

Abstract
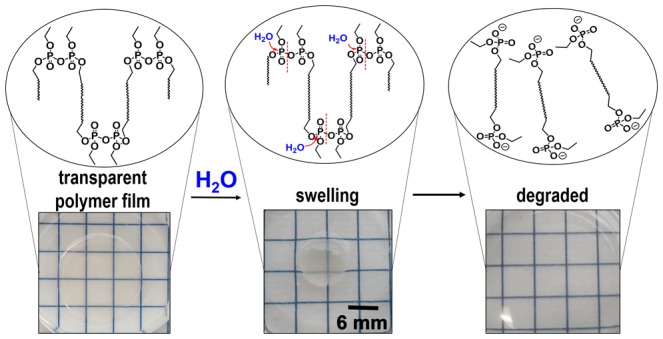
Biodegradable polyethylene mimics have been synthesized by the introduction of pyrophosphate groups into the polymer backbone, allowing not only hydrolysis of the backbone but also further degradation by microorganisms. Because of cost, low weight, and good mechanical properties, the use of polyolefins has increased significantly in the past decades and has created many challenges in terms of disposal and their environmental impact. The durability and resistance to degradation make polyethylene difficult or impossible for nature to assimilate, thus making the degradability of polyolefins an essential topic of research. The biodegradable polypyrophosphate was prepared via acyclic diene metathesis polymerization of a diene monomer. The monomer is accessible via a three-step synthesis, in which the pyrophosphate was formed in the last step by DCC coupling of two phosphoric acid derivatives. This is the first report of a pyrophosphate group localized in an organic polymer backbone. The polypyrophosphate was characterized in detail by NMR spectroscopy, size exclusion chromatography, FTIR spectroscopy, differential scanning calorimetry, and thermogravimetry. X-ray diffraction was used to compare the crystallization structure in comparison to analogous polyphosphates showing poly(ethylene)-like structures. In spite of their hydrophobicity and water insolubility, the pyrophosphate groups exhibited fast hydrolysis, resulting in polymer degradation when films were immersed in water. Additionally, the hydrolyzed fragments were further biodegraded by microorganisms, rendering these PE mimics potential candidates for fast release of hydrophobic cargo, for example, in drug delivery applications.
Introduction
Polyethylene (PE) is the most produced commodity polymer today.1 With the invention of Karl Ziegler in the past century,2 perfectly linear PE was accessible that changed our everyday life drastically.3 PE and other polyolefins are resistant to most chemicals and environmental conditions; they do not degrade hydrolytically or by enzymes.4 The durability of PE is an attractive feature and has driven its success not only in lightweight materials and packaging but also in biomedical applications, for example, for long-term implants.5 However, if littered in nature, PE plastic parts do not degrade over a period of decades to centuries.4
Biodegradable alternatives to commodity polymers are discussed today heavily for packaging applications.6 In contrast, in the biomedical field, degradable polymers have found already several applications if degradation of the device is desired, for example, for sustained drug release,7 temporary prostheses,8 or tissue engineering.9 For such uses, polyesters, polyamides, or polysaccharide derivatives and others have been applied, which can be degraded by different enzymes in vivo.10 Furthermore, the biocompatibility not only of the polymer but also of its degradation products are crucial for the use in the biomedical field.11
Combining properties of PE, such as crystallinity and thermoplasticity, with controlled degradation would be a desirable platform for biomedicine and the plastic sector. Others and we have prepared potentially degradable PE mimics, which are polymers based on long aliphatic alkyl chains, which are spaced by heteroatom linkages that may allow degradation into smaller fragments.12 For full biomineralization, these smaller fragments should eventually be resorbed by the body or degraded by microorganisms.13,14 Such potentially degradable groups, esters, and acetals, for example, have been installed into PE mimics already,15,16 but polymers do not or only very slowly degrade.16 Recently, we have prepared PE mimics with phosphoesters/amide linkages between the aliphatic chains, since their degradation rate might be adjusted by variation of the binding motif.17 Polyphosphorodiamidates were found to be much more hydrolysis-labile than polyphosphoesters.18 However, in such nonpolar PE mimics, degradation under physiological or environmental conditions is very slow, rendering them as too stable for certain applications, in which degradation is demanded.19 Recently, Mecking and co-workers synthesized long-chain polyphosphoesters by polyesterification and reported that a low number of pyrophosphate linkages were formed during the process. These bonds—undesired in polyphosphoesters (PPEs)—were however responsible for a partial degradation of the materials at mild hydrolysis conditions. Upon immersing polymer films in basic or acidic conditions, cleavage occurred selectively in anhydride groups, and the phosphoester bonds remained intact during the course of their study.19 To date, no polypyrophosphates have been reported as degradable mimics for PE with a potentially very fast hydrolysis.
Herein, we present a biodegradable aliphatic polypyrophosphate PE mimic. The hydrophobic and semicrystalline PE-like material exhibited fast hydrolysis rates under mild conditions. Additionally, we proved that microorganisms occurring in activated sludge from the sewage plant further degraded the degradation products after hydrolysis. The readily cleavable pyrophosphate group was introduced to the monomer, which was polymerized by acyclic diene metathesis polycondensation without cleaving of the pyrophosphate bond. Hydrogenation of the unsaturated product from the ADMET polycondensation was performed without any further catalyst addition by a modified Grubbs catalyst to produce the saturated PE mimic. This is the first example of a PE mimic with very fast hydrolysis conditions of polymer films. The degradation products are soluble in neutral and basic aqueous solution and undergo further microbial degradation, which renders polypyrophosphates an interesting class of polymers for degradation on demand in biomedical or other packaging applications where fast dissolution of the matrix is desired.
Results and Discussion
Monomer Synthesis
We prepared a novel pyrophosphate monomer (1) for the ADMET polycondensation via a three-step synthesis (Scheme 1). The first step was the esterification of ethyl dichlorophosphate with 1 equiv of 10-undecen-1-ol in the presence of triethylamine (Et3N) to produce ethylundec-10-en-1-yl phosphorochloridate (1a). The resulting 1a was then hydrolyzed to the free phosphoric acid derivative (1b). The A2-type pyrophosphate monomer was obtained by coupling of two molecules of 1b with dicyclohexylcarbodiimide similar to a literature protocol20 to give 1 as an off-white oil (Scheme 1).
Scheme 1. Synthesis of Monomer 1 and Its Subsequent Polymerization by Acyclic Diene Metathesis (to P1) and Hydrogenation (to P1-H).

The monomer was obtained in high purity after column chromatography. The 1H NMR spectrum of 1 shows at 5.7 and 4.9 ppm the signals for the terminal double bond and at 4.2 ppm the protons next to the pyrophosphate. The upfield signals at 2.0, 1.6, and 1.3 ppm result from the protons of the β-carbon of the pyrophosphate, the protons adjacent to the double bond, and the remaining protons of the alkyl groups (Figure 1a). 31P NMR spectroscopy proved the formation of the pyrophosphate as a single resonance at −12.95 ppm, which is a typical chemical shift for aliphatic pyrophosphates (Figure 1a).21 For comparison, we prepared the phosphate analogue bis(undec-10-en-1-yl) ethyl phosphate (2) and polymerized it according to the literature.22
Figure 1.

Characterization data of pyrophosphate monomer and polymers. (a) 1H and 31P (inset) NMR spectra of 1, P1, and P1-H with peak assignments (measured in CDCl3 or CD2Cl2, 300 MHz, 298 K). (b) IR spectra of 1 and P1-H. (c) GPC elugrams of P1 and P1-H in THF (RI detection).
ADMET Polymerization
During the past years, acyclic diene metathesis (ADMET) was established as a reliable method for the synthesis of PPEs, allowing the adjustment of main and side chains with the high functional group tolerance of olefin metathesis with modern Grubbs-type catalysts.23,24 We polymerized 1 and 2 via ADMET polycondensation with Grubbs catalyst first to the respective unsaturated polymers (P1 and P2; characterization data of P2 can be found in the Supporting Information). After polymerization, the addition of ethyl vinyl ether did not only terminate the reaction but also generated an active Ru catalyst for hydrogenation.25 The hydrogenation was performed without any further addition of catalyst at a hydrogen pressure of 70 bar for ca. 24 h to yield the hydrogenated polymers P1-H and P2-H (Scheme 1 and Scheme S2). Successful polymerization was proven by 1H NMR spectroscopy with the formation of the internal double bonds in the backbone at 5.3 ppm and the disappearance of the terminal double bonds at 4.9 and 5.7 ppm (Figure 1a). GPC showed an apparent molecular weight of Mn = 13000 g mol–1 (Mw/Mn = 1.9) for P1. Additionally, the NMR spectra prove that the pyrophosphate group remained untouched during the ADMET procedure and the subsequent hydrogenation: The resonances for the methylene groups next to the pyrophosphate at 4.2 ppm in the 1H NMR spectra remain unchanged. In addition, no change of the 31P NMR resonance at −12.95 ppm after polymerization and hydrogenation was detected (Figure 1a). After hydrogenation, the double-bond resonances disappeared from the 1H NMR spectra, and only the protons around the pyrophosphate at 4.2 and 1.7 ppm and the protons from the alkyl chains at 1.2 ppm remained; GPC detected an apparent molecular weight of Mn = 9000 g mol–1 (Mw/Mn = 1.9) for P1-H.
Solid-State Characterization
The thermal stability of both synthesized polymers was examined by thermal gravimetric analysis (TGA). The polypyrophosphate (P1-H) proved a distinctively lower thermal stability compared to the polyphosphate (P2-H). P1-H showed an onset of degradation at ca. 280 °C, while P2-H started to decompose at ca. 320 °C. The char yields of both polymers are in agreement with the phosphate weight content.
The crystallinity as an important factor for many properties of PE in dependency to the molecular weight has been reported.26,27 The crystallinity of P1-H and P2-H was analyzed by DSC; both polymers are solids at room temperature. P1-H exhibited a melting endotherm at Tm(P1-H) = 38 °C with a melting enthalpy of ΔHm = −67 J g–1, while P2-H showed a distinct higher melting event at Tm of 51 °C with a similar melting enthalpy of ΔHm = −69 J g–1, indicating a similar degree of crystallinity. Compared to 100% crystalline polyethylene with ΔHm = 293 J g–1, the crystallinity of P1-H and P2-H was determined to be 23% and 24%, respectively. However, the lower melting point of P1-H is appointed to the size of the pyrophosphate group compared to the phosphate group, which both act as defects during the crystallization of the polymers. The crystallization behavior of PPEs has already been studied in our group and has been compared to polyethylene.28 PPEs with such a long alkyl chain are considered to have a similar lamellar crystalline structure as polyethylene with a pseudohexagonal crystal structure. In agreement with the results by DSC measurement, the XRD measurement showed a similar degree of crystallinity and a similar crystal structure between both polymers (Figure 2c) and toward PE. In detail, a lattice spacing of 0.43 and 0.41 nm can be found by XRD with the peak positions at 2Θ of 21.32° and 21.70° for P1-H and P2-H, respectively. The appearance of the pseudohexagonal phase indicated that the crystal lattices of P1-H and P2-H are slightly inflated compared to the orthorhombic phase of PE, which exhibits an XRD peak at 2Θ = 20.5°.29
Figure 2.
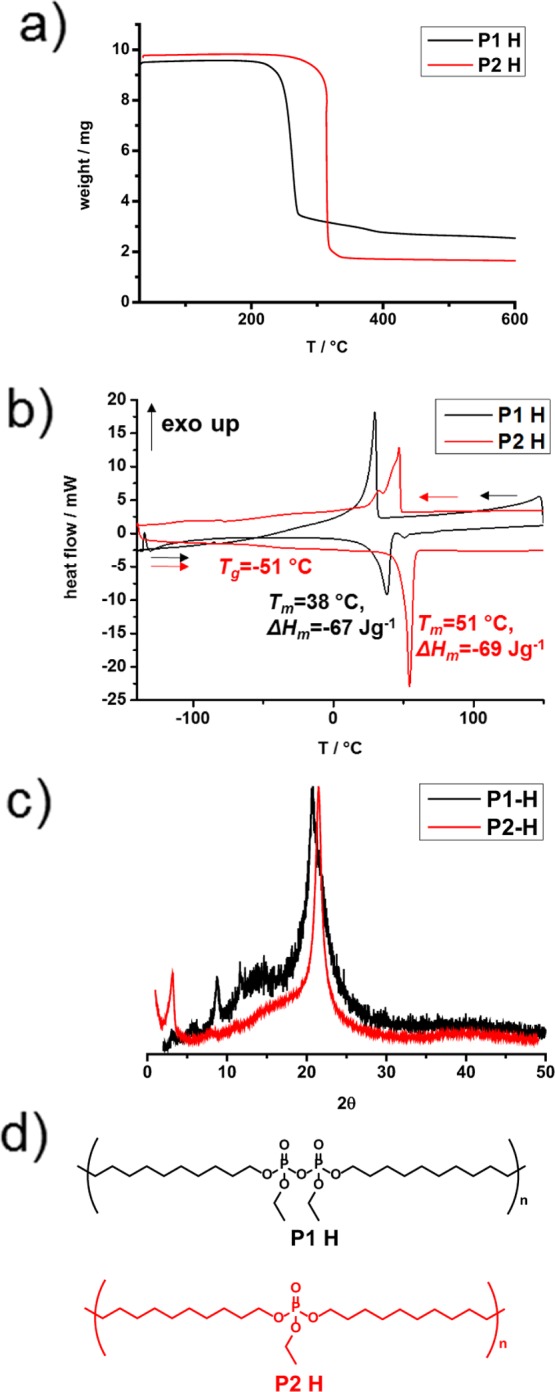
Bulk properties of polypyrophosphates and analogue polyphosphates. (a) TGA of P1-H and P2-H with a heating rate of 10 K min–1. (b) DSC of P1-H and P2-H with a heating (second run) and cooling rate of 10 K min–1. (c) X-ray diffractograms of P1-H and P2-H. (d) Structures of P1-H and P2-H.
Polymer Degradation
PPEs are potential candidates for the development of degradable materials as they can be degraded either hydrolytically or in the presence of enzymes, for example, by phosphodiesterase I.30−33 The mechanism of hydrolysis of PPEs depends on their structure, and either random hydrolysis of the main and side chain esters or a backbiting mechanism was reported.34
To evaluate hydrolysis of the PPPs, we cast polymer films, immersed them into aqueous buffer solutions with different pH values, and analyzed their weight loss. Molecular information during the degradation was collected from the 31P NMR spectra after dissolving the dried films after certain incubation time in CDCl3. The degradation of P1-H and the analogue P2-H was studied at basic, acidic, and neutral conditions at 37 °C (Figure 3). Fastest degradation and dissolution of the P1-H films occurred under basic conditions (pH = 13); a rapid weight loss resulted in the complete dissolution of the film after 2 h (Figure 3c,d). When P1-H films were immersed into a PBS solution at pH 7.0, the polymer films initially swelled, and an increased mass was obtained. After 4 h, however, a rapid weight loss was detected until total dissolution after 7 h. This rapid weight loss might be rationalized with the degradation products dissolving over time, where the swollen polymer film reaches a state not stable enough to keep the integrity of the film, followed by quick disassembly. 31P NMR of the dried films clearly proved the hydrolysis of the pyrophosphate bond during the overall investigation. In addition, the degradation time from the 31P NMR spectra for full hydrolysis of the pyrophosphate bonds was comparable to the full weight loss of the P1-H films. When the films were immersed into an acidic solution (pH = 0), no weight loss of the P1-H films could be measured, and the film became opaque during the treatment (Figure 3b). 31P NMR spectra of the dried polymer films confirmed, however, a decreasing pyrophosphate signal and an increasing phosphoric acid signal over time (Figures S9 and S10). In addition, the degradation rate was slower compared to basic or neutral conditions (Figure 3d). We assume that under acidic conditions the water-insoluble and partially protonated degradation product was formed, which exhibits higher crystallinity and turned the film opaque. DSC measurement from the dried film after degradation confirmed a higher crystallinity compared to the initial casted polymer film (Figure 3c). P1-H films before degradation exhibited several melting events between 35 and 60 °C (and ΔH = −74 J g–1, measured from the first heating run), while the degraded film melted at 102 °C with a higher melting enthalpy of ΔH = 147 J g–1, indicating the formation of crystals of the P1-H degradation products.
Figure 3.

Degradation studies of P1-H. (a) Scheme of P1-H hydrolysis. (b) P1-H film before and after neutral, acidic, and basic hydrolysis. (c) DSC of P1-H film before and after acidic degradation. (d) Mass loss of P1-H films under acidic, basic, and neutral conditions (black) and 31P NMR intensity of pyrophosphate P1-H films under acidic and neutral conditions (blue). (e) Degradation of P1-H in 0.5 mL of THF-d8 with 0.1 mL of D2O and 0.05 mL of TFA or DIPEA. (f) Biodegradation of P1-H, P2-H, and starch in aqueous conditions with microorganisms from activated sludge.
Interestingly, in organic solution, the degradation of P1-H was at least 1 order of magnitude slower than in the films. We dissolved P1-H in 0.5 mL/0.1 mL THF-d8/D2O and added 0.05 mL of trifluoroacetic acid or diisopropylethylamine as a base (Figure 3e). Degradation was studied over a period of several months. Under acidic conditions, P1-H degraded completely after 2 months (t1/2 = 7.6 days). Under basic conditions, a t1/2 = 17.6 days under these conditions was determined. In addition, an equilibrium between pyrophosphate and phosphate was reached after 3 months (Figure 3e). Even if pH values are hard to compare between water and an organic solvent, the slower degradation kinetics of P1-H in solution compared to the polymer film might be rationalized by aggregation of the polymer in solution. The solution of P1-H in THF/water was a clear solution, but dynamic light scattering proved the formation of aggregates (>1 μm), in which the polar pyrophosphate might be protected against hydrolysis, while in the crystal lattice they are exposed to the surface and the solvent.
Polymer films of P2-H did not show any degradation under neutral conditions at 37 °C. In addition, no weight loss was recorded when P2-H was immersed at pH 13 and 37 °C. To collect data about molecular degradation, the 1H–31P HMBC NMR spectrum of P2-H film after degradation at pH 13 was collected and showed more than 70% hydrolysis to the diester.
As the polyprophosphate PE mimics undergo rapid backbone degradation by hydrolysis, we evaluated the biodegradability of P1-H (as powder) in an aqueous environment with activated sludge from the local sewage plant (kindly provided from the “Wirtschaftbetrieb Mainz” and the plant in Mainz-Mombach, Germany). As the medium possessed a pH value of 7.4, we expected a fast polymer backbone hydrolysis under these conditions. The resulting fragments should be biodegradable and ensure a full biomineralization of P1-H. Figure 3f shows the biodegradation of P1-H compared to P2-H, which does not undergo a backbone degradation at pH 7.4 (and acts as a negative control) and starch (as a positive control). The biodegradation was measured following the OECD 301F guideline with the Oxitop system measuring the biochemical oxygen demand (BOD) over a period of 28 days.35 All tests were performed in duplicate, and the maximum error was ±6%. The BOD of starch after 28 days reached a degradation of 73%. The BOD determined for P1-H reached a degradation of 81% after 28 days, which resembles the material as “readily biodegradable”, a term considered for chemicals with >60% biodegradation after 28 days.35 Initially, a lag time was detected, which might be due to hydrolysis of the polymer and consumption of other nutrients in the mixture by the microorganisms. When P1-H was hydrolyzed to smaller fragments, the microorganisms were able to further mineralize them, resulting in an increase of the BOD value. Under the same conditions, P2-H showed negligible biodegradation with 3% which is in the error value, which is probably attributed to the phosphoester bond, which does not undergo hydrolysis at neutral conditions during 30 days. As the polymer itself is very hydrophobic, degradation by microorganisms from the sewage plant seems to be very low.
Summary
We prepared the first pyrophosphate-containing polyethylene mimic that is readily biodegradable. The polymer was prepared by acyclic diene metathesis polycondensation of 1, followed by hydrogenation. The saturated semicrystalline polymer (P1-H) is water-insoluble but undergoes rapid backbone hydrolysis under neutral, basic, or acidic conditions when polymer films were immersed in water. The resulting degradation products were further identified as readily biodegradable by microorganisms according to the OECD 301F guideline. P1-H is the first long-chain polyphyrophosphate that resembles the crystallinity of polyethylene but is readily degraded. These materials might be interesting for a quick release under mild conditions in the biomedical field or also for advanced packaging. In addition, they broaden the field of phosphorus-containing polymers with a very labile linkage in the main chain compared to structurally very similar polyphosphoesters. A combination of pyrophosphate- and phosphate-based polymers might also be used to control degradation and/or release rates.
Acknowledgments
The authors thank Prof. Dr. Katharina Landfester (MPIP, Mainz, Germany) for continuous support. F.R.W. and H.T.T. thank Heraeus Medical (Wehrheim, Germany) for support. We thank the “Wirtschaftsbetrieb Mainz” (Mainz, Germany) for providing us with the activated sludge. The authors thank the German Ministry for Education and Research (BMBF) for their support of the program “Research for sustainable development (FONA)”, “PlastX - Plastics as a systemic risk for social-ecological supply systems” (grant number: 01UU1603A).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.8b02474.
Synthesis and characterization methods, monomer and polymer syntheses, degradation studies, NMR spectra and GPC elugrams (DOCX)
The authors declare no competing financial interest.
Supplementary Material
References
- Türk O.Biogene Polyolefine: Polyethylen aus biogenem Ethanol. In Stoffliche Nutzung nachwachsender Rohstoffe; Springer: 2014; pp 431–438. [Google Scholar]
- Wilke G.Karl Ziegler—The Last Alchemist. In Ziegler Catalysts; Springer: 1995; pp 1–14. [Google Scholar]
- Galli P.; Vecellio G. J. Polym. Sci., Part A: Polym. Chem. 2004, 42 (3), 396–415. 10.1002/pola.10804. [DOI] [Google Scholar]
- Austin R.Degradation studies of polyolefins. In Degradable Materials; CRC Press: 2018; pp 209–236. [Google Scholar]
- AlMa’adeed M. A.-A.; Krupa I.. Polyolefin Compounds and Materials; Springer: 2016. [Google Scholar]
- Haider T.; Völker C.; Kramm J.; Landfester K.; Wurm F. R. Plastics of the future? The impact of biodegradable polymers on the environment and on society. Angew. Chem., Int. Ed. 2019, 58, 50. 10.1002/anie.201805766. [DOI] [PubMed] [Google Scholar]
- Jeong B.; Bae Y. H.; Lee D. S.; Kim S. W. Biodegradable block copolymers as injectable drug-delivery systems. Nature 1997, 388 (6645), 860. 10.1038/42218. [DOI] [PubMed] [Google Scholar]
- Vert M. Polymeric biomaterials: strategies of the past vs. strategies of the future. Prog. Polym. Sci. 2007, 32 (8–9), 755–761. 10.1016/j.progpolymsci.2007.05.006. [DOI] [Google Scholar]
- Rezwan K.; Chen Q.; Blaker J.; Boccaccini A. R. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials 2006, 27 (18), 3413–3431. 10.1016/j.biomaterials.2006.01.039. [DOI] [PubMed] [Google Scholar]
- Nair L. S.; Laurencin C. T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32 (8–9), 762–798. 10.1016/j.progpolymsci.2007.05.017. [DOI] [Google Scholar]
- Luckachan G. E.; Pillai C. K. S. Biodegradable Polymers- A Review on Recent Trends and Emerging Perspectives. J. Polym. Environ. 2011, 19 (3), 637–676. 10.1007/s10924-011-0317-1. [DOI] [Google Scholar]
- Bauer K. N.; Tee H. T.; Velencoso M. M.; Wurm F. R. Main-chain poly(phosphoester)s: History, syntheses, degradation, bio-and flame-retardant applications. Prog. Polym. Sci. 2017, 73, 61–122. 10.1016/j.progpolymsci.2017.05.004. [DOI] [Google Scholar]
- Amass W.; Amass A.; Tighe B. A review of biodegradable polymers: uses, current developments in the synthesis and characterization of biodegradable polyesters, blends of biodegradable polymers and recent advances in biodegradation studies. Polym. Int. 1998, 47 (2), 89–144. 10.1002/(SICI)1097-0126(1998100)47:2<89::AID-PI86>3.0.CO;2-F. [DOI] [Google Scholar]
- Lucas N.; Bienaime C.; Belloy C.; Queneudec M.; Silvestre F.; Nava-Saucedo J.-E. Polymer biodegradation: Mechanisms and estimation techniques – A review. Chemosphere 2008, 73 (4), 429–442. 10.1016/j.chemosphere.2008.06.064. [DOI] [PubMed] [Google Scholar]
- Caire da Silva L.; Rojas G.; Schulz M. D.; Wagener K. B. Acyclic diene metathesis polymerization: History, methods and applications. Prog. Polym. Sci. 2017, 69, 79–107. 10.1016/j.progpolymsci.2016.12.001. [DOI] [Google Scholar]
- Stempfle F.; Ortmann P.; Mecking S. Long-Chain Aliphatic Polymers To Bridge the Gap between Semicrystalline Polyolefins and Traditional Polycondensates. Chem. Rev. 2016, 116 (7), 4597–4641. 10.1021/acs.chemrev.5b00705. [DOI] [PubMed] [Google Scholar]
- Cankaya A.; Steinmann M.; Bülbül Y.; Lieberwirth I.; Wurm F. R. Side-chain poly(phosphoramidate)s via acyclic diene metathesis polycondensation. Polym. Chem. 2016, 7 (31), 5004–5010. 10.1039/C6PY00999A. [DOI] [Google Scholar]
- Steinmann M.; Wagner M.; Wurm F. R. Chem. - Eur. J. 2016, 22 (48), 17329–17338. 10.1002/chem.201603990. [DOI] [PubMed] [Google Scholar]
- Busch H.; Majumder S.; Reiter G.; Mecking S. Semicrystalline Long-Chain Polyphosphoesters from Polyesterification. Macromolecules 2017, 50 (7), 2706–2713. 10.1021/acs.macromol.7b00243. [DOI] [Google Scholar]
- Khorana H. G.; Todd A. R. 465. Studies on phosphorylation. Part XI. The reaction between carbodi-imides and acid esters of phosphoric acid. A new method for the preparation of pyrophosphates. J. Chem. Soc. 1953, 0, 2257–2260. 10.1039/jr9530002257. [DOI] [Google Scholar]
- Zhou Y.; Yin S.; Gao Y.; Zhao Y.; Goto M.; Han L.-B. Selective P–P and P–O–P Bond Formations through Copper-Catalyzed Aerobic Oxidative Dehydrogenative Couplings of H-Phosphonates. Angew. Chem., Int. Ed. 2010, 49 (38), 6852–6855. 10.1002/anie.201003484. [DOI] [PubMed] [Google Scholar]
- Bauer K. N.; Tee H. T.; Lieberwirth I.; Wurm F. R. In-Chain Poly(phosphonate)s via Acyclic Diene Metathesis Polycondensation. Macromolecules 2016, 49 (10), 3761–3768. 10.1021/acs.macromol.6b00366. [DOI] [Google Scholar]
- Marsico F.; Turshatov A.; Peköz R.; Avlasevich Y.; Wagner M.; Weber K.; Donadio D.; Landfester K.; Baluschev S.; Wurm F. R. Hyperbranched unsaturated polyphosphates as a protective matrix for long-term photon upconversion in air. J. Am. Chem. Soc. 2014, 136 (31), 11057–11064. 10.1021/ja5049412. [DOI] [PubMed] [Google Scholar]
- Steinbach T.; Alexandrino E. M.; Wurm F. R. Unsaturated poly (phosphoester) s via ring-opening metathesis polymerization. Polym. Chem. 2013, 4 (13), 3800–3806. 10.1039/c3py00437f. [DOI] [Google Scholar]
- Ortmann P.; Heckler I.; Mecking S. Physical properties and hydrolytic degradability of polyethylene-like polyacetals and polycarbonates. Green Chem. 2014, 16 (4), 1816–1827. 10.1039/c3gc42592d. [DOI] [Google Scholar]
- Richards R. B. Polyethylene-structure, crystallinity and properties. J. Biochem. Toxicol. 1951, 1 (8), 370–376. 10.1002/jbt.2570010812. [DOI] [Google Scholar]
- Tung L.; Buckser S. The Effects of Molecular Weight on the Crystallinity of Polyethylene. J. Phys. Chem. 1958, 62 (12), 1530–1534. 10.1021/j150570a015. [DOI] [Google Scholar]
- Zheng Y.-R.; Tee H. T.; Wei Y.; Wu X.-L.; Mezger M.; Yan S.; Landfester K.; Wagener K.; Wurm F. R.; Lieberwirth I. Morphology and Thermal Properties of Precision Polymers: The Crystallization of Butyl Branched Polyethylene and Polyphosphoesters. Macromolecules 2016, 49 (4), 1321–1330. 10.1021/acs.macromol.5b02581. [DOI] [Google Scholar]
- Uehara H.; Kanamoto T.; Kawaguchi A.; Murakami S. Real-Time X-ray Diffraction Study on Two-Stage Drawing of Ultra-High Molecular Weight Polyethylene Reactor Powder above the Static Melting Temperature. Macromolecules 1996, 29 (5), 1540–1547. 10.1021/ma951222y. [DOI] [Google Scholar]
- Wolf T.; Steinbach T.; Wurm F. R. A Library of Well-Defined and Water-Soluble Poly(alkyl phosphonate)s with Adjustable Hydrolysis. Macromolecules 2015, 48 (12), 3853–3863. 10.1021/acs.macromol.5b00897. [DOI] [Google Scholar]
- Wang Y.-C.; Tang L.-Y.; Li Y.; Wang J. Thermoresponsive block copolymers of poly (ethylene glycol) and polyphosphoester: thermo-induced self-assembly, biocompatibility, and hydrolytic degradation. Biomacromolecules 2009, 10 (1), 66–73. 10.1021/bm800808q. [DOI] [PubMed] [Google Scholar]
- Wang Y.-C.; Tang L.-Y.; Sun T.-M.; Li C.-H.; Xiong M.-H.; Wang J. Self-assembled micelles of biodegradable triblock copolymers based on poly (ethyl ethylene phosphate) and poly (ϵ-caprolactone) as drug carriers. Biomacromolecules 2008, 9 (1), 388–395. 10.1021/bm700732g. [DOI] [PubMed] [Google Scholar]
- Appukutti N.; Serpell C. J. High definition polyphosphoesters: between nucleic acids and plastics. Polym. Chem. 2018, 9 (17), 2210–2226. 10.1039/C8PY00251G. [DOI] [Google Scholar]
- Bauer K. N.; Liu L.; Wagner M.; Andrienko D.; Wurm F. R. Mechanistic study on the hydrolytic degradation of polyphosphates. Eur. Polym. J. 2018, 108, 286–294. 10.1016/j.eurpolymj.2018.08.058. [DOI] [Google Scholar]
- OECD Guidelines for the Testing of Chemicals; Organization for Economic: 1994. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.