template plasmid that enters the target cells will confer antibiotic resistance without successful recombination. See Support Protocol 1 for details on how to purify the targeting cassette and remove template plasmid.
-
29.
In order to screen for successful recombinants, use the sequencing primers from step 4 to perform colony PCR as described in Support Protocol 2.
-
30.Once a successful recombinant is identified, confirm it by submitting the corresponding colony PCR product for Sanger sequencing (Metzker, 2005) using the sequencing primers. Each sequencing primer should be used for a separate Sanger sequencing reaction when submitted to the sequencing facility.Targeting primers and targeting cassette PCR products often contain random mutations, so it is important to obtain full sequencing coverage of the final recombineering product to ensure it is free of undesired mutations (Thomason et al., 2014). Order additional sequencing primers if necessary for large modifications, as most Sanger sequencing reads are less than 1000 bp. Ordering sequencing primers spaced out every 500 bp over a region of interest generally provides reliable coverage with overlapping reads. For example, a 5000 bp insertion should have about 8 equally spaced sequencing primers throughout the insertion in addition to the standard sequencing primers that flank the insertion.
-
31.After sequence confirmation, if the E. coli genome itself was modified, return to step 23 to conduct additional genetic modifications or conduct Support Protocol 3 to remove the psc101-gbaA plasmid from the cell if there are no further modifications to be made. However, if a target BAC was modified, it is important to ensure that it is not a heteromultimer, meaning a fusion of parent and recombinant BACs, regardless of whether further modifications are being made or not. For determining if a target BAC is a heteromultimer, proceed to step 32.The E. coli genome has its own dedicated system for resolving genome dimers involving FtsK and xerCD, which splits chromosome dimers back into monomers before the cell divides (Bigot, Sivanathan, Possoz, Barre, & Cornet, 2007). Therefore, it is generally not necessary to check for heteromultimerization when modifying the E. coli genome.When modifying a BAC, colony PCR and sequencing can confirm that the modification is present, but they are generally unreliable techniques for identifying heteromultimers. Theoretically, heteromultimers should yield two different colony PCR bands on a gel with two different sizes, but in practice the smaller PCR product generally amplifies more efficiently than the larger product. Sometimes the larger product is not even visible, despite actually being present.
-
32.
Use the successful recombinant to inoculate a 5 mL culture of LB with 5 μL of 1000X kanamycin stock solution in a sterile 14 mL culture tube. Incubate the culture overnight at 30 °C, shaking at 250 r.p.m. for ~16 hours.
-
33.
Thoroughly mix 500 μL of the overnight culture with 500 μL of sterile 50% glycerol in a sterile 1.7 mL microcentrifuge tube (without a hole in the lid) and store immediately at –80 °C to recombineer the target BAC with other targeting cassettes in the future.
-
34.
To isolate the target BAC DNA, follow steps 35–44 to perform isopropanol precipitation, as adapted from previous work (Warming, Costantino, Court, Jenkins, & Copeland, 2005).
-
35.
Pellet the rest of the overnight culture at 4,500 × g for 5 minutes. Then resuspend the pellet in 250 μL of Solution I from the Omega Bio-tek Plasmid Mini Kit and transfer to a 1.7 mL microcentrifuge tube.
-
36.
Add 250 μL of Solution II from the Omega Bio-tek Plasmid Mini Kit to the resuspended cells, mix by inversion, and incubate at room temperature for 3 minutes exactly to lyse the cells. The mixture should appear more transparent after adding Solution II.
-
37.
Add 250 μL of Solution III from the Omega Bio-tek Plasmid Mini Kit to the lysed cells and mix by inversion immediately to precipitate cellular debris, which appears as white chunks.
-
38.
Incubate the lysed cells on ice for 5 minutes, then pellet the cellular debris at ≥16,000 × g for 5 minutes.
-
39.
Transfer the clear supernatant to a new 1.7 mL microcentrifuge tube and pellet any remaining debris with a second spin at ≥16,000 × g for 5 minutes.
-
40.
Transfer the clear supernatant to another new 1.7 mL microcentrifuge tube. Add 750 μL of isopropanol, mix thoroughly by inversion, and incubate on ice for 10 minutes. The solution should start to turn cloudy as the DNA precipitates.
-
41.
Pellet the DNA at ≥16,000 × g for 10 minutes, then remove the supernatant without disturbing the white pellet at the bottom of the tube.
-
42.Add 400 μL of 70% ethanol to the tube and gently pipette up and down several times to wash the pellet without dislodging it.If the pellet becomes dislodged, spin at ≥16,000 × g for 1 minute to secure it back to the bottom of the tube.
-
43.
Remove the 70% ethanol and air dry the pellet with the lid open for 10 minutes.
-
44.
Resuspend the pellet in 50 μL of molecular biology-grade water.
-
45.Choose one or two restriction endonucleases that, when used to digest the target BAC, will yield different fragmentation patterns on an agarose gel depending on whether the BAC is the desired recombinant or the unmodified parent BAC. The simplest way to identify an enzyme that will give distinguishable patterns is to simulate the fragmentation patterns on DNA editing software, such as Snapgene or ApE, for both the unmodified parent BAC and the desired recombinant BAC (Fig. 4A) and to ensure that each pattern has at least one band that is unique to the parent BAC or the modified BAC. Aim to choose one or two restriction endonucleases that together: 1) cut the BAC 2–3 times and 2) have at least one recognition site that disappears or appears upon modification.Bear in mind that the psc101-gbaA plasmid has a copy number of about 10 plasmids per cell, and will also have a fragmentation pattern that will be much brighter and appear on top of the target BAC fragmentation pattern (Fig. 4B). Choose an enzyme that cuts the psc101-gbaA plasmid at least once, because uncut psc101-gbaA normally yields a complex pattern. If the chosen enzyme that cuts the target BAC does not cut psc101-gbaA, add a second enzyme to digest the psc101-gbaA plasmid. However, remember to account for how the second enzyme might affect the target BAC pattern.
-
46.
Digest 25 μL of the isolated target BAC DNA using the selected endonucleases following the manufacturer’s instructions. Additionally, digest 25 μL of the unmodified parent BAC as a negative control.
Figure 4.
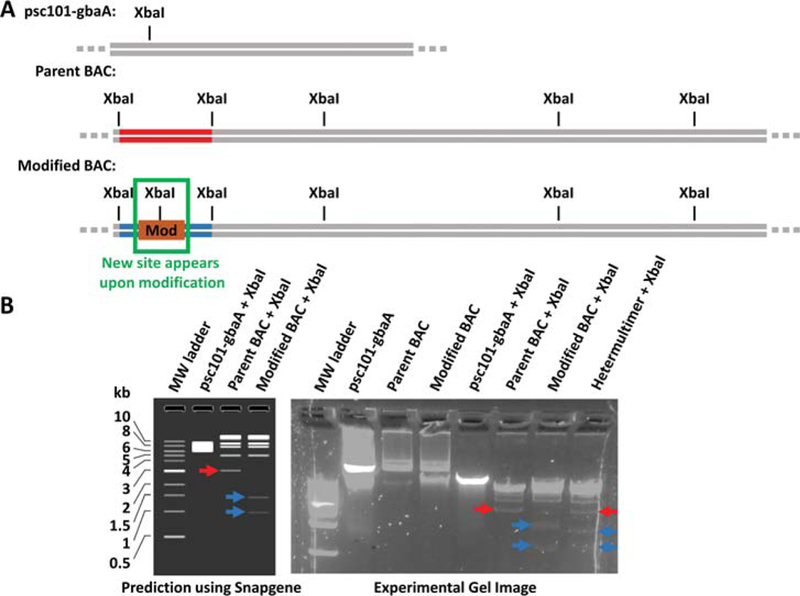
Example strategy to identify heteromultimers using restriction enzyme digestion patterns. (A) Simplified plasmid maps are shown with the predicted sites of cleavage by the XbaI restriction endonuclease. XbaI is chosen in this example because it cleaves the psc101-gbaA recombineering plasmid at least once and because the desired modification (Mod) introduces a new XbaI site. (B) (Left) XbaI yields a predicted fragmentation pattern of the parent BAC that is distinct from that of the modified BAC. In particular, the parent BAC pattern has a unique band (red arrow) that is not present in the modified BAC pattern, and the modified BAC pattern has unique bands (blue arrows) that are not present in the parent BAC pattern. Furthermore, these unique, identifying bands are not obscured by the psc101-gbaA digestion pattern or other bands in the digestion patterns. The experimental gel image (Right) for this example shows that the hetermultimer contains both the unique parent BAC band (red arrow) and the unique modified BAC bands (blue arrows). The pure modified BAC pattern lacks the unique parent BAC band. The brightness and contrast in the gel image has been adjusted to render the faint bands more visible.
Preparing and running the agarose gel to obtain digestion patterns
-
47.Add 500 mg of agarose to an Erlenmeyer flask followed by 50 mL of 0.5X TBE buffer. Plug the top of the flask with a wad of paper towel and microwave on high power for about 60 seconds, or until the agarose is dissolved.Monitor the agarose while microwaving to ensure that it does not boil over. Use oven mitts or paper towels when handling the hot flask.
-
48.
Add 5 μL of GelGreen dye to the molten agarose, swirl the flask to distribute the dye, then pour into a tray in a gel caster with an 8-well comb. Allow the gel to cool and solidify for 15–30 minutes.
-
49.
Carefully remove the comb and place the solidified gel into an electrophoresis chamber filled with 0.5X TBE buffer. Ensure that there is sufficient TBE buffer to submerge the entire gel.
-
50.
To check the digestion product, mix 10 μL of the digestion reaction with 2 μL of 6X gel loading dye, then load it into one of the wells of the agarose gel next to another well that contains 5 μL of Quick-Load Purple 1 kb DNA Ladder.
-
51.Run the gel at 150 volts for 30 minutes, then visualize on a blue LED transilluminator.If the bands are not sufficiently separated, continue to run the gel for additional 10 minute increments until separation is sufficient.
-
52.
If the fragmentation pattern unambiguously matches the expected pattern for the recombinant BAC and not the pattern for the unmodified BAC, return to step 23 to continue with further modifications as required. If there are no further modifications to make to the target BAC, see Support Protocol 3 to remove the psc101-gbaA plasmid from the cell. If the fragmentation pattern corresponds to the expected pattern for the recombinant BAC overlaid on top of the pattern for the unmodified BAC, or if there is any doubt, proceed to Support Protocol 4 to “remonomerize” the target BAC and obtain a pure monomer of the desired recombinant BAC.
Because of the low copy number of BACs per cell, it can sometimes be quite difficult to isolate enough DNA to analyze by gel electrophoresis. If the bands of the restriction digestion pattern are too faint to confidently identify successful recombinants or heteromultimers, grow a 25 mL culture of each target BAC in question and isolate the DNA using the ZymoPURE II Plasmid Midiprep Kit D4200, following the manufacturer’s instructions. Midiprep kits, while more expensive, isolate a larger quantity of DNA, which can make visualizing digestion patterns easier when isopropanol precipitation is insufficient.
BASIC PROTOCOL 2
DIRex RECOMBINEERING FOR SEAMLESS DELETIONS OR SMALL INSERTIONS
If many sequential modifications need to be made in the target or, as is the case for point mutations, the modification must be made without leaving an antibiotic resistance gene behind, then Basic Protocol 2 or 3 should be used. Further, if the modification is a point mutation, deletion, or a small insertion/replacement (≤30 bp), DIRex recombineering is the most rapid approach to introduce these modifications (Näsvall, 2017). First, a kanamycin resistance gene (kanR) and a ccdB gene, which is a counterselection gene that is lethal only under certain conditions, replace or are inserted into the target region that will be modified (Fig. 5A). Subsequently, kanR+ccdB-containing intermediates are selected for using kanamycin and arabinose, which induces expression of the ccdA antitoxin that renders ccdB non-lethal (H. Wang et al., 2014). The kanR and ccdB genes are flanked by inverted repeats, which are in turn flanked by homology arms that contain a direct 30 bp repeat centered around the modification. The architecture of the indirect and direct repeats promotes spontaneous excision of the kanR gene, the ccdB gene, the inverted repeats, and one of the direct repeats, thereby leaving behind a single copy of the modification (Fig. 5B). The excision event is thought to occur through strand slippage and mispairing during replication (Bzymek & Lovett, 2001; Näsvall, 2017). Successful recombinants are then selected for by removing arabinose such that ccdA is no longer expressed and the ccdB toxin becomes lethal. Any cells that have not excised ccdB to yield the final recombinant, which will be the vast majority of cells, will not survive this selection pressure. We note that there are many potential options for counterselection genes available, but the protocols in this article utilize ccdB because in our experience it functions quickly and reliably and without requiring the use of minimal media plates that slow the growth of cells. Finally, we note that although various scales can be used, we strongly recommend (and describe below) performing the protocol on a 1 mL scale for ease of sample processing and using heat blocks, as described in previous work (H. Wang et al., 2014).
Figure 5.
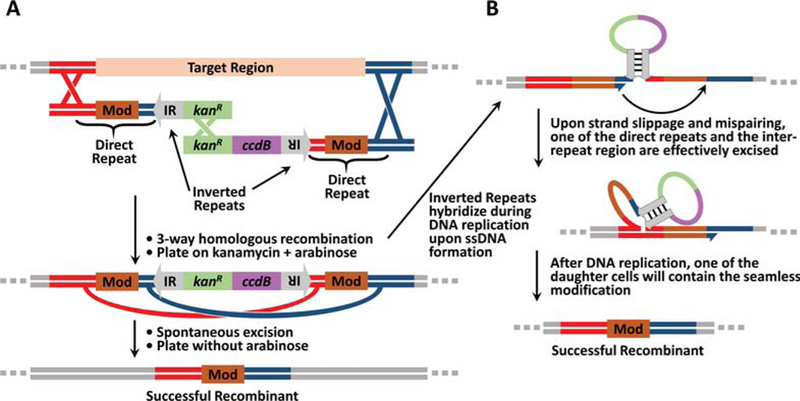
Schematic of DIRex recombineering. (A) In DIRex recombineering, intermediates containing the conditionally-lethal gene (ccdB) are first selected for using kanamycin. The modification (Mod) is directly repeated in the homology arms. Thus, the direct and inverted repeats (IR) promote spontaneous excision to yield the final recombinant. Successful recombinants are selected for by removing arabinose. In the absence of arabinose, the ccdA antitoxin is no longer expressed and ccdB then kills unmodified cells. (B) DIRex is hypothesized to promote spontaneous excision through hybridization between the two inverted repeats to form a hairpin during replication that brings the direct repeats into close proximity (Bzymek & Lovett, 2001; Näsvall, 2017). The direct repeats can then promote strand slippage during synthesis that results in excision of one of the direct repeats and everything between the direct repeats.
Materials
Target BAC (supplied by user)
psc101-gbaA plasmid (see Table 1)
Targeting primers (designed and ordered by user)
Sequencing primers (designed and ordered by user)
R6K-KCA plasmid (see Table 1)
R6K-AKC plasmid (see Table 1)
1.7 mL microcentrifuge tubes (e.g., VWR 87003–294)
Bunsen burner and gas line
18-gauge needles (e.g., Becton Dickinson 305195)
Sterile 10 μL pipette tips (e.g., VWR 89079–466)
Sterile 200 μL pipette tips (e.g., VWR 89079–460)
Sterile 1000 μL pipette tips (e.g., VWR 89079–472)
Lysogeny broth media (see recipe)
1000X streptomycin stock solution (see recipe)
DH10B E. coli cells (Invitrogen 18297010) or other target E. coli strain
50 mL conical tubes (e.g., Cellstar 227261)
0.1 cm electroporation cuvettes (e.g., Bio-Rad 1652083)
Kimwipes (Kimberly-Clark 34155)
Super optimal broth with catabolic repression media (see recipe)
LB agar plates with 1X chloramphenicol and 1X tetracycline (see recipe)
9” soda-lime glass Pasteur pipettes (e.g., VWR 14672–380)
1000X tetracycline stock solution (see recipe)
1000X chloramphenicol stock solution (see recipe)
Sterile 50% glycerol (see recipe)
10% rhamnose stock solution (see recipe)
10% arabinose stock solution (see recipe)
LB agar plates with 1X kanamycin and 0.2% w/v arabinose (see recipe)
DNA Editing Software (e.g., Snapgene, ApE, Serial Cloner)
P10 pipettor (e.g., VWR 89079–962)
P200 pipettor (e.g., VWR 89079–970)
P1000 pipettor (e.g., VWR 89079–974)
Shaking heat block for 1.5 mL tubes (e.g., Eppendorf Thermomixer F1.5 5384000020)
UV-Vis spectrometer (e.g., Thermo Fisher Nanodrop 2000c)
Ice maker and ice buckets
Refrigerated microcentrifuge ≥16,000 × g(e.g., Thermo Fisher Heraeus Fresco 21 75002426)
Electroporator (Bio-Rad Micropulser 1652100)
30 °C incubator (e.g., VWR Gravity convection incubator 89511–422)
Design targeting primers and create the targeting cassette
First, choose appropriate homology arms to make the desired modification. Immediately to the left of the region to be deleted or replaced, highlight 50 bp, not including the region to be deleted or replaced, and copy the sequence from the top strand (in the 5′ to 3′ direction) for the left homology arm (Figs. 6A–B).
Immediately to the right of the region to be deleted or replaced, highlight 50 bp, not including the region to be deleted or replaced, and copy or mark the sequence from the BOTTOM strand for the right homology arm (Fig. 6B). Ensure that the sequence copied and designated as the right homology arm is the bottom strand in the 5′ to 3′ direction (i.e., the reverse complement of the TOP strand). In the case of an insertion, the 3′ ends of the left homology arm and the right homology arm will be adjacent to each other if the homology arms are properly designed. In the case of a point mutation, the base to be modified should lie be between the 3′ ends of the homology arms (Fig. 7).
- To create targeting primers and targeting cassettes for:
- A deletion: Take the reverse complement of the last 15 bp of the right homology arm and append it to the 3′ end of the left homology arm. Take the reverse complement of the last 15 bp of the left homology arm—not including the bases appended from the right homology arm—and append it to the 3′ end of the right homology arm. The resulting 65 bp homology arms complement each other for the last 30 bp of their 3′ ends, which will generate a 30 bp direct repeat of the deletion junction near each end of the targeting cassette. Append each of these homology arms to the DIRexPrimer sequence in Table 2 to create the left and right targeting primers.
- A small insertion or replacement 30 bp or less: Append the insertion to the 3′ end of the left homology arm and append the reverse complement of the insertion to the 3′ end of the right homology arm. If the insertion is less than 30 bp, let N = 15 – ((the size of the insertion)/2). Take the reverse complement of the last N bp of the right homology arm and append it to the 3′ end of the left homology arm after the insertion. Take the reverse complement of the last N bp of the left homology arm—not including the insertion or bases appended from the right homology arm—and append it to the 3′ end of the right homology arm. The resulting 65 bp homology arms complement each other for the last 30 bp of their 3′ ends (Fig. 6C), which will generate a 30 bp direct repeat of the insertion and flanking regions near each end of the targeting cassette. Append each of these homology arms to the DIRexPrimer sequence in Table 2 to create the left and right targeting primers (Fig 6D). Double-check the orientation of the insertion; the direct repeat will become the final product.Note that the target region to be replaced can be large or small, it is only the new insertion that is replacing the target region that is limited to ~30 bp. The size of the insertion is only limited by the size of the DNA oligos that can be purchased. Most vendors currently limit standard oligo sizes to 100 or 120 bp.
-
A point mutation: Append the mutant base to the 3′ end of the left homology arm and append the complement of the mutant base to the 3′ end of the right homology arm. Take the reverse complement of the last 15 bp of the right homology arm—not including the mutant base—and append it to the 3′ end of the left homology arm after the mutant base. Take the reverse complement of the last 15 bp of the left homology arm—not including the mutant base or bases appended from the right homology arm—and append it to the 3′ end of the right homology arm. The resulting 66 bp homology arms complement each other for the last 31 bp of their 3′ ends, which will generate a 31 bp direct repeat—containing the point mutation in the middle—near each end of the targeting cassette. Append each of these homology arms to the DIRexPrimer sequence in Table 2 to create the left and right targeting primers.Order the left and right targeting primers, then use them to create the targeting half-cassettes. Generate the left half-cassette using the left targeting primer and DIRexKan.Rev primer from Table 2 in conjunction with R6K-AKC as the PCR template (see Support Protocol 1). Generate the right half-cassette using the right targeting primer and the DIRexKan.For primer from Table 2 in conjunction with R6K-KCA as the PCR template (see Support Protocol 1).
Because the direct repeats in the overhangs of the left and right targeting primers need to flank an inverted repeat, the inverted repeat-binding site at the 3′ end of the left and right target primers is identical. If the left and right targeting primers were used simultaneously in the same PCR reaction to generate the targeting cassette, there would be a mixture of PCR products that have only the left homology arms, only the right homology arms, or both homology arms. By designing the targeting cassette with slightly overlapping halves, each targeting cassette will necessarily contain one left homology arm and one right homology arm with a defined orientation. The half-cassettes will be electroporated together and joined by homologous recombination inside the cell (Fig. 5A). Design and order sequencing primers as in Basic Protocol 1, step 4 that are 200 bp to the left of the left homology arm and 200 bp to the right of the right homology arm.
Figure 6.
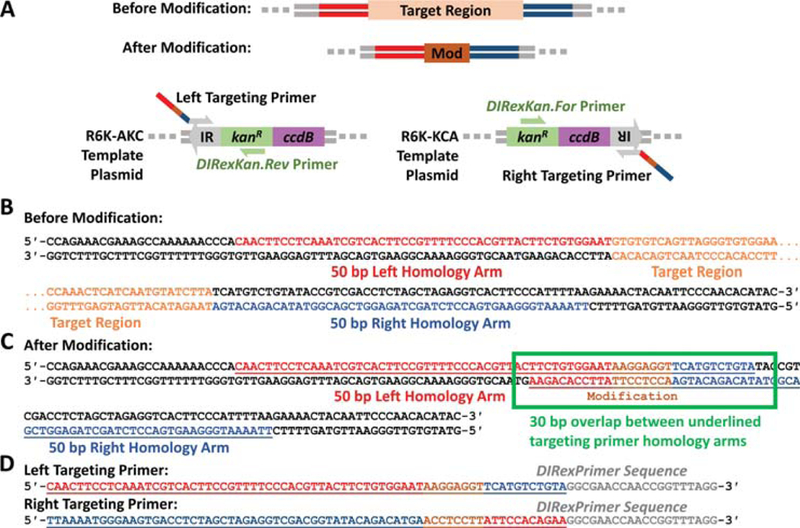
Primer design example for a small seamless insertion via DIRex recombineering. (A) The left and right targeting primers are used to amplify the left and right targeting half-cassettes from the R6K-AKC and R6K-KCA template plasmids (Table 1), respectively. The overhangs of the left and right targeting cassettes contain the left (red) or right (blue) homology arms that flank the target region to be replaced, respectively, along with a direct repeat that contains the small insertion (Mod) and a small portion of the other homology arm. (B) The left homology arm is copied from the top strand to the left of the target region (red color), and the right homology is copied from the bottom strand to the right of the target region (blue color). (C) A simple strategy to design the full homology arms with the direct repeat is to generate the desired sequence with the modification (brown) between the left and right homology arms from B. The full homology arm of the left targeting primer is shown as the underlined region of the top strand and the full homology arm of the right targeting primer is shown as the underlined region of the bottom strand. The 3′ ends of these full homology arms overlap by 30 bp, with the modification in the center of the overlap. In other words, each homology arm contains the modification on the 3′ end followed by N bases of the complement of the other homology arm, where N = 15 – ((the size of the insertion)/2). (D) Finally, the full left and right homology arms are appended to the 5′ ends of the DIRexPrimer sequence (Table 2) to form the full left and right targeting primers.
Figure 7.
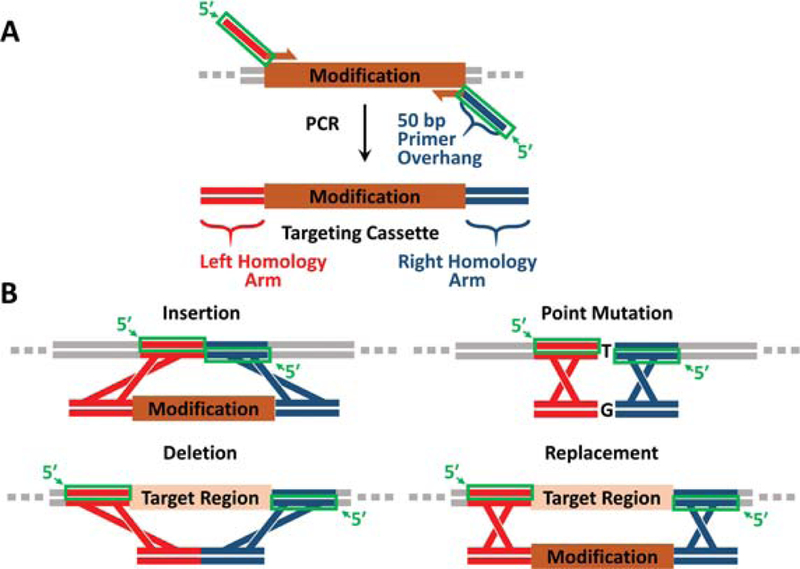
Guide for homology arm design. (A) Homology arms (red and blue) are introduced as 5′ overhangs on primers (green boxes) that amplify the targeting cassette via PCR. (B) Homology arm overhangs for insertions, point mutations, deletions, and replacements are highlighted as green boxes. The modification targeting cassettes for point mutations and deletions using two-step recombineering (Basic Protocol 3) are small and can be generated by annealing oligos rather than by PCR.
Recombineering with the targeting cassette
-
5.
If the target BAC and the psc101-gbaA plasmid are already both present in DH10B cells, or if the intent is to engineer the E. coli genome and the psc101-gbaA plasmid is already present in the strain to be engineered, proceed to step 6. Otherwise, carry out Basic Protocol 1, steps 5–22.
-
6.Start a new 1 mL culture of LB media with 1 μL of 1000X chloramphenicol stock solution and 1 μL of 1000X tetracycline stock solution in a 1.7 mL microcentrifuge tube with a hole in the lid. Inoculate the culture with a colony or a stab from a glycerol stock of a DH10B strain containing the target BAC and the psc101-gbaA plasmid. Place the tube in a shaking heat block set to 30 °C and 1000 r.p.m. and incubate overnight for ~16 hours.Note, when engineering the E. coli genome, omit the addition of chloramphenicol to all agar plates and media in this step and all subsequent steps.
-
7.Use 40 μL of the overnight culture, which should now appear opaque, to inoculate a fresh 1.7 mL microcentrifuge tube with 1 mL LB media containing 1X chloramphenicol and 1X tetracycline and a hole in the lid. Incubate this fresh culture at 30 °C for exactly 2.0 hours. While the culture is growing, chill 50 mL of molecular biology-grade water in a 50 mL conical tube on ice.When preparing or manipulating bacterial cultures in general, use sterile technique to prevent contamination. Keep tubes, bottles, and tips boxes closed and only open momentarily as needed. Everything that comes in contact with the culture must be sterile. Perform any manipulations that require sterility near the base of a Bunsen burner or inside a biosafety cabinet to minimize the chance of airborne contamination.
-
8.After 2.0 hours, add 20 μL of 10% rhamnose stock solution and 20 μL of 10% arabinose stock solution to the culture and increase the temperature of the heat block to 37 °C. Incubate the culture for exactly 40 more minutes.Rhamnose induces the expression of the lambda phage proteins that carry out recombineering (Red αβγ) from the psc101-gbaA plasmid. The arabinose induces the ccdA antitoxin that binds the ccdB toxin and renders it non-lethal. The short period of growth at 37 °C assists the expression of these proteins. After 40 minutes, the culture should be in mid-log phase and slightly cloudy.
-
9.Carry out Basic Protocol 1, steps 10–18, but in this case to electroporate ~100 ng each of the left and right targeting half-cassettes instead of the target BAC or recombineering plasmid. After resuspending the electroporated cells in 1 mL of SOC media, return them to the original 1.7 mL microcentrifuge tube, then add 20 μL of 10% arabinose stock solution to the media. Incubate the culture in the heat block at 30 °C for 2 hours at 1,000 r.p.m.Do not forget to add arabinose to ensure that ccdB does not kill the cells.
-
10.
After the 2-hour recovery period, pellet the cells at 10,000 × g for 1 minute. Resuspend the pellet in 50 μL of LB media.
-
11.Streak the resuspended cells on an LB agar plate with 1X kanamycin and 0.2% w/v arabinose to select for those rare recombinants that have incorporated the targeting cassette.Do not forget to add arabinose to ensure that ccdB does not kill the cells.
-
12.After the plate has dried, meaning there is no more running liquid on top of the agar, incubate it at 30 °C for ~18 hours or until colonies appear.There should be a few dozen to ~100 colonies on the plate.
-
13.To identify the rare cells that have excised the inverted repeats, one of the direct repeats, the kanR gene, and the ccdB gene to yield a single copy of the direct repeat (Fig. 5B), pick at least three colonies and streak them onto an LB agar plate with 1X chloramphenicol and 1X tetracycline. This step counterselects against the ccdB gene.Because there is no arabinose on the plate and thus no expression of the ccdA antitoxin, only those rare cells that have undergone spontaneous excision of ccdB will survive. Split the plate into thirds and streak each of the 3 colonies in a zig-zag pattern across a third of the plate. Three colonies are picked because 30–50% of colonies will be heteromultimers of unmodified parent and kanR+ccdB intermediate BACs. Heteromultimers easily undergo intramolecular recombination to yield parent BAC and survive counterselection (Fig. 8) at a frequency much higher than spontaneous excision. Therefore, it is important to pick several colonies to increase the chances of choosing a monomer. Note that it is simpler and faster to just streak several colonies rather than to screen for monomers of the kanR+ccdB intermediate.Ensure that chloramphenicol is present in the plates. Without chloramphenicol, cells can survive counterselection by losing the ccdB-containing target BAC, which happens at a frequency much greater than the frequency of successful recombination. Chloramphenicol will kill any cells that lose the target BAC.
-
14.Incubate the plate overnight at 30 °C for ~16 hours or until colonies appear.When a monomer kanR+ccdB intermediate is streaked on a plate in the absence of arabinose, a dozen or a few dozen isolated colonies should appear. These colonies will have undergone spontaneous excision to yield the final recombinant. If a heteromultimer is streaked on a plate, a lawn of bacteria will grow. These bacteria have recombined to yield unmodified parent BAC. In further steps, only screen colonies from streaks that have yielded just a few dozen isolated colonies.If every colony chosen, rather than just 30–50% of the colonies, behaves as a heteromultimer, it could be that the parent target BAC was a homomultimer prior to the insertion of the kanR+ccdB cassette (Fig. 9). In such a situation, nearly 100% of kanR+ccdB intermediates will be heteromultimers. To address this issue, the target BAC must be remonomerized using Support Protocol 4 before reattempting this protocol.
-
15.
In order to screen for successful recombinants, use the sequencing primers from step 4 to perform colony PCR as described in Support Protocol 2.
-
16.Once a successful recombinant is identified, confirm it by submitting the corresponding colony PCR product for Sanger sequencing (Metzker, 2005) using the sequencing primers. Each sequencing primer will be used for a separate Sanger sequencing reaction.Targeting primers and targeting cassette PCR products often contain random mutations, so it is important to get full sequencing coverage of the final recombineering product to ensure it is free of undesired mutations (Thomason et al., 2014). Order additional sequencing primers if necessary for large modifications as most Sanger sequencing reads are less than 1000 bp. Ordering sequencing primers spaced out every 500 bp over a region of interest generally provides reliable coverage with overlapping reads. For example, a 5000 bp insertion should have about 8 equally spaced sequencing primers throughout the insertion in addition to the standard sequencing primers that flank the insertion.
-
17.
After sequence confirmation, return to step 5 to introduce further modifications as needed. If there are no additional modifications to make, see Support Protocol 3 to remove the psc101-gbaA plasmid from the cell.
Figure 8.
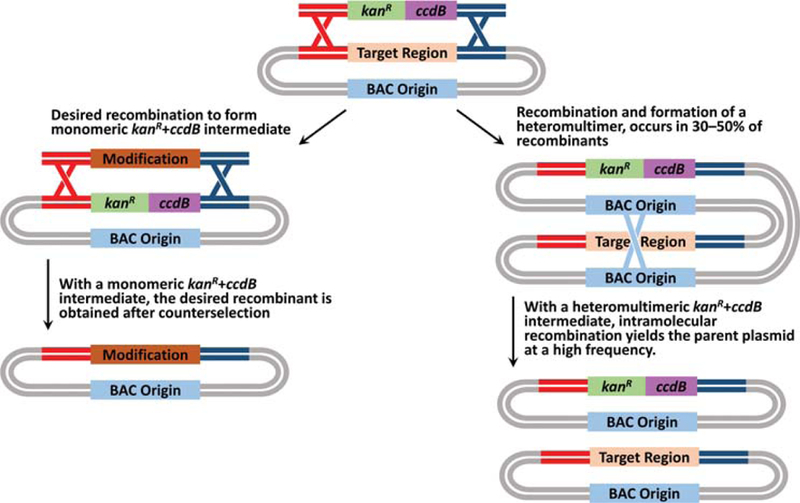
Schematic showing the outcomes of recombineering with monomeric kanR+ccdB intermediates (left) compared to recombineering with heteromultimers of kanR+ccdB intermediate and unmodified parent (right). The monomeric intermediate must undergo recombineering with the targeting cassette to eliminate the toxic ccdB gene. However, the heteromultimer can undergo intramolecular recombination to eliminate the toxic ccdB gene, yielding unmodified parent BAC at a frequency much higher than successful recombineering.
Figure 9.
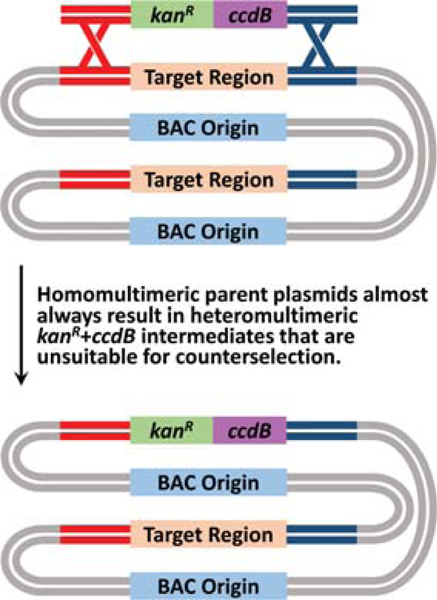
Schematic showing the outcome of recombineering with a homomultimer. Target BACs that are homomultimeric will almost always form heteromultimeric kanR+ccdB intermediates, which are unsuitable for counterselection owing to the high rate of intramolecular recombination to yield unmodified parent BAC.
We note that during counterselection the toxic ccdB gene selects against any potential heteromultimers that form via fusion of a kanR+ccdB intermediate BAC and successfully modified recombinant BAC. Heteromultimers of unmodified parent BAC and a kanR+ccdB intermediate BAC almost always yield parent BAC after counterselection, so a heteromultimer of unmodified parent BAC and successfully modified recombinant BAC is very unlikely. Therefore, unlike in Basic Protocol 1, it is unlikely that the final recombinant is a heteromultimer. Hence, it is generally not necessary to screen for heteromultimers. If in doubt, carry out Basic Protocol 1, steps 32–52 to ensure that the final recombinant BAC is not a heteromultimer.
BASIC PROTOCOL 3
TWO-STEP RECOMBINEERING FOR LARGE, SEAMLESS INSERTIONS OR FOR GENE REPLACEMENTS
For large (>30 bp), seamless insertions or replacements, a standard two-step recombineering protocol should be used (H. Wang et al., 2014). We note that two-step recombineering can also be used to make deletions, point mutations and small insertions/replacements, but it is an unnecessarily laborious way to do so compared to Basic Protocol 2. For the first step of Basic Protocol 3, a kanR gene and a ccdB gene replace or are inserted into the target region that will be modified (Fig. 10). KanR+ccdB-containing intermediates are selected for by streaking on plates containing kanamycin and arabinose, which induces expression of the ccdA antitoxin that renders ccdB non-lethal. In the second step, the kanR and ccdB genes in the intermediate are replaced by a targeting cassette that introduces the final modification. Successful recombinants are selected for by removing arabinose, such that ccdB is rendered lethal and unmodified cells do not survive (H. Wang et al., 2014). We note that there are many potential options for counterselection genes available. The protocols in this article utilize ccdB because, in our experience, it functions quickly and reliably and without requiring the use of minimal media plates that slow the growth of cells. Finally, we note that although various scales can be used, we strongly recommend (and describe below) performing the protocol on a 1 mL scale for ease of sample processing and using heat blocks, as described in previous work (H. Wang et al., 2014).
Figure 10.
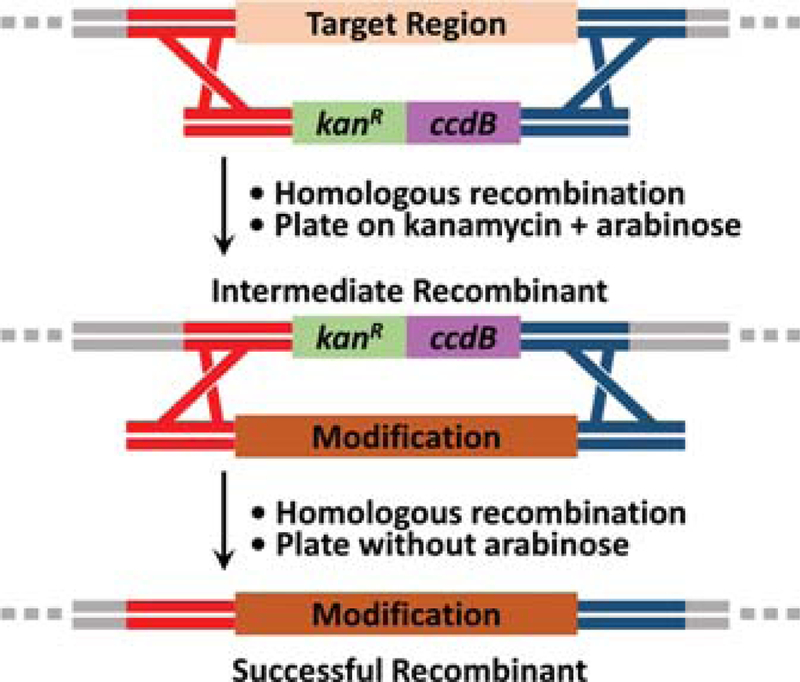
Schematic of two-step recombineering. First, an intermediate containing a conditionally lethal gene (ccdB) is selected for using kanamycin. Second, the ccdB gene in the intermediate is replaced with the desired modification, which is selected for by removing arabinose. In the absence of arabinose, the ccdA antitoxin is no longer expressed and ccdB kills unmodified intermediate cells, leaving only the successful recombinant cells that have the final modification.
Materials
Target BAC (supplied by user)
psc101-gbaA plasmid (see Table 1)
Targeting primers (designed and ordered by user)
Sequencing primers (designed and ordered by user)
R6K-kan-ccdB plasmid (see Table 1)
Template plasmid (for insertions, supplied by user)
1.7 mL microcentrifuge tubes (e.g., VWR 87003–294)
Bunsen burner and gas line
18-gauge needles (e.g., Becton Dickinson 305195)
Sterile 10 μL pipette tips (e.g., VWR 89079–466)
Sterile 200 μL pipette tips (e.g., VWR 89079–460)
Sterile 1000 μL pipette tips (e.g., VWR 89079–472)
Lysogeny broth media (see recipe)
1000X streptomycin stock solution (see recipe)
DH10B E. coli cells (Invitrogen 18297010) or other target E. coli strain
50 mL conical tubes (e.g., Cellstar 227261)
0.1 cm electroporation cuvettes (e.g., Bio-Rad 1652083)
Kimwipes (Kimberly-Clark 34155)
Super optimal broth with catabolic repression media (see recipe)
LB agar plates with 1X chloramphenicol and 1X tetracycline (see recipe)
9” soda-lime glass Pasteur pipettes (e.g., VWR 14672–380)
1000X tetracycline stock solution (see recipe)
1000X chloramphenicol stock solution (see recipe)
Sterile 50% glycerol (see recipe)
10% rhamnose stock solution (see recipe)
10% arabinose stock solution (see recipe)
LB agar plates with 1X kanamycin and 0.2% w/v arabinose (see recipe)
DNA Editing Software (e.g., Snapgene, ApE, Serial Cloner)
P10 pipettor (e.g., VWR 89079–962)
P200 pipettor (e.g., VWR 89079–970)
P1000 pipettor (e.g., VWR 89079–974)
Shaking heat block for 1.5 mL tubes (e.g., Eppendorf Thermomixer F1.5 5384000020)
UV-Vis spectrometer (e.g., Thermo Fisher Nanodrop 2000c)
Ice maker and ice buckets
Refrigerated microcentrifuge ≥16,000 × g (e.g., Thermo Fisher Heraeus Fresco 21 75002426)
Electroporator (Bio-Rad Micropulser 1652100)
30 °C incubator (e.g., VWR Gravity convection incubator 89511–422)
Design targeting primers and create the targeting cassette
First, choose appropriate homology arms to make the desired modification. Immediately to the left of the region to be deleted or replaced, highlight 50 bp, not including the region to be deleted or replaced, and copy the sequence from the top strand (in the 5′ to 3′ direction) for the left homology arm.
Immediately to the right of the region to be deleted or replaced, highlight 50 bp, not including the region to be deleted or replaced, and copy or mark the sequence from the BOTTOM strand for the right homology arm. Ensure that the sequence copied and designated as the right homology arm is the bottom strand in the 5′ to 3′ direction (i.e., the reverse complement of the TOP strand). In the case of an insertion, the 3′ ends of the left homology arm and the right homology arm will be adjacent to each other if the homology arms are properly designed. In the case of a point mutation, the base to be modified should lie between the 3′ ends of the homology arms (Fig. 7).
- Basic Protocol 3 requires separate targeting cassettes for the first and second steps. To design targeting primers and targeting cassettes for the first step, append the left homology arm sequence to the Kan.For primer sequence and append the right homology arm to the KanccdB.Rev primer sequence in Table 2. Obtain these primers, and then use them to generate the kanR+ccdB targeting cassette using R6K-kan-ccdB as a PCR template (see Support Protocol 1). To create targeting primers and targeting cassettes for the second step in the case of:
- A deletion: Append the left homology arm sequence to the reverse complement of the right homology arm sequence to create the forward modification oligo. Append the right homology arm to the reverse complement of the left homology arm sequence to create the reverse modification oligo. Anneal the forward and reverse modification oligos to generate the modification targeting cassette according to Support Protocol 1.In the case of deletions, the modification targeting cassettes for the second step are so small that they are most easily made by ordering the entire top and bottom strands of the cassette as oligos and annealing them according to Support Protocol 1.
- A large >30 bp insertion or replacement: Create or obtain a plasmid that has the genes or region to be inserted into the target. Design primers that have melting temperatures (Tm) of ~60 °C and that amplify the entire region to be inserted (Sawitzke et al., 2013). In the primer design, append the left homology arm to one primer and the right homology arm to the other primer. Note that the choice of which homology arm to append to which primer will determine the ultimate orientation of the insertion. Once the primers are obtained, use the primers in conjunction with the template plasmid to create the targeting cassette via PCR (see Support Protocol 1).
- A point mutation or a small <30 bp insertion: Append the modified base or small insertion to the 3′ end of the left homology arm sequence, followed by the reverse complement of the right homology arm sequence to create the forward modification oligo. Append the reverse complement of the modified base or small insertion to the right homology arm sequence, followed by the reverse complement of the left homology arm sequence to create the reverse modification oligo. Anneal the forward and reverse modification oligos to generate the modification targeting cassette according to Support Protocol 1.
In the case of point mutations or small insertions, the modification targeting cassettes for the second step are so small that they are most easily made by ordering the entire top and bottom strands of the cassette as oligos and annealing them. The size of the homology arms can be reduced to meet oligo manufacturing size limits, but can be no smaller than 35 bp. Bear in mind that smaller homology arms will result in reduced recombineering efficiency (Yu et al., 2000). Also, be sure to double-check the orientation of the insertion. Design and order sequencing primers as in Basic Protocol 1, step 4 that are 200 bp to the left of the left homology arm and 200 bp to the right of the right homology arm.
Recombineering with the targeting cassettes
-
5.
If the target BAC and the psc101-gbaA plasmid are already both present in DH10B cells, or if the intent is to engineer the E. coli genome and the psc101-gbaA plasmid is already present in the strain to be engineered, proceed to step 6. Otherwise, perform Basic Protocol 1, steps 5–22.
-
6.Start a new 1 mL culture of LB media with 1 μL of 1000X chloramphenicol stock solution and 1 μL of 1000X tetracycline stock solution in a 1.7 mL microcentrifuge tube with a hole in the lid. Inoculate the culture with a colony or a stab from a glycerol stock of a DH10B strain containing the target BAC and the psc101-gbaA plasmid. Place the tube in a shaking heat block set to 30 °C and 1000 r.p.m. and incubate overnight for ~16 hours.Note that, if engineering the E. coli genome, omit the addition of chloramphenicol to all agar plates and media in this step and all subsequent steps.
-
7.Use 40 μL of the overnight culture, which should now appear opaque, to inoculate a fresh 1 mL LB culture with 1X chloramphenicol and 1X tetracycline and a hole in the lid. Incubate this fresh culture at 30 °C and 1,000 r.p.m. for exactly 2.0 hours. While the culture is growing, chill 50 mL of molecular biology-grade water in a 50 mL conical tube on ice.When preparing or manipulating bacterial cultures in general, use sterile technique to prevent contamination. Keep tubes, bottles, and tips boxes closed and only open momentarily as needed. Everything that comes in contact with the culture must be sterile. Perform any manipulations that require sterility near the base of a Bunsen burner or inside a biosafety cabinet to minimize the chance of airborne contamination.
-
8.After 2.0 hours, add 20 μL of 10% rhamnose stock solution and 20 μL of 10% arabinose stock solution to the culture and increase the temperature of the heat block to 37 °C. Incubate the culture for exactly 40 more minutes at 1,000 r.p.m.Rhamnose induces the expression of the lambda phage proteins that carry out recombineering (Red αβγ) from the psc101-gbaA plasmid. The arabinose induces the ccdA antitoxin that binds the ccdB toxin and renders it non-lethal. The short period of growth at 37 °C assists the expression of these proteins. After the 40 minutes, the culture should be in mid-log phase and slightly cloudy.
-
9.Carry out Basic Protocol 1, steps 10–18, but in this case to electroporate ~200 ng of the kanR+ccdB targeting cassette instead of the target BAC or recombineering plasmid. After resuspending the electroporated cells in 1 mL of SOC media, return them to the 1.7 mL original microcentrifuge tube, then add 20 μL of 10% arabinose stock solution to the media. Incubate the culture in the heat block at 30 °C for 2 hours at 1,000 r.p.m.Arabinose needs to always be present at concentration of 0.2% v/v in the media or in agar plates from this point forward to ensure that ccdB does not kill the cells. If you forget to add arabinose in any of the subsequent steps, with the exception of the molecular biology-grade water washes before the second electroporation, you must restart the protocol. While the culture may eventually grow in the absence of arabinose, it will consist of cells that have inactivated ccdB via random mutations, thus rendering counterselection impossible in the second step. Do not omit arabinose until explicitly instructed to do so at the end of the protocol.
-
10.
After the 2-hour recovery period, pellet the cells at 10,000 × g for 1 minute. Resuspend the pellet in 50 μL of LB media.
-
11.Streak the resuspended cells on an LB agar plate with 1X kanamycin and 0.2% w/v arabinose to select for those rare recombinants that have incorporated the targeting cassette.Do not forget to add arabinose to ensure that ccdB does not kill the cells.
-
12.After the plate has dried, meaning there is no more running liquid on top of the agar, incubate it at 30 °C for ~18 hours or until colonies appear.There should be a few dozen to ~100 colonies on the plate.
-
13.Pick three colonies from the plate and use them to inoculate fresh 1 mL cultures of LB media with 1 μL of 1000X kanamycin stock solution and 20 μL of 10% arabinose stock solution in 1.7 mL microcentrifuge tubes with holes in the lids. Incubate these fresh cultures in a shaking heat block set to 30 °C and 1000 r.p.m. overnight for ~16 hours.Three colonies are picked because 30–50% of colonies will be heteromultimers of unmodified parent and kanR+ccdB intermediate BACs. Heteromultimers easily undergo intramolecular recombination to yield parent BAC to survive counterselection (Fig. 8) at a frequency much higher than successful recombineering. Therefore, it is important to pick several colonies to increase the chances of choosing a monomer. Note that it is simpler and faster to just pick several colonies to recombineer in the second step rather than to screen for monomers of the kanR+ccdB intermediate.
-
14.Use 40 μL of each overnight culture, which should now appear opaque, and use it to inoculate a fresh 1 mL LB cultures with 1 μL of 1000X kanamycin stock solution and 20 μL of 10% arabinose stock solution. Incubate these fresh cultures at 30 °C for exactly 2.0 hours. While the cultures are growing, chill 50 mL of molecular biology-grade water in a 50 mL conical tube on ice.Optionally, thoroughly mix 500 μL of each overnight culture with 500 μL of sterile 50% glycerol in a sterile 1.7 mL microcentrifuge tube (without a hole in the lid) and store immediately at –80°C. Once a kanR+ccdB intermediate is confirmed to be monomeric via successful recombineering, it can be used again in the future to introduce modifications into the same location. This intermediate can be used to make any other modification in the region as long as the homology arms straddle the kanR+ccdB genes. Furthermore, when generating a set of different modifications in the same location, we note that it is useful to pilot just one modification of a set to obtain a confirmed kanR+ccdB intermediate monomer. The confirmed monomer can then be used to complete the set of modifications in parallel rather than using three different intermediates. This approach reduces the number of samples that need to be processed by a factor of three.
-
15.After 2.0 hours, add 20 μL of 10% rhamnose stock solution to each culture and increase the temperature of the heat block to 37 °C. Incubate the cultures for exactly 40 more minutes.Rhamnose induces the lambda phage proteins that carry out recombineering. Arabinose should already be present in the culture to maintain ccdA antitoxin expression.
-
16.Carry out Basic Protocol 1, steps 10–18, but in this case to electroporate ~200 ng of the modification targeting cassette instead of the target BAC or recombineering plasmid. After resuspending the electroporated cells in 1 mL SOC media, return them to the original 1.7 mL microcentrifuge tubes, then add 20 μL of 10% arabinose stock solution to each culture. Incubate the cultures in the heat block at 30 °C for 2 hours at 1,000 r.p.m.It is particularly important that the recovery time is at least 2 hours at this step so that the cells divide enough times to isolate successful recombinants from any unmodified kanR+ccdB intermediates (Warming et al., 2005). During mid-log phase, there are usually the equivalent of ~4–8 copies of the genome, and presumably of BACs as well, in a single cell (Åkerlund, Nordström, & Bernander, 1995). It is likely that only one of these will be modified in a cell where successful recombination has occurred. Only one copy of the ccdB gene is required to kill a cell. Thus, it is critical that any successful recombinant genomes or BACs have time to segregate into daughter cells away from any copies of ccdB before arabinose is removed and ccdA antitoxin production ceases.
-
17.After the 2-hour recovery period, pellet the cells at 10,000 × g for 1 minute. Resuspend the pellet in 1 mL of LB media.It is important to remove the arabinose from the cells in this step by pelleting and resuspending in 1 mL LB media. In the absence of arabinose, ccdB will kill any cells that still carry the gene, thus counterselecting against unmodified cells and selecting for successful recombinants.
-
18.Streak 50 μL of the 1 mL of resuspended cells onto one side of an LB agar plate with 1X chloramphenicol and 1X tetracycline. Next, use the wet glass hockey stick and spread the solution onto the other half of the plate as a means of quickly diluting the cells. Alternatively, plate 5 μL of the resuspended cells onto one side of the plate, then 50 μL of the resuspended cells onto the other half. Ensure that there is no arabinose in the plates.Do not plate all the cells from the 1 mL culture onto the plate. The ccdB gene does not kill cells immediately and the cells will divide for several rounds before dying (Jaffé, Ogura, & Hiraga, 1985). If too many cells are plated at once, they will grow into a lawn before dying. This lawn obscures successful recombinants and prevents them from forming distinguishable colonies. 50 μL out of the 1 mL culture (1/20th of the cells) on one half of a 10 cm plate generally yields good results, but sometimes still forms a lawn. Therefore, it is also useful to plate a more dilute sample on the other half of the plate.Ensure that chloramphenicol is present in the plates. Without chloramphenicol, cells can survive counterselection by losing the ccdB-containing target BAC, which occurs at a frequency much greater than the frequency of successful recombination. Chloramphenicol will kill any cells that lose the target BAC.
-
19.After the plate has dried, meaning there is no more running liquid on top of the agar, incubate it at 30 °C for ~18 hours or until colonies appear.Either the more concentrated or more dilute half of the plate should have a few dozen distinguishable colonies. Plates with hundreds or thousands of colonies are likely the result of heteromultimers that recombined to yield unmodified parent BAC and are thus not worth screening. If, after attempting the protocol a few times, every plate consistently has hundreds or thousands of colonies, it could be that the parent target BAC was a homomultimer prior to the insertion of the kanR+ccdB cassette (Fig. 9). In such a situation, nearly 100% of kanR+ccdB intermediates will be heteromultimers, rather than just 30–50%. To address this issue, the target BAC must be remonomerized using Support Protocol 4 before reattempting this protocol.
-
20.In order to screen for successful recombinants, use the sequencing primers from step 4 to perform using colony PCR as described in Support Protocol 2.Insertions of ~2 kb and larger undergo recombineering quite inefficiently (Kuhlman & Cox, 2010). In such a case, many of the colonies on the plate will be false positives in which the ccdB gene has acquired a mutation that renders it inactive and/or non-lethal. Inactivation of ccdB is an event that occurs at a frequency similar to recombineering events, so it is especially important to screen many colonies (24–96 colonies) when recombineering with large cassettes.
-
21.Once a successful recombinant is identified, confirm it by submitting the corresponding colony PCR product for Sanger sequencing (Metzker, 2005) using the sequencing primers. Each sequencing primer will be used for a separate Sanger sequencing reaction.Targeting primers and targeting cassette PCR products often contain random mutations, so it is important to get full sequencing coverage of the final recombineering product to ensure it is free of undesired mutations (Thomason et al., 2014). Order additional sequencing primers if necessary for large modifications as most Sanger sequencing reads are less than 1000 bp. Ordering sequencing primers spaced out every 500 bp over a region of interest generally provides reliable coverage with overlapping reads. For example, a 5000 bp insertion should have about 8 equally spaced sequencing primers throughout the insertion in addition to the standard sequencing primers that flank the insertion.
-
22.
After sequence confirmation, return to step 5 to introduce further modifications as needed. If there are no additional modifications to make, see Support Protocol 3 to remove the psc101-gbaA plasmid from the cell.
The glycerol stock of the kanR+ccdB intermediate from step 14 that yielded a successful recombinant should be saved as a “confirmed monomer”. We note that during counterselection the toxic ccdB gene selects against any potential heteromultimers that form via fusion of a kanR+ccdB intermediate BAC and successfully modified recombinant BAC. Heteromultimers of unmodified parent BAC and a kanR+ccdB intermediate BAC almost always yield parent BAC after counterselection, so a heteromultimer of unmodified parent BAC and successfully modified recombinant BAC is very unlikely. Therefore, unlike in Basic Protocol 1, it is unlikely that the final recombinant is a heteromultimer. Hence, it is generally not necessary to screen for heteromultimers. If in doubt, carry out Basic Protocol 1, steps 32–52 to ensure that the final recombinant BAC is not a heteromultimer.
ALTERNATE PROTOCOL 1
LARGER SCALE CULTURES FOR INEFFICIENT RECOMBINEERING
Basic Protocols 1–3 use 1 mL of mid-log cultures to generate electrocompetent cells. This scale is convenient to process and is generally sufficient for routine recombineering (H. Wang et al., 2014). However, increasing the number of electrocompetent cells for difficult, inefficient recombineerings—such as for large inserts or replacements—can increase the chances of obtaining a successful recombinant when 1 mL scale recombineering has failed. This protocol describes how to perform Basic Protocol 3 on a 25 mL scale rather than a 1 mL scale, as adapted from previous work (Warming et al., 2005).
Materials
Target BAC-kanR+ccdB intermediate (from Basic Protocol 3, steps 6–13)
Modification targeting cassette (from Basic Protocol 3, step 3)
Sterile 10 μL pipette tips (VWR 89079–466)
Sterile 200 μL pipette tips (VWR 89079–460)
Sterile 1000 μL pipette tips (VWR 89079–472)
10 mL serological pipette (Cellstar 607180)
Sterile 50 mL baffled flasks (Wheaton 354235)
Aluminum foil
Lysogeny broth media (see recipe)
1000X kanamycin stock solution (see recipe)
10% arabinose stock solution (see recipe)
50 mL conical tubes (Cellstar 227261)
Molecular biology-grade water (Corning 46-000-CM)
0.1 cm electroporation cuvettes (Bio-Rad 1652083)
Kimwipes (Kimberly-Clark 34155)
Super optimal broth with catabolic repression media (see recipe)
14 mL sterile culture tubes (VWR 60818-703)
15 mL conical tube (Cellstar 188271)
DNA Editing Software (e.g Snapgene, ApE, Serial Cloner)
P10 pipettor (e.g. VWR 89079-962)
P200 pipettor (e.g. VWR 89079-970)
P1000 pipettor (e.g. VWR 89079-974)
Pipette gun (e.g. Drummond Portable Pipet-Aid XP 4-000-101)
30 °C shaking incubator (e.g. Thermo Fisher MaxQ 4000 SHKE4000)
Ice maker and ice buckets
UV-Vis spectrometer (e.g. Thermo Fisher Nanodrop 2000c)
Shaking water bath (e.g. Shel Lab SWBR17)
Refrigerated centrifuge capable of 4,500 × g with rotor and adapters for 15 mL and 50 mL conical tubes (e.g. Beckman Coulter Avanti J-E 369003 with JA-10 rotor 369687)
Electroporator (Bio-Rad Micropulser 1652100)
Refrigerated microcentrifuge (e.g. Thermo Fisher Heraeus Fresco 21 75002426)
Recombineering on a 25 mL scale
Design and create targeting primers and cassettes as described in Basic Protocol 3, steps 1–4.
If target BAC and the psc101-gbaA plasmid are already both present in DH10B cells, or if the intent is to engineer the E. coli genome and the psc101-gbaA plasmid is already in the strain to be engineered, proceed to step 6. Otherwise, carry out Basic Protocol 1, steps 5–22.
- Carry out Basic Protocol 3, steps 6–13 to generate the kanR+ccdB intermediate.The insertion of the kanR+ccdB cassette is usually efficient enough to carry out on a 1 mL scale rather than a 25 mL scale. Ideally, perform large scale recombineering with a confirmed kanR+ccdB intermediate monomer when possible to reduce the number of large-volume samples that need to be processed. Otherwise, perform this protocol with three kanR+ccdB intermediates if there is no confirmed monomer available.
- Use 500 μL of the overnight culture, which should now appear opaque, to inoculate a fresh 25 mL LB culture with 25 μL of 1000X kanamycin stock solution and 500 μL of 10% arabinose stock solution in a 50 mL sterile baffled flask covered with aluminum foil. Incubate the fresh culture at 30 °C for about 1.5 hours in a shaking incubator at 250 r.p.m. After 1.5 hours, check the OD600 every 20 minutes until the culture has reached an OD600 of ~0.35. While the culture is growing, chill 50 mL of molecular biology-grade water in a 50 mL conical tube on ice.Sterilize the 50 mL glass baffled flasks by rinsing thoroughly with deionized water, covering the opening with aluminum foil to serve as a cap, and autoclaving.
- After the culture attains an OD600 of ~0.35, add 500 μL of 10% rhamnose stock solution to the culture and move the culture to a 37 °C shaking water bath. Incubate the culture at 150 r.p.m. for exactly 40 more minutes.Air incubators do not efficiently heat larger cultures in short periods of time, so it is important to use a shaking water bath. Alternatively, if a shaking water bath is not available the flasks can be swirled by hand in a 37 °C bath.
After incubation at 37 °C, the culture should have an OD600 of ~0.6. Place the culture on ice to arrest growth. Next, transfer the cells to a 50 mL conical tube and pellet at 4,500 × g in a refrigerated centrifuge at 4 °C for 5 minutes. Carefully remove the media with a 20 mL pipette to avoid disturbing the pellet.
Gently resuspend the pellet in 10 mL ice-cold molecular biology-grade water by gently pipetting several times. Next, pellet again at 4,500 × g in a refrigerated centrifuge at 4 °C for 5 minutes.
- Repeat step 7.After the first wash, the pellet becomes very loose and easy to disturb. Slowly remove media with the pipette gun from the top down. Do not pour off the media because this risks losing a significant quantity of the cells.
- Resuspend the pellet in ~1 mL of molecular biology-grade water and transfer to an ice-cold, sterile 1.7 mL microcentrifuge tube.Often there is ~1 mL of residual water around the pellet in the bottom of the Falcon tube, in which case it is unnecessary to add additional water. In the next step, the cells are concentrated further.
Pellet the cells at 10,000 × g in a refrigerated microcentrifuge at 4 °C for 1 minute.
- Resuspend the pellet in a final volume of ~30–50 μL of molecular biology-grade water. Keep the tube on ice. These cells are now “electrocompetent”.Often there is ~20 μL of residual water around the pellet, in which case it is unnecessary to add additional water to the tube. The resuspended cells should appear opaque. Because this batch of electrocompetent cells will be more concentrated, ensure there is enough water to thoroughly resuspend the pellet with no clumps.
Add ~200 ng of the modification targeting cassette to the electrocompetent cells and mix gently by pipetting a few times. Keep the tube on ice.
Before electroporating, set up all of the necessary electroporation materials. Plug in the Bio-Rad micropulser and turn it on. The screen should display the default setting “Ec1”, which is the proper setting to use. Press the measurement button twice until a light appears next to the “ms” label and the screen reads “0.00”. Set a bottle of SOC media close to the electroporator with the cap completely loose for quick access. Ensure that a box of Kimwipes is nearby.
Add the electrocompetent cells that have been mixed with DNA into the 0.1 cm gap between the metal plates of a pre-chilled electrocuvette. Next, quickly place the cap back onto the electrocuvette.
Wipe any condensation or ice off the outside of the electrocuvette, then place it into the sample holder with the notch facing forward. Gently slide the electrocuvette into the electroporation chamber until the electrocuvette “clicks” between the flexible metal electrodes in the electroporation chamber.
- Press the “Pulse” button. After about a second, a buzzer should indicate that the electroporation is complete and a number representing the pulse length will appear on the screen. Quickly remove the cuvette from the sample holder and resuspend the cells in 1 mL of SOC media.If the pulse length is less than 4.80 ms or if the screen says “Arc”, repeat the electroporation with new electrocompetent cells and add a smaller quantity of DNA. It may be necessary to desalt the DNA again or wash the cells an extra time. It is often useful to prepare a few batches of electrocompetent cells in parallel in case the electroporation needs to be repeated a few times before it is successful.
Add the resuspended cells to 4 mL of SOC media in a sterile 14 mL culture tube and add 100 μL of 10% arabinose stock solution. Incubate the culture at 30 °C for 2 hours at 250 r.p.m in a shaking incubator.
After recovery, pellet the cells in a 15 mL conical tube at 4,500 × g for 5 minute. Resuspend the pellet in 1 mL of LB media.
Carry out Basic Protocol 3, steps 18–22.
SUPPORT PROTOCOL 1
CREATING TARGETING CASSETTES
The quality of targeting cassette DNA is paramount to the success of recombineering. It is essential to obtain low-salt, high-concentration DNA samples that are relatively free of PCR side products or other unwanted DNA, such as template plasmids that can result in false positives upon selection. Cassettes of 100 bp or smaller can easily be generated by purchasing the top and bottom strand as separate oligos and annealing them to yield cassettes with high-concentration and purity, as described in previous work (Warming et al., 2005). Targeting cassettes too large to order as oligos can be generated by PCR using the targeting primers and a PCR template. However, it is important to be aware if two important issues when generating targeting cassettes by PCR:
PCR often generates a series of truncated side products from non-specific primer binding. These side products are sometimes, but not always, visible on an agarose gel as faint bands or smears below the expected band. Because smaller targeting cassettes recombine with a much higher efficiency than larger cassettes, these truncated side products often recombine into the region of interest much more often than the main desired cassette. This problem is particularly significant when the desired cassette is very large. Therefore, it is wise to carefully purify cassettes larger than 2 kb via gel extraction to remove these truncation products.
In cases where the PCR template plasmid encodes an antibiotic resistance gene or some other gene that will be applied to select for successful recombinants, it is absolutely critical to inactivate or completely remove the PCR template plasmid from the final targeting cassette product. If any intact PCR template plasmid enters the cells that are being recombineered, it will result in colonies after selection that have not actually undergone recombination. These false positives have simply taken up template plasmid leftover from the PCR reaction. If there is even minimal intact template plasmid contamination, these ‘cheaters’ will greatly outnumber any rare, successful recombinants. The most efficient approach to prevent false positives resulting from template plasmid uptake is to use DNA templates that cannot replicate in DH10B E. coli. Otherwise, the PCR product must be fully digested with endonucleases that destroy only the template plasmid, and/or be purified by gel extraction.
Materials
Targeting primers or oligos (designed and ordered by user)
R6K-kan-ccdB plasmid (see Table 1)
R6K-KCA plasmid (see Table 1)
R6K-AKC plasmid (see Table 1)
Molecular biology-grade water (e.g., Corning 46–000-CM)
1.7 mL microcentrifuge tubes (e.g., VWR 87003–294)
Sterile 10 μL pipette tips (e.g., VWR 89079–466)
Sterile 200 μL pipette tips (e.g., VWR 89079–460)
Sterile 1000 μL pipette tips (e.g., VWR 89079–472)
Q5 Reaction Buffer (New England Biolabs B9027S)
Lid lock (e.g., VWR 14229–941)
3 M sodium acetate (see recipe)
70% ethanol (e.g., Koptec V1401)
100% ethanol (e.g., Koptec V1001)
8-well PCR tube strips (e.g., VWR 20170–004)
OneTaq Quick-Load 2X Master Mix with Standard Buffer (New England Biolabs M0486L)
Agarose (e.g., Lonza 50004)
0.5X TBE buffer (see recipe)
Paper towels
GelGreen Nucleic Acid Gel Stain (Biotium 41005)
Quick-Load Purple 1 kb DNA Ladder (New England Biolabs N0552S)
6X Gel Loading Dye (New England Biolabs B7024S)
10X Cutsmart Buffer (New England Biolabs B7204S)
DpnI endonuclease (New England Biolabs R0176S)
Industrial razor blades (e.g., VWR 55411–050)
Gel extraction kit (e.g., Omega Bio-tek Gel Extraction Kit D2500–01)
PCR clean-up kit (e.g., Omega Bio-tek Cycle Pure Kit D6492–01)
Refrigerated microcentrifuge (e.g., Thermo Fisher Heraeus Fresco 21 75002426)
500 mL glass beaker (e.g., Corning 1003–600)
Magnetic stir bar (e.g., VWR 58947–132)
Foam float for 1.7 mL tubes (e.g., VWR 82017–634)
Stirring hot plate with temperature probe (e.g., Corning PC-420D 6795–420KIT)
Thermal cycler (e.g. Bio-Rad T100 Thermal Cycler 1861096)
Precision balance (e.g. Mettler Toledo ME1002TE 30216559)
250 mL glass Erlenmeyer flask (e.g., VWR 10536–914)
Agarose gel electrophoresis system (e.g. Bio-Rad Mini-Sub Cell GT System 1664401)
P10 pipettor (e.g., VWR 89079–962)
P200 pipettor (e.g., VWR 89079–970)
P1000 pipettor (e.g., VWR 89079–974)
Microwave oven
Mini-gel caster (e.g., Bio-Rad 1704422)
Blue LED transilluminator (e.g., Maestrogen LED Transilluminator SLB-01W)
37 °C incubator (e.g., VWR Gravity convection incubator 89511–422)
60 °C heat block (e.g., Corning LSE Digital Dry Bath 6875-SB with 24 × 1.5 mL block 480119)
UV-Vis spectrometer (e.g., Thermo Fisher Nanodrop 2000c)
Annealing oligos to generate small targeting cassettes ≤100bp
Order oligos as desalted 100 μM stock solutions or as dry desalted oligos. Dissolve dry desalted oligos in molecular biology-grade water to yield 100 μM stock solutions. Be sure to thoroughly mix by vortexing the capped tube for ~1 minute, followed by spinning the tube at 10,000 × g for 1 minute.
Add the following reagents to a sterile 1.7 mL microcentrifuge tube: 70 μL of molecular biology-grade water, 20 μL of 5X Q5 Reaction Buffer, 3 μL of 100 μM top strand oligo stock solution, and 3 μL of 100 μM bottom strand oligo stock solution. Close the tube and secure the lid with a lid lock.
- Bring 300 mL of water in a 500 mL beaker to a rolling boil while stirring with a magenetic stir bar on a hot plate set to 105 °C.Cover the beaker with an aluminum foil lid to prevent heat loss and accelerate heating.
Place the tube from step 2 in a foam float and float it on the boiling water for 5 minutes.
- After 5 minutes, set the beaker of boiling water on a bench. Allow the water to cool slowly to room temperature with the tube still floating on the water. This step generally requires 30–60 minutes.For proper annealing, it is important to cool the samples slowly from boiling to room temperature over the course of at least 30 minutes. Do not just boil the tube then place it at room temperature. Use a sufficient volume of water to ensure slow cooling.
- After the beaker has cooled to room temperature, remove the tube from the water and add 10 μL of 3 M sodium acetate to the annealed oligos, followed by 250 μL of 100% ethanol. Mix the contents by pipetting up and down.The solution should quickly become slightly cloudy as a precipitate forms.
- Spin the tube at ≥10,000 × g for 10 minutes at room temperature.A white pellet should be clearly visible at the bottom of the tube after centrifugation. This pellet contains the annealed oligos.
Remove the supernatant, being careful to not disturb the white pellet at the bottom of the tube.
- Add 400 μL of 70% ethanol to the tube and gently pipette up and down a few times. Discard the 70% ethanol.The pellet may dislodge, so be extra careful to not aspirate and discard the pellet.
- Spin the tube at ≥10,000 × g for 1 minute at room temperature.This extra spin shifts residual ethanol and any dislodged pieces of the pellet back down to the bottom of the tube.
Carefully remove any residual ethanol from around the pellet with a 10 μL pipette. Allow the contents of the open tube to air dry for ~10 minutes.
Resuspend the pellet in 100 μL of molecular biology-grade water to obtain a targeting cassette solution. The resulting DNA concentration will be about ~200 ng/μL.
Generating large targeting cassettes by PCR
- Set up the following 50 μL PCR reaction in one tube of an 8-well PCR tube strip: 25 μL of molecular biology-grade water, 25 μL of Q5 Hi-Fidelity 2X Master Mix, 1 ng of template DNA, and 2.5 μL of a 10 μM stock of each targeting primer.Dilute a portion of the 100 μM primer stocks to 10 μM before adding to the PCR reaction. Also, only add a very small amount of template DNA (~1 ng) so that it is easier to remove the template in later steps.
Calculate the annealing temperature for the primers using the New England Biolabs Tm Calculator (https://tmcalculator.neb.com/#!/main). Ensure the polymerase option is set to “Q5 Hi-Fidelity 2X Master Mix”. Determine the expected size of the PCR product using DNA editing software.
-
Run the PCR reaction in a thermal cycler using the following conditions:
98 °C 30 seconds
98 °C 10 seconds
Annealing Temp. 30 seconds
72 °C 30 seconds per kb of expected PCR product
Return to step 2 for 35 cycles
72 °C 2 minutes
4 °C hold
- Add 500 mg of agarose to an Erlenmeyer flask, followed by 50 mL of 0.5X TBE buffer. Plug the top of the flask with a wad of paper towel and microwave on high power for about 60 seconds, or until the agarose is dissolved.Monitor the agarose while microwaving to ensure that it does not boil over. Use oven mitts or paper towels when handling the hot flask.
- Add 5 μL of GelGreen dye to the molten agarose, swirl the flask to distribute the dye, then pour the solution into a tray in a gel caster with an 8-well comb. Allow the gel to cool and solidify for 15–30 minutes.GelGreen can be visualized using a blue LED transilluminator, which does not damage DNA. If possible, do not use ethidium bromide dye to visualize the PCR products, because it must be visualized using an ultraviolet transilluminator. Ultraviolet light will cause damage to the DNA products and increase the incidence of unwanted mutations.
Carefully remove the comb and place the solidified gel into an electrophoresis chamber filled with 0.5X TBE buffer. Ensure that there is sufficient buffer to submerge the entire gel.
To check the PCR product, mix 3 μL of PCR reaction with 1 μL of 6X gel loading dye. Next, load the resulting solution into one of the wells of the agarose gel next to another well that has 5 μL of Quick-Load Purple 1 kb DNA Ladder.
Run the gel at 150 volts for 20 minutes, then visualize on a blue LED transilluminator.
- If there is a bright band corresponding to the expected size of the PCR product, proceed to step 10. If there is no band, the band is faint, the band is clearly the wrong size, or there are many bands/a smear, the PCR reaction must be optimized. Return to step 1 and run eight parallel reactions in the 8-well PCR tube strips. Run the reactions with a gradient of annealing temperatures from 62–72 °C.If none of the temperatures in the annealing temperature gradient work, redo the gradient, but replace 10 μL of molecular biology-grade water with 10 μL of Q5 High GC Enhancer per reaction. Alternatively, try extending the 72 °C extension step to 60 seconds per kb of expected PCR product. If the PCR reaction still fails, try redesigning the primers to minimize primer-dimer formation. Visit https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/multiple-primer-analyzer.html to predict primer-dimer propensity.
- If the PCR product is less than 2 kb and was amplified from template DNA that is not expected to interfere with selection or counterselection, such as templates for Basic Protocol 3 second step modification cassettes or templates that cannot replicate in DH10B E. coli, purify the PCR product using the Omega Bio-tek Cycle Pure Kit following the manufacturer’s instructions. Critically, however, be sure to elute in 30 μL molecular biology-grade water or 0.2X elution buffer to obtain the final targeting cassette. Measure the resulting DNA concentration using a UV-Vis spectrometer. For products larger than 2 kb and/or products that were amplified from plasmids that could interfere with selection, proceed to step 11.As a general rule, use a PCR clean-up kit, such as the Omega Bio-tek Cycle Pure Kit, to purify the DNA whenever possible, instead of purifying DNA from the PCR reaction by gel extraction. Gel extraction generally provides very poor DNA yields, so it should only be used when absolutely necessary. Basic Protocols 1–3 use R6K-kan-ccdB, R6K-AKC, or R6K-KCA as template plasmids. These plasmids only have R6K origins that cannot replicate in DH10B cells and can only replicate in cells expressing the pi protein (Kolter, Inuzuka, & Helinski, 1978), such as Pir2 E. coli cells. If the <2 kb PCR cassette was amplified from one of these three R6K plasmids, purify using the Omega Bio-tek Cycle Pure Kit. When there is a choice, always choose or design template vectors that are replication-incompetent in the strain to be used for recombineering. The R6K origin provides one simple and commonly applied option (J. Wang et al., 2006).
- If the PCR product is larger than 2 kb, but the template plasmid is not expected to interfere with selection, skip to step 12. Otherwise, add 5 μL of 10X Cutsmart buffer and 1 μL of DpnI endonuclease per 50 μL PCR reaction. Mix thoroughly by pipetting up and down. Incubate the reaction at 37 °C overnight for ~16 hours, then proceed to step 12.E. coli that have an intact dam methylase methylate the adenine of every GATC sequence that occurs inside the cell. GATC is a very common sequence that occurs multiple times in most plasmids. DpnI is an endonuclease that can cut GATC only when the adenine is methylated. Adding DpnI digests the E. coli-derived template plasmid but leaves the newly synthesized, unmethylated PCR product intact. DpnI digestion will not be successful if the template is not methylated, such as when it is derived from E. coli that have lost dam methylase function, as indicated by the letters “dam” in the strain genotype. If DpnI will not work, use DNA editing software to choose other restriction endonucleases that cut the template plasmid, but do not cut the PCR product. The combination of using a small amount of template plasmid, endonuclease digestion, and gel extraction minimizes the amount of intact template plasmid that can interfere with downstream recombineering and selection (Sawitzke et al., 2013).
- Cast another gel by repeating steps 4–6. Mix the PCR reaction with 10 μL of 6X gel loading dye. Distribute this solution across 6 wells, ~10 μL per well. Add 5 μL of Quick-Load Purple 1 kb DNA Ladder next to the wells containing the PCR reaction.In order to obtain a good gel extraction yield, it is essential to keep the band sharp so that the DNA stays concentrated in a small volume of gel. If the well of an 8-well comb is filled higher than 10 μL, the band can become warped and diffuse. It is better to split the sample across more wells than to overfill a smaller number of wells.
- Run the gel at 120 volts for 15 minutes, then visualize on a blue LED transilluminator.Only run the gel until the desired band is separated enough from any visible side products for excision. Running the gel too long can cause the bands to become warped and diffuse. If the desired band is still too close to easily excise from side products, keep running the gel in 10 minute increments until there is sufficient separation.
- Excise the desired band using a clean, new razor blade. Cut a strip across all 6 lanes and remove as much excess agarose as possible from the sides, tops and bottoms of the bands. The gel slice should ideally weigh less than 300 mg.Use a blue LED transilluminator while cutting bands out of the gel; do not use an ultraviolet transilluminator, if at all possible.
Purify the targeting cassette from the gel slice using the Omega Bio-tek Gel Extraction Kit following the manufacturer’s instructions, but elute in 30 μL molecular biology-grade water or 0.2X elution buffer to yield the final targeting cassette. Measure the concentration with a UV-Vis spectrometer and ensure that the concentration is at least 10 ng/μL. Otherwise, repeat steps 1–15. We typically observe yields of 20–50 ng/μL using this protocol.
Eluting with molecular biology-grade water gives the lowest possible salt concentration. However, eluting with unbuffered water could cause lower yields because elution of DNA from silica columns is sensitive to pH. Using 0.2X elution buffer keeps the salt concentration low and also stabilizes the pH to ensure good DNA yields.
SUPPORT PROTOCOL 2
SCREENING FOR RECOMBINANTS BY COLONY PCR
After selection or counterselection, not all colonies on the plate will be successful recombinants. Many of the colonies will be “false positives” that have only incorporated a truncated portion of the recombineering cassette, that are unmodified parent resulting from a heteromultimer intramolecular recombination (Fig. 8), or that have an inactive, mutant copy of the ccdB gene. Often, when trying to incorporate a particularly large targeting cassette, a large majority of the colonies will be false positives. Colony PCR—in which diagnostic PCR reactions are carried out on individual colonies from the plate—is the quickest and easiest way to identify successful recombinants amongst the false positives. Successful deletion or insertion recombinants can be identified by amplifying the target region with the sequencing primers and looking for an expected size change relative to the umodified parent target region or kanR+ccdB intermediate in the PCR product on an agarose gel. If the expected size change is too small to confidently identify on an agarose gel—generally 200 bp or smaller—successful recombinants can also be identified by amplifying the expected target region with one of the targeting primers and one of the sequencing primers that faces that targeting primer (Sawitzke et al., 2013; Thomason et al., 2014). In this case, a PCR product will only be generated if the targeting cassette has been incorporated. Successful recombinants can be identified by evaluating whether there is a band corresponding to the expected PCR product or whether there is no band. For point mutations, the previous two methods will not work, owing to the lack of size change or change in primer binding sites upon recombination. The simplest way to identify successful point mutations is to amplify the target region with the sequencing primers and then Sanger sequence the PCR product (Sawitzke et al., 2013; Thomason et al., 2014).
Materials
Sequencing primers (designed and ordered by user)
1.7 mL microcentrifuge tubes (e.g., VWR 87003–294)
Sterile 10 μL pipette tips (e.g., VWR 89079–466)
Sterile 200 μL pipette tips (e.g., VWR 89079–460)
Sterile 1000 μL pipette tips (e.g., VWR 89079–472)
8-well PCR tube strips (e.g., VWR 20170–004)
OneTaq Quick-Load 2X Master Mix with Standard Buffer (New England Biolabs M0486L)
Molecular biology-grade water (e.g., Corning 46–000-CM)
Lysogeny broth media (see recipe)
Agarose (e.g., Lonza 50004)
0.5X TBE buffer (see recipe)
Paper towels
GelGreen Nucleic Acid Gel Stain (Biotium 41005)
Quick-Load Purple 1 kb DNA Ladder (New England Biolabs N0552S)
PCR clean-up kit (e.g., Omega Bio-tek Cycle Pure Kit D6492–01)
P10 pipettor (e.g., VWR 89079–962)
P200 pipettor (e.g., VWR 89079–970)
P1000 pipettor (e.g., VWR 89079–974)
Thermal cycler (e.g., Bio-Rad T100 Thermal Cycler 1861096)
Precision balance (e.g., Mettler Toledo ME1002TE 30216559)
250 mL glass Erlenmeyer flask (e.g., VWR 10536–914)
Agarose gel electrophoresis system (e.g., Bio-Rad Mini-Sub Cell GT System 1664401)
Microwave oven
Mini-gel caster (e.g., Bio-Rad 1704422)
Blue LED transilluminator (e.g., Maestrogen LED Transilluminator SLB-01W)
Performing Colony PCR
- Create the PCR master mix by adding 300 μL of molecular biology-grade water, 300 μL of OneTaq Quick-Load 2X Master Mix with Standard Buffer, and 1.2 μL of each 100 μM primer stock to a 1.7 mL microcentrifuge tube. Mix the contents thoroughly by pipetting up and down.For recombinants that will have a detectable size change relative to the unmodified parent or kanR+ccdB intermediate, use the sequencing primers to screen the colonies. Otherwise, use one of the targeting primers and the sequencing primer that faces that targeting primer, such that the PCR will only occur if recombination was successful.
- Distribute 22 μL of PCR master mix to each tube of three sterile, 8-well PCR tube strips—24 tubes total. Distribute 50 μL of LB media to each tube of another set of three sterile 8-well PCR tube strips.This protocol describes how to screen 24 colonies because it is the maximum number of colonies that can be conveniently screened on one agarose gel with two 15-well combs. However, the protocol can be scaled up or down accordingly. Based on our experience, it is advisable to screen at least 8 colonies.
- Pick a colony for screening with a sterile 10 μL pipette, then dip the tip into the first PCR tube of PCR mastermix and swirl it around. Next, take the same tip and swirl it around in the first PCR tube of LB media. Discard the tip and repeat the process until all the PCR tubes have been inoculated with unique colonies. It is often helpful to include an unmodified parent control and/or a kanR+ccdB intermediate control as one of the 24 samples to more easily identify size changes in the PCR product.The tubes of LB media serve to store the colonies in an organized manner so that colonies that are identified as successful recombinants can later be retrieved and grown. The colonies can be stored in the LB media in the PCR tubes at 4 °C for at least a week.
Calculate the annealing temperature for the primers using the New England Biolabs Tm Calculator (https://tmcalculator.neb.com/#!/main). Ensure the polymerase option is set to “OneTaq Quick-Load 2X Master Mix with Standard Buffer”. Determine the expected size of the PCR product using DNA editing software.
-
Run the PCR reactions in a thermal cycler using the following conditions:
94 °C 5 minutes
94 °C 15 seconds
Annealing Temp. 15 seconds
68 °C 60 seconds per kb of expected PCR product
Return to step 2 for 30 cycles
68 °C 5 minutes
4 °C hold
Note that the reaction conditions for the OneTaq polymerase mixture are different than those for the Q5 polymerase mixture in Support Protocol 1. Along with different temperatures and times, the initial denaturation in step 1 of the program is extended to 5 minutes to lyse the bacteria and release template DNA.
Identifying PCR products by gel electrophoresis
-
6.Add 500 mg of agarose to an Erlenmeyer flask followed by 50 mL of 0.5X TBE buffer. Plug the top of the flask with a wad of paper towel and microwave on high power for about 60 seconds, or until the agarose is dissolved.Monitor the agarose while microwaving to ensure that it does not boil over. Use oven mitts or paper towels when handling the hot flask.
-
7.
Add 5 μL of GelGreen dye to the molten agarose, swirl the flask to distribute the dye, then pour into a tray in a gel caster with two 15-well combs. Allow the gel to cool and solidify for 15–30 minutes.
-
8.
Carefully remove the comb and place the solidified gel into an electrophoresis chamber filled with 0.5X TBE buffer. Ensure that there is sufficient buffer to submerge the entire gel.
-
9.Add 3 μL of Quick-Load Purple 1 kb DNA Ladder to the first well in each row. Check all of the colony PCR products by loading 2 μL of each reaction into a well.The OneTaq Quick-Load 2X Master Mix already contains a green loading dye and a density reagent, so the PCR reaction can just be directly loaded into the well.
-
10.
Run the gel at 150 volts for 20 minutes, then visualize on a blue LED transilluminator to search for successful recombinants. If the modification is a point mutation, identify several colonies that have PCR products with the expected size and that have bright bands, purify the PCR products using the Omega Bio-tek Cycle Pure Kit, and submit the purified DNA samples for Sanger sequencing using the sequencing primers and following the service provider’s instructions.
-
11.
Once a successful recombinant is identified, either by visually inspecting the colony PCRs on a gel and/or sequencing, return to the PCR tube containing that colony in LB media and grow it up for the next step.
While it is absolutely necessary to sequence the colony PCR products when identifying successful point mutations, it is a good idea to fully sequence other types of modifications once a successful recombinant is identified and chosen by visual inspection of colony PCR products on a gel. Targeting cassettes will sometimes contain random mutations introduced in oligo manufacturing or in PCR reactions that can end up in the final recombinant. It is important to ensure that the final recombinant is free of these unwanted, potentially-deleterious mutations (Thomason et al., 2014).
SUPPORT PROTOCOL 3
CURING THE RECOMBINEERING PLASMID
After a target BAC or strain is modified, the recombineering plasmid is no longer needed and can potentially interfere with downstream experiments if other psc101 plasmids, other tetracycline-resistant plasmids, arabinose induction, or rhamnose induction are used. The psc101 origin of the recombineering plasmid used in these protocols is temperature-sensitive, and can thus be removed or “cured” from the strain by growing at higher temperatures (H. Wang et al., 2014). Alternatively, in the case of target BACs, the mixture of target BAC DNA and recombineering plasmid can be transformed into empty E. coli cells. Colonies from the transformation can then be screened for cells that received the target BAC but not the recombineering plasmid, simply by ensuring that the cells grow in chloramphenicol but not in tetracycline.
Materials
LB agar plates with 1X chloramphenicol (see recipe)
Sterile 10 μL pipette tips (e.g., VWR 89079–466)
Sterile 200 μL pipette tips (e.g., VWR 89079–460)
Sterile 1000 μL pipette tips (e.g., VWR 89079–472)
Lysogeny broth media (see recipe)
10 mL serological pipette (e.g., Cellstar 607180)
1000X chloramphenicol stock solution (see recipe)
1000X tetracycline stock solution (see recipe)
14 mL sterile culture tubes (e.g., VWR 60818–703)
Sterile 50% glycerol (see recipe)
1.7 mL microcentrifuge tubes (e.g., VWR 87003–294)
42 °C incubator (e.g., VWR Gravity convection incubator 89511–422)
P10 pipettor (e.g., VWR 89079–962)
P200 pipettor (e.g., VWR 89079–970)
P1000 pipettor (e.g., VWR 89079–974)
30 °C shaking incubator (e.g., Thermo Fisher MaxQ 4000 SHKE4000)
Pipette gun (e.g., Drummond Portable Pipet-Aid XP 4–000-101)
Curing the recombineering plasmid at high temperature
- Streak a colony or glycerol stock of the strain containing the target BAC and the recombineering plasmid on an LB agar plate with 1X chloramphenicol. If curing the recombineering plasmid from a strain with a modified genome, streak onto a plate with strain-compatible antibiotics or no antibiotics for this step and for all subsequent steps in this protocol.Ensure that the plate does not contain tetracycline. For curing the recombineering plasmid from strains that do not carry BACs, do not use chloramphenicol plates. Instead, look for antibiotics that the strain is resistant to or, less preferably, use plates that have no antibiotic. Strain genotypes that have the letters “gyrA96” are usually resistant to 30 μg/mL nalidixic acid. Strain genotypes that have the letters “rpsL” are usually resistant to 100 μg/mL streptomycin.
Incubate the plate at 42 °C overnight for ~16 hours or until colonies appear.
Select any single colony from the plate and streak it onto a new LB agar plate with 1X chloramphenicol.
- Incubate the plate at 42 °C overnight for ~16 hours or until colonies appear.Occasionally, after the first streak at 42 °C (step 1), the colonies that appear will still contain a few cells that retain the recombineering plasmid. Streaking a second time at 42 °C (step 3) ensures that the resulting colonies are pure and do not contain the recombineering plasmid.
- Select any single colony from the plate from step 3 and use it to inoculate 5 mL of LB media with 1X chloramphenicol. Use the same colony to inoculate another separate culture with 5 mL of LB media with 1X tetracycline. Incubate both cultures at 30 °C in a shaking incubator at 250 r.p.m. overnight for ~16 hours.Pick the colony with a sterile pipette tip. Swirl the tip in the 1X chloramphenicol culture first, then use the same tip and swirl it in the 1X tetracycline culture. The tetracycline culture serves as a test to see if the recombineering plasmid is actually cured.
If the chloramphenicol-containing culture grows but the tetracycline culture does not, thoroughly mix 500 μL of the chloramphenicol culture with 500 μL of sterile 50% glycerol in a sterile 1.7 mL microcentrifuge tube (without a hole in the lid) and store immediately at –80 °C. This glycerol stock is the pure target BAC without the recombineering plasmid. If the tetracycline culture did grow, return to step 5 and test several other colonies.
SUPPORT PROTOCOL 4
REMONOMERIZING HETEROMULTIMERS OR HOMOMULTIMERS
Plasmids are prone to forming multimers during recombineering (Thomason et al., 2007). Multimers can interfere with counterselection or result in undesired mixed products. We note that this issue does not occur when targeting the E. coli genome, as E. coli have a system to prevent the formation of multimeric genomes (Bigot et al., 2007). Using the particular protocols described above, we have found that 30–50% of recombinant colonies are heteromultimers resulting from the fusion of unmodified parent BACs and kanR+ccdB intermediate BACs. These heteromultimers are unsuitable for counterselection because they recombine to eliminate the toxic ccdB gene and yield unmodified parent BAC at a frequency much higher than the desired recombineering event (Fig. 8). Furthermore, Basic Protocols 2 and 3 become exceedingly inefficient if the target BAC is a homomultimer because heteromultimeric kanR+ccdB intermediates will then represent nearly 100% of recombinant colonies after the introduction of the kanR+ccdB targeting cassette (Fig. 9). Heteromultimers can be reliably detected by endonuclease digestion, as described in Basic Protocol 1, steps 34–52. Homomultimers are very difficult to reliably detect with simple methods, as they will appear normal via a PCR- or digestion-based test. Furthermore, it is challenging to ascertain the size of very large, intact plasmids without applying specialized separation techniques, such as pulsed-field gel electrophoresis. The simplest method to detect a homomultimer is to attempt recombineering and then observe that all of the recombinants are heteromultimeric. If heteromultimers or homomultimers are suspected, the issue can be resolved by digesting the BAC with an endonuclease that cuts once, then religating to yield a monomeric species that can be retransformed into DH10B cells (Thomason et al., 2007).
Materials
Target BAC or Strain with psc101-gbaA plasmid (supplied by user)
Sterile 10 μL pipette tips (e.g., VWR 89079–466)
Sterile 200 μL pipette tips (e.g., VWR 89079–460)
Sterile 1000 μL pipette tips (e.g., VWR 89079–472)
10 mL serological pipette (e.g., Cellstar 607180)
14 mL sterile culture tubes (e.g., VWR 60818–703)
Lysogeny broth media (see recipe)
1000X chloramphenicol stock solution (see recipe)
Midiprep DNA isolation kit (e.g., ZymoPURE II Plasmid Midiprep Kit D4200)
Restriction endonucleases (selected by user, e.g., New England Biolabs)
PCR clean-up kit (e.g., Omega Bio-tek Cycle Pure Kit D6492–01)
T4 DNA ligase (New England Biolabs M0202S)
New England Biolabs 10-beta Competent E. coli cells (New England Biolabs C3019H)
LB agar plates with 1X chloramphenicol (see recipe)
DNA Editing Software (e.g., Snapgene, ApE, Serial Cloner)
P10 pipettor (e.g., VWR 89079–962)
P200 pipettor (e.g., VWR 89079–970)
P1000 pipettor (e.g., VWR 89079–974)
Pipette gun (e.g., Drummond Portable Pipet-Aid XP 4–000-101)
30 °C shaking incubator (e.g., Thermo Fisher MaxQ 4000 SHKE4000)
Thermal cycler (e.g., Bio-Rad T100 Thermal Cycler 1861096)
42 °C water bath (e.g., VWR General Purpose Water Bath 89501–460)
30 °C incubator (e.g., VWR Gravity convection incubator 89511–422)
Remonomerizing heteromultimers or homomultimers
Inoculate a 5 mL culture of LB containing 5 μL of 1000X chloramphenicol stock solution stock solution with the suspected multimer in a sterile 14 mL culture tube. Incubate the culture overnight at 30 °C, shaking at 250 r.p.m. for ~16 hours
Isolate the target BAC DNA using a midiprep DNA isolation kit, following the manufacturer’s instructions.
Using DNA editing software, identify a restriction endonuclease that only cuts the target BAC once. Ideally, the endonuclease should leave a >1 bp overhang.
Digest ~4 μg of the isolated target BAC DNA with the chosen restriction endonuclease, following the manufacturer’s instructions. Ideally, digest the DNA overnight at the appropriate temperature for the enzyme used to ensure that there will be very little uncut multimer remaining in the sample.
After digestion, remove the restriction endonuclease by purifying the DNA with the Omega Bio-tek Cycle Pure Kit, following the manufacturer’s instructions. Elute the DNA into 30 μL of molecular biology-grade water or into 0.2X elution buffer.
- Mix 9 μL of the elution with 1 μL of 10X T4 ligase buffer in a PCR tube. Add 1 μL of T4 ligase and mix the solution thoroughly by pipetting up and down. Incubate the ligation reaction at room temperature overnight for ~16 hours. In addition, set up a control reaction without T4 ligase.Large vectors can be difficult to religate and transform. Incubating the ligation reaction for extended periods of time helps to maximize the number of religated monomers available for transformation.
After incubation, heat inactivate the ligation reaction at 65 °C for 10 minutes in a thermal cycler or heat block.
- Transform 5 μL of the ligation into 50 μL of New England Biolabs 10-beta Competent E. coli cells, following the manufacturer’s instructions.Transforming large vectors can be very difficult and inefficient. We recommend using commercially available high-efficiency competent cells, and transforming them via heat shock. Electroporation would require purifying the ligation to remove salt, which is generally not worth the loss of religated DNA that occurs during purification.
After recovery, pellet the cells at 10,000 × g for 1 minute. Resuspend the pellet in 50 μL of LB media.
Streak the resuspended cells on an LB agar plate with 1X chloramphenicol. Incubate the plate at 30 °C overnight for ~16 hours or until colonies appear.
If the ligase plate has more colonies than the no-ligase control plate, select several colonies and check if they are monomeric or heteromultimeric, as described in Basic Protocol 1, steps 34–52. If there is no difference between the ligase plate and the no-ligase control plate, redo the protocol.
There is no easy way to distinguish homomultimers from monomers. Therefore, when attempting to remonomerize a suspected homomultimer, select a few colonies from the ligase plate and reattempt recombineering with these colonies in parallel. Remonomerization can be challenging for large BACs >10 kb. Homomultimers can be an issue particularly when making a series of modifications on the same target, and oftentimes it is easier to return to previous recombineering steps and obtain a new target BAC that is monomeric, rather than trying to remonomerize a homomultimer that has formed in the middle of a series of modifications.
REAGENTS AND SOLUTIONS
Lysogeny broth media
Dissolve 25 g of Dehydrated LB Broth, Miller (e.g., Difco 244620) in 1000 mL of milli-Q water and sterilize by autoclaving. Store at room temperature. Lysogeny broth is sometimes referred to as “Luria-Bertani broth”, “Luria broth”, or just “LB media.”
1000X streptomycin stock solution
Dissolve 1 g of streptomycin sulfate (e.g., MP Biomedicals 100556) in 10 mL of molecular biology-grade water (e.g., Corning 46–000-CM). Sterilize using a 10 mL syringe (e.g., Becton Dickinson 309604) to pass the solution through a 0.2 μm cellulose acetate syringe filter (e.g., VWR 28145–477) into a sterile 15 mL conical tube (e.g., Cellstar 188271). Store at –20 °C.
1000X chloramphenicol stock solution
Dissolve 250 mg of chloramphenicol (e.g., Sigma-Aldrich C0378) in 10 mL of 100% ethanol (e.g., Koptec V1001) in a sterile 15 mL conical tube (e.g., Cellstar 188271). Store at –20 °C.
1000X tetracycline stock solution
Dissolve 100 mg of tetracycline hydrochloride (e.g., Calbiochem 58346) in molecular biology-grade water (e.g., Corning 46–000-CM). Sterilize using a 10 mL syringe (e.g., Becton Dickinson 309604) to pass the solution through a 0.2 μm cellulose acetate syringe filter (e.g., VWR 28145–477) into a sterile 15 mL conical tube (e.g., Cellstar 188271). Protect from light by wrapping the tube in aluminum foil. Store at −20 °C.
1000X kanamycin stock solution
Dissolve 500 mg of kanamycin monosulfate (e.g., Alfa Aesar J61272) in 10 mL of molecular biology-grade water (e.g., Corning 46–000-CM). Sterilize using a 10 mL syringe (e.g., Becton Dickinson 309604) to pass the solution through a 0.2 μm cellulose acetate syringe filter (e.g., VWR 28145–477) into a sterile 15 mL conical tube (e.g., Cellstar 188271). Store at –20 °C.
10% arabinose stock solution
Dissolve 1 g of L-arabinose (e.g., Chem-Impex 01654) in 10 mL of molecular biology-grade water (e.g., Corning 46–000-CM). Sterilize using a 10 mL syringe (e.g., Becton Dickinson 309604) to pass the solution through a 0.2 μm cellulose acetate syringe filter (e.g., VWR 28145–477) into a sterile 15 mL conical tube (e.g., Cellstar 188271). Store at –20 °C.
10% rhamnose stock solution
Dissolve 1 g of L-rhamnose (e.g., MP Biomedicals 102809) in 10 mL of molecular biology-grade water (e.g., Corning 46–000-CM). Sterilize using a 10 mL syringe (e.g., Becton Dickinson 309604) to pass the solution through a 0.2 μm cellulose acetate syringe filter (e.g., VWR 28145–477) into a sterile 15 mL conical tube (e.g., Cellstar 188271). Store at –20 °C.
Super optimal broth with catabolic repression media
Dissolve 3.2 g of Dehydrated SOC broth (e.g., Teknova S0225) in 100 mL of milli-Q water and sterilize by autoclaving. Store at room temperature.
LB agar plates
Dissolve 25 g Dehydrated LB Broth, Miller (e.g., Difco 244620) in 1000 mL of milli-Q water. Next, add 15 g of agar (e.g., Sigma A1296), which will not dissolve until melted in the autoclave. Sterilize by autoclaving, then retrieve the bottle shortly after the autoclave cycle is finished and before the agar solidifies. Allow the bottle to cool with occasional swirling until it is still hot but able to be held by hand for ~15 seconds (~50 °C). Add 1 mL of 1000X antibiotic stock solution(s) and/or 20 mL of 10% arabinose stock solution. Mix the contents by vigorously swirling the bottle, then quickly distribute 15 mL of the molten agarose solution into each 10 cm petri dish (e.g., VWR 25384–094) using a 50 mL serological pipette (e.g., Cellstar 768180) and pipette gun (e.g., Drummond Portable Pipet-Aid XP 4–000-101). Perform the distribution near the base of a Bunsen burner for sterility. Be careful not to introduce bubbles into the plates, as bubbles will make it challenging to reliably identify small bacterial colonies. Allow the plates to solidify at room temperature for ~30 minutes, then return them to the original plastic sleeve and store at 4 °C. Wrap the sleeves of plates in aluminum foil or store them in a dark place if the antibiotic(s) added are light-sensitive.
0.2X elution buffer
Add 2 mL of elution buffer (e.g., Omega Bio-tek PD089; excess buffer is often included with kits) to 8 mL of molecular biology-grade water (e.g., Corning 46–000-CM) in a sterile 15 mL conical tube (e.g., Cellstar 188271). Store at room temperature.
Sterile 50% glycerol
Mix 25 mL of molecular biology-grade water (e.g., Corning 46–000-CM) with 25 mL of glycerol (e.g., Sigma-Aldrich G7893) in a 50 mL conical tube (e.g., Cellstar 227261). Sterilize by autoclaving. Store at room temperature.
3 M sodium acetate
Dissolve 2.5 g of sodium acetate (e.g., Macron 7372) in 10 mL of molecular biology-grade water (e.g., Corning 46–000-CM).
0.5X TBE buffer
Dilute 200 mL of 5X TBE buffer (e.g., Biotium 41006) into 900 mL of milli-Q water. Store at room temperature.
COMMENTARY
Background Information
The lambda phage red recombination pathway was first characterized using various recombination deficient lambda phage mutants—hence red αβγ (Signer, 1971). To briefly summarize the proposed functions of these genes during recombineering (Murphy, 2016):
Red α encodes a 5′→3′ exonuclease, commonly referred to as exo or lambda exonuclease, that degrades one strand of DNA and exposes regions of the formerly double-stranded targeting cassette as single-stranded DNA.
Red β encodes an annealase—commonly referred to as beta, bet, or β—that coats single-stranded DNA in the targeting cassette and anneals it with homologous single-stranded DNA that is exposed in the target DNA, usually in Okazaki gaps formed during target DNA replication.
The γ gene encodes a protein, commonly referred to as gam or gamma, that inhibits the E. coli recBCD exonuclease so that it does not destroy both strands of the linear targeting cassette.
While it was known since the 1960s that these genes could mediate general lambda phage recombination, their ability to promote efficient gene replacement using linear PCR cassettes in E. coli was only demonstrated decades later (Murphy, 1998). Shortly thereafter, it was shown that homology arms as short as 42 bp could be used to replace genes using the recET recombination system (Zhang, Buchholz, Muyrers, & Stewart, 1998), which functions similarly to the lambda red system. It was later shown that the more efficient lambda red system could also use short homology arms (Muyrers, 1999). This development was followed by the rapid publication of several related recombinogenic engineering or “recombineering” techniques (Copeland, Jenkins, & Court, 2001; Datsenko & Wanner, 2000; Yu et al., 2000). Because the lambda red and recET systems can utilize such short homology arms, targeting cassettes can be generated via PCR using 5′ primer overhangs, rather than having to laboriously clone the large, 500–1000 bp homology arms that are necessary for other homologous recombination methods in E. coli (Hamilton et al., 1989; Kong et al., 1999; Winans et al., 1985). Since the inception of recombineering, many improvements and variations of the method have been developed, including the incorporation of new counterselection schemes, such as ccdA/ccdB (H. Wang et al., 2014), and the development of even faster recombineering methods, such as DIRex (Näsvall, 2017). For a more comprehensive review of the history, proposed mechanisms, and variations of recombineering, see (Mosberg, Lajoie, & Church, 2010; Murphy, 2016; Poteete, 2008). The protocols described here are based on reports of others (Datsenko & Wanner, 2000; Näsvall, 2017; Sawitzke et al., 2013; Thomason et al., 2014; H. Wang et al., 2014; Warming et al., 2005), but with modifications that we have found improve the efficiency of the technique and with detailed commentary to assist a novice lab (such as our own when we first began using these approaches) in successfully choosing and applying recombineering methods.
Critical Parameters
While recombineering is quite robust and reliable when performed correctly, there are numerous pitfalls that can hinder success for a new user. In particular, attention must be paid to the quality of targeting cassette DNA, the formation of multimeric plasmids, and the architecture of the target. Many of these parameters are directly addressed in the annotations below the protocols steps.
Targeting cassette DNA quality
If the targeting cassette DNA concentration is too low, it will be difficult to transform sufficient DNA to attain efficient recombineering (Yu et al., 2000). The DNA must also be pure and desalted. Otherwise, the sample can conduct significant current or arc during electroporation and kill the bacterial cells. For DNA cassettes larger than ~2 kb, it is important to remove unwanted DNA truncation products via gel extraction. Shorter truncation products will undergo recombination at a much higher rate than the large desired cassette (Kuhlman & Cox, 2010). Finally, it is essential for the purified targeting cassette DNA to be free of any template plasmid used during the PCR that could cause false positives upon transformation.
Multimerization
Multimers can easily form during recombineering (Thomason et al., 2007), which can then hinder future recombineering steps or applications. One-step recombineering (Basic Protocol 1) does not select against heteromultimers. It is therefore important to verify whether or not the final recombinant is heteromultimeric. In Basic Protocols 2 and 3, heteromultimeric kanR+ccdB intermediates can form that will generate many false positives during counterselection via intramolecular recombination (Fig. 8). In general, the time and effort required to screen for monomeric intermediates are not justified, as it is easier to simply proceed with three or more colonies with the expectation that at least one will yield successful recombinants. However, if only heteromultimeric kanR+ccdB intermediates form, the parent target BAC is likely a homomultimer (Fig. 9). The homomultimer will either need to be remonomerized (Support Protocol 4), or replaced with a different monomeric clone. It is often easier to find other monomeric clones of the parent when possible, such as when the parent BAC is an intermediate in a series of modifications to be made, than it is to attempt in vitro remonomerization (Support Protocol 4).
Target sequence and repetition
It is critical to have an accurate sequence of the targeting cassette and target BAC/genome so that correct primers and homology arms can be designed. An accurate and complete sequence is also important for identifying any repeats or repetitive regions that can interfere with recombineering in the following ways:
-
1)
If one or both of the targeting cassette homology arms is targeted to a region that is repeated in the target, such as commonly used promoters, terminators or polyadenylation signals, a mixture of products can result from the cassette landing in different iterations of the repeat or spanning and thus deleting repeats.
-
2)
Repetition in the target can lead to intramolecular recombination that eliminates the ccdB gene without undergoing the desired recombination (Bird et al., 2011).
-
3)
Unintentional homology between the middle of the targeting cassette and the target can cause recombination with only a fragment of the targeting cassette, especially because shorter cassettes recombine more efficiently (Kuhlman & Cox, 2010; Lim, Min, & Jung, 2008).
-
4)
If there are repeated regions within the targeting cassette itself, these regions can undergo recombination and cause deletions or duplications before the cassette is incorporated into the target.
When there is a choice, avoid targeting cassettes with repetition or targeting repetitive regions. Syntenic dot-plots map the regions of homology between two sequences in a 2D plot, and are a useful approach to easily identify repetition and homology (Noé & Kucherov, 2005). To search for problematic repetition, visit https://bioinfo.lifl.fr/yass/index.php (use default settings except E-value threshold = 0.1, window range = 10 to 20000 bp, window incr = 0) to create and examine syntenic dot-plots of:
-
1)
The target versus itself: Avoid choosing homology arms within or between repeats. Altenatively, engineer each repeat on separate BACs and assemble the BACs after editing using restriction endonuclease cloning.
-
2)
The targeting cassette versus itself: Avoid introducing repetitive elements with the targeting cassette.
-
3)
The proposed targeting cassette versus the target: If there is unintended homology, attempt to eliminate or reduce it from the proposed targeting cassette.
When problematic repetition is unavoidable, many colonies will need to be screened during recombineering. Alternatively, traditional methods such as restriction endonuclease cloning will need to be used. Reducing the induction time of the recombineering enzymes has also been shown to reduce unwanted recombination between repeats (Narayanan, 2008).
Troubleshooting
For a summary of troubleshooting tips from throughout this article, see Table 3.
Table 3.
Troubleshooting table
| Problem | Potential Cause | Diagnosis | Solution |
|---|---|---|---|
| Too many colonies on the
plate and/or Recombineering yields unmodified target |
Targeting cassette contaminated with template plasmid | Perform colony PCR with template-specific primers or examine restriction endonuclease fragmentation patterns for template-specific patterns. | Digest the targeting cassette with template-specific endonucleases and gel extract the cassette; or switch to a template with an R6K origin. |
| Heteromultimeric kanR+ccdB intermediate | Examine restriction endonuclease pattern for overlap between intermediate and parent. | Pick a new kanR+ccdB intermediate or remonomerize using Support Protocol 4. | |
| Homomultimeric target | All resulting kanR+ccdB intermediates are heteromultimeric. | Pick a new target BAC or remonomerize using Support Protocol 4. | |
| No antibiotic to maintain target BAC | Examine restriction endonuclease pattern for presence of BAC or attempt to grow in LB + 1X chloramphenicol. | Remake agar plates and ensure that chloramphenicol is present. | |
| Recombination between repeats in target excise kanR+ccdB intermediate | Examine syntenic dot-plot of target vs target for repeats of the target regions or repeats that flank the target region. | If possible, move the region to be targeted or delete all repeats except one. Otherwise, vary induction times, increase scale, and screen many colonies. | |
| Final recombinants contain truncated targeting cassettes | Targeting cassette contaminated with PCR truncation products | Run PCR product on an agarose gel and look for faint bands or smears. They may not be visible. | Gel extract the PCR product and/or optimize the PCR conditions. |
| Targeting cassette contains repeats | Examine syntenic dot-plot of cassette vs cassette for repeats within the cassette. | If possible, eliminate the repeats from the cassette Otherwise, vary induction times, increase scale, and screen many colonies. | |
| Targeting cassette contains unintended internal homology | Examine syntenic dot-plot of cassette vs target for homologies other than the homology arms. | If possible, eliminate the internal homology from the cassette. Otherwise, vary induction times, increase scale, and screen many colonies. | |
| No colonies
obtained and/or Counterselection only yields kanR+ccdB with inactivated ccdB |
Too few cells plated or inefficient recombineering | Culture not cloudy or pellet very small before streaking on plate or sequencing reveals a mutated ccdB gene. | Redo the recombineering on a larger scale with more concentrated DNA and/or increase homology arm length to 1 kb to increase efficiency (Yu et al., 2000). |
| Wrong antibiotic or concentration | Double-check resistance cassette. Attempt streaking on a gradient of plates with different antibiotic concentrations. | Remake plates with a lower, but still selective, antibiotic concentration, or use the correct antibiotic. | |
| Incorrect homology arms | Double-check the homology arm sequences and whether the correct strand—top or bottom—was used. Sanger sequence the target region to ensure sequence file is correct. | Reorder targeting primers with correct homology arms. Ensure that the sequence file for the target is correct via sequencing—sequence files are sometimes inaccurate. |
Understanding Results
With the protocols described here, one can expect a successful recombination frequency of roughly 1 in 10,000–100,000 cells (Datta et al., 2008). Colonies that appear after counterselection are often either successful recombinants or false positives with inactivated ccdB, with the ratio depending on the efficiency of recombineering. With less efficient large cassettes, a larger proportion of colonies will be false positives with inactivated ccdB. We have observed that, with large cassettes, a successful recombinant can generally be identified after screening 8–24 colonies. When recombineering, one can expect to observe dozens to ~100 colonies after selection or after counterselection. Note that it is normal to observe a thin lawn of bacteria around the colonies when higher concentrations of cells are spread on the plate, which is not an issue. If there are hundreds to thousands of colonies on the plate after selection or counterselection, troubleshooting as described in Table 3 is likely required.
Time Considerations
A few days are generally required to plan and prepare for a modification or a series of modifications. Large primers greater than 50 bp sometimes require extra time to manufacture, so they should be ordered well ahead of time. Once a target BAC or target strain is acquired, the targeting cassettes are synthesized, and the psc101-gbaA plasmid and target BAC are transformed together into DH10B cells, recombineering can be performed on a predictable time table. From growing up a colony or glycerol stock of a target BAC+psc101-gbaA strain to identifying successful recombinants on a plate, each iteration of Basic Protocol 1 requires ~3 days excluding any time required for remonomerization, Basic Protocol 2 requires 4 days, and Basic Protocol 3 requires 5 days. Most days involve just a few minutes to inoculate cultures or streak colonies, or ~30 minutes to set up a colony PCR screen that requires 1–2 hours to run in the thermal cycler. However, days that involve electroporation will require one to dedicate 7–8 hours and to adhere to a strict time table, although there will be several growth periods within that time that require less researcher attention.
Significance Statement.
Recombineering is a valuable technique for editing DNA plasmids when traditional methods, such as restriction endonuclease cloning or Gibson assembly, are not feasible or provide inadequate precision owing to target DNA size or the lack of unique restriction endonuclease sites. Recombineering is also the premier technique for editing the Escherichia coli genome. Instead of cutting DNA or assembling fragments of DNA in vitro, recombineering uses homologous recombination to replace DNA segments with PCR products in vivo. The technique can enable facile engineering of large DNA or even intact genomes. It can also be a complex and challenging technique to learn, with many pitfalls. Here, we provide simple guidelines for users choosing from the vast menu of recombineering options and introduce the experimental method via three detailed, easy-to-follow protocols.
ACKNOWLEDGEMENT
The authors acknowledge funding from the NIH Director’s New Innovator Award Grant 1DP2GM119162 and NIAMS Grant R01AR071443 (to M.D.S.) and the National Science Foundation Graduate Research Fellowships under Grant No. 1122374 (to L.J.P.).
Footnotes
INTERNET RESOURCES
http://www.biotec.tu-dresden.de/research/stewart/group-page.html
The Sewart Lab website. Source of the psc101-gbaA plasmid and a rich resource for more tips on recombineering and other variations of the recombineering protocol.
https://redrecombineering.ncifcrf.gov/court-lab.html
The Court Lab website. A rich resource for more tips on recombineering and other variations of the recombineering protocol.
https://bioinfo.lifl.fr/yass/yass.php
YASS. A user-friendly online tool for generating and examining syntenic dot-plots. These plots are useful for identifying regions of homology between two sequences in order to spot problematic repeats or unintended homologies.
http://jorgensen.biology.utah.edu/wayned/ape/
The website for downloading “A plasmid Editor” or “ApE”, a free DNA editing software.
https://tmcalculator.neb.com/#!/main
New England Biolabs Tm calculator. An online tool from New England Biolabs for calculating primer annealing temperatures for New England Biolabs PCR products.
Multiple Primer Analyzer. An online tool from Thermo Fisher Scientific for predicting the formation of primer dimers, which can inhibit PCR reactions. For difficult PCRs, primers should be redesigned when possible to prevent primer dimer formation.
LITERATURE CITED
- Åkerlund T, Nordström K, & Bernander R (1995). Analysis of cell size and DNA content in exponentially growing and stationary-phase batch cultures of Escherichia coli. J. Bacteriol, 177(23), 6791–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker O, Tsurkan S, Fu J, Klink B, Rump A, Obst M, … Stewart AF (2017). The contribution of homology arms to nuclease-assisted genome engineering. Nucleic Acids Res, 45(13), 8105–8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman C, Papa LJ III, Hendel SJ, Moore CL, Suen PH, Weickhardt AF, … Shoulders MD (2018). An adaptable platform for directed evolution in human cells. J. Am. Chem. Soc, 140, 18093–18103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigot S, Sivanathan V, Possoz C, Barre FX, & Cornet F (2007). FtsK, a literate chromosome segregation machine. Mol. Microbiol, 64(6), 1434–1441. [DOI] [PubMed] [Google Scholar]
- Bird AW, Erler A, Fu J, Hériché JK, Maresca M, Zhang Y, … Stewart AF (2011). High-efficiency counterselection recombineering for site-directed mutagenesis in bacterial artificial chromosomes. Nat. Methods, 9(1), 103–109. [DOI] [PubMed] [Google Scholar]
- Bzymek M, & Lovett ST (2001). Evidence for two mechanisms of palindrome-stimulated deletion in Escherichia coli: single-strand annealing and replication slipped mispairing. Genetics, 158(2), 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvin NM, & Hanawalt PC (1988). High-efficiency transformation of bacterial cells by electroporation. J. Bacteriol, 170(6), 2796–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Fockler C, Barnes WM, & Higuchi R (1994). Effective amplification of long targets from cloned inserts and human genomic DNA. Proc. Natl. Acad. Sci. U.S.A, 91(12), 5695–5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland NG, Jenkins NA, & Court DL (2001). Recombineering: a powerful new tool for mouse functional genomics. Nat. Rev. Genet, 2(10), 769–779. [DOI] [PubMed] [Google Scholar]
- Coren JS (2017). Retrofitting the BAC cloning vector pBeloBAC11 by the insertion of a mutant loxP site. BMC Res Notes, 10(1), 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, & Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A, 97(12), 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Costantino N, Zhou X, & Court DL (2008). Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc. Natl. Acad. Sci. U.S.A, 105(5), 1626–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dower WJ, Miller JF, & Ragsdale CW (1988). High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res, 16(13), 6127–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton CM, Aldea M, Washburn BK, Babitzke P, & Kushner SR (1989). New method for generating deletions and gene replacements in Escherichia coli. J. Bacteriol, 171(9), 4617–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P, Rivera-Pérez J, & Bradley A (1991). The length of homology required for gene targeting in embryonic stem cells. Mol. Cell Biol, 11(11), 5586–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Nojima H, & Okayama H (1990). High efficiency transformation of Escherichia coli with plasmids. Gene, 96(1), 23–28. [DOI] [PubMed] [Google Scholar]
- Jaffé A, Ogura T, & Hiraga S (1985). Effects of the ccd function of the F plasmid on bacterial growth. J. Bacteriol, 163(3), 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter R, Inuzuka M, & Helinski DR (1978). Trans-complementation-dependent replication of a low molecular weight origin fragment from plasmid R6K. Cell, 15(4), 1199–1208. [DOI] [PubMed] [Google Scholar]
- Kong Y, Yang T, & Geller AI (1999). An efficient in vivo recombination cloning procedure for modifying and combining HSV-1 cosmids. J. Virol. Methods, 80(2), 129–136. [DOI] [PubMed] [Google Scholar]
- Kuhlman TE, & Cox EC (2010). Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res, 38(6), e92.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SI, Min BE, & Jung GY (2008). Lagging strand-biased initiation of red recombination by linear double-stranded DNAs. J. Mol. Biol, 384(5), 1098–1105. [DOI] [PubMed] [Google Scholar]
- Medaney F, Dimitriu T, Ellis RJ, & Raymond B (2016). Live to cheat another day: bacterial dormancy facilitates the social exploitation of beta-lactamases. ISME J, 10(3), 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker ML (2005). Emerging technologies in DNA sequencing. Genome Res, 15(12), 1767–1776. [DOI] [PubMed] [Google Scholar]
- Moore CL, Papa LJ III, & Shoulders MD (2018). A processive protein chimera introduces mutations across defined DNA regions in vivo. J. Am. Chem. Soc, 140(37), 11560–11564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosberg JA, Lajoie MJ, & Church GM (2010). Lambda red recombineering in Escherichia coli occurs through a fully single-stranded intermediate. Genetics, 186(3), 791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KC (1998). Use of bacteriophage λ recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol, 180(8), 2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KC (2016). λ recombination and recombineering. EcoSal Plus, 7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyrers J (1999). Rapid modification of bacterial artificial chromosomes by ET- recombination. Nucleic Acids Res, 27(6), 1555–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K (2008). Intact recombineering of highly repetitive DNA requires reduced induction of recombination enzymes and improved host viability. Anal. Biochem, 375(2), 394–396. [DOI] [PubMed] [Google Scholar]
- Narayanan K, & Chen Q (2011). Bacterial artificial chromosome mutagenesis using recombineering. J. Biomed. Biotechnol, 971296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näsvall J (2017). Direct and Inverted Repeat stimulated excision (DIRex): Simple, single-step, and scar-free mutagenesis of bacterial genes. PLoS One, 12(8), e0184126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noé L, & Kucherov G (2005). YASS: enhancing the sensitivity of DNA similarity search. Nucleic Acids Res, 33, W540–W543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteete AR (2008). Involvement of DNA replication in phage lambda Red-mediated homologous recombination. Mol. Microbiol, 68(1), 66–74. [DOI] [PubMed] [Google Scholar]
- Sawitzke JA, Thomason LC, Bubunenko M, Li X, Costantino N, & Court DL (2013). Recombineering: using drug cassettes to knock out genes in vivo. Methods Enzymol, 533, 79–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer E (1971). Chapter 7 General Recombination In Hershey AD (Ed.), The Bacteriophage Lambda (pp. 139–174). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Siguret V, Ribba AS, Chérel G, Meyer D, & Piétu G (1994). Effect of plasmid size on transformation efficiency by electroporation of Escherichia coli DH5α. BioTechniques, 16(3), 422–426. [PubMed] [Google Scholar]
- Thomason LC, Costantino N, Shaw DV, & Court DL (2007). Multicopy plasmid modification with phage λ Red recombineering. Plasmid, 58(2), 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason LC, Sawitzke JA, Li X, Costantino N, & Court DL (2014). Recombineering: genetic engineering in bacteria using homologous recombination. Curr. Protoc. Mol. Biol, 106, 1.16.11–1.16.39. [DOI] [PubMed] [Google Scholar]
- Uil TG, Vellinga J, de Vrij J, van den Hengel SK, Rabelink MJWE, Cramer SJ, … Hoeben RC (2011). Directed adenovirus evolution using engineered mutator viral polymerases. Nucleic Acids Res, 39(5), e30.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bian X, Xia L, Ding X, Müller R, Zhang Y, … Stewart AF (2014). Improved seamless mutagenesis by recombineering using ccdB for counterselection. Nucleic Acids Res, 42(5), e37.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Sarov M, Rientjes J, Fu J, Hollak H, Kranz H, … Zhang Y (2006). An improved recombineering approach by adding recA to λ red recombination. Mol. Biotechnol, 32(1), 43–54. [DOI] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, & Copeland NG (2005). Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res, 33(4), e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenberg M, Bamps S, Soedling H, Hope IA, & Dolphin CT (2010). Escherichia coli MW005: lambda Red-mediated recombineering and copy-number induction of oriV-equipped constructs in a single host. BMC Biotechnol, 10, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild J, Hradecna Z, & Szybalski W (2002). Conditionally amplifiable BACs: switching from single-copy to high-copy vectors and genomic clones. Genome Res, 12(9), 1434–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winans SC, Elledge SJ, Krueger JH, & Walker GC (1985). Site-directed insertion and deletion mutagenesis with cloned fragments in Escherichia coli. J. Bacteriol, 161(3), 1219–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MY, Doan ND, DiChiara AS, Papa LJ III, Cheah JH, Soule CK, … Shoulders MD (2018). A high-throughput assay for collagen secretion suggests an unanticipated role for hsp90 in collagen production. Biochemistry, 57(19), 2814–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, & Court DL (2000). An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A, 97(11), 5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Buchholz F, Muyrers JP, & Stewart AF (1998). A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet, 20(2), 123–128. [DOI] [PubMed] [Google Scholar]
KEY REFERENCES
- Murphy KC (2016). λ recombination and recombineering. EcoSal Plus, 7(1).An excellent, extensive review that provides in-depth histories, proposed mechanisms and variations of recombineering. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, & Copeland NG (2005). Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res, 33(4), e36..Describes doing recombineering on a 25 mL scale. Also describes a highly popular recombineering method that uses galK selection and counterselection and a defective lambda prophage to induce the red system rather than a psc101 plasmid. Alternative Protocol 1 adapts this protocol to kanamycin selection and ccdB counterselection using the psc101-gbaA plasmid to induce the red system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näsvall J (2017). Direct and Inverted Repeat stimulated excision (DIRex): Simple, single-step, and scar-free mutagenesis of bacterial genes. PLoS One, 12(8), e0184126.Describes the development of DIRex recombineering and how to design DIRex primers in-depth. This article uses chloramphenicol selection and sacB counterselection rather than kanamycin selection and ccdB counterselection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bian X, Xia L, Ding X, Müller R, Zhang Y, Fu J, Stewart AF (2014). Improved seamless mutagenesis by recombineering using ccdB for counterselection. Nucleic Acids Res, 42(5), e37..This article describes the development of recombineering with ccdB counterselection and performing recombineering on a 1 mL scale. [DOI] [PMC free article] [PubMed] [Google Scholar]