

Abstract
Lipoxygenases (LOX) form a family of lipid peroxidizing enzymes, which have been implicated in a number of physiological processes and in the pathogenesis of inflammatory, hyperproliferative and neurodegenerative diseases. They occur in two of the three domains of terrestrial life (bacteria, eucarya) and the human genome involves six functional LOX genes, which encode for six different LOX isoforms. One of these isoforms is ALOX15, which has first been described in rabbits in 1974 as enzyme capable of oxidizing membrane phospholipids during the maturational breakdown of mitochondria in immature red blood cells. During the following decades ALOX15 has extensively been characterized and its biological functions have been studied in a number of cellular in vitro systems as well as in various whole animal disease models. This review is aimed at summarizing the current knowledge on the protein-chemical, molecular biological and enzymatic properties of ALOX15 in various species (human, mouse, rabbit, rat) as well as its implication in cellular physiology and in the pathogenesis of various diseases.
Keywords: eicosanoids, lipoxygenase, leukotrienes, evolution, enzymology, lipid metabolism
Graphical Abstract
1. Introduction
Lipoxygenases (LOXs) are non-heme iron-containing fatty acid dioxygenases (Brash, 1999; Ivanov et al., 2010; Haeggstrom and Funk, 2011; Kuhn et al., 2014) that catalyze the dioxygenation of polyunsaturated fatty acids to the corresponding hydroperoxy derivatives (Fig. 1). For a long time it was believed that true LOX enzymes only occur in plants but in 1974 an arachidonic acid 12-lipoxygenating enzyme was discovered in human platelets (Hamberg and Samuelsson, 1974) and this enzyme was named platelet type 12-LOX (ALOX12). Some months later a different LOX-isoenzyme was reported in the lysate of immature red blood cells (Schewe et al., 1975), which was capable of oxidizing membrane lipids. This enzyme was named reticulocyte-type 15-LOX (the rabbit ortholog of human ALOX15). Since then a large number of LOX-isoforms exhibiting different enzymatic properties have been described in various species. Completion of the human genome project revealed that the human genome contains 6 functional LOX genes (ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, ALOXE3) encoding for 6 different LOX-isoforms (Funk et al., 2002). In most mammalian cells linoleic acid (C18:∆2, n-6), alpha- (C18:∆3, n-3) and gamma- (C18:∆3, n-6), linolenic acid, arachidonic acid (C20:∆4, n-6), eicosapentaenoic acid (C20:∆5, n-3) and docosahexaenoic acid (C22:∆6, n-3) are the most abundant polyenoic fatty acids serving as substrates for the different mammalian LOX-isoforms. Mammalian LOXs prefer free fatty acids as substrate but the cellular concentration of free fatty acids is rather low. Thus, to initiate the formation of LOX products in cellular systems polyenoic fatty acids must be liberated from the cellular ester lipids by the catalytic activity of ester lipid hydrolyzing enzymes, preferentially by cytosolic phospholipase A2 (Mancini and Di Battista, 2011). The hydroperoxy fatty acids formed by the different LOX isoforms are subsequently converted to a large array of bioactive lipid mediators, which include leukotrienes (Savari et al., 2014), lipoxins (Romano, 2010), hepoxilins (Pace-Asciak, 2009), eoxins (Sachs-Olsen et al., 2010), resolvins (Yoo et al., 2013), protectins (Serhan and Petasis, 2011) and others. However, some LOX isoforms, in particular the ALOX15 orthologs of rabbits (Schewe et al., 1975), pigs (Takahashi et al., 1993) and rats (Pekarova et al., 2015) are capable of oxygenating complex ester lipids even if they are constituents of complex lipid-protein assemblies, such as biomembranes (Kuhn et al., 1990b), and lipoproteins (Belkner et al., 1993).
Fig. 1. Catalytic activities of ALOX15 orthologs.
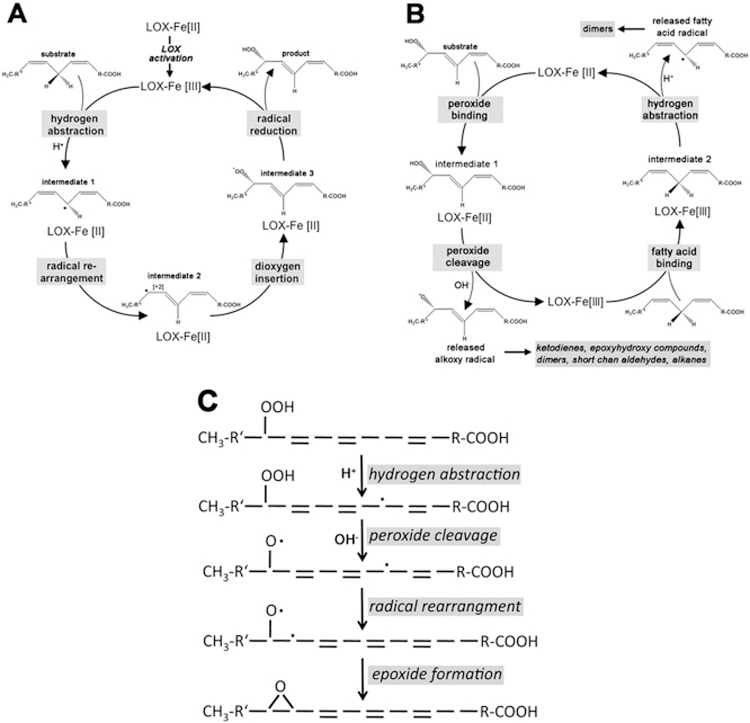
A) The lipoxygenase reaction consists of 4 elementary reactions (hydrogen abstraction, radical rearrangement, dioxygen insertion, peroxy radical reduction). To initiate the reaction the ferrous LOX is first activated by peroxide-dependent oxidation to a ferric form [modified from (Ivanov et al., 2010)]. B) The lipohydroperoxidase activity is initiated when a lipid hydroperoxide (ROOH) is bound at the active site of the enzyme. The enzyme then catalyzes a homolytic cleavage of the hydroperoxy bond, which leads to the formation of an oxygen-centered alkoxy radical, a hydroxyl and oxidizes the ferrous iron to a ferric form. Then the enzyme binds a linoleic acid molecule (or an alterative reductant such as guaiacol) and releases a carbon-centered linoleic radical. This reaction reduces the ferric LOX back to its ferrous form to start the next catalytic cycle. The released radical intermediates may then initiate free radical secondary reactions leading to the formation of mixed oxygenated and non-oxygenated linoleic acid dimers. C) The leukotriene synthase activity of various LOX-isoforms involved a homolytic cleavage of the hydroperoxy group and a hydrogen abstracton from a bisallylic methylene. These consecutive reaction steps lead to the formation of a fatty acid biradical, which stabilizes by epoxide formation.
Among the six different human LOX-isoforms ALOX5 (Radmark et al., 2015) and ALOX15 (Ivanov et al., 2010) are probably the best characterized isoenzymes and the biological relevance of these mammalian LOX-isoforms has recently been reviewed (Kuhn et al., 2014). This paper is aimed at summarizing our current knowledge on the protein-chemical, molecular-biological and enzymatic properties of ALOX15 orthologs in various mammalian species and at critically evaluating the experimental data characterizing the physiological and patho-physiological roles of this particular LOX-isoform. Among the ALOX15 orthologs rabbit ALOX15 has been characterized most comprehensively because of its early discovery and its long lasting history. Although there are considerable species-specific differences between the rabbit enzyme and the corresponding orthologs of other mammalian species rabbit ALOX15 is frequently considered as suitable model for ALOX15 orthologs of other species including humans. Writing a review about a well-characterized enzyme, which has been discovered more than 40 years ago, is always very selective and strongly depends on the perspective of the authors. Although we did our best to balance this selection we might have overlooked important contributions and we apologize to those distinguished colleagues who significantly contributed to the field but whose work could not be referenced because of space limitations.
2. Lipoxygenase family and human lipoxygenase isoforms
2.1. Lipoxygenase distribution in terrestrial life
As indicated above the human genome involves six functional LOX genes and evolution of this class of enzymes recently became a matter of discussion (Hansen et al., 2013; Horn et al., 2014). In viruses functional LOXs haven not been characterized although LOX-like sequences have been deposited in publically available sequence databases (Horn et al., 2014). LOX occur in two (bacteria, eukarya) of the three domains of terrestrial life and although LOX-like sequences have been reported in various archeae there are no convincing data suggesting expression of functional LOX-isoforms in these microorganisms (Horn et al., 2014). In bacteria LOXs do occur (Hansen et al., 2013) but they are not widely distributed (Horn et al., 2014). Among the bacterial genomes sequenced so far (~13,000, Aug. 2014) some 60 LOX-like sequences have been identified on the basis of amino acid comparison. These bacterial species include firmicutes, different types of proteobacteria, cyanobacteria, actinobacteria and representatives of the CFB group (Horn et al., 2014). Although the functionality of most bacterial LOXs has not been characterized, the observation that less than 0.5 % of the bacterial genomes contain potential LOX genes, suggest that these enzymes only sporadically occur in bacteria. In fact, most human pathogenic bacteria including E. coli (bacterial model organism) do not carry LOX genes (Horn et al., 2014). In eucarya LOXs are more widely distributed and functional enzymes have been characterized in algae, fungi, protists, plants as well as in lower and higher animals (Horn et al., 2014). However, well-characterized eucaryotic model organisms representing lower evolutionary stages of terrestrial life such as Saccharomyces cerevisiae, Drosophila melanogaster and Caenorhabditis elegans do not carry LOX genes. In mosses (Physcomitrella patens) and higher plants (Glycine max, Oryza sativa, Zea mays) a large number of LOX isoforms have been detected indicating wide spread occurrence of these enzymes in plants. When we searched the ENSEMBL (http://metazoa.ensembl.org) and the NCBI (www.ncbi.nim.gov) protein databases for Placozoa, Porifera, Coelenterata, Platyhelminthes, Nematoda, Mollusca, Annelida, Echinodermata, Chelicerata, Cephalochordata, Tunicata with the key word “lipoxygenase” we found that functional LOXs occur in selected invertebrates and this conclusion is consistent with the previous characterization of several enzymes in corals (Brash et al., 1996), mussels (Hada et al., 1997; Coffa and Hill, 2000), sea urchins (Hawkins and Brash, 1987) and others. On the other hand, more detailed database searches indicated that most invertebrates do not carry functional LOX genes and a rough estimate suggested that among the more than one million invertebrate species on earth less than 1% carry LOX-like sequences (Horn et al., 2014). In lower cordates (Branchiostoma floridae, Ciona intestinalis) LOX sequences have been identified but more detailed characterization of the corresponding enzymes is still pending. In higher cordates, particularly in vertebrates including mammals, LOX sequences are more common. The genomes of the elephant shark (Callorhinchus milii; model organism for cartilaginous fish), the zebrafish (Danio rerio; model organism for bony fish), the western clawed frog (Xenopus Silurana tropicalis; model organism for amphibia), the american alligator (Alligator missisippiensis; model organism for reptilia) and chicken (Gallus gallus; model organism of aves) contain LOX sequences and although little functional data are currently available for these animal species, the sequence data suggest a wide distribution of functional LOX in higher vertebrates. However, subclassification of these enzymes on the basis of their sequence homology and their assignment to either of the human isoforms is complicated since most of these isoenzymes only share a low degree of sequence similarity (20–30%) with any human LOX isoform. Thus, it can hardly be predicted, which of the LOX isoforms present in lower animals is the functional equivalent (ortholog enzyme) or the immediate precursor of human ALOX15.
The mouse genome (Mus musculus; a model organism for mammals) involves seven functional LOX genes (alox5, alox15, alox15b, alox12, alox12b, aloxe3, aloxe12). In humans all mouse LOX genes except for the aloxe12 gene are well conserved as functional gene (Funk et al., 2002). The mouse aloxe12 gene is present in the human genome as corrupted and functionless pseudogene. The question, which of the mouse LOX genes might constitute the functional equivalent of human ALOX15, has been discussed controversially. Since mouse alox15 converts arachidonic acid to 12-H(p)ETE it has been suggested that mouse alox15 may functionally be more closely related to human ALOX12. However, this would only be the case if ALOX15 orthologs exhibit their bioactivity via the formation of arachidonic acid oxygenation products. In contrast, when ALOX15 orthologs exhibit their biological function by oxygenating of complex lipid-protein assemblies (lipoproteins, biomembranes) there is hardly any functional similarity between mouse alox15 and human ALOX12. ALOX15 orthologs including mouse alox15 can oxidize complex lipid-protein complexes whereas the ALOX12 orthologs of humans and mice are not capable to do so. Genomic sequence alignments, chromosomal localization and comparison of the enzyme properties strongly suggest that the mouse leukocyte-type 12-LOX (old nomenclature) and the human reticulocyte-type 12/15-LOX (old nomenclature) are orthologous enzymes. Usually, enzyme orthologs fullfill similar functions in different organisms and thus mouse alox15 may constitute the functional equivalent of human ALOX15 despite their different reaction specificity of arachidonic acid oxygenation. A similar situation was observed in rats. Here again, the alox15 ortholog is an arachidonic acid 12-lipoxygenating enzyme species and is also capable of oxygenating membrane phospholipids (Watanabe and Haeggstrom, 1993; Pekarova et al., 2015).
2.2. LOX classification systems
Traditionally, animal LOXs have been classified according to their reaction specificity using arachidonic acid as model substrate. When oxygen is introduced at carbon atom 5 of the fatty acid backbone the corresponding enzyme was called 5-LOX. If the substrate is oxygenated at carbon 15 a 15-LOX was predicted as catalyst. This nomenclature is simple and straightforward but it does not consider the evolutionary relatedness of the enzymes. Moreover, it leads to confusions since LOX-isoforms, which share a high degree of evolutionary relatedness, might exhibit different reaction specificities. This is, for instance, the case for human ALOX15, which oxygenates arachidonic acid at carbon 15 (Sloane et al., 1991a), and mouse alox15, which catalyses arachidonic acid 12-lipoxygenation (Sun and Funk, 1996). Similarly, human ALOX15B introduces dioxygen at carbon 15 of the model substrate (Brash et al., 1997) whereas the corresponding murine ortholog (alox15b) is an 8-lipoxygenating enzyme species (Jisaka et al., 2000). Even more confusing was the observation that except for alox5 and aloxe3 all other murine LOX isoforms are 12-lipoxygenating enzymes and, thus should be classified together despite their structural and functional differences. To avoid such confusions the simple specificity related nomenclature should not be used any more. In recent years newly discovered LOX-isoforms are frequently classified according to their sequence similarity with any of the human isoforms. This classification concept works well for most mammalian LOX-isoforms but because of the low degree of sequence conservation problems may arise when LOX-isoforms of evolutionary more distant species are to be classified. For instance, in zebrafish a number of LOX transcripts originating from several genes have been identified, but neither of them shares a high degree of amino acid conservation with human ALOX15 (Haas et al., 2011; Jansen et al., 2011).
3. Enzymology of ALOX15
3.1. Multiple catalytic activities of ALOX15 (moonlighting character)
Various LOX-isoforms including ALOX15 (Schewe, 2002) exhibit multiple catalytic activities. They oxygenate polyenoic fatty acids to hydroperoxy derivatives but also exhibit a lipohydroperoxidase activity (sometimes also called hydroperoxide isomerase activity), which converts lipid hydroperoxides to secondary lipid peroxidation products. As indicated in Fig. 1A the lipid oxygenase activity involves as initial reaction a hydrogen abstraction from a bisallylic methylene of the fatty acid substrate. In contrast, the lipohydroperoxidase activity is initiated by a homolytic cleavage of the hydroperoxy bond, which formally leads to the formation of alkoxy and hydroxy radicals (Fig. 1B). In addition to lipoxygenase and lipohydroperoxidase activity ALOX15- orthologs exhibit a leukotriene synthase activity converting hydroperoxy fatty acids containing a conjugated diene system to epoxyeicosanoids carrying conjugated trienes (Bryant et al., 1985; Brash et al., 1989; Schewe, 2002).
3.1.1. Lipoxygenase activity
The lipoxygenase activity is initiated by hydrogen abstraction from a bisallylic methylene and leads to the formation of hydroperoxy lipids (Fig. 1A). For ALOX15 this activity is not restricted to free polyenoic fatty acids since phospholipids and even biomembranes and lipoproteins are ALOX15 substrates. The mechanistic details for the oxygenase reaction with different substrates are given in chapters 3.2., 3.3., 3.4.
3.1.2. Lipohydroperoxidase activity
Under certain reaction conditions (anaerobiosis, hypoxia, limited fatty acid supply) LOXs are capable of catalyzing the secondary conversion of hydroperoxylipids to an array of secondary lipid peroxidation produts. This catalytic property, which was first described for soybean LOX1 (Garssen et al., 1971), was called lipohydroperoxidase activity and the product mixture involved ketodienes, epoxy hydroxy compounds, short chain aldehydes, volatile hydrocarbons (pentane) and mixed oxygenated and non-oxygenated fatty acid dimers (Garssen et al., 1972; de Groot et al., 1973). Mechanistic experiments indicated that this reaction did also proceed under aerobic conditions when linoleic acid was replaced with guaiacol serving as artificial electron donor (Streckert and Stan, 1975). The reaction sequence of the lipohydroperoxidase activity resembles that of the lipoxygenase reaction and its catalytic cycle involves a valency change of the nonheme iron (Fig. 1B). A similar anaerobic lipohydroperoxyidase activity has later been reported for rabALOX15 (Hartel et al., 1982; Salzmann et al., 1984). Interestingly, for this enzyme the lipohydroperoxidase activity was not restricted to anaerobic conditions but was also observed in hypoxia (Kuhn et al., 1986a). From this data it was concluded that the ratio between oxygenase vs. lipohydroperoxidase activity of a given LOX-isoform depends on both, the enzyme properties and the oxygen pressure in the assay sample. Later experiments with recombinant human ALOXE3 suggested that its oxygenase activity is largely suppressed under normoxic conditions but that it exhibits a strong lipohydroperoxidase (hydroperoxide isomerase) activity (Zheng and Brash, 2010a; Zheng and Brash, 2010b). The lipohydroperoxidase activity may not be considered as in vitro artifact but has been implicated in the formation of the water barrier of human and mouse epidermis (Munoz-Garcia et al., 2014).
Hepoxilins (Pace-Asciak, 2009; Pace-Asciak, 2015) are epoxy hydroxy eicosanoids formed from 12-HpETE via the lipohydroperoxidase activity of ALOX12 orthologs. The epoxide ring can be hydrolyzed, which leads to the formation of trihydroxy eicosanoids (trioxilins). There are two classes of hepoxilins (A and B), which differ from each other by the relative positions of the two functional groups (hydroxy group and the epoxide ring). When 15-HpETE is converted by the lipohydroperoxidase activity of ALOX15 hepoxins [11S-hydroxy-14S,15S-epoxy-5Z,8Z,12E-eicosatrienoic acid (14,15-HXA3) and 13R-hydroxy-14S,15S-epoxy-5Z,8Z,11Z-eicosatrienoic acid (HXB3)] are formed. Although the detailed mechanism of hepoxilin and hepoxin signaling have not been clarified, a large number of bioactivities has been described for these lipohydroperoxidase products (Nigam et al., 2007; Zafiriou et al., 2011; Gregus et al., 2013; Krieg et al., 2013; Munoz-Garcia et al., 2014; Pace-Asciak, 2015).
3.1.3. Leukotriene synthase activity
ALOX5 and ALOX15 orthologs are capable of converting hydroperoxy fatty acids such as 5- and 15-HpETE to epoxy leukotrienes (Bryant et al., 1985; Ueda et al., 1986; Brash et al., 1989). Although several aspects of the leukotriene synthase activity of LOX-isoforms have not been explored, the catalytic cycle involves hydrogen abstraction from a bisallylic methylene and homolytic cleavage of the hydroperoxy group forming a fatty acid biradical. This biradical is then stabilized via expoxide formation (Fig. 1C). Thus, formally, the leukotriene synthase activity of LOX-isoforms may be considered as combination of its oxygenase (hydrogen abstraction from a bisallylic methylene) and its lipohydroperoxidase (hemolytic cleavage of the peroxy group) activity.
3.2. Reaction kinetics with polyenoic fatty acids
Purified native and recombinant rabbit ALOX15 (Ludwig et al., 1987) and its recombinant human ortholog (Kühn et al., 1993) exhibit non-linear reaction kinetics. A typical kinetic progress curve exhibits sigmoid shape (Fig. 2). It starts with a kinetic lag-phase, in which the catalytic activity is increased reaching the maximal turnover rate. This lag-phase is followed by a more or less linear part, in which the reaction rate is not altered but then the activity is decreasing owing to suicidal inactivation.
Fig. 2. Kinetic progress curve of arachidonic oxygenation by pure rabbit ALOX15.
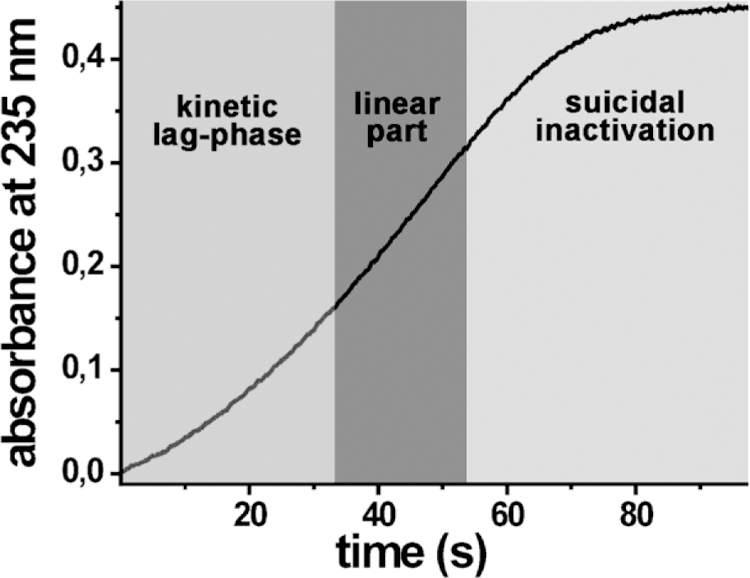
When peroxide-free fatty acids are used as substrates the kinetic progress curve of the ALOX15 reaction can be separated in three periods. i) Kinetic lag-phase: The oxygenation reaction starts with a kinetic lag-phase, in which product formation increases with time. ii) Linear phase: The lag phase is followed by a more or less linear part of the progress curve, in which the reaction rate does not change. iii) Suicidal inactivation phase: During the final part of the progress curve the reaction rate decreases with time, which has been related to suicidal inactivation of the enzyme.
3.2.1. Kinetic lag-phase and autocatalytic activation
The catalytic cycle of the LOX reaction (Fig. 1) consists of four stereochemically controlled elementary reactions (hydrogen abstraction, radical rearrangement, oxygen insertion and product dissociation) and involves a valence shuttling of the non-heme iron (Kuhn et al., 1986b; Rickert and Klinman, 1999; Lehnert and Solomon, 2003). When isolated from native and/or recombinant sources ALOX15 is present as catalytically silent ferrous enzyme. To initiate fatty acid oxygenation, the enzyme must first be oxidized to a ferric form capable of initiating hydrogen abstraction (Fig. 1). Unfortunately, single activation of the enzyme is not sufficient to keep it running, since during catalysis small quantities of radical intermediates escape from the active site (Ludwig et al., 1987; Schilstra et al., 1994) leaving the enzyme in an inactive ferrous (Fe2+) form. To keep the reaction at quasi-stationary levels, repeated enzyme activation is required and the primary oxygenation products appear to serve as enzyme activators. In this sense, the LOXs exhibit autocatalytic properties. Studying the oxygenation of 15S-HETE by pure rabbit ALOX15, it was found that the corresponding oxygenation product(s) did not activate the enzyme (Kuhn et al., 1986c). When activated with 13S-HpODE the reaction proceeded for up to 2 min but then ceased. Subsequent addition of 13S-HpODE restarted the reaction at the initial rate and this could be repeated several times. Quantitative evaluation of the kinetic progress curves of 15S-HETE oxygenation suggested that 1 mole of exogenous 13S-HpODE is sufficient for the oxygenation of 9–10 moles of 15S-HETE (Kuhn et al., 1986c). The chemistry of peroxide dependent LOX-activation appears to be more complex than simple oxidation of the ferrous non-heme iron, since it depends on the presence of molecular dioxygen. The kinetic progress curve for ALOX15 catalyzed oxygenation of (19R/S,5Z,8Z,11Z,14Z)-19-hydroxyeicosa-5,8,11,14-tetraenoic acid (19-hydroxy-5Z,8Z,11Z,14Z-eicosatetraenoic acid) under normoxic conditions was characterized by an extensive (more than 30 min) kinetic lag phase (Ivanov et al., 2005). However, under hyperoxic conditions a much shorter lag-phase was observed, suggesting an oxygen dependence of the activation process. Thus, molecular dioxygen serves not only as a lipoxygenase substrate, but also impacts peroxide-dependent enzyme activation (Ivanov et al., 2005). A similar oxygen dependence of the LOX activation was recently reported for ALOXE3 (Zheng and Brash, 2010b).
3.2.2. Suicidal inactivation
ALOX15 undergo suicidal inactivation during the oxygenation of polyenoic fatty acids (Hartel et al., 1982), but the molecular basis for this enzyme inactivation remains unclear. Initially, it has been suggested that hydroperoxy fatty acids may oxidize catalytically relevant amino acids at the active site. In fact, treatment of pure rabbit ALOX15 with 13S-HpODE induced selective oxidation of a methionine residue (Rapoport et al., 1984). However, site-directed mutagenesis of this methionine to an oxidation resistant alanine did not reduce the degree of suicidal inactivation (Gan et al., 1995). Covalent modification of rabbit ALOX15 was reported when the enzyme was incubated with 15S-HpETE (Wiesner et al., 2003) and separation of proteolytic cleavage peptides by two-dimensional-gel electrophoresis confirmed this hypothesis (Kuhn et al., 2005b). Despite these descriptive experimental data the molecular basis for suicidal enzyme inactivation remains unclear.
3.2.3. Temperature- and pH dependence of fatty acid oxygenation
Although pH alterations frequently occur in vivo, little is known on their impact on ALOX15 activity. ALOX15 catalyzed oxygenation of fatty acids was strongly pH-dependent and optimal enzymatic activity was observed between pH 7.0 and 7.4 in the absence of detergents. However, addition of surface-active compounds modified the pH optimum. In ALOX15 four histidine residues are 1st order iron ligands (Gillmor et al., 1997; Kuban et al., 1998) and thus pH alterations might impact their iron liganding properties. When the native rabbit ALOX15 was incubated at different pH (6.0, 7.4, and 9.0) its iron content remained unchanged (85–90%). These unpublished data suggest that the iron is stably liganded and that there is no pH-dependent loss in iron content under near-physiological conditions.
For human ALOX15 a broad temperature optimum (20° C - 35° C) was observed for linoleic acid oxygenation (Segraves and Holman, 2003) and these results correlated with data obtained in thermal stability assays of the enzyme (Mei et al., 2008). In the absence of substrate, rabbit ALOX15 is stable over long time intervals only at lower temperatures (<10° C). At higher temperatures (> 20° C) it undergoes structural fluctuations and loses catalytic activity (Mei et al., 2008). However, refolding experiments (changes in the CD-spectra determined as readout parameter) indicated that structural alterations induced by short-time exposure to 30° C were completely reversible. In contrast, further temperature elevations (45° C) induced irreversible changes (Mei et al., 2008). Similar results were obtained when the catalytic activity was assayed and additional experiments at other temperatures suggested that the threshold temperature (loss of reversibility) was around 35° C for this enzyme (Mei et al., 2008).
3.2.4. Activation of ALOX15 by membrane binding
In vitro membrane binding studies and membrane oxygenase activity assays indicated that rabbit ALOX15 binds to different types of biomembranes, such as plasma membranes, mitochondrial membranes and endoplasmic membranes (Kuhn et al., 1990a; Kuhn et al., 1990b; van Leyen et al., 1998; Walther et al., 2002; Walther et al., 2004) and immunohistochemical staining (Kühn, unpublished data) did not provide any evidence for preferential binding to any type of subcellular membranes. This is a marked difference to human ALOX5, which preferentially binds to the nuclear envelope (Radmark et al., 2015). Membrane binding of ALOX15 proceeds in a calcium dependent manner (Brinckmann et al., 1998; Walther et al., 2004) and strongly augments the specific fatty acid oxygenase activity of the enzyme without impacting its reaction specificity (Lankin et al., 1985; Brinckmann et al., 1998). In contrast, to ALOX5, for which specific calcium binding sites have been identified (Bindu et al., 2004; Schroder et al., 2014), ALOX15 does not carry such high affinity calcium binding sites. In vitro membrane binding assays and site directed mutagenesis of surface exposed hydrophobic amino acids, which have been mapped to both, the N-terminal ß-barrel (Tyr15, Phe70, Leu71) and the catalytic domain (Trp181, Leu195) suggested their importance for reversible membrane binding (Walther et al., 2004). The role of calcium needed as essential cofactor for membrane binding remains unclear but it was hypothesized that the positively charged calcium ions might neutralize negative charges of the membrane phospholipids, reducing electrostatic repulsive forces, which may counteract membrane binding (Walther et al., 2004).
3.3. Substrate specificity
3.3.1. Free polyenoic fatty acids as ALOX15 substrates
Most LOX-isoforms strongly prefer free polyenoic fatty acids as substrates and this is also the case for ALOX15 orthologs. Among the naturally occurring polyenoic fatty acids linoleic acid, alpha- and gamma-linolenic acid, arachidonic acid, eicosapentaenoic acid and docosahexaenoic acid are well accepted. Monoenoic fatty acids (oleic acid) and saturated fatty acids of comparable chain length (stearic acid, arachidic acid) are not oxygenated as indicated by oxygraphic measurements. However, they function as weak competitive inhibitors. The molar turnover numbers of purified rabbit and human ALOX15 vary between 5–50 s−1 depending on the quality of enzyme preparations. Polyenoic fatty acids carrying a hydrophilic group close to the omega-end are not well oxidized by rabbit ALOX15 (Ivanov et al., 1998; Walther et al., 2001). Although there are no direct structural data for ALOX15-substrate complexes currently available, models for ALOX15-fatty acid complexes have recently been worked out (Toledo et al., 2010; Toledo et al., 2011) on the basis of the X-ray coordinates (Gillmor et al., 1997). According to these models the substrate fatty acid slides into the hydrophobic pocket with its methyl end ahead so that the hydrogen to be abstracted during the initial elementary reaction, is located in close proximity to the catalytic non-heme iron (Kuhn et al., 1986b; Rickert and Klinman, 1999). According to recent MD simulations, linoleic acid and arachidonic acid share common overall orientation at the active site. However, arachidonic acid is bound closer to the active site helix α18 and has a limited degree of motional freedom. In contrast, the tail of linoleic acid fluctuates more freely and adopts a number of energetically similar conformations at the active site (Toledo et al., 2010; Toledo et al., 2011).
Site directed mutagenesis on human ALOX15 suggested that Arg403 may interact with the negatively charged carboxylate of the substrate fatty acid (Gan et al., 1996). Mutagenesis studies revealed that Arg403Leu exchange induces strong impairment of the catalytic activity for free fatty acids (Gan et al., 1996) and similar results have later been obtained for the rabbit ortholog (Di Venere et al., 2013). However, interpretation of these data remains controversial since catalytic inactivity can have a number of mechanistic reasons.
Although polyunsaturated fatty acids appear to penetrate the 15-LOXs active site with their methyl end ahead, other substrates were suggested to bind in an inverted orientation (Van Os et al., 1981; Kuhn et al., 1986b). Initial evidence for such an inverse, head-to-tail substrate orientation was provided when the oxygenation of 15S-HpETE by soybean lipoxygenase-1 was studied (Van Os et al., 1981). The major reaction products were identified as (8S,15S,5Z,9E,11Z,13E)-8,15-dihydroperoxy-5,9,11,13-eicosatetraenoic acid (8S,15S-DiHpETE) and (5S,15S,6E,8Z,11Z,13E)-5,15-dihydroperoxy-6,8,11,13-eicosatetraenoic acid (5S,15S-DiHPETE) and the stereochemistry of the reaction was compatible with an inverse substrate orientation. To provide evidence for a similar inverse substrate alignment the oxygenation of 15S-HETE by rabbit ALOX15 was explored (Schwarz et al., 1998). This substrate was oxygenated at carbon 5 and carbon 14 of the fatty acid backbone suggesting the possibility of simultaneous straight (oxygenation at C14) and inverse (oxygenation at C5) substrate alignment. Methylation of the substrates carboxylic group strongly augmented the reaction rate and shifted the product pattern almost completely to 5S-lipoxygenation. Introduction of a bulky glycerol moiety reversed the kinetic effects of methylation and induced preferential C14-oxygenation (Schwarz et al., 1998). These data are consistent with the concept of an inverse substrate orientation. With a defined substrate there may be a binding equilibrium between “normal” (methyl terminus ahead) and inverse alignment (carboxylate ahead). This binding equilibrium may be influenced by functional groups on either end of the fatty acid backbone (Schwarz et al., 1998).
3.3.2. Phospholipids and cholesterol esters are ALOX15 substrates
Purified rabbit ALOX15 oxygenates phospholipids (Schewe et al., 1975) and cholesterol esters (Belkner et al., 1991) containing polyunsaturated fatty acids. In fact, rabbit ALOX15 has first been discovered because of its phospholipid oxidizing capability (Schewe et al., 1975). Although the reaction rates of phospholipid oxidation are at least one order of magnitude lower then the rate of free polyenoic fatty acid oxygenation, specific reaction products (Table 1) have been analyzed for different phospholipids and cholesterol esters. These data suggest that the enzyme tightly controls the oxygenation reaction. On the other hand, oxygenation of phospholipids by ALOX15 is somewhat surprising since in silico docking studies indicated that binding of a phospholipid molecule in the substrate binding pocket is hardly possible without major rearrangement of the active site structure. The volume of the substrate-binding pocket is simply not big enough to accommodate a complete phospholipid molecule without steric clashes (Fig. 3). Thus, specific oxygenation of phospholipids by rabbit ALOX15 can only be explained if the enzyme exhibits a high degree of motional flexibility allowing substantial rearrangement of the active site to enable phospholipid binding. This is apparently not the case for the secretable LOX of Pseudomonas aeruginosa (Lu et al., 2013). The active site of this enzyme is big enough to bind a phospholipid molecule. In fact, the enzyme was crystallized with a phospholipid molecule bound in the substrate-binding pocket (Garreta et al., 2013). We recently compared rabbit ALOX15 and P. aeruginosa LOX site-by-site and found similar phospholipid oxygenase activities for both enzymes when normalized to their arachidonic acid oxygenase activity (Banthiya, unpublished data).
Table 1.
Major oxygenation products of ester lipid oxygenation by pure rabbit ALOX15
| Lipid class | lipid substrate | main product |
|---|---|---|
| Phospholipds | 1-palmitoyl,2-linoleoyl phosphatidyl choline | 13S-HpODE |
| 1-palmitoyl,2-arachidonyl phosphatidyl choline | 15S-HpETE | |
| 1-palmitoyl,2-eicosapentaenoyl phosphatidyl choline | 15S-HpEPE | |
| 1-palmitoyl,2-docosahexaenoyl phosphatidyl choline | 17S-HpDHE | |
| diacylglyceroles | 1-stearyl,2-arachidonyl glycerole | 15-HETE |
| 1-stearyl,2-linoleoyl glycerole | 13S-HpODE | |
| Cholesterol esters | cholesteryl linoleate | 13S-HpODE |
| cholesteryl linolinate | 13-HpOTE | |
| cholesteryl arachidonate | 15S-HpETE |
Fig. 3. Molecular docking studies of a phospholipid molecule at the active site of rabbit ALOX15.

To construct these images the following sets of X-ray coordinates (PDB entries) were employed: rabbit ALOX15 (2POM), soybean LOX-1 (1YGE), and phospholipid (4G32). The GOLD program with default parameters was used for docking the phospholipid into the active sites of rabbit ALOX15 conformers and soybean LOX1. For preparation of images the Accelrys Discovery Studio 4.0 Visualizer was employed. The amino acids labeled represent examples for steric clashes with the phospholipid substrate. A) rabbit ALOX15 (non-liganded conformer), B) rabbit ALOX15 (liganded conformer, C) soybean LOX1. The docking studies were carried out by Kumar Reddy Kakularam from the Department of Animal Sciences, School of Life Science, University of Hyderabad (India) and the National Institute of Animal Biotechnology, Hyderabad (India). Permission for publication was granted.
3.3.3. Biomembranes und lipoproteins as ALOX15 substrates
The ALOX15 orthologs of rabbits (Kuhn et al., 1990b), humans (Kühn et al., 1993) and pigs (Takahashi et al., 1993) are capable of directly oxygenating complex lipid protein assemblies such as biomembranes and lipoproteins. ALOX15 catalyzed oxygenation of membrane lipids was implicated in the maturational breakdown of mitochondria during reticulocyte–erythrocyte transition and inhibition of ALOX15 delayed organelle degradation in rabbit reticulocytes (Schewe et al., 1975; Schewe et al., 1986; Grullich et al., 2001). Specific ALOX15 products have been detected in membranes of rabbit reticulocytes (Kuhn and Brash, 1990) and the oxidation degree of membrane lipids in mitochondria was significantly higher then in the plasma membranes (Kuhn et al., 1990a) suggesting a preferential in vivo activity of the enzyme on mitochondrial membranes. Addition of rabbit ALOX15 to purified rat liver mitochondrial membranes in vitro induced disruption of the organelle, inactivation of the respiratory chain and the release of matrix enzymes (Rapoport and Schewe, 1986). In other in vitro models of ALOX15-membrane interaction the enzyme integrated into the membranes of various organelles, allowing release of proteins from the organelle lumen and access of proteases to both, lumenal and integral membrane proteins (van Leyen et al., 1998). Taken together, the catalytic activity of ALOX15 on biomembranes provides a mechanism, by which the maturational degradation of cellular organelles and restructuring of biomembranes can be explained (Rapoport and Schewe, 1986; van Leyen et al., 1998).
The oxidative hypothesis of atherosclerosis (Witztum and Steinberg, 1991) suggests that oxidative modification of lipoproteins plays a major role in the pathogenesis of this disease. According to this scenario low density lipoprotein is oxidized by enzymatic and non-enzymatic reactions to an atherogenic species, which is rapidly taken up by monocytes/macrophages via scavenger receptor mediated pathways. Since these pathways are not feedback-controlled, excessive uptake of oxidized low-density lipoproteins renders monocytes/macrophages to lipid-laden foam cells, which accumulate in the subendothelial space of the arteries forming fatty streaks that constitute early atherosclerotic lesions (Libby, 2012). In reconstituted molecular systems, purified ALOX15 is capable of oxidizing the ester lipids of low-density lipoproteins to specific ALOX15 products (Belkner et al., 1993) and as dominant substrates the cholesterol esters of the lipoprotein core have been identified. The ALOX15 derived hydroperoxy lipids subsequently induce free-radical mediated secondary reactions, which render the product pattern more unspecific at longer incubation periods (Upston et al., 1997; Belkner et al., 1998). The possible involvement of ALOX15 in the formation of oxidized low-density lipoprotein was the basis for the pro-atherogenic character of the enzyme and expression silencing studies in mice confirmed this activity in various mouse atherosclerosis models (Cyrus et al., 1999; George et al., 2001; Zhao et al., 2005). In human atherosclerotic lesions ALOX15 is expressed only at low levels and thus, the patho-physiological role of the enzyme was challenged (Spanbroek et al., 2003; Gertow et al., 2011).
3.4. Product specificity
The LOX reaction constitutes a special type of lipid peroxidation and differs from non-enzymatic reactions in several respects, such as higher reaction rate, limited substrate selectivity, mechanisms of regulatory interference and the high product specificity. Non-enzymatic lipid peroxidation converts a given substrate to a complex array of primary oxygenation products (mixture of various positional and optical isomers) whereas LOXs usually generate a single product isomer.
3.4.1. Product specificity with polyenoic acids
ALOX15 oxygenates the naturally occurring polyenoic fatty acids predominantly at the n-6 position and this reaction requires initial hydrogen abstraction from the n-8 carbon atom. Linoleic acid is converted to 13-HpODE, whereas arachidonic acid is oxidized at C15 forming 15S-HpETE. Interestingly, small amounts (ranging from 3–10% of the total product mixture) of 12S-HpETE are also formed. When first described, this dual positional specificity (Bryant et al., 1982) of rabbit ALOX15 was quite surprising since singular positional specificity has been hypothesized for all LOX-isoforms. However, later on dual positional specificity was confirmed for the recombinant ALOX15 orthologs of men (Sloane et al., 1991a; Kühn et al., 1993), mice (Bürger et al., 2000), rats (Watanabe and Haeggstrom, 1993) and orangutans (Vogel et al., 2010) and a mechanistic scenario explaining the dual positional specificity was provided. According to this concept fatty acid substrates are aligned at the active site of LOX in such a way that hydrogen abstraction from two different bisallylic methylenes (C13 and C10 of arachidonic acid) is possible (Kuhn et al., 1990c; Kuhn et al., 1991; Ivanov et al., 2010).
For most LOXs initial hydrogen abstraction and subsequent oxygen insertion proceed in an antarafacial manner but for other fatty acid oxygenases a suprafacial relation was determined (Maas and Brash, 1983; Garscha et al., 2007; Wennman et al., 2014). However, the molecular basis for this property is unclear and remains a matter of discussion. Another unsolved problem of the ALOX15 reaction is the direction of the radical rearrangement (2nd elementary reaction in Fig. 1). During non-enzymatic peroxidation of linoleic acid the carbon centered fatty acid radical formed by initial hydrogen abstraction can rearrange in two opposite directions ([+2] rearrangement leading to linoleic acid 13-lipoxygenation vs. [−2] rearrangement leading to 9-lipoxygenation), but for ALOX15 only [+2] rearrangement occurs. Quantum chemical calculations using a completely solvated model of rabbit ALOX15 suggested that both [+2] and [−2] rearrangements are similarly likely (Suardiaz et al., 2013). However, there appear to be differences in the energy barriers for oxygen insertion at the [+2] vs. the [−2] position. It was concluded that oxygen insertion at the [n-2] position (C9 of linoleic acid) may sterically be hindered by surrounding amino acids Leu597, Gln548 and Phe175 (Suardiaz et al., 2013; Suardiaz et al., 2014a; Suardiaz et al., 2014b) so that only C13 oxygenation is possible.
3.4.2. Product specificity with complex substrates
Esterified polyenoic fatty acids (phospholipids, cholesterol esters) are oxygenated with a lower degree of specificity (Kuhn et al., 1990b). Although 13S-HpODE and 15S-HpETEare the major oxygenation products formed when rabbit ALOX15 was incubated in vitro with mitochondrial membranes, there is a significant share of unspecific side products, which together may account for up to 30 % of the total product mixture. These side products include (9E,11E)-13-hydroperoxy-9,11-octadecadienoic acid [13-HpODE(E,E)], (10E,12Z)-9-hydroperoxy-10,12-octadecadienoic acid [(9-HpODE)] and (10E,12E)-9-hydroperoxy-10,12-octadecadienoic acid [(9-HpODE(E,E)]. It should be stressed at this point that highly specific product patterns were only observed at short (5–15 min) incubation periods (Belkner et al., 1998). At longer incubation times the product specificity is markedly decreased (Upston et al., 1996; Belkner et al., 1998; Heydeck et al., 2001). The mechanistic basis for the time dependent alterations in product specificity has not been explored in detail but two experimental details need to be considered: i) ALOX15 undergoes suicidal inactivation and long-term incubations in the presence of substrate completely inactivates the enzyme. ii) The hydroperoxy lipids formed by ALOX15 during early stages of long-term incubations may induce free radical mediated secondary reactions leading to an unspecific product pattern. To initiate such secondary reactions redox-active catalysts (metal ions, vitamine E) are required which are constituents of biomembranes and lipoproteins.
3.4.3. Alteration of product specificity by substrate modification
The reaction specificity of ALOX15 is not an absolute enzyme property but depends on the chemistry of the substrate, which impacts substrate alignment at the active site. For instance, 15S-HETE is oxygenated mainly at C15 of the substrates carbon backbone, but methylation of the carboxylic group strongly favors oxygen insertion at C5 (Schwarz et al., 1998). A similar effect was observed when a bulky group (phenyl, t-butyl) was introduced into the methyl tail of the substrate (Walther et al., 2001).
When arachidonic acid isomers differing from each other by the position of their double bonds (4,7,10,13-eicosatetraenoic acid, 5,8,11,14-eicosatetraenoic acid , 6,9,12,15-eicosatetraenoic acid , 7,10,13,16-eicosatetraenoic acid , 8,11,14,17-eicosatetraenoic acid ) were used as substrate for rabbit ALOX15 (Fig. 4) variable product patterns were analyzed (Kuhn et al., 1990c) and the results of these experiments can be summarized as follows: i) 4,7,10,13-eicosatetraenoic acid was oxygenated with a singular positional specificity and oxygen was almost exclusively introduced at C14. ii) 5,8,11,14-eicosatetraenoic acid (native arachidonic acid) was oxygenated with dual positional specificity and 15S-HpETE and 12S-HpETE were the major oxygenation products in a ratio of about 85:15. iii) 6,9,12,15-eicosatetraenoic acid was oxygenated with a pronounced dual positional specificity since oxygen was introduced in similar quatities at C14 (46%) and at C11 (54%). iv) As 4,7,10,13-eicosatetraenoic acid 7,10,13,16-eicosatetraenoic acid was oxidized with singular positional specificity and oxygen was again introduced only at C14. v) As for 5,8,11,14-eicosatetraenoic acid a dual positional specificity was observed for 8,11,14,17-eicosatetraenoic acid oxygenation and oxygen insertion proceeded at C15 (80%) and C10 (20%). This data clearly indicate that the reaction specificity depends on the fatty acid structure, which impacts substrate alignment at the active site (Kuhn et al., 1990c).
Fig. 4. Reaction specificity of rabbit ALOX15 with arachidonic acid isomers.

The different arachidonic acid isomers are aligned at the active site of rabbit ALOX15 in such a way that different bisallylic methylenes are located in close proximity to the enzyme bound non-heme iron so that hydrogen abstraction from these carbon atoms is possible. For instance, for the 4,7,10,13-isomer (ω−7) hydrogen is abstracted only from C12 and oxygen is inserted only at C14 (n+2 radical rearrangement). With this substrate rabbit ALOX15 exhibits a singular positional specificity as indicated by product analysis (GC/MS). With arachidonc acid (5,8,11,14-isomer, ω−6) the iron is located between the bisallylic methylenes C13 and C10 (but closer to C13) and thus, hydrogen can be abstracted from both carbon atoms with strong preference of the C13 bisallylic methylene. Oxygen is then preferentially (85%) inserted at C15 but to a lesser extent (15%) also at C12 (n+2 radical rearrangement in both cases). With this substrate the enzyme exhibits a dual positional specificity. For the 6,9,12,15-isomer (ω−5) an even more pronounced dual positional specificity was observed since oxygen was inserted in similar quantities at C17 and C14.
3.4.4. Alteration of product specificity by site-directed mutagenesis
3.4.4.1. Triad concept of reaction specificity
If the structure of the enzyme substrate complex is important for the reaction specificity, it should be possible to specifically modify the enzyme structure and thus, induce alterations in the reaction specificity. Multiple amino acid sequence alignments of 12- and 15-lipoxygenating LOXs suggested that the amino acids at the positions 416, 417 and 418 of human ALOX15 might be important for the reaction specificity. When smaller residues were introduced at these positions of human ALOX15 12-lipoxygenating enzymes were created (Sloane et al., 1991b). From their data the authors concluded that this region of the primary structure may impact substrate alignment at the active site. Later on similar strategies were employed for rabbit, mouse and rat ALOX15 (Borngraber et al., 1996; Borngraber et al., 1999; Pekarova et al., 2015) and the triad concept of positional specificity of ALOX15 orthologs was developed (Fig. 5). This concept suggests that Phe353, Ile418/Met419 and Ile593 form the bottom of the substrate-binding pocket and that the geometry of their side chains determine how deep a fatty acid may slide into the active site (Ivanov et al., 2010). Alanine-scan mutagenesis of the four candidate specificity determinants (Phe353, Ile418, Met419, and Ile593) indicated that Ile418 and Phe353 are most important for the positional specificity of rabbit ALOX15. In contrast, Ile593 and Met419 are only of minor importance (Borngraber et al., 1999). On the basis of mutagenesis data three regions of the primary structure are important for the positional specificity of ALOX15 orthologs: (i) The region around Ile418 and Met419 (Sloane determinant) (Sloane et al., 1991b; Sloane et al., 1995); (ii) The region around F353 (Borngräber 1 determinant) (Borngraber et al., 1996); and (iii) The region around I593 (Borngräber 2 determinant) (Borngraber et al., 1999). Site-directed mutagenesis studies on 12- and 15-lipoxygenating ALOX15 orthologs of men (Sloane et al., 1995), rabbits (Borngraber et al., 1999), rhesus monkeys (Vogel et al., 2010), orangutans (Vogel et al., 2010), rats (Pekarova et al., 2015) and pigs (Suzuki et al., 1994) support the triad concept. The relative importance of the triad constituents varies for different isoenzymes. For human and orangutan ALOX15 orthologs Phe353 and Ile418 (numbering according to the rabbit ALOX15) play a major role since single mutations of these amino acids to less-space filling residues convert the LOX to an almost completely 12-lipoxygenating enzyme. Consequently, these residues are considered first-order determinants for these enzyme orthologs.
Fig. 5. Triad concept of positional specificity of ALOX15 orthologs.

For ALOX15 orthologs arachidonic acid slides into the substrate-binding pocket with its methyl end ahead and is bound at the active site by hydrophobic interactions and probably by π-π-interactions of the substrates double bonds with aromatic active site amino acids. The amino acids, which align with Phe353, Ile418 and Ile593 of the rabbit enzyme, form the bottom of the substrate binding pocket and the methyl terminus of the fatty acid substrate interacts with the side chains of these amino acids. For the 15-lipoxygenating rabbit ALOX15 these positions are occupied by bulky residues (Phe353, Ile418, Ile593) so that the substrate fatty acid does not penetrate as deep into the substrate-binding pocket (left side of the images). Thus, the bisallylic methylene C13 of the arachidonic acid is bound in close proximity to the catalytic non-heme iron and this alignment results in major 15-lipoxygenation. In 12-lipoxygenating ALOX15 orthologs (mouse, rats, pigs, cattle) either of these positions is occupied by a less space-filling amino acid, which allows the substrate fatty acid to penetrate deeper into the active site (right side of the image) and the black arrows indicate the direction of substrate movement. This movement approaches the bisallylic methylene C10 of arachidonc acid to the non-heme iron so that hydrogen abstraction from C10 becomes possible. In the 12-lipoxygenating panel (right side of the image) the amino acid exchanges are indicated, which lead to alterations in the reaction specificity during in vitro mutagenesis.
3.4.4.2. A-vs-G concept of reaction specificity
Multiple amino acid alignments of various LOXs with known reaction specificity indicate that most S-LOXs contain an Ala at a critical position, whereas R-LOXs contain a Gly instead (Coffa and Brash, 2004; Coffa et al., 2005b; Schneider et al., 2007). Mutagenesis studies on different S-LOXs indicated that an Ala-to-Gly exchange increases the share of R-HETE isomers (Coffa and Brash, 2004; Coffa et al., 2005a). The molecular basis for the observed specificity alterations is not entirely clear, but an impact of the amino acid side chain geometry on intra-enzyme oxygen movement was suggested (Schneider et al., 2007; Newcomer and Brash, 2015). When similar mutagenesis studies were carried out on ALOX15 orthologs form rabbits, men, rhesus monkeys, orangutans and mice only minor alterations in the reaction specificity were observed (Jansen et al., 2011). The major arachidonic acid oxygenation product of the Ala404Gly mutants remained 15S-H(p)ETE, whereas 11R-H(p)ETE only contributed smaller amounts to the product mixture. The zebrafish expresses an unusual 12-lipoxygenating LOX-isoform, which carries a Gly at this critical position. Thus, the enzyme was predicted to exhibit R-lipoxygenating activity. However, when it was expressed as recombinant protein arachidonic acid 12S-lipoxyenation was observed (Haas et al., 2011; Jansen et al., 2011). These data suggest that this LOX-isoform does not follow the Ala-vs-Gly concept.
4. Molecular biology of ALOX15
4.1. Structure of mammalian ALOX15 genes
The human ALOX15 gene is located on the short arm (p13.3) of chromosome 17 in a gene cluster, which also contains the genes encoding for the other LOX isoforms except for ALOX5 (the ALOX5 gene is localized on the long arm of chromosome 10). It spans more than 11 kbp and consists of 14 exons and 13 introns. The corresponding mouse gene (alox15) is localized in a synthenic region on chromosome 11. The promoter region of the human ALOX15 gene was cloned and potential binding sites for transcription factors were identified (Kelavkar et al., 1998). Interestingly, the promoter region does not carry a TATA- nor a CAAT box suggesting a housekeeping character for this gene. However, the tissue-specific expression pattern and the tightly controlled expression regulation on transcriptional and post-transcriptional levels are hardly consistent with this conclusion. When we searched the 5’-flanking region of the human ALOX 15 gene with the Champion ChiP Transcription Factor Database program (http://www.sabiosciences.com/chipqpcrsearch.php?app=TFBS) for potential transcription factor binding sites, we found that 36 of such sites were localized between 20 kb upstream and 10kb downstream of the transcription initiation site. A similar search for the 5’-flanking region of mouse and rat alox15 genes only revealed 10 potential binding sites. Interestingly, these sites were conserved between the two rodents. When we compared the lists of potential transcription factor binding sites in the promoter region of the human, mouse and rat LOX15 gene we found that the following transcription factor binding sites were shared by the three genes: GCNF, GCNF-1, GCNF-2, Ahr, NF1. However, it remains unclear whether these binding sites are of any functional relevance.
4.2. Genetic variability of human ALOX15 gene
Completion of the 1000 human genome project (www.1000genomes.org) revealed 78 single nucleotide polymorphisms (global allele frequency >1%) in the ALOX15 gene. Considering the fact that the ALOX15 gene consists of about 11,000 base pairs, an average SNP frequency of one SNP per 150 base pairs was calculated. A genome wide comparison of the SNP frequencies for human genes indicates the average occurrence of one SNP per 100–300 base pairs. Thus, the human ALOX15 gene is characterized by an average genetic variability.
Human ALOX15 consists of 662 amino acids and 94 non-synonymous coding variations are described in the 1000 Human genome database. In addition, eight nonsense mutations have been identified, which are likely to lead to catalytically silent truncated enzyme variants (Horn et al., 2013). His360, His365, His540, His544 and the C-terminal Ile662 function as direct ligand for the catalytic non-heme iron (Gillmor et al., 1997) but neither of these residues was affected by genetic variations. Human ALOX15 exhibits dual positional specificity (see 3.3.1) and the side chains of Phe352, Ile417, Ile592 play a major role for this enzyme property (Ivanov et al., 2010). Analyzing the data obtained in the 1000 Genome database we found a rare variation at Phe352, in which the bulky Phe is exchanged to a less space-filling Leu (rs143365387), which is present at this position in the mouse ortholog. According to the triad concept (Ivanov et al., 2010), this enzyme variant should be a 12-lipoxygenating enzyme species and previous mutagenesis studies confirmed this conclusion (Borngraber et al., 1999). Variations at the other two positions of the triad determinants have not been found (Horn et al., 2013).
Among the 94 non-synonymous coding variations listed in the 1000 Human genome database only two (Pro617Ser, Thr560Met) have a global allele frequency of > 1 % and are therefore classified as SNPs (Horn et al., 2013). In addition to these SNPs three selected rare mutant enzyme variants (Arg205Gln, Gly422Glu, Gly422Arg) have been characterized with respect to their functional properties (Horn et al., 2013). Thr560 and Gly422 are localized inside the core of the protein and the Thr560Met and Gly422Glu mutants are catalytically inactive. In contrast, Arg205 and Pro617 are surface exposed and the Arg205Gln and the Pro617Ser mutants exhibit similar reaction kinetic properties as the wildtype enzyme. Membrane binding upregulates the catalytic activity of ALOX15 but is also required for oxygenation of the membrane lipids (Lankin et al., 1985; Brinckmann et al., 1998). Surface exposed hydrophobic amino acids such as Tyr15, Leu70, Leu71, Lys180 and Leu194 have been implicated in membrane binding (Walther et al., 2002; Walther et al., 2004) and rare naturally occurring mutants of these residues have been described in the 1000 Human genome project (Horn et al., 2013). In silico docking studies and molecular dynamic simulations with different fatty acid substrates suggested that Arg402 and Phe414 (numbering for human ALOX15) might be involved in substrate binding. For Arg402 two rare genetic variations have been identified in the 1000 Genome database [Arg402Gln (TMP_ESP_17_4536752) and Arg402Trp (rs144038526)]. We found that Arg402Trp has a reduced catalytic activity (36 % residual activity) while its reaction specificity was not affected.
4.3. Tissue specific expression of ALOX15 and transcriptional expression regulation
In humans ALOX15 is constitutively expressed at high levels in immature red blood cells, in eosinophils and in airway epithelial cells (Nadel et al., 1991). Lower expression levels have been reported for polymorphonuclear leukocytes of different species (Narumiya et al., 1982; Vanderhoek and Bailey, 1984), alveolar macrophages (Levy et al., 1993), vascular cells (Takayama et al., 1987), uterus (Lei and Rao, 1992), the male reproductive system (Fischer et al., 2005), various parts of the brain (van Leyen et al., 2006; Han et al., 2015) and in atherosclerotic lesions (Yla-Herttuala et al., 1990).
Erythrocytes of various species (men, rabbits, mice, rats) do not express ALOX15. However, during experimental and natural anemia expression of the enzyme is upregulated in immature red cells (Ludwig et al., 1988; Kroschwald et al., 1989; Schewe et al., 1990). In rabbits, anemia-induced ALOX15 expression is not restricted to red cells but was also detected in peripheral monocytes, lung, spleen, kidney and liver, but not in skeletal muscle and various parts of the brain (Trebus et al., 2002). Although the mechanism of this systemic ALOX15 induction remains unclear, anemia induced cytokines might be involved. Erythropoietin, which is strongly induced during experimental anemia, might be a suitable candidate but in vitro incubation of human peripheral monocytes with erythropoietin did not induce ALOX15 expression (Kuhn et al., unpublished data).
Human peripheral blood monocytes do not express ALOX15. However, the Th2-cytokines interleukin-4 and interleukin-13 (IL4, IL13) (Conrad et al., 1992; Nassar et al., 1994) strongly upregulate ALOX15 expression in these cells. In fact, microarray experiments indicated that the ALOX15 gene is the most strongly upregulated gene product of the IL4 response in human peripheral monocytes (Chaitidis et al., 2005). Although the mechanism of IL4 induced upregulation of ALOX15 expression is not completely understood, several constituents of the signal transduction cascade have been identified. Competition assays with an IL4 receptor antagonist suggested involvement of the IL-4/13 cell surface receptor (Brinckmann et al., 1996). Moreover, phosphorylation and acetylation of the transcription factor STAT6 by histone acetyltransferase CREB-binding protein/p300 has been implicated (Shankaranarayanan et al., 2001). The ALOX15 promoter involves putative STAT6 binding sites (Liu et al., 2012) and serial promoter deletion studies and STAT6 binding site mutations suggested their functionality. Additional regulatory events include phosphorylation of Jak2 and Tyk2, p38 MAPK induced phosphorylation of STAT1 and STAT3 and activation of PKCd (Roy and Cathcart, 1998; Xu et al., 2003; Xu et al., 2004). More recently, ERK½ protein kinase as well as the transcription factors Elk1, Egr-1 and CREB have been implicated in the IL13 induced signaling cascade (Bhattacharjee et al., 2013). Although the IL4- and IL13-induced signaling cascades leading to increased ALOX15 expression share common elements, the signaling pathways are distinct (Bhattacharjee et al., 2013). Interestingly, IL4 does not induce ALOX15 expression in all peripheral monocytes since 10–40% of cells remain ALOX15 negative (Kuhn and O’Donnell, 2006). The reasons for this heterogeneity are unclear, but may be related to the maturation stage of the cells and/or their metabolic states (Tsao et al., 2014). IL4 does also induce upregulation of ALOX15 expression in A549 airway epithelial cells (Brinckmann et al., 1996) and orbital fibroblasts (Chen et al., 2006). In A549 cells, the Ku antigen, which is induced in response to IL4/13 stimulation, binds to the ALOX15 promoter and induces expression of ALOX15 (Kelavkar et al., 2000). However, this is clearly not the only mechanism of IL4/13 induced transcriptional upregulation of ALOX15. In a recent study (Han et al., 2014) a role of histone H3 methylation was suggested. Following IL4 stimulation demethylation of H3 was observed and this reaction was catalyzed by the H3K27me2/3-specific demethylase UTX. In fact, siRNA induced expression silencing of UTX attenuated IL4-induced ALOX15 expression. These data indicate that epigenetic processes are involved in IL4-induced expression regulation of the ALOX15 gene.
ALOX15 expression is silenced on transcriptional levels in cancer cells and can be reactivated by histone deacetylase inhibitors (Zuo et al., 2009). Although the molecular basis for transcriptional repression is not entirely clear here again histone modification has been implicated (Zuo et al., 2008). More detailed studies on the underlying mechanism suggested that the nucleosome remodeling and histone deacetylase repression complex (NuRD) may play a critical role. In cancer cells NuRD is recruited to the ALOX15 promoter and expression silencing of NuRD components activated ALOX15 expression. Thus, ALOX15 expression can be silenced on epigenetic levels and histone deacetylases can activate transcription of the ALOX15 gene by interfering with NuRD recruitment (Zuo et al., 2009).
The tissue specific expression of the mouse ALOX15 ortholog has not been well characterized. The major cellular sources of this enzyme are residential mouse peritoneal macrophages. Interestingly, thioglycollate elicitation in vivo decreased the share of alox15 positive cells to about 10% (Kuhn and O’Donnell, 2006). On the other hand, murine peripheral monocytes, alveolar macrophages and bone marrow derived macrophages express alox15 only at low levels (Kuhn and O’Donnell, 2006). These data suggest that alox15 might selectively be induced by mechanisms specific for the peritoneal cavity. On the other hand, in human peritoneal macrophages, which were prepared from human ascitis puncture fluid, we did not obtain any evidence for dominant ALOX15 expression (Kuhn, unpublished data). In humans (Nadel et al., 1991) and cattle (De Marzo et al., 1992) ALOX15 is high level expressed in bronchoepithelial cells but in mice alox15 expression in the airway epithelium is much lower. Thus, there are clearly species-specific differences in the tissue-specific expression patterns of ALOX15 orthologs but the mechanistic details for the differences remain elusive.
4.4. ALOX15 mRNA
In 1987 an initial report partly characterizing the cDNA of rabbit ALOX15 was published (Thiele et al., 1987) and its complete primary structure was released in 1989 (Fleming et al., 1989). The sequence comprises some 3600 bases and involves an open reading frame, which encodes for 663 amino acids. Like the human ALOX5 cDNA (Matsumoto et al., 1988) the human and rabbit ALOX15 messengers contain a rather short (28 bases) 5’-untranslated region (5’-UTR). In contrast, the 3’-UTR is much longer (almost 1600 bases for the rabbit enzyme) and involves a cytidine-rich repetitive motif (ten consecutive copies with the consensus sequence C4PuC3TCTTC4AAG) localized in close proximity to the stop codon. This sequence motif was named differentiation control element (DICE) since it has been implicated in maturation-dependent expression regulation of the enzyme (Reimann et al., 2002; Messias et al., 2006).
The human ALOX15 mRNA (Table 2) comprises some 2700 bases and thus, is considerably shorter than the rabbit messenger. The major reason for this is the relatively short (704 bases) 3’-UTR, which only contains a truncated version of the DICE sequence. The open reading frame of human ALOX15 mRNA encodes for 662 amino acids. The difference in the amino acid sequence between rabbit and human ALOX15 is due to an insertion of a Glu residue in the rabbit sequence, which is localized in the unstructured loop region interconnecting the two domains of the enzyme (see 5.2.). As for the rabbit ortholog the 5’-UTR is rather short (15 bases). For the human enzyme an alternative transcript has been suggested (Table 2), which encodes for a N-terminally elongated variant of the protein. On the genomic level the coding information for this N-terminal extra peptide is localized in the short 5’-UTR of the normal transcript and in an additional exon (exon 0) localized some 540 base pairs upstream from the original CAP-site. The biological relevance of this alternative transcript and of the corresponding protein has not been explored.
Table 2.
Structural comparison of rabbit, human, mouse and rat ALOX15 cDNA.
| Parameter | Rabbit | Human | Mouse | Rat |
|---|---|---|---|---|
| cDNA size (bases) | 3614 | 2707 | 2414 | 2707 |
| Encoded amino acids | 663 | 662 | 663 | 663 |
| Amino acid identity (%) | 81 | 100 | 74 | 75 |
| 5’-UTR (bases) | 28 | 15 | 26 | 6 |
| 3’-UTR (bases) | 1595 | 704 | 396 | 218 |
| Elongated transcript | − | + | − | − |
| Presence of DICE | ++ | + | − | − |
The murine alox15 messengers (mouse, rat) are very similar to each other and resemble the human ALOX15 mRNA in size and composition. The 3’-UTR is even smaller than that of the human messenger and the DICE element is structurally not conserved. The degree of amino acid identity to the human ALOX15 is only 75% and some of the observed amino acid differences are responsible for the different reaction specificity of the murine enzymes.
4.5. Translational regulation of ALOX15 expression
ALOX15 mRNA is present in young rabbit reticulocytes but no functional enzyme can be detected in these cells (Thiele et al., 1982). As mechanistic basis for this unusual observation regulatory proteins have been suggested, which bind to the DICE sequence localized in the 3’-UTR of the ALOX15 mRNA. In vitro translation assays indicated that protein binding of regulatory proteins to DICE prevents translation of the mRNA (Ostareck-Lederer et al., 1994) and the regulatory proteins have been identified as hnRNP K and hnRNP E1 (Ostareck et al., 1997). Transfection of the two proteins into HeLa cells silenced the translation of reporter mRNAs carrying the repetitive element of the rabbit ALOX15 mRNA in their 3’-untranslated region (Ostareck et al., 1997). Silenced LOX mRNA specifically co-immunoprecipitated with hnRNP K and addition of recombinant hnRNP K and/or hnRNP E1 causes inhibition of 80S ribosome assembly on the ALOX15 mRNA. These data suggest a specific cytoplasmic function for hnRNPs as translational suppressor proteins in early rabbit reticulocyte development. In later stages of red cell maturation these regulatory proteins may then be degraded proteolytically and functional ALOX15 is expressed. Similar translational control mechanisms have recently been described for lipopolysaccharide induced toll-like receptor 4 signaling (Liepelt et al., 2014). For a long time it remained unclear whether similar translational regulation may also occur in human systems since the DICE element is only present as 4-fold repetitive version in the 3’-UTR of human ALOX15 mRNA. However, more recent mechanistic studies confirmed translational regulation of human ALOX15 mRNA in the erythroid cell line K562 (Naarmann et al., 2008; Naarmann et al., 2010), which involves the DEAD-box RNA helicase 6 (DDX6). This RNA helicase specifically interacts with hnRNP K/E1 in a DICE-dependent manner and was co-localized with ALOX15 mRNA to P-body-like RNP granules. These data suggest that in premature human erythroid cells translational silencing of ALOX15 mRNA is maintained by DDX6 mediated storage in ribonuclear protein granules. Similar translational control mechanisms may be assumed for IL4-treated human umbilical vein endothelial cells (HUVECs). In vitro cultured HUVECs do not express ALOX15. However, after 3 days of IL4 exposure ALOX15 mRNA was detected (RT-PCR) but no functional ALOX15 protein could be found (Lee et al., 2001).
Multiple nucleotide alignments of the 3’-UTR of rabbit, mouse and rat ALOX15 suggested absence of the DICE element in the murine messengers and this opens the question whether translational expression regulation of murine alox15 orthologs follows similar maturation dependent kinetics as shown for rabbit and human orthologs. More recent binding studies exploring the minimal consensus sequence required for hnRNP K/E binding to RNA indicated that the complete DICE sequence is not needed for efficient binding (Moritz et al., 2014). Instead, 3–6 repetitive CCCC or UCCC elements appear to function as minimal binding sequence with binding constants in the nM range. When we inspected the 3’-UTRs of mouse and rat alox15 mRNA we confirmed the existence of several CCCC and UCCC repeats in the two messengers (Fig. 6) but it remains to be explored whether these repetitive sequences are of functional relevance.
Fig. 6. Presence of 4-fold C repeats in the 3’-UTR of murine ALOX15 mRNA.

The murine alox15 mRNAs do not contain the repetitive DICE element, which has been implicated in translational regulation of the rabbit and human ALOX15 mRNA. Instead on the genomic level various 4-fold C-repeats (CCCC or TCCC) are present and these sequences may functionally substitute for the lacking DICE element.
5. Structural biology of ALOX15 and comparison with other LOX isoforms
5.1. Protein-chemical properties of ALOX15
5.1.1. Amino acid composition, molecular weight and isoelectric point
Human ALOX15 consist of a single polypeptide chain (662 amino acids) and has a molecular weight of ~75 kDa. The enzyme contains 11 cysteine residues but no disulfide bridge. The primary structure of the human enzyme shares a high degree (75–85%) of sequence identity with the corresponding orthologs of other mammals, which includes the archaic human subspecies H. neandertalensis and H.denisovan (Chaitidis et al., 2013; Adel et al., 2015). For native and recombinant rabbit ALOX15 an isoelectric point (pI) of 5.50 (Rapoport et al., 1979) was determined but the human ortholog has an experimental pI of 5.85 (Kühn et al., 1993).
5.1.2. Iron content, iron ligand sphere and functional role of non-heme-iron
As fatty acid dioxygenases ALOX15 orthologs contain one mole non-heme iron per mole enzyme. During the catalytic cycle the iron shuttles between its ferric and ferrous form. The iron ligand sphere consists of 1st- and 2nd order ligands. In rabbit ALOX15 four histidines (His361, His366, His541, His545), the C-terminal Ile and a water molecule (alternatively a hydroxyl ion) constitute the primary iron ligands (Fig. 7A) and mutagenesis studies suggested their functionality. Extended X-ray absorption fine structure spectroscopy suggested a distorted octahedral iron ligand sphere for rabbit ALOX15 (Kuban et al., 1998), which was consistent with the crystal structure (Gillmor et al., 1997; Choi et al., 2008). According to these data His361 and the C-terminal Ile663 determine octahedron’s longitudinal axis, whereas His366, His541, His545 and a water molecule (hydroxyl ion) may constitute the edges of the octahedron’s ground square. The second order iron ligands, particularly Glu357 and Gln548 hydrogen-bridge the first order ligands. Gln548Leu exchange, which disrupts the hydrogen bond network, induced a loss in catalytic activity suggesting that this mutation might alter the structure of the iron cluster (Ivanov et al., 2004).
Fig. 7. Structural properties of rabbit ALOX15.

(A) Iron ligand sphere of rabbit ALOX15. Four histidines (His361, His366, His541, His545), the N-terminal Ile/663 and a water molecule are the 1st order iron ligands of rabbit ALOX15 (B). Overlay of the two structures (ligand-free conformer, ligand-bound conformer) of the rabbit ALOX15. Ligand-free conformer A is indicated in grey and ligand-bound structure (conformer B) in yellow. The non-heme iron is also shown. It can be seen that helix 2 is strongly dislocated upon ligand binding by about 12 Å. Rotation of the active site helix 18 can also be seen. (C) Crystal structure of rabbit ALOX15 dimers. In the crystals rabbit ALOX15 forms heterodimers consisting of a ligand-free (conformer A) and a ligand-bound (conformer B) monomer. Inset: The residues contributing to the interaction between the two monomers are indicated and a number of leucine and tryptophane residues contribute. The program VMD 1.4.8 version (University of Illinois) and the coordinates of rabbit LOX complex (PDB code: 2P0M) were used to create these images.
When LOXs are isolated from native and/or recombinant sources the iron is present as ferrous ion and in this configuration the enzyme is catalytically inactive. To initiate fatty acid oxygenation the enzyme must be activated to a ferric form (probably a Fe3+-OH−-complex), which is capable of catalyzing hydrogen abstraction from a bisallylic methylene group of the fatty acid substrate. During this reaction the hydroxyl anion abstracts a hydrogen atom from the fatty acid. However, the electron is not tightly bound at the proton but tunnels to the ferric iron in a concerted proton tunneling-electron tunneling process (Lehnert and Solomon, 2003).
5.1.3. Lack of post-translational modification and proteolysis resistance
When rabbit ALOX15 was first purified from immature red blood cells it was suggested that the enzyme contains 5% of its molecular weight as carbohydrates (Rapoport et al., 1979), but this conclusion could not be confirmed in later experiments. The recombinant enzyme expressed in E. coli is fully active and these data suggest that glycosylation may not be required for its catalytic activity. Although the primary structure contains a number of potential phosphorylation sites there is no evidence that protein phosphorylation/dephosphorylation constitutes a regulatory element of cellular ALOX15 activity. ALOX15 is capable of binding to biomembranes, but there is no lipid anchor attached to the protein. There is no experimental evidence for sizeable N-myristoylation, S-palmitoylation, farnesylation or geranylation of the enzyme.
Purified rabbit ALOX15 is surprisingly stable when digested with proteases in vitro. Even long-term incubations (up to two hours) of purified rabbit ALOX15 with 0.5% trypsin did only lead to minor impairment of the catalytic activity with absolute conservation of the product specificity (Wiesner and Kuhn, unpublished data). On the other hand, SDS-PAGE of the cleavage mixture indicated almost complete disappearance of the native enzyme and the formation of a large number of proteolytic cleavage peptides indicating a high degree of proteolysis. These data suggest that the enzyme rapidly undergoes proteolysis but that the 3D-structure is sufficiently stabilized by non-covalent interactions to allow specific fatty acid oxygenation.
5.2. Crystal structure of ALOX15
5.2.1. Overall shape and global structure of rabbit ALOX15
Crystallization of the rabbit ALOX15 has already been reported in 1990 (Sloane et al., 1990), but its 3D-structure was only solved 7 years later as enzyme inhibitor complex (Gillmor et al., 1997). Although an overall resolution of 2.4 Å was reached important structural elements have not been specified in the original electron density map. More recent re-evaluation of the original X-ray coordinates suggested a mixture of two structurally distinct conformers: i) A ligand-free conformer (conformer A), in which the central cavity of the enzyme harboring the non-heme iron was empty. ii) A ligand-bound conformer (conformer B), in which the central cavity of the enzyme was occupied by the exogenous inhibitor (Choi et al., 2008). Monomeric rabbit ALOX15 has a cylindrical shape (height of 10 nm) with an elliptic ground square (longer diameter 6.1 nm, shorter one of 4.5 nm).
5.2.2. N-terminal β -barrel domain
The single polypeptide chain of rabbit ALOX15 folds into a two-domain structure: a small N-terminal β-barrel domain and a larger mostly helical catalytic domain. The small N-terminal domain comprises 110 amino acids and is composed of 8 β-sheets. Its size and structure are similar to the C-terminal β-barrel domains of mammalian lipases (Winkler et al., 1990), which have been implicated in membrane binding of these enzymes (May et al., 2000). Gene technical deletion of the β-barrel domain significantly impaired but did not abolish the membrane binding capacity of the recombinant enzyme (Walther et al., 2002). More detailed site-directed mutagenesis suggested that surface-exposed hydrophobic amino acids in both domains are involved in membrane binding (Walther et al., 2004; Walther et al., 2011). The N-terminal ß-barrel domain of ALOX15 has not only been implicated in membrane binding but may also constitute a regulator of the catalytic activity. Gene technical truncation of this structural subunit of the rabbit ALOX15, human ALOX15 and other relevant ortologs resulted in reduction of the catalytic efficiency of arachidonic acid oxygenation and more rapid suicidal inactivation of the mutants. These data suggested a potential role of the N-terminal β-barrel domain in the regulation of catalytic turnover (Romanov et al., 2006; Walther et al., 2011). The two structural subunits of ALOX15 are covalently interconnected by a flexible oligopeptide, which might allow interdomain movement.
5.2.3. C-terminal catalytic domain
The C-terminal catalytic domain of ALOX15 (residues 114–663) consists of 21 helices, which are interrupted by a small β-sheet sub-domain (Gillmor et al., 1997). The center of the C-terminal domain involves two long helices, which carry four of the five protein iron ligands. The putative substrate-binding pocket is a boot-shaped cavity, which is accessible from the protein surface. Arg403 lines the entrance into the substrate-binding pocket and the side chains of Phe353, Ile418, and Ile593 define the bottom of the active site. The walls of the substrate-binding cavity are lined by 23 predominantly hydrophobic amino acids from six different helices (α2, α7, α9, α10, α16, and α18) and by the loop connecting the helices α9 and α10. In the ligand-free conformer (conformer A), the side chain of Leu597, which is localized at the C-terminus of helix α18 protrudes into the substrate-binding cavity limiting its depth and volume. According to the X-ray data (Choi et al., 2008) ligand binding at the active site induces two major structural alterations: i) The external helix α2 is dislocated (Fig. 7B). ii) Helix α18 including Leu597 retreats from the cavity enlarging the volume of the substrate-binding pocket. Unfortunately, for rabbit and human ALOX15 the functional importance of Leu597 has not been studied in detail. Combined quantum mechanics/molecular mechanics calculations as well as molecular dynamics simulations for the Leu597Ala mutant suggested the possibility of an alternative binding mode of the substrate (arachidonic acid), which was associated with an inversion of the stereochemistry of 15-lipoxygenation from S to R (Suardiaz et al., 2014b), However, preliminary site-directed mutagenesis studies did not confirm this conclusion (Ivanov, unpublished data). In fact, in these experiments the major lipoxygenation product of arachidonic acid was 15S-H(p)ETE.
5.2.4. Intraenzyme oxygen movement
Dioxygen is a small apolar molecule, which freely penetrates biomembranes. However, its water solubility of rather low and thus, complex living organisms require special oxygen carrier proteins for effective oxygen transport. For a long time it was believed that because of its chemical properties (small size, hydrophobic character) oxygen is more or less uniformly distributed in proteins (Calhoun et al., 1983). However, more recent studies suggested asymmetric oxygen distribution inside a number of oxygen metabolizing enzymes (Scott and Gibson, 1997; Chu et al., 2000; Ostermann et al., 2000) including soybean LOX1 (Minor et al., 1996; Knapp et al., 2001; Knapp and Klinman, 2003) and the existence of preformed oxygen diffusion channels. To identify potential routes for oxygen diffusion in rabbit ALOX15 an experimental strategy involving structural modeling, molecular dynamics simulations, site-directed mutagenesis and kinetic measurements were applied (Saam et al., 2007). For the substrate free enzyme a high oxygen affinity area (global energetic minimum of the probability of oxygen occupancy) was identified and this region was localized opposite to the non-heme iron. Interestingly, this area was localized in close proximity to carbon atom 15 of arachidonic acid in a three-dimensional model of the enzyme-substrate complex. The energetic computations revealed that the probability of oxygen occupancy in this high oxygen affinity area is 7-fold higher than in the surrounding of carbon 11 of the arachidonic acid backbone, which is consistent with preferential arachidonic acid 15-lipoxygenation. Three major channels interconnecting the protein surface with the high oxygen affinity area have been suggested for the substrate-free protein. The first channel starts at the bottom of the substrate-binding pocket and is completely closed in the enzyme–substrate complex. The second channel follows the substrate-binding pocket and is also blocked upon substrate binding. The third channel interconnects the opposite side of the protein surface with the high oxygen affinity area and remains open after substrate binding. To provide experimental evidence for the functionality of this potential oxygen access channel, Leu367, which appears to be critical for oxygen conductivity of this path, was mutated to a more space filling Phe. For the Leu367Phe mutant a 10-fold increased Michaelis constant for oxygen and 20-fold reduction in the catalytic efficiency (kcat/KMO2) of the enzyme was observed but there was no alteration in the reaction specificity (Saam et al., 2007). These data suggest that oxygen penetration from the protein periphery into the high oxygen affinity area may be impaired by Leu367Phe exchange but that the mutant enzyme completely controlled the stereochemistry of the reaction. The steric effects of Leu367Phe exchange together with a reorientation of the hydrogen-bonding network of associated water molecules have been suggested as molecular basis for impaired oxygen conductivity of this channel (Saam et al., 2007). Although oxygen access channels have been suggested for other LOX-isoforms (Knapp et al., 2001; Newcomer and Brash, 2015) more work in needed to precisely define them.
5.3. Structural flexibility of rabbit ALOX15
X-ray crystallography provides valuable information on the overall structure of biomacromolecules but in crystals the motional flexibility is limited. On the other hand, in aqueous solutions the macromolecules are more flexible and this allows structural rearrangement in response to alterations of the external milieu (temperature, pH, protein concentration) and/or in response to presence or absence of effector molecules (substrates, inhibitors, activators). A number of spectroscopic studies, such as small angle X-ray scattering (Hammel et al., 2004; Shang et al., 2011), dynamic fluorescence studies and fluorescence resonance energy transfer measurements (Mei et al., 2008; Di Venere et al., 2013), as well as various computational methods, such as translation, libration and screw rotation motion detection (Shang et al., 2011), molecular dynamics simulations and quantum mechanics/molecular mechanics calculations (Suardiaz et al., 2014b) have been applied to explore the motional flexibility ALOX15. Taken together, the results suggest a high degree of structural dynamics for rabbit ALOX15 in aqueous solutions. Three elements, which contribute to this structural flexibility (rearrangement upon lgand binding, interdomain movement, protein dimerization) are briefly discussed below.
5.3.1. Structural rearrangements upon substrate/ligand binding
Ligand binding at the active site appears to lead to structural rearrangement of the enzyme. Comparison of the crystal structures of conformer A (ligand free ALOX15) and conformer B (inhibitor RS7 bound at the active site) of rabbit ALOX15 (Choi et al., 2008) suggested that inhibitor binding induces conformational alterations. Helices α2- and α18- are particularly affected. The α18-helix contains Leu597, the side chain of which has been suggested to control the shape and the size of the cavity (Gillmor et al., 1997). In the inhibitor-bound form the side chain is displaced providing space for the ligand. Substrate docking studies and molecular dynamics simulations suggested that displacement of α18-helix is required for proper substrate binding (Toledo et al., 2011). According to these docking studies linoleic acid and arachidonic acid share a similar overall alignment at the active site, but the degree of motional flexibility of the methyl tail is higher for linoleic acid. For both fatty acids molecular dynamics simulations suggested a correlation between motional flexibility of helix α18 and substrate fatty acid binding (Toledo et al., 2010) but experimental confirmation of this hypothesis is pending.
5.3.2. Interdomain movement
The two domains of rabbit ALOX15 are covalently interconnected by a flexible linker peptide, which does not fold into a stable secondary structure. In addition to this covalent linkage the two domains contact each other via a 1600 Å2 interdomain contact plane and the majority of the amino acids present on both sites of the contact plane carry hydrophobic side chains. Initial small angle X-ray scattering (SAXS) measurements on aqueous solutions of wild-type rabbit ALOX15 and its N-terminal truncation mutant, which lacks the N-terminal ß-barrel domain, suggested a high degree of interdomain movement (Hammel et al., 2004). In fact, superposition of the crystal structure with the low-resolution SAXS model showed almost perfect alignment of the catalytic domain but revealed a mismatch in the region of the N-terminal β-barrel domain. Such mismatch was not observed for the N-terminal ß-barrel truncation mutant. Since SAXS data may be interpreted in different ways (Putnam et al., 2007) and since no interdomain movement was observed for other LOX isoforms (Dainese et al., 2005) the initial SAXS measurements on rabbit ALOX15 were repeated under variable experimental conditions and the following results were obtained (Shang et al., 2011): i) At pH 6.8 and in the absence of salt interdomain movement was largely repressed and the low resolution SAXS structure did match the crystal structure of the ligand-free conformer. ii) At pH 8.0 and in the presence of salt (200 mM NaCl), the N-terminal domain appears to swings away from the catalytic domain resulting in significant expansion of the molecule. More recent molecular dynamics simulation (Moin et al., 2011) and site directed mutagenesis studies at the interdomain interface (Ivanov et al., 2012) confirmed the principal possibility of interdomain motion.
5.3.3. Dimerization
In the crystal structure of the rabbit ALOX15–inhibitor complex (PDB 2P0M entry) the enzyme was present as protein dimer, in which the hydrophobic Leu179, Leu183, Leu188, and Leu192 form a cluster, which resembles a leucine-zipper like motif (Alber, 1992). In addition, Trp181 of the α2-helix, which has been previously identified as a key membrane-binding determinant of the C-terminal domain (Walther et al., 2002) and His585 of helix α18 contribute to the adhesive forces between two monomers (Fig. 7C). Modelling (Shang et al., 2011) of the monomer-monomer interface revealed that association of conformer A (unliganded) with conformer B (liganded) has a solvation free energy of −25 kcal/mol and hence, is thermodynamically favored over other monomer combinations (conformer A + conformer A, conformer B + conformer B).
To explore whether ALOX15 dimers may also occur in aqueous solutions SAXS measurements were carried out under different experimental conditions. Although at low protein concentrations (< 1mg/ml), at low ionic strength and low pH (6.8) ALOX15 is mainly present as hydrated monomer, SAXS experiments suggested a monomer–dimer equilibrium (Fig. 8). At higher protein concentrations (>1 mg/ml), in the presence of salt (200 mM NaCl) and at higher pH (8.0) the monomer-dimer equilibrium was strongly shifted toward ALOX15 dimers (Shang et al., 2011). In addition, the impact of an active site ligand (13S-HODE) on the monomer-dimer equilibrium was tested (Fig. 8). When we compared the experimental SAXS patterns obtained for ligand-free and ligand-containing rabbit ALOX15 with theoretical patterns calculated from the crystal structures, we observed significant discrepancies (Ivanov et al., 2012), which allowed the following conclusions: (i) Ligand-free rabbit ALOX15 is present in aqueous solutions predominantly (85%) as protein monomers. ii) Addition of 13S-HODE shifted the monomer-dimer equilibrium strongly towards protein dimers. In fact, in the presence of 13S-HODE 95% of the enzyme was present as dimers. (iii) In the refined dimer model, the N-terminal domain appears to swing away from catalytic subunit. Thus, interdomain movement might be considered a prerequisite for enzyme dimerization. (iv) In contrast to wild-type ALOX15 the presence of 13S-HODE did hardly induce dimerization for the Trp181Glu+His585Glu double mutant. With this enzyme variant oligomer formation was observed (Fig. 8). These data suggest the importance of Trp181 and His585 for intermonomer interaction.
Fig. 8: Ligand induced oligomerization of rabbit ALOX15 and impact of interdomain interface mutants on enzyme oligomerization.

Low-resolution models of rabbit ALOX15 were calculated on the bases of small angle X-ray scattering data in the absence (upper panel) and presence (lower panel) of a 10-fold molar excess of 13S-HODE as active site ligand. Structural models of ALOX15 dimers were generated by rigid body refinement applying P2 symmetry on conformers A keeping the intermonomer interface as shown in the crystal structure (PDB code: 2P0M). For the tetramer model of the Leu183Glu+Leu192Glu mutants in complex with 13(S)-HODE P222 symmetry was considered. The four catalytic domains are shown in gray, N-terminal domains in red and the α2 helixes in orange. The tetrameric structure shows significant difference to the dimers with P2 symmetry. Images were modified according to (Ivanov et al., 2012).
6. Biological role of ALOX15
6.1. Physiological roles of ALOX15
6.1.1. Principal mechanisms for exhibiting bioactivity
The classical concept of the arachidonic acid cascade suggests that eicosanoid synthesizing enzymes such as ALOX5 and COX-isoforms exhibit their biological functions via the formation of bioactive signaling molecules (prostaglandins, leukotrienes, lipoxins etc.). This may also be the case for ALOX15 since a number of biological activities have been reported for products of the ALOX15 pathway (Kuhn, 1996; Powell and Rokach, 2015). However, there are at least two additional scenarios, by which ALOX15 orthologs may exhibit their bioactivities (Fig. 9A): i) Structural and functional modification of complex lipid-protein-assemblies such as biomembranes and lipoproteins. Since ALOX15 orthologs are capable of oxidizing polyenoic fatty acids containing ester lipids even if they are incorporated in biomembranes and lipoproteins (see 3.2.3.) the enzymes have been implicated in the process of restructuring cellular organelles and in the metabolism of lipoproteins. ii) As intracellular lipid peroxidizing enzyme ALOX15 is involved as pro-oxidant in the regulation of the cellular redox equilibrium. Since the cellular redox state strongly impacts the activity of redox dependent transcription factors catalytic activity of ALOX15 orthologs must be considered as regulator of cellular gene expression and thus, their catalytic activity may alter the cellular phenotype. In fact, transfection-induced overexpression of ALOX15 in U937 cells profoundly alters the gene expression pattern of these cells (data have been deposited in the NCBI GEO database, accession number GSE8173). More detailed information on the biological relevance of ALOX15 orthologs is provided in Fig. 9B and each of these topics is briefly discussed below.
Fig. 9. Biological relevance of ALOX15.
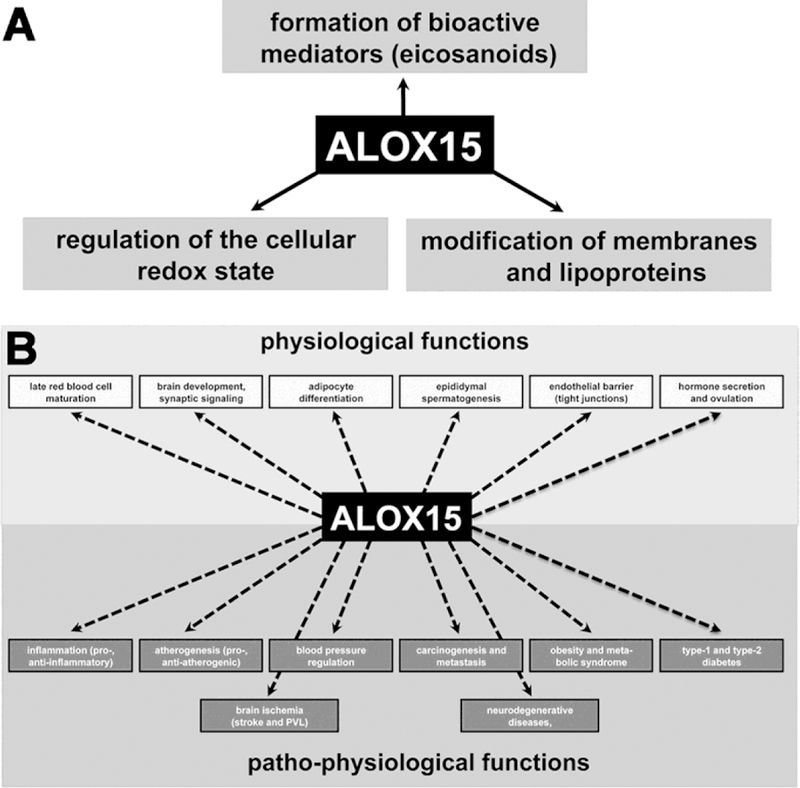
(A) Principle mechanisms, by which ALOX15 orthologs exhibit their bioactivity. (B) Physiological and patho-physiological processes, in which ALOX15 orthologs have been implicated.
6.1.2. Red cell maturation
Normal erythrocytes do not contain sizable amounts of ALOX15. However, when erythropoiesis is challenged (Rapoport and Schewe, 1986) by repeated bleeding or by forced hemolysis (phenylhydrazine injection) immature red blood cells (reticulocytes) express large amounts of ALOX15. In fact, rabbit stress reticulocytes are the richest natural source of ALOX15 and model calculations suggested that up to 4 mg of ALOX15 protein is present in 1 ml of packed rabbit reticulocytes (Rapoport et al., 1979). Interestingly, the enzyme is almost undetectable in young stress reticulocytes but during in vitro maturation of these cells expression of the enzyme parallels the maturational decline of cellular respiration (Hohne et al., 1983). These anti-parallel biological dynamics (increase in ALOX15 expression vs. decrease in cellular respiration) and the observation that isolated ALOX15 in vitro induces structural decomposition of rat liver mitochondria (Schewe et al., 1977) implicated ALOX15 in maturational breakdown of mitochondria during late erythopoiesis. Consistent with this hypothesis, oxidation products formed by ALOX15 were found in reticulocyte membranes (Kuhn and Brash, 1990). In vitro studies with the isolated rabbit ALOX15 showed that the enzyme does not just bind to mitochondrial and other organelle membranes and oxidizes the membrane lipids (Kuhn et al., 1990b), but also directly permeabilizes them, forming pores in the membrane (van Leyen et al., 1998). Freshly isolated reticulocytes matured in vitro degrade their mitochondria more slowly in the presence of a LOX inhibitor (Rapoport and Schewe, 1986; Grullich et al., 2001; Blanc et al., 2007). However, functional inactivation of the alox15 gene in mice did not lead to major functional defects in erythropoiesis (Sun and Funk, 1996). This negative outcome of the expression silencing strategy may be explained by the fact that in addition to ALOX15-dependent intracellular degradation (Rapoport and Schewe, 1986), there are at least two alternative mechanisms for mitochondrial degradation in erythroid cells (Gronowicz et al., 1984): i) engulfment and digestion within autophagic vacuoles (Kent et al., 1966), and ii) exocytosis of mitochondria as exosomes (Griffiths et al., 2012). More detailed studies on erythropoiesis under both stressed and non-stressed conditions are needed to explore the relative contribution of each of the mitochondrial degradation pathways.
6.1.3. Brain development and synaptic signaling
ALOX15 orthologs are expressed in both rat (Watanabe et al., 1993) and canine brain (Nishiyama et al., 1992) but the expression levels are rather low. 12-H(p)ETE, the major arachidonic acid oxygenation product of the murine alox15 orthologs, has been implicated as signaling mediator in axon guidance suggesting a direct function of the enzyme in brain development (de La Houssaye et al., 1999; Ross et al., 2000; Mikule et al., 2002; Nishiyama et al., 2003). 12-H(p)ETE functions as second messenger (Piomelli, 1991), modulating the signals of other stimuli, such as the axon guidance molecule semaphorin 3A (Pekcec et al., 2013).
In addition to these effects in brain development ALOX15 has been implicated in synaptic signaling modulating long-term depression (Normandin et al., 1996; Feinmark et al., 2003; DeCostanzo et al., 2010) and long-term potentiation (Piomelli et al., 1987a; Piomelli et al., 1987b), which are key elements of interneuronal communication. However, alox15 knockout mice do not show any behavioral defects (Sun and Funk, 1996), suggesting that either these effects can be bypassed, or the knockouts have found ways to compensate the alox15 defect.
6.1.4. Adipocyte differentiation
White adipocytes, the major cell type of adipose tissue, differentiate from mesenchymal stem cells and a complex network of regulatory mediators controls adipogenesis (Gustafson et al., 2015). Initial evidence for the involvement of LOX pathways in adipocyte differentiation originated from in vitro maturation experiments of primary rat preadipocytes (Shillabeer et al., 1998). In these in vitro maturation experiments inhibitor studies suggested that metabolites of both, the cyclooxygenase and lipoxygenase pathways, regulate preadipocyte differentiation. However, it remained unclear which of the different LOX-isoforms were involved and the mechanistic basis for LOX-involvement has not been explored (Shillabeer et al., 1998). 3T3-L1 cells are frequently employed as cellular model of adipocyte differentiation (Madsen et al., 2003) and the ALOX15 inhibitor baicalein inhibits adipocyte maturation of 3T3-L1 cells in vitro (Hallenborg et al., 2010). Although the mechanism of this effect has not been studied in detail peroxisome proliferation activating receptor gamma (PPARγ) has been implicated. PPARgamma plays an important role in this regulatory network of adipogenesis (Lefterova et al., 2014) and co-activators of this nuclear receptor play a major role in adipogenesis. 13-HODE and 15-HETE, the major oxygenation products of ALOX15 catalyzed oxygenation of linoleic acid and arachidonic acid respectively, have been suggested as PPAR-gamma co-activators (Huang et al., 1999; Shappell et al., 2001). PPAR gamma agonists prevented baicalein-induced inhibition of adipogenesis (Hallenborg et al., 2010) and IL4-dependent induction of ALOX15 expression upregulated the cellular activity of PPAR gamma (Huang et al., 1999). These findings suggested a physiological role of ALOX15 in the generation of endogenous PPAR-gamma ligands. Although the required co-activator concentrations are rather high and the specificity of this effect has not been studied in detail, these findings suggest a physiological role of ALOX15 in PPAR-gamma dependent adipocyte maturation. Moreover, 15-HETE induced angiogenesis in adipose tissue has been implicated in tissue growth (Soumya et al., 2013).
In addition to ALOX15 other LOX-isoforms, such as ALOXE3, ALOX12 and ALOX5 have been implicated in adipogenesis (Cole et al., 2013). For instance, forced expression of ALOXE3 or addition of ALOXE3 products (hepoxilins) stimulated adipogenesis and RNAi-mediated expression knockdown prevented adipocyte differentiation (Hallenborg et al., 2010). Although these data need to be confirmed for in vivo adipogenesis the results suggested that ALOXE3 might constitute an important player in this process and that specific ALOXE3 inhibitors might be useful to interfere with this process.
6.1.5. Epididymal spermatogenesis and fertilization
Rat testes microsomes oxidize linoleate and arachidonate derivatives and the corresponding enzyme has been purified to apparent electrophoretic homogeneity (Shahin et al., 1978). More detailed analysis of the reaction products indicated the formation of a 2:1 mixture of 13- and 9-HODE as well as 13-hydroxy-12-oxo-octadec-cis-9-enoic acid. This product mixture suggested the presence of LOX and a hydroperoxide isomerase in the microsomes (Grossman et al., 1979). Although the functional relevance of these products has not been explored they have been implicated in sperm development. Spermatogenesis proceeds according to a complex developmental program in which a diploid progenitor germ cell transforms into highly specialized spermatozoa (Keber et al., 2013). Mammalian spermatozoa complete their morphogenesis in the epididymis and a prominent hallmark of epididymal sperm maturation is the proximal-distal migration down the sperm flagellum of the cytoplasmic droplet. Since alox15 is present in the cytoplasmic droplet the enzyme has been implicated in spermatogenesis (Fischer et al., 2005; Moore et al., 2010). In boars ALOX15 has even been suggested as fertility marker (Lovercamp et al., 2007). Compared with wildtype mice spermatozoa of ALOX15 deficient animals retained the cytoplasmatic droplet in the epididymis suggesting defective sperm maturation. Aberrant epididymal sperm maturation might contribute to the reduced fertility and smaller litter size of alox15 deficient mice (Moore et al., 2010). Except for ALOX15 a secretable phosphoplipase A2 (Pla2g3) has been implicated in epididymal sperm maturation (Sato et al., 2010). Although testicular spermatogenesis in Pla2g3-deficent mice was normal, epididymal spermatozoa displayed hypomotility and their ability to fertilize intact oocytes was markedly impaired. Moreover, the gonads of Pla2g3−/− mice contained less alox15 metabolites when compared with wildtype controls suggesting a concerted action of Pla2g3 and alox15 during late sperm maturation (Sato et al., 2010).
In addition to epididymal sperm maturation alox15 has also been implicated in the degradation of paternal mitochondria after fertilization (Sutovsky et al., 2004). Maternal inheritance of mitochondrial DNA has long been considered a paradox in embryogenesis but recent data clearly document that paternal mitochondria enter oocytes during fertilization but are then targeted for degradation. In this degradation process the ubiquitin-proteasome system and alox15 have been implicated, but alox15 appears not to be essential (Sutovsky et al., 2004).
When a sperm approaches an oocyte during fertilization, the membrane surounding the acrosome (cap-like structure in the anterior half of the sperm’s head) fuses with the plasma membrane of the sperm. This membrane fusion exposes the content of the acrosome releasing a number of enzymes capable of breaking through the egg’s coating envelope (zona pelucida). The acrosome reaction initiates a number of restructuring events within the zona pelucida preventing the penetration of additional sperms. During in vitro fertilization the acrosome reaction can be induced by arachidonic acid and two different unspecific LOX-inhibitors (NDGA, ETYA) partially prevented this reaction. In contrast, inhibitors of the COX pathway were ineffective. This data suggested the involvement of ALOX15 in the acrosome reaction and the formation of 15-HETE was consistent with this conclusion (Lax et al., 1990). However, later experiments did not confirm this hypothesis (Mack et al., 1992).
6.1.6. Regulation of sexual hormone secretion and ovulation
Leydig cells are the main testosterone-producing cell in the male gonades and the luteinizing hormone (LH) formed in the pituitary gland constitutes the major endocrine stimulus of testosterone biosynthesis (Ezcurra and Humaidan, 2014). In vitro studies on testosterone biosyntheseis by isolated rat testis Leydig cells indicated that different LOX-inhibitors (NDGA, ETYA, caffeic acid, esculetin) inhibited LH-stimulated steroid synthesis in a dose-related manner suggesting the involvement of the LOX-pathway in testosterone production (Mele et al., 1997). Although these results confirmed previous data obtained in related experimental setups (Cooke et al., 1984; Dix et al., 1984; Dix et al., 1985) the identity of the implicated LOX-isoforms and the underlying mechanism has not been explored. Similarly, LOX inhibitors prevent the release of LH and prolactin from the anterior pituitary gland suggesting a possible involvement of the lipoxygenase pathway (Conte et al., 1986; Kiesel et al., 1987) but here again, the chemical identity of the LOX-isoform has not been tested. However, prolactin release from pituitary cells was induced by 15-HETE suggesting the involvement of ALOX15 (Rabier et al., 1987; Rabier et al., 1988). In porcine anterior pituitary cells the 12-lipoxygenating porcine ALOX15 is expressed and imunohistochemical double staining suggested a co-localization of the enzyme with luteinizing hormone and follicle-stimulating hormone (Ikawa et al., 1996). When primary cultures of porcine anterior pituitary cells were incubated with 12-HpETE or 13-HpODE a significant increase in the release of these two hormones was observed. Interestingly, 12-HETE and 13-HODE were ineffective suggesting that the cellular redox state may be important for hormone release (Ikawa et al., 1996).
Eicosanoids have been implicated as regulators in ovulation and inhibition of their synthesis results in ovulatory failure. The ALOX15 product 15-HETE was significantly elevated prior to ovulation and systemic administration of the ALOX15 inhibitor NDGA suppressed this process (Downey et al., 1998). Synthesis of 15-HETE by cultured granulosa and theca interna cells was reduced by the presence of the ALOX15 inhibitor NDGA. These results suggest ALOX15 metabolites of arachidonic acid may be involved in ovulation.
6.1.7. Epithelial and endothelial barrier function
The permeability of the arterial endothelium is important for the pathogenesis of vascular diseases and ALOX15 has been implicated in its regulation (Kundumani-Sridharan et al., 2013). 15S-HETE, the major ALOX15 metabolite of arachidonic acid oxygenation, induced endothelial barrier permeability via Src and Pyk2-dependent tyrosine phosphorylation of zonula occludens 2 proteins and their dissociation from the tight junction complexes. Ex vivo studies revealed that exposure of arteries from wildtype mice to arachidonic acid led to Src-Pyk2-dependent zonula occludens-2 tyrosine phosphorylation, tight junction disruption, and macrophage adhesion, whereas the arteries from alox15 knockout mice were protected from these effects. Moreover, high-fat diet induced arterial expression of alox15 led to tight junction disruption and macrophage adhesion but functional inactivation of the alox15 gene prevented these effects. These findings implicated alox15 in high-fat diet-induced endothelial tight junction disruption suggesting a role of the enzyme in arterial barrier function (Kundumani-Sridharan et al., 2013). More detailed mechanistic studies indicated that 15S-HETE, the major alox15 oxygenation product of arachidonic acid, disrupts endothelial tight junctions by stimulating tyrosine phosphorylation of zona occludens-2 proteins, which initiates dissociation from claudins 1/5 (Chattopadhyay et al., 2014). In addition, 15S-HETE enhances phosphorylation of zona occludens-1 proteins phosphorylation via PKCε-mediated MEK1-ERK½ activation, which also causes dissociation from occludin, disrupting vascular endothelial tight junctions (Chattopadhyay et al., 2014). Taken together, these data suggest that alox15 products induce phosphorylation of zonula occludens-1 and −2, which leads to impairment of arterial tight junctions formation disrupting the endothelial barrier function. As alternative mechanism for alox15 dependent induction of endothelial dysfunction phosphorylation of junction adhesion molecule A at Tyr164, Tyr218, and Tyr280 was suggested (Chattopadhyay et al., 2015). This phosphorylation, which involves activation of Src and Pyk2 protein kinases, as well as enhanced expression of xanthinoxidase, induces dissociation of the junction adhesion molecule A from occludin causing disruption of the tight junction (Chattopadhyay et al., 2015). Similar observations were made for retinal endothelial cells (Othman et al., 2013). In this model alox15 metabolites induce endothelial cell barrier dysfunction via NADPH-oxidase dependent mechanisms, which involve suppression of protein tyrosine phosphatase and activation of the VEGF-receptor 2 signaling pathway. Taken together, the involvement of three different oxidizing enzymes (ALOX15, xanthinoxidase, NADPH-oxidase) in vascular endothelial tight junction formation suggest the importance of the cellular redox state for the endothelial barrier function but the detailed mechanisms remain to be explored.
6.1.8. Autophagy
Autophagy is catabolic pathway responsible for the degradation of dysfunctional cellular components via lysosomal degradation (Glick et al., 2010; Zhang, 2013; Lemasters, 2014). The breakdown of cellular components is locally destructive but promotes cellular survival under certain conditions. Autophagy allows simultaneous degradation and recycling of cellular components. During autophagy cytoplasmic constituents are isolated from the rest of the cell within a double-membraned vesicle (autophagosome), which subsequently fuses with a lysosome. There are different forms of autophagy such as macroautophagy, microautophagy and chaperone-mediated autophagy. In the context of disease, autophagy has been considered an adaptive response to stress promoting survival but may also promote cell death and morbidity. Recently, it was shown that peritoneal macrophages prepared from ALOX15 deficient mice contain defective mitochondria and numerous cytoplasmic vacuoles containing electron dense material (Morgan et al., 2015). These data suggest defects in the autophagic pathway or in membrane processing in these cells. If these data can be reproduced in other cells and tissues ALOX15 may play a more general role in autophagy.
6.2. Pathological roles of ALOX15
6.2.1. Inflammation
Of the six human LOX-isoforms ALOX5 (Radmark et al., 2015) and ALOX15 (Kuhn and O’Donnell, 2006) have been implicated in the pathogenesis of inflammation. ALOX5 is involved in the biosynthesis of pro-inflammatory leukotrienes (Liu and Yokomizo, 2015) and ALOX5 inhibitors (Berger et al., 2007) and leukotriene receptor antagonists (Zhang et al., 2014) are currently available for prescription for treatment of bronchial asthma. For ALOX15 both, pro- and anti-inflammatory activities have been reported (Kuhn and O’Donnell, 2006; Kuhn et al., 2014) in various animal inflammation models but its role in different kinds of human inflammation remains to be explored.
6.2.1.1. Pro-inflammatory properties of ALOX15
The major arachidonic acid oxygenation products of ALOX15 (15-HETE, 12-HETE, 13-HODE) exhibit pro-inflammatory activities in various inflammation models (Kuhn, 1996). For instance, in rabbit skin 15-HpETE was reported to induce inflammation (Higgs et al., 1981). The pathophysiological responses to nasal antigen challenge were also related to an increased release of 15-HETE (Ramis et al., 1991). For isolated human polymorphonuclear leukocytes the ALOX15 product 13-HODE was shown to exhibit chemotactic activity at concentrations as low as 10−10 M (Henricks et al., 1991). As compared with leukotriene B4, 13-HODE was about half as potent. 5-Oxo-15-HETE, a more complex ALOX15 metabolite, also exhibited chemotactic activity (Schwenk et al., 1992).
Incubation of human eosinophils, which express ALOX15 at high levels, with arachidonic acid leads to the formation of conjugated trienes carrying a glutathione residue (Feltenmark et al., 2008). Because of its cellular origin this metabolite, which constitutes the 14,15-equivalent of leukotriene C4, was named eoxin C4. In analogy to leukotriene C4 eoxin C4 is further metabolized to 14,15-eoxin D4 and eoxin E4 by consecutive cleavage of the glutathionyl moiety. In vitro, eoxins induce increased permeability of endothelial cell monolayers suggesting pro-inflammatory activity. In this cellular inflammation model eoxins were 100 times more potent than histamine and almost equally potent as cysteinyl leukotrienes (Feltenmark et al., 2008). These ALOX15 metabolites have later been implicated as pro-inflammatory mediators in airway inflammation and Hodgkin lymphoma (Claesson, 2009; Sachs-Olsen et al., 2010).
The pro-inflammatory effects of ALOX15 may not be restricted to the formation of mediators increasing the inflammatory properties of immune competent cells. The effects of ALOX15 metabolites on the arterial barrier function (Kundumani-Sridharan et al., 2013; Othman et al., 2013; Chattopadhyay et al., 2015) are also of pro-inflammatory character and here alterations in the cellular redox state might be considered as mechanistic link (see 6.1.7.). Moreover, disruption of the alox15 gene protects hyperlipidemic mice from nonalcoholic fatty liver disease (Martinez-Clemente et al., 2010). In this model alox15 deficient mice developed a lower degree of hepatic steatosis and these alterations were paralleled by a decrease in hepatic inflammatory markers such as reduced macrophage infiltration, decreased levels of tumor necrosis factor α, monocyte chemoattractant protein-1 and interleukin-6 and −18 expression (Martinez-Clemente et al., 2010). However, it remains unclear whether the primary ALOX15 products induce all these pro-inflammatory alterations.
6.2.1.2. Anti-inflammatory properties of ALOX15
Inflammatory resolution is an active process aimed at reestablishing normal tissue homeostasis (Freire and Van Dyke, 2013). It is initiated by alterations of the cellular composition in the inflamed tissue and by alterations in the pattern of inflammatory mediators. Formation of pro-inflammatory leukotrienes is downregulated whereas biosynthesis of anti-inflammatory mediators is switched on. These anti-inflammatory hormones include a number of ALOX15 products such as lipoxins (Sachs-Olsen et al., 2010), resolvins (Spite et al., 2014), protectins (Serhan and Petasis, 2011) and maresins (Serhan et al., 2012). These mediators induce a number of basic anti-inflammatory elementary processes such as reduction of leukocyte migration (Fierro et al., 2003), normalization of vascular permeability (Ereso et al., 2009), apoptosis of pro-inflammatory neutrophils (El Kebir and Filep, 2013) and differentiation of anti-inflammatory M2 macrophages capable of phagocytosing apoptotic neutrophils, bacterial remnants and necrotic debris (Ohira et al., 2010). Pro-resolving eicosanoids and docosanoids are multiple oxygenation products of arachidonic acid, eicosapentaenoic acid and docosahexaenoic acid and their biosynthesis involves ALOX5, ALOX12, ALOX15 as well as aspirin modified COX-isoforms (Sala et al., 2010). However, the anti-inflammatory properties of ALOX15 should not be limited to their involvement in the biosynthesis of lipoxins, resolvins, maresins and protectins. The primary products of linoleic and arachidonic acid oxygenation [13S-H(p)ODE, 15S-H(p)ETE] do also exhibit anti-inflammatory activities in various inflammation models (Kuhn, 1996). Moreover, ALOX15 products activate PPAR signaling (Altmann et al., 2007; Limor et al., 2008), which stimulates anti-inflammation via alternative mechanisms (Martin, 2010). Oxidized phospholipids, which may be formed by ALOX15 catalyzed oxygenation of membrane lipids (Kuhn et al., 1990b), are capable of preventing the binding of agonists to toll-like receptors and thus, prevent activation of the innate immune response (Oskolkova et al., 2010).
Unilateral somatic gene transfer of an ALOX15 minigene in an experimental model of glomerulonephritis suppresses inflammation and preserved kidney function in the transfected kidney (Munger et al., 1999). Although the mechanism of this effect has not been explored in detail the data are consistent with an anti-inflammatory effect of ALOX15. Functional silencing of the ALOX15 induced uncontrolled inflammation and tissue damaging in two different experimental models of arthritis and these data are consistent with an anti-inflammatory and tissue-protective role of the enzyme (Kronke et al., 2009a). Although peritoneal macrophages of these animals produced significantly reduced levels of lipoxin A4 it remains unclear whether the formation of these pro-resolving mediators is the major reason for the anti-inflammatory effect. Alternatively, it was suggested that ALOX15 may play an important role in development of osteoclasts but here again the molecular mechanisms are not well understood (Kronke et al., 2009b).
6.2.2. Cardio-vascular diseases
6.2.2.1. Blood pressure regulation
ALOX15 has been implicated in the regulation of vascular tone and thus, may play a role in blood pressure regulation and hypertension (Nasjletti, 1998; Chawengsub et al., 2009; Zhu and Ran, 2012). Arachidonic acid induces endothelium-dependent relaxation of rabbit aorta and this effect was blocked by the LOX inhibitor NDGA (Pfister and Campbell, 1992). Similar effects have been reported for bovine coronary arteries (Rosolowsky and Campbell, 1993) and the inducing metabolites have been identified as the ALOX15 products 11,14,15- and 11,12,15-trihydroxyeicosatrienoic acids (Pfister et al., 1998). Chronic hypoxia and hypercholesterolemia enhanced ALOX15 mediated vasorelaxation in rabbit arteries (Aggarwal et al., 2008; Aggarwal et al., 2009). More direct evidence for the in vivo relevance of ALOX15 in blood pressure regulation was recently provided by experiments with alox15 knockout mice (Kriska et al., 2012). Although systolic blood pressures did not differ between these mice and wild-type controls alox15−/−-mice exhibited higher resistance towards L-NAME- and high-salt-induced hypertension than corresponding controls. The alox15 inhibitor nordihydroguaiaretic acid attenuated this resistance suggesting the involvement of lipid peroxidation. The molecular basis for this effect has not been explored and it remains unclear of whether or not it is related to the vasomotor properties of alox15 products. Interestingly, injection of wild-type peritoneal macrophages, which are a major source of alox15 in mice, into alox15 knockout animals abolished their resistance toward L-NAME-induced hypertension. Inversely, wildtype mice acquired resistance to L-NAME-induced hypertension after depletion of macrophages by clodronate injection (Kriska et al., 2012).
6.2.2.2. Atherogenesis
In the early 1990s the hypothesis of oxidative modification of low density lipoprotein (LDL) was introduced (Witztum and Steinberg, 1991; Chisolm and Steinberg, 2000) and the updated version of this theory was critically reviewed more recently (Steinberg, 2009). This hypothesis suggested that oxidized LDL exhibits strong pro-atherogenic activities because it is rapidly taken up by macrophages via scavenger receptor mediated pathways. Since these pathways are not feedback-controlled excessive intercellular lipid deposition may occur and macrophages develop into lipid-laden foam cells. These cells then accumulate in the subendothelial space of the arteries to form fatty streaks which are considered early atherosclerotic lesions (Perrotta, 2013). ALOX15 is capable of oxidizing LDL (Belkner et al., 1998) and other lipoproteins (Pirillo et al., 2006). In atherosclerotic lesions of rabbits (Kuhn et al., 1994) and humans (Folcik et al., 1995; Kuhn et al., 1997) esterified 13S-HODE has been detected but the biosynthetic origin of this compound has not been clarified. Several studies employing alox15 knockout mice supported a pro-atherogenic role of alox15 (Cyrus et al., 1999; Cyrus et al., 2001; George et al., 2001; Huo et al., 2004; Zhao et al., 2005; Poeckel et al., 2009). On the other hand, overexpression of the enzyme in two rabbit and one mouse atherosclerosis models suggested an anti-atherogenic effect of the enzyme (Shen et al., 1996; Trebus et al., 2002; Merched et al., 2008). In the transgenic mouse model it was suggested that alox15 activity in the local milieu afforded atheroprotection via the formation of pro-resolving mediators (Merched et al., 2008) and this was later on suggested as more general paradigm (Hersberger, 2010). Taken together, the role of ALOX15 in atherosclerosis remains controversial (Kuhn et al., 2005a; Wittwer and Hersberger, 2007) and the contradicting data may be related to differences in the various animal atherosclerosis models and/or to the different dietary supplementation strategies employed. Systemic stem cell knockout of the alox15 gene may not necessarily lead to an inverse effect as macrophage specific overexpression of the enzyme.
In advanced human atherosclerotic lesions ALOX15 is only expressed at low levels (Spanbroek et al., 2003; Gertow et al., 2011). However, these data do not necessarily exclude involvement of the enzyme in atherogenesis because of the following reasons: i) If ALOX15 is involved in maturation and differentiation of macrophages it might contribute to atherogenesis without being expressed in the lesion. ii) If the enzyme is involved in early stages of macrophage differentiation functionally different macrophages are generated and thus, lesional foam cell formation may be impacted. iii) If the enzyme is expressed in cells not present in the lesions it might contribute to systemic LDL oxidation, which is considered a risk factor for atherogenesis. iv) If ALOX15 is only involved in early stages of lesion formation (Kuhn et al., 1994; Kuhn et al., 1997) it may be absent in advanced lesions but still might contribute to early stages of lesion development. In all these cases expression silencing and pharmacological intervention with the ALOX15 pathway may impact lesion formation without expression of ALOX15 in advanced lesions.
ALOX15 is not only capable of oxidizing LDL to an atherogenic species but it also oxygenates high-density lipoproteins (HDL). ALOX15 mediated oxidation of HDL3 impairs activation of endothelial nitric oxide synthase (Cutuli et al., 2014). Moreover, ALOX15 oxidized HDL upregulates expression of the lectin-like oxidized low-density lipoprotein receptor 1 in human endothelial cells (Pirillo et al., 2012) and both effects may be considered pro-atherogenic. On the other hand, in vitro ALOX15-modified HDL3 failed to inhibit the TNF-alpha-induced inflammatory response in human endothelial cells (Pirillo et al., 2008). HDL is an anti-atherogenic lipoprotein since it is involved in reverse cholesterol transport. Forced ALOX15 expression in mouse macrophages (J774 cells) increases the degradation of macrophage ATP-binding cassette transporter G1, which has been implicated in reverse cholesterol, transport. These findings provide evidence that ALOX15 may contribute to atherogenesis by impairing the cholesterol efflux from lipid laden foamy macrophages (Nagelin et al., 2008). On the other hand, similar experiments with ALOX15 overexpression in RAW macrophages induced increased cholesterol mobilization and augmented reverse cholesterol transport (Weibel et al., 2009). This anti-atherogenic effect was paralleled by an increased expression of ABC-transporters, which play an important role in reversed cholesterol transport. These data suggest that overexpression of human ALOX15 in RAW macrophages promotes reversed cholesterol transport, which is an anti-atherogenic effect.
6.2.3. Carcinogenesis and metastasis
ALOX15 and ALOX15B have been implicated in many aspects of carcinogenesis, such as angiogenesis, inflammation and metastasis and this was shown for solid tumors and hematologic malignancies (Pidgeon et al., 2007; Klil-Drori and Ariel, 2013). However, the precise roles of the two enzymes have not been clarified since tumor-promoting and tumor suppressing activities have been reported (Klil-Drori and Ariel, 2013). Transgenic mice overexpressing ALOX15 in endothelial cells (Harats et al., 2005) under the regulation of the murine preproendothelin-1 promoter were protected from tumor growth and metastasis in two different cancer models (mammary gland and Lewis lung carcinoma). This inhibition was concomitant with a higher number of apoptotic cells in the transgenic mice and with impaired neoangiogenesis in the tumor tissue (Harats et al., 2005). The anti-apoptotic enzyme glutathione peroxidase 4 (GPx4) is an endogenous inhibitor of ALOX15 (Schnurr et al., 1996) and inactivation of GPx4 caused rapid cell death. In vitro, GPx4 deficient fibroblasts form tumor spheroids and subcutaneous implantation of these spheroids induced solid tumors in mice, which were characterized by an increase in microvessel density (Schneider et al., 2010). Pharmacological inhibition of alox15 successfully reversed tumor development and normalized the vessel architecture suggesting alox15 as regulator of tumor angiogenesis.
Transfection of HCT116 colon carcinoma cells with ALOX15 induced activation of the ERK protein kinase, which increased the rate of cell proliferation. These data suggest a pro-carcinogenic activity of the enzyme (Yoshinaga et al., 2004). Treatment of these cells with NDGA (non-specific LOX inhibitor with antioxidant properties) appeared to block ERK activation, which is consistent with the pro-carcinogenic activity of ALOX15 (Yoshinaga et al., 2004). However, since cell cycle regulation is redox sensitive (Chiu and Dawes, 2012) the observed effect of NDGA might not directly be related to ALOX15 inhibition. In other cellular models of colorectal carcinoma (HCT116, HT29) ALOX15 exhibited anti-carcinogenic properties, which was related to inhibition of the anti-apoptotic effect of the inflammatory transcription factor nuclear factor kappa B (Cimen et al., 2011). Here again, the molecular basis for the observed anti-carcinogenic affect is not completely understood but overexpression of ALOX15 inhibited the degradation of the inhibitor of kappa B, impaired nuclear translocation of p65 and p50, decreased DNA binding in the nucleus and reduced the transcriptional activity of NF-κB (Cimen et al., 2009; Cimen et al., 2011).
Unresolved chronic inflammation is a key process in tumor progression (Janakiram and Rao, 2014) and thus, pro-resolving lipid mediators (eicosanoids and related metabolites of other polyenoic fatty acids) such as lipoxins (Ryan and Godson, 2010), resolvins (Lee and Surh, 2012) and maresins (Dalli et al., 2013) have been implicated in carcinogenesis (Janakiram et al., 2011). Resolving eicosanoids are generally believed to exhibit anti-tumor activities (Wendel and Heller, 2009). Chronic inflammation of colonic mucosa creates a pro-carcinogenic milieu and patients suffering from ulcerative colitis exhibit defective lipoxin biosynthesis (Mangino et al., 2006). Thus, the lack of pro-resolving mediators may drive malignant transformation of normal epithelial cells during chronic inflammation. On the other hand, under certain conditions these mediators may also act in a pro-carcinogenic manner. For instance, depletion of regulatory T cells induced by cyclophosphamide treatment of patients with large established tumors caused significant tumor progression and this effect was suggested to be mediated by an increase in lipoxin A4 levels (Zhang et al., 2010).
6.2.4. Metabolic disorders
6.2.4.1. Obesity and metabolic syndrome
ALOX15 has been implicated in adipocyte differentiation (see 6.1.4.) and thus, the enzyme may play a role in obesity and in the pathogenesis of metabolic syndrome. In mice a lipid rich-diet increased the number of macrophages in the visceral adipose tissue (Nunemaker et al., 2008). However, this increase was significantly lower in alox15 deficient animals when compared with wildtype controls. Moreover, the pancreatic islets of alox15 knockout mice were protected from diet-induced hyperplasia and from reduced glucose-stimulated insulin secretion when compared with wildtype controls (Nunemaker et al., 2008). When we performed similar feeding experiments with our colony of alox15 knockout mice we were unable to show that systemic alox15 deficiency protects mice from the development of insulin resistance of peripheral tissues (skeletal muscle, visceral adipose tissue).
Gene expression profiles indicated expression of various LOX-isoforms in human white adipose tissue (Lieb et al., 2014). Interestingly, high-level expression of ALOX15 was only found in the omental adipose tissue whereas only small amounts of ALOX15 mRNA and protein were detected in subcutaneous fat (Dobrian et al., 2010). Immunohistochemical stainings indicated that the major cellular sources of ALOX15 were not the adipocytes but rather the stromal vasculature (Dobrian et al., 2010). These data are consistent with previous observation suggesting that invaded macrophages are the major source of ALOX15 in mouse visceral adipose tissue (Sears et al., 2009). Western-type high fat diet induces latent adipose tissue inflammation in wildtype mice, which is indicated inter alia by a massive infiltration of macrophage into the visceral adipose tissue. In contrast, in alox15 knockout mice the degree of macrophage infiltration was significantly reduced. These data implicate alox15 in high fat diet induced adipose tissue inflammation, which appears to be important for the pathogenesis of obesity and for the development of insulin resistance of visceral adipose tissue (Sears et al., 2009). Despite the observation that alox15 expression is limited in adipocytes fat specific knockdown of alox15 expression significantly reduced high fat diet induced inflammation of pancreatic islets (Cole et al., 2012).
Although the underlying mechanisms remain unclear these results suggest a crosstalk between alox15 expression in adipose tissue and inflammation of pancreatic islets. From these data it was concluded that inhibition of alox15 expression in adipose tissue might offer systemic protection from obesity-induced consequences and that blocking alox15 activity in adipose tissue might constitute a novel therapeutic principle for the treatment of type 2 diabetes (Cole et al., 2012).
Nonalcoholic fatty liver disease is a major hepatic consequence of the metabolic syndrome. Alox15 mRNA is upregulated in apoE-deficient mice, which are frequently employed as model for this disease (Ferre et al., 2009). However, the detailed role of alox15 in nonalcoholic fatty liver disease remains unclear. Adipose tissue and adipocytes from obese Zucker rats, which is a frequently employed rat model for the metabolic syndrome, show increased expression of alox15 (Chakrabarti et al., 2011) but the patho-physiological role of the enzyme has not been explored. An important question, which has not been answered conclusively, is how the alox15 expression is upregulated in the diet induced obesity models. Th2 cytokines induce expression of ALOX15 in various cell types (Heydeck et al., 1998; Schnurr et al., 1999) and the transcription factor PPAR-gamma has also been implicated. Since PPAR-gamma is activated by ALOX15 metabolites there may exist damaging feed forward mechanisms (Huang et al., 1999).
6.2.4.2. Diabetes mellitus
12- and 15-lipoxygenating LOX-isoforms including ALOX15 have been implicated in the pathogenesis of diabetes (Natarajan et al., 1993; Bleich et al., 1998; Bleich et al., 1999; Laybutt et al., 2002; Chen et al., 2005; Nunemaker et al., 2008; Sears et al., 2009; Ma et al., 2010) but the molecular mechanisms for its involvement are not completely understood. ALOX15 products might function as signaling molecules but alternative mechanisms have also been discussed. Since ALOX15 is a pro-oxidative enzyme producing hydroperoxy lipids, ALOX15-induced oxidative stress and subsequent mitochondrial dysfunction might account for increased pathology detected in diabetic cardiomyopathy and other vascular disorders (Boudina and Abel, 2007). ALOX15 expression is upregulated in both, cell culture and animal models of diabetes (Dobrian et al., 2011). Insulin secretion of cultured human islet cells was reduced by nanomolar concentrations of 12S-HETE and 12S-HpETE and increased levels of 12S-HETE were linked to coronary artery disease in type 2 diabetic patients (Zhang et al., 2012). Although 12S-HpETE is not the major product of human ALOX15 it is formed by the enzyme in smaller amounts (see 3.3.1.). However, in murine models 12S-HpETE is the dominant alox15 product. Hepoxilin A3, a secondary arachidonic acid oxygenation product generated by 12-lipoxygenating LOX-isoforms including human and murine ALOX15 (Nigam et al., 2007), induce insulin secretion in pancreatic beta cells and islets (Pace-Asciak, 2015). Furthermore, HXA3 protects the rat insulinoma cell line RINm5F against oxidative stress-induced cell death, although the mechanism needs further investigation (Zafiriou et al., 2011).
For type-1 diabetes only scattered information are currently available as to the pathological role of ALOX15. Female nonobese diabetic (NOD) mice are a suitable model for this disease (Zafiriou et al., 2011). Remarkably the NOD-ALOX15null strain, in which alox15 is absent, is almost completely protected from developing diabetic symptoms (McDuffie et al., 2008). These results suggest that alox15 contributes to the pathology and mechanistically, this may be related to effects of alox15 on islet cell and/or macrophage functionality (Green-Mitchell et al., 2013). Similarly, Alox15-deficient mice are more resistant to induction of type 1 diabetes by streptozotocin when compared with corresponding alox15 expressing controls (Bleich et al., 1999).
6.2.5. Neurological disorders
Neurons are especially vulnerable to oxidative stress, and oxidative stress-related pathology is a hallmark of several CNS diseases, including stroke, Parkinson’s and Alzheimer’s Disease. 12- and 15-lipoxygenating LOX-isoforms have been linked to apoptotic cell death of in vitro cultured primary neurons (Canals et al., 2003; Khanna et al., 2003; Zhang et al., 2004) as well as in several other brain-derived cells (Lovat et al., 2002; Lovat et al., 2003) and in mouse hippocampal cell lines (Pallast et al., 2009; Pallast et al., 2010). Similar apoptotic effects can be elicited in vivo by direct injection of arachidonic acid into the brain, which causes edema (Chan and Fishman, 1978; Chan et al., 1983).
6.2.5.1. Periventricular leukomalacia (PVL)
Periventricular leukomalacia (PVL) is a white matter injury in infants that is the dominant pathological factor for determining long-term cognitive and motoric deficits in premature infants. ALOX15 expression is increased in the brains of PVL patients (Haynes and van Leyen, 2013) and several cell types including microglia and oligodendrocyte precursor cells were affected. Some of these cells were TUNEL-positive, suggesting that ALOX15 might contribute to disease pathology. This hypothesis is supported by cell culture studies, in which oligodendrocyte precursors were vulnerable to an ALOX15-dependent cell death when cultured in the absence of cysteine (Wang et al., 2004; van Leyen et al., 2008). It will be interesting to see if isoform-specific ALOX15 inhibitors are protective in animal models of PVL.
6.2.5.2. Stroke
The strongest evidence for any LOX isoform causing injury to the CNS exists in stroke (van Leyen, 2013). Arachidonic acid, which is increasingly liberated from phospholipids by cytosolic phospholipase A2 (cPLA2) under stroke conditions, provides the substrate and thus, may activate the ALOX15 pathway. Moreover, intracellular calcium rises under stroke conditions, which favors membrane binding and augments the catalytic activity of ALOX15 (Brinckmann et al., 1998). In addition, the protein levels of ALOX15 increase specifically in the penumbra region surrounding the core infarct. This brain region is vulnerable to ischemia-induced delayed cell death (van Leyen et al., 2006). The factors leading to transcriptional up-regulation of ALOX15 expression in the ischemic brain have not been identified, but may include members of the STAT family of transcriptional activators, which regulate expression of the ALOX15 gene in several other cell types (Conrad and Lu, 2000). Increased ALOX15 in the ischemic cortex is accompanied by increased pro-apoptotic AIF, in both human stroke patients (Yigitkanli et al., 2013), as well as mouse models of stroke (Pallast et al., 2010). ALOX15 also co-localizes with MDA2, an antibody that recognizes malonedialdehyde-modified lysine residues suggesting increased lipid peroxidation in the penumbra area (Yigitkanli et al., 2013). Taken together, these findings document that ALOX15 is part of a major cell death pathway that is activated in the ischemic regions. Functional inactivation of the alox15 gene protected mice against stroke (Khanna et al., 2005; van Leyen et al., 2006), reduced leakage of the blood-brain barrier and minimized edema formation (Jin et al., 2008). These protective effects are replicated by pre-treatment of the animals with LOX inhibitors. Since many of the conventional LOX inhibitors exhibit strong antioxidant activity newer inhibitors with low antioxidant activity (van Leyen et al., 2008; Rai et al., 2014) have been more recently applied and these compounds were protective even when given four hours after stroke induction (Yigitkanli et al., 2013).
In addition to the damaging effects of ALOX15 in the penumbra area the restorative potential of ALOX15-derived mediators including lipoxins and protectins has been explored. For example, the neuroprotective effects of rosiglitazone have been related to the formation of lipoxin A4 (Sobrado et al., 2009) and an agonist at the lipoxin A4 receptor induced neurovascular protection in a rat model of ischemic stroke (Hawkins et al., 2014). Since 12- and 15-lipoxygenating LOX have been implicated in lipoxin formation ALOX15 orthologs might be involved. Moreover, neuroprotectins, specifically neuroprotectin D, reduce tissue damage in animal models of stroke (Bazan, 2009). Since ALOX15 has been implicated in biosynthesis of neuroprotectin D1 from docosahexaenoic acid the enzyme may be involved in this protective effect. Interestingly, infusion of docosahexaenoic acid was also protective in experimental stroke (Belayev et al., 2011).
6.2.5.3. Alzheimer’s disease
ALOX15 expression is increased in the brains of Alzheimer’s patients (Pratico et al., 2004) and increased levels of 12- and 15-HETE were found in the cerebrospinal fluid of patients with Alzheimer’s pathology (Yao et al., 2005). Consistent with a damaging function of ALOX15 in Alzheimer’s disease the extent of the degenerative defects in a transgenic Alzheimer’s mouse model (tg2576) was reduced when alox15 was absent (Yang et al., 2010). In humans elevated ALOX15 expression was found when brains of Alzheimer’s patients were compared those with no pathology (Rao et al., 2011). In contrast, ALOX15 expression was reduced in the hippocampus of Alzheimer’s patients and reduced neuroprotectin D1 levels paralleled this effect (Lukiw et al., 2005). There are several possible reasons for these discrepancies, which may be related to the complexity of disease progression. Further studies are needed to get a clearer picture of differential LOX expression and its consequences in Alzheimer’s brains.
In cell culture models, an amyloid beta-derived peptide was found to cause cell death in primary neurons. This effect was blocked by LOX inhibition and similar protective effects were described when an antisense oligonucleotide targeting alox15 expression was employed (Lebeau et al., 2001; Lebeau et al., 2004). In contrast, expression of miRNA125b, a micro-RNA silencing translation of ALOX15 mRNA, was increased in Alzheimer’s patients. When primary neuroglia cells were treated with amyloid ß1–42 protein increased levels of miRNA125b led to down-regulation of ALOX15 expression. Silencing the expression of miRNA125b restored ALOX15 expression and protected these cells from damage (Zhao et al., 2013). Unfortunately, it remains unclear whether there is a direct causative link of these cellular in vitro studies to Alzheimer’s pathology.
6.2.5.4. Multiple sclerosis and other neurodegenerative diseases
As indicated above ALOX15 metabolites have been identified as co-activators of PPAR-gamma and agonists of this signaling pathway reduce clinical severity of experimental allergic encephalomyelitis, an animal model of human multiple sclerosis. In alox15 deficient mice the pathology of experimental allergic encephalomyelitis was significantly worsened implicating this enzyme as protective regulator in pathogenesis of this disease (Emerson and LeVine, 2004). We repeated these experiments with our strain of alox15-deficient mice and obtained comparable results (Fig. 11). On the other hand, pharmacological interference with an ALOX15 inhibitor (systemic application of baicalein) attenuated the clinical symptoms of experimental autoimmune encephalomyelitis in mice (Xu et al., 2013). Baicalein treatment reduced activation of microglia, suppressed glial phagocytosis and impaired the cerebral production of proinflammatory cytokines. Mechanistically, baicalein treatment did not affect ALOX15 expression but significantly reduced the formation of ALOX15 metabolites and led to increased expression of PPARβ/δ in microglia. These data suggest that functional inactivation of the ALOX15 pathway activates PPARβ/δ signaling, which is protective for autoimmune encephalomyelitis in mice. Thus, pharmacological interference of the ALOX15 pathway might be developed as innovative therapeutic concept in the treatment of multiple sclerosis (Xu et al., 2013).
In other degenerative diseases of the central nervous system, much remains to be studied about possible ALOX15 involvement in the respective pathology. There is little information about whether ALOX15 may play a role in pathogenesis of amyelotrophe lateral sclerosis, Parkinson’s and Huntington’s disease. Although oxidative stress and increased lipid peroxidation has been implicated in the pathogenesis of virtually all neurodegenerative disorders (Mhatre et al., 2004) it remains unclear whether or not ALOX15 may contribute. Pharmacological interference studies suggested a role of lipid peroxidation in the pathogenesis of Huntington’s disease (Kumar et al., 2011) and an inhibitor of COX1, COX2 and ALOX5 pathways induced protective effects in an animal model of Huntington’s disease (Lee et al., 2011). However, a possible role for ALOX15 has not been explored.
After spinal cord injury, ALOX15 was increased 25-fold in rats, compared to a 1.7-fold upregulation detected for COX-2 (Di Giovanni et al., 2003). But whether or not this increase contributes to the injury is presently unknown.
7. Evolutionary aspects of ALOX15
As indicated above LOXs occur in two of the three domains of terrestrial life (see 2.1.) but they rarely occur in lower single- and multi-cellular organisms (Horn et al., 2014). In highly developed plants and animals occur LOXs more frequently and most mammalian species express various LOX-isoforms (Horn et al., 2014).
7.1. Mammalian ALOX15 orthologs
In mammals two subtypes of ALOX15 orthologs can be differentiated according to their enzymatic properties. Lower mammals such as mice (Sun and Funk, 1996), pigs (Yoshimoto et al., 1990) and cattle (De Marzo et al., 1992) express 12-lipoxygenating ALOX15 orthologs whereas humans (Sloane et al., 1991a) and orangutans (Vogel et al., 2010) express 15-lipoxygenating enzyme species. The structural basis for the variable positional specificity of ALOX15 orthologs has been explored and the triad concept (Ivanov et al., 2010) suggests that Phe353, Ile418/Met419 and Ile593 (numbering for rabbit ALOX15) form the bottom of the substrate binding pocket. If these positions are occupied by amino acids carrying bulky side chains (Phe353+Ile418/Met419+Ile593) arachidonic acid 15-lipoxygenation is catalyzed. In contrast, if one or more of these positions is occupied by a less bulky residue (Leu353+Val418Met419+Val593 for mouse alox15; Leu353+Ala418/Met419+Val593 for rat alox15; Phe353+Val418/Val419+Ile593 for pig ALOX15; Phe353+Val418/Val419+Ile593 for macaca ALOX15) arachidonic acid 12-lipoxygenation results. The naturally occurring variability in reaction specificity of ALOX15 orthologs (Horn et al., 2014) can simply be mimicked by Ile418Ala exchange in human ALOX15. Wildtype human ALOX15 oxygenates arachidonic acid mainly at C15 but its Ile418Ala mutant mainly catalyzed arachidonic acid 12-lipoxygenation [Adel et al., 2015 submitted]. Recent MD simulations have confirmed this experimental finding for rabbit ALOX15 and the substrate fatty acid appears to slide deeper into the substrate-binding pocket of the mutant enzyme. However, these spatial alterations may not be the only reason for the drastic changes in reaction specificity but energetic aspects (lower energetic barriers for the formation of the transition states) may also contribute [Adel et al., 2015 submitted].
7.2. Change in reaction specificity of ALOX15 orthologs during primate evolution
ALOX15 orthologs of higher mammals [H. sapiens (Sloane et al., 1991a), H. neandertals (Chaitidis et al., 2013), H. denisovans (Adel et al., 2015), P. pygmeus (Vogel et al., 2010)] mainly catalyze arachidonic acid 15 lipoxygenation. In contrast, lower mammals [M. musculus (Sun and Funk, 1996), R. norwegicus (Pekarova et al., 2015), Sus scrofa (Yoshimoto et al., 1990)] and even lower primates (Vogel et al., 2010) exhibit arachidonic acid 12-lipoxygenation activity. This data suggested that the reaction specificity of ALOX15 orthologs might have systematically been changed from 12- to 15-lipoxygenation during late primate evolution and that this evolutionary change might have happened between macaca and orangutans (Adel et al., 2015 submitted). Unfortunately, the positional specificity of ALOX15 orthologs of chimpanzee, gorilla, gibbon and baboon remains to be determined experimentally but arachidonic acid 15-lipoxygenation can be predicted on the basis of their primary structure. In contrast, 12-lipoxygenation may be predicted for baboon ALOX15. For gibbon ALOX15 prediction of the reaction specificity is not possible but preliminary data from our lab suggest almost equal amounts of 12- and 15-HpETE. If our predictions can be confirmed by more detailed experimental data one may conclude that in lower mammals (mice, rats, pigs, baboon, macaca) 12-lipoxygenating ALOX15 enzymes are expressed. In contrast, in higher mammals (gorilla, orangutan, chimpanzee, human subspecies) 15-lipoxygenating ALOX15 isoforms are present (Johannesson et al., 2010) and gibbons may constitute a transition species. The only known exception from this rule are rabbits but here 12- and 15-lipoxygenating ALOX15 species are expressed in a tissue specific manner (Berger et al., 1998). Unfortunately, the biological relevance of this evolutionary switch in the reaction specificity still remains unclear.
8. Isoform-specific ALOX15 inhibitors
Lipoxygenase inhibitors have frequently been used to characterize the biological role of these enzymes in various animal disease models. However, in the absence of supporting results obtained with alternative loss-of-function (knockout mice or siRNA-induced expression knockdown) and/or gain-of-function (LOX-overexpressing transgenics or LOX transfectants) strategies these results should be interpreted with care because of two reasons:
Isoform-specificity: Many LOX inhibitors used in the literature (NDGA, CDC, AA861, baicalein, PD146176) do not exhibit a pronounced isoform-specificity. Most of them inhibit several ALOX-isoforms and thus, it is hardly possible to conclude on the basis of the outcome of inhibitor studies, which LOX-isoform is involved in a biologically relevant process. On the other hand, testing the isoform-specificity of a LOX inhibitor is of major biological relevance. For instance, an ALOX5 inhibitor, which will be developed as anti-asthmatic drug (Hofmann and Steinhilber, 2013), should not significantly impact the activity of ALOX12B. Otherwise problems with skin development may occur (Epp et al., 2007; Krieg et al., 2013) and the drug is likely to have unwanted side effects. Most experts are well aware of this problem but scientists not so experienced in LOX biology do sometimes not appreciate this problem. A second issue related to isoform-specificity is the way how isoform specificity of a LOX inhibitor is determined. In the past the assay systems employed to test isoform specificity were not strictly comparable and this may lead to misinterpretations. Lysates of rat basophilic leukemia cells were frequently used as ALOX5 source and platelet lysates were employed for testing ALOX12 activity (Sendobry et al., 1997). These assay systems are not strictly comparable since the inhibitor may differently be metabolized in the two assay systems and foreign proteins may bind the inhibitors in different ways. To avoid such problems inhibitor studies should always be carried out with purified recombinant enzyme preparations (Rai et al., 2010; Kenyon et al., 2011). Alternatively, the different LOX isoforms of one species (human, mouse, rats) should be expressed in a single eucaryotic overexpression system (such as COS or HEK cells) and the cellular lysate may be employed as enzyme source. Recently, a systematic study was carried out, in which all 12-lipoxygenating rat LOX isoforms were overexpressed in HEK cells and an array of commercially available LOX inhibitors was tested using the cell lysates (Gregus et al., 2013). The data obtained indicate that these commercial LOX-inhibitors (NDGA, CDC, AA861, baicalein, PD146176) only exhibited a low degree of isoform specificity although some of them have previously been suggested as isoform-specific inhibitors. Most surprisingly, PD146176, which has been employed in experimental strategies as ALOX15 specific inhibitor (Sendobry et al., 1997) did not at all inhibit rat alox15 (Gregus et al., 2013). For the time being, it remains unclear why this compound effectively inhibits rabbit and human ALOX15 but not the rat ortholog. The reasons may be related to the different positional specificities of the two ALOX15 orthologs but more detailed experiments are needed to support this conclusion. Irrespective of the lacking mechanistic data the results clearly indicate the possibility that an inhibitor that effectively interferes with human ALOX15 may not inhibit orthologous enzymes of other species (species-specificity). To avoid misinterpretations the inhibitory potency of ALOX15 inhibitors used as probes to test the role of this LOX isoforms in a biological process should always be confirmed in the corresponding assay system. A good example for an experimental strategy, which follows these specificity criteria was the recent report by Rai and co-workers (Rai et al., 2014), which indicated that certain oxazole-4-carbonitrile based LOX inhibitors share a high inhibitory potency for human and mouse ALOX15 but hardly inhibit other mammalian LOX-isoforms.
Off-target effects of LOX-inhibitors: Some frequently employed ALOX15 inhibitors (NDGA, gallic acids) exhibit anti-oxidative properties and thus, may directly impact the cellular redox homeostasis. Since the cellular redox state is important for regulating the gene expression pattern on genetic (Brune et al., 2013) and epigenetic (Goswami, 2013; Kim et al., 2013) levels, it is difficult to discriminate which of the two functions (LOX inhibition vs. redox activity) is the major reason for an observed biological effect. Consequently, results obtained with these types of ALOX15 inhibitors must be interpreted with care and to avoid misinterpretation, the inhibitor studies should always be confirmed by alternative loss-of-function strategies. Characterizing the redox activity of a potential ALOX15 inhibitor is not a trivial task and depends on the experimental assay systems. There is a large number of enzymatic and non-enzymatic oxidation systems and a given compound might be effective in one of them but ineffective in other systems. Since it is impossible to test the anti-oxidative properties of a potential ALOX15 inhibitor in all of these assay systems the employed systems must clearly be specified.
9. Open questions and perspectives
In recent years the structural biology of LOX became a quickly developing area in LOX research and crystal structures for a number of LOX-isoforms have been solved during the past 10 years (Newcomer and Brash, 2015). However, there is only scattered direct structural information for LOX-substrate complexes. A phosphorylation-mimicking mutant of a stabilized version of human ALOX5 was crystallized with arachidonic acid bound at the catalytic center (Gilbert et al., 2012) and the X-ray coordinates suggested an inverse head-to-tail substrate alignment, which explains the 15-lipoxygenating activity of this enzyme variant. Unfortunately, 15-lipoxygenating activity of phosphorylation mimicking mutants of ALOX5 orthologs of different vertebrate species could not be confirmed (Adel et al., 2014) so that it remains unclear whether the X-ray data represent a catalytically productive structure. More recently, the crystal structure of the coral 8R-LOX-arachidonic acid complex was solved at 2.0 Å resolution and the X-ray coordinates suggested a catalytically competent conformation (Neau et al., 2014). Unfortunately, for the time being no crystal structure is currently available for ALOX15-substrate complexes. However, the recent methodological advances in LOX crystallization suggest that corresponding X-ray data will soon become available for wildtype ALOX15 orthologs and relevant mutants with altered positional specificity.
Another unsolved problem in structural ALOX15 research is the capability of ALOX15 orthologs to specifically oxygenate complex ester lipids, such as phospholipids and cholesterol esters (Kuhn et al., 1990b; Belkner et al., 1991; Takahashi et al., 1993). Recent molecular docking studies suggested that binding of a phospholipid molecule at the active site of rabbit ALOX15 is sterically impossible without major rearrangements in the active site architecture. Rabbit ALOX15 exhibits a high degree of motional flexibility (Shang et al., 2011) but whether this flexibility is high enough to allow proper binding of phospholipids at the active site remains to be explored. A similar problem exists for the 15-lipoxygenating soybean LOX1, which appaers to be structurally more stable than the rabbit enzyme (Dainese et al., 2005). This enzyme is also capable of specifically oxidizing phospholipids (Brash et al., 1987) but here again docking studies indicate steric clushes for phospholipid binding at the active site. Direct X-ray data on an ALOX15-phospholipid complex would shed light on the structural rearrangements that are required for ALOX15-catalyzed specific phospholipid oxygenation.
Although LOXs are oxygen-metabolizing enzymes there is apparently no targeted oxygen binding at the substrate free enzyme. In silico studies (Saam et al., 2007) suggested a region of high oxygen occupancy probability and several paths via which molecular dioxygen may reach this area. Although mutagenesis data support this concept there is no direct experimental proof for heterogeneous oxygen distribution within the LOX protein and for a targeted intra-enzyme oxygen movement. To obtain such data X-ray crystallographic studies under high oxygen or xenon concentrations should be carried out (Svensson-Ek et al., 2002; Cohen et al., 2006).
Because of their lipohydroperoxidase activity LOXs have been implicated in the formation of free radicals and the LOX-superoxide connection has been a matter of discussion for several years. Back in the mid 1970 it was reported that superoxide dismutase effectively inhibited linoleic acid oxygenation by soybean LOX1 (Richter et al., 1975). Moreover, a soybean LOX-superoxide complex has been implicated in the oxidation of furan derivatives by this enzyme (Boyer et al., 1979) but the detailed mechanism of superoxide production was not explored. In another experimental approach it was suggested (Roy et al., 1995) that LOXs are capable of cooxidizing glutathione in the presence of linoleic acid and simultaneously generate superoxide anions. However, here again the molecular basis for superoxide production has not been explored. More recently, a number of inhibitors have been developed which inhibit different LOX-isoforms but also function as superoxide scavengers (Pontiki and Hadjipavlou-Litina, 2007; Cretu et al., 2013; Ben-Nasr et al., 2015). Although these data suggest that there might be a connection between the lipoxygenase/lipohydroperoxidase reaction and superoxide formation, the mechanistic basis for this connection remains elusive. According to our current understanding of the lipoxygenase reaction these enzymes do not activate molecular dioxygen (Fig. 1A) and the radical intermediartes formed remain largely enzyme bound. Thus, a direct formation of superoxide during the lipoxygenase reaction is rather unlikely. However, the lipohydroperoxidase activity of LOXs involves the formation of fatty acid radicals (Fig. 1B), which may induce secondary superoxide production. It would be worth to further explore under which experimental conditions LOXs may serve as effective sources for free radicals.
Inhibitor studies and characterization of the performance of alox15 deficient mice in a number of mouse disease models provided valuable information on the physiological and patho-physiological role of ALOX15. The recent observation that alox15 may play a role in vascular endothelial tight-junction formation (Kundumani-Sridharan et al., 2013; Chattopadhyay et al., 2014) implicates the enzyme in regulation of vascular permeability, which is of biological relevance for the pathogenesis of inflammatory diseases. Unfortunately, it has not been explored whether ALOX15 might also be involved in epithelial tight-junction formation. In contrast to the vascular endothelium, epithelial cells (airway epithelium, enteral epithelium) have more pronounced tight junctions, which contribute to the primary defense mechanism preventing the penetration of pathogens or microparticles. Moreover, these tight junctions counteract the release of dedifferentiated epithelial cells, which is an important step of metastasis. If ALOX15 is also involved in epithelial tight-junction formation the enzyme may play a role in the pathogenesis of inflammatory bowel and lung diseases and in metastasis of enteral and pulmonal carcinoma.
The biological relevance of variable positional specificity of ALOX15 orthologs (major 15-lipoxygenating vs. major 12-lipoxygenating ALOX15 orthologs) remains a matter of discussion. Since orthologous enzymes should have the similar functions in different species one may conclude that ALOX15 orthologs do not exhibit their biological activity via the formation of signaling molecules (see 6.1.1.). However, the recent observation of a systemic change in positional specificity of ALOX15 orthologs during late primate evolution (see 6.1.2.) suggests that this change might constitute a targeted evolutionary response. 12- and 15-lipoxygenating enzymes have been implicated in the biosynthesis of proresolving mediators such as lipoxins (Hu et al., 2012), resolvins (Spite et al., 2014) and maresins (Serhan et al., 2012; Serhan et al., 2015). However, it has not been explored whether 12-lipoxygenating or 15-lipoxygenating ALOX15 orthologs exhibit a higher biosynthetic capacity for these pro-resolving mediators. If, for instance, 15-lipoxygenating ALOX15 orthologs exhibit an augmented lipoxin biosynthesizing capacity the evolutionary switch in positional specificity of ALOX15 orthologs might be regarded as optimizing strategy for inflammatory resolution. In other words, lower mammals expressing a 12-lipoxygenating ALOX15 should have a lower resolution capacity when compared with highly developed species expressing 15-lipoxygenating ALOX15.
The development of isoform-specific ALOX15 inhibitors would clearly advance the research on the biological role of ALOX15 orthologs since reliable pharmacological interference studies could be carried out with such substances. Unfortunately, for the time being comprehensively validated isoform-specifc ALOX15 inhibitors are not commercially available. There are LOX inhibitors, for which isoform specificity have been claimed, but the experimental data indicating isoform-specificity are sometimes not well documented. Moreover, possible non-target effects of these compounds have not been well explored.
Fig. 10. Alox15-deficient mice suffer from more severe experimental autoimmune encephalitis when compared with alox15 sufficient controls.
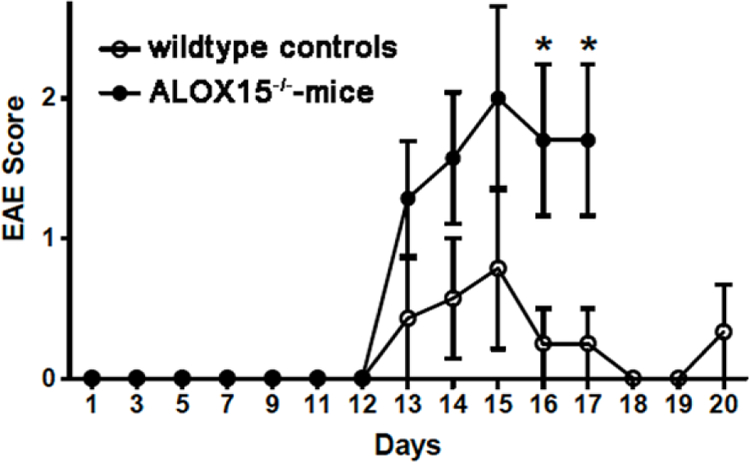
Experimental autoimmune encephalitis (EAE) was induced in 8–10 week old female alox15-deficient mice (LOX-KO; n=7) and corresponding wildtype controls (WT; n=7) by subcutaneous immunization with 200 µg MOG35–55 peptide (purity >95%, Pepceuticals, Leicester, UK) emulsified in an equal volume of PBS and complete Freund’s adjuvant containing 6 mg/ml Mycobacterium tuberculosis H37Ra (Difco, FranklinLakes, NJ). Bordetella pertussis toxin (200 ng, PTX, List Biological Laboratories, Campbell, CA) was administered intraperitoneally at day 0 and 2 post-immunization. Mice were weighed and scored daily as follows: 0 = no disease; 1 = complete tail paralysis; 2 = abnormal gait, hindlimb paresis; 3 = hindlimb plegia; 4 = paraplegia and forelimb weakness; 5 = moribund or death due to EAE. Mann-Whitney-U-test *=p<0.05. The experiments were carried out by Silvina Romero-Suarez and Carmen Infante-Duarte at the Institute for Medical Immunology, Charité Universitätsmedizin Berlin. Data were kindly provided and publication was allowed by the authors.
Highlights.
the human genome involves six lipoxygenase genes including ALOX15
ALOX15 specifically oxygenates phospholipids and biomembranes
ALOX15 exhibits a high degree of structural flexibility
ALOX15 plays a role in differentiation and in pathogenesis of various diseases
evolutionary change in reaction specificity optimizes membrane oxygenase activity.
Achnowledgement
This review and the corresponding Gene Wiki article are written as part of the Gene Wiki Review series--a series resulting from a collaboration between the journal GENE and the Gene Wiki Initiative. The Gene Wiki Initiative is supported by National Institutes of Health (GM089820). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. The authors would like to thank Deutsche Forschungagemeinschaft (Ku961/11–1) for providing financial support. The corresponding Gene Wiki entry for this review can be found here: https://en.wikipedia.org/wiki/ALOX15.
List of non-standard abbreviations
- LOX
lipoxygenase
- AA
arachidonic acid
- EPA
5,8,11,14,17-eicosapentaenoic acid
- DHA
4,7,10,13,16,19-docosahexaenoic acid
- 13S-H(p)ODE
(13S,9Z,11E)-13-hydro(pero)xyoctadeca-9,11-dienoic acid
- 15S-H(p)ETE
(15S,5Z,8Z,11Z,13E)-15-hydro(pero)xyeicosa-5,8,11,13-tetraenoic acid
- 12S-H(p)ETE
(12S,5Z,8Z,10E,14Z)-12-hydro(pero)xyeicosa-5,8,10,14-tetraenoic acid
- 11R-H(p)ETE
(11R,5Z,8Z,12E,14Z)-11-hydro(pero)xyeicosa-5,8,12,14-tetraenoic acid
- IL4(13)
interleukin-4(13)
- DDX6
DEAD-box RNA helicase 6 (DDX6)
- hnRNP K/E1
heterogeneous ribonucleoprotein particle K/E1
- DICE
differentiation control element
- SAXS
small angle X-ray scattering
- PPAR
peroxisome proliferation activating receptor
- LH
luteinizing hormone
- NDGA
nordihydroguaiaretic acid
- LDL
low density lipoprotein
- HDL
high density lipoprotein
- GPx4
glutathione peroxidase-4
- CDC
cinnamyl-3,4-dihydroxy-α-cyanocinnamide
- GC/MS
gas chromatography/mass spectrometry
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adel S, Hofheinz K, Heydeck D, Kuhn H and Häfner AK, 2014. Phosphorylation mimicking mutations of ALOX5 orthologs of different vertebrates do not alter reaction specificities of the enzymes. Biochim Biophys Acta 1841, 1460–1466. [DOI] [PubMed] [Google Scholar]
- Adel S, Kakularam KR, Horn T, Reddanna P, Kuhn H and Heydeck D, 2015. Leukotriene signaling in the extinct human subspecies Homo denisovan and Homo neanderthalensis. Structural and functional comparison with Homo sapiens. Arch Biochem Biophys 565, 17–24. [DOI] [PubMed] [Google Scholar]
- Aggarwal NT, Pfister SL and Campbell WB, 2008. Hypercholesterolemia enhances 15-lipoxygenase-mediated vasorelaxation and acetylcholine-induced hypotension. Arterioscler Thromb Vasc Biol 28, 2209–15. [DOI] [PubMed] [Google Scholar]
- Aggarwal NT, Pfister SL, Gauthier KM, Chawengsub Y, Baker JE and Campbell WB, 2009. Chronic hypoxia enhances 15-lipoxygenase-mediated vasorelaxation in rabbit arteries. Am J Physiol Heart Circ Physiol 296, H678–88. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Alber T, 1992. Structure of the leucine zipper. Curr Opin Genet Dev 2, 205–10. [DOI] [PubMed] [Google Scholar]
- Altmann R, Hausmann M, Spottl T, Gruber M, Bull AW, Menzel K, Vogl D, Herfarth H, Scholmerich J, Falk W and Rogler G, 2007. 13-Oxo-ODE is an endogenous ligand for PPARgamma in human colonic epithelial cells. Biochem Pharmacol 74, 612–22. [DOI] [PubMed] [Google Scholar]
- Bazan NG, 2009. Neuroprotectin D1-mediated anti-inflammatory and survival signaling in stroke, retinal degenerations, and Alzheimer’s disease. J Lipid Res 50 Suppl, S400–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Khoutorova L, Atkins KD, Eady TN, Hong S, Lu Y, Obenaus A and Bazan NG, 2011. Docosahexaenoic Acid therapy of experimental ischemic stroke. Transl Stroke Res 2, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkner J, Stender H and Kuhn H, 1998. The rabbit 15-lipoxygenase preferentially oxygenates LDL cholesterol esters, and this reaction does not require vitamin E. J Biol Chem 273, 23225–32. [DOI] [PubMed] [Google Scholar]
- Belkner J, Wiesner R, Kuhn H and Lankin VZ, 1991. The oxygenation of cholesterol esters by the reticulocyte lipoxygenase. FEBS Lett 279, 110–4. [DOI] [PubMed] [Google Scholar]
- Belkner J, Wiesner R, Rathman J, Barnett J, Sigal E and Kuhn H, 1993. Oxygenation of lipoproteins by mammalian lipoxygenases. Eur J Biochem 213, 251–61. [DOI] [PubMed] [Google Scholar]
- Ben-Nasr S, Aazza S, Mnif W and Mda G. Miguel, 2015. Antioxidant and anti-lipoxygenase activities of extracts from different parts of Lavatera cretica L. grown in Algarve (Portugal). Pharmacogn Mag 11, 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M, Schwarz K, Thiele H, Reimann I, Huth A, Borngraber S, Kuhn H and Thiele BJ, 1998. Simultaneous expression of leukocyte-type 12-lipoxygenase and reticulocyte-type 15-lipoxygenase in rabbits. J Mol Biol 278, 935–48. [DOI] [PubMed] [Google Scholar]
- Berger W, De Chandt MT and Cairns CB, 2007. Zileuton: clinical implications of 5-Lipoxygenase inhibition in severe airway disease. Int J Clin Pract 61, 663–76. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Shukla M, Yakubenko VP, Mulya A, Kundu S and Cathcart MK, 2013. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic Biol Med 54, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindu PH, Sastry GM and Sastry GN, 2004. Characterization of calcium and magnesium binding domains of human 5-lipoxygenase. Biochem Biophys Res Commun 320, 461–7. [DOI] [PubMed] [Google Scholar]
- Blanc L, Barres C, Bette-Bobillo P and Vidal M, 2007. Reticulocyte-secreted exosomes bind natural IgM antibodies: involvement of a ROS-activatable endosomal phospholipase iPLA2. Blood 110, 3407–16. [DOI] [PubMed] [Google Scholar]
- Bleich D, Chen S, Gu JL and Nadler JL, 1998. The role of 12-lipoxygenase in pancreatic -cells (Review). Int J Mol Med 1, 265–72. [PubMed] [Google Scholar]
- Bleich D, Chen S, Zipser B, Sun D, Funk CD and Nadler JL, 1999. Resistance to type 1 diabetes induction in 12-lipoxygenase knockout mice. J Clin Invest 103, 1431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borngraber S, Browner M, Gillmor S, Gerth C, Anton M, Fletterick R and Kuhn H, 1999. Shape and specificity in mammalian 15-lipoxygenase active site. The functional interplay of sequence determinants for the reaction specificity. J Biol Chem 274, 37345–50. [DOI] [PubMed] [Google Scholar]
- Borngraber S, Kuban RJ, Anton M and Kuhn H, 1996. Phenylalanine 353 is a primary determinant for the positional specificity of mammalian 15-lipoxygenases. J Mol Biol 264, 1145–53. [DOI] [PubMed] [Google Scholar]
- Boudina S and Abel ED, 2007. Diabetic cardiomyopathy revisited. Circulation 115, 3213–23. [DOI] [PubMed] [Google Scholar]
- Boyer RF, Litts D, Kostishak J, Wijesundera RC and Gunstone FD, 1979. The action of lipoxygenase-1 on furan derivatives. Chem Phys Lipids 25, 237–46. [DOI] [PubMed] [Google Scholar]
- Brash AR, 1999. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem 274, 23679–82. [DOI] [PubMed] [Google Scholar]
- Brash AR, Boeglin WE and Chang MS, 1997. Discovery of a second 15S-lipoxygenase in humans. Proc Natl Acad Sci U S A 94, 6148–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash AR, Boeglin WE, Chang MS and Shieh BH, 1996. Purification and molecular cloning of an 8R-lipoxygenase from the coral Plexaura homomalla reveal the related primary structures of R- and S-lipoxygenases. J Biol Chem 271, 20949–57. [DOI] [PubMed] [Google Scholar]
- Brash AR, Ingram CD and Harris TM, 1987. Analysis of a specific oxygenation reaction of soybean lipoxygenase-1 with fatty acids esterified in phospholipids. Biochemistry 26, 5465–71. [DOI] [PubMed] [Google Scholar]
- Brash AR, Yokoyama C, Oates JA and Yamamoto S, 1989. Mechanistic studies of the dioxygenase and leukotriene synthase activities of the porcine leukocyte 12S-lipoxygenase. Arch Biochem Biophys 273, 414–22. [DOI] [PubMed] [Google Scholar]
- Brinckmann R, Schnurr K, Heydeck D, Rosenbach T, Kolde G and Kuhn H, 1998. Membrane translocation of 15-lipoxygenase in hematopoietic cells is calcium-dependent and activates the oxygenase activity of the enzyme. Blood 91, 64–74. [PubMed] [Google Scholar]
- Brinckmann R, Topp MS, Zalan I, Heydeck D, Ludwig P, Kuhn H, Berdel WE and Habenicht JR, 1996. Regulation of 15-lipoxygenase expression in lung epithelial cells by interleukin-4. Biochem J 318 ( Pt 1), 305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune B, Dehne N, Grossmann N, Jung M, Namgaladze D, Schmid T, von Knethen A and Weigert A, 2013. Redox control of inflammation in macrophages. Antioxid Redox Signal 19, 595–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant RW, Bailey JM, Schewe T and Rapoport SM, 1982. Positional specificity of a reticulocyte lipoxygenase. Conversion of arachidonic acid to 15-S-hydroperoxy-eicosatetraenoic acid. J Biol Chem 257, 6050–5. [PubMed] [Google Scholar]
- Bryant RW, Schewe T, Rapoport SM and Bailey JM, 1985. Leukotriene formation by a purified reticulocyte lipoxygenase enzyme. Conversion of arachidonic acid and 15-hydroperoxyeicosatetraenoic acid to 14, 15-leukotriene A4. J Biol Chem 260, 3548–55. [PubMed] [Google Scholar]
- Bürger F, Krieg P, Marks F and Fürstenberger G, 2000. Positional- and stereo-selectivity of fatty acid oxygenation catalysed by mouse (12S)-lipoxygenase isoenzymes. Biochem J 348 Pt 2, 329–35. [PMC free article] [PubMed] [Google Scholar]
- Calhoun DB, Vanderkooi JM, Woodrow GV 3rd and Englander SW, 1983. Penetration of dioxygen into proteins studied by quenching of phosphorescence and fluorescence. Biochemistry 22, 1526–32. [DOI] [PubMed] [Google Scholar]
- Canals S, Casarejos MJ, de Bernardo S, Rodriguez-Martin E and Mena MA, 2003. Nitric oxide triggers the toxicity due to glutathione depletion in midbrain cultures through 12-lipoxygenase. J Biol Chem 278, 21542–9. [DOI] [PubMed] [Google Scholar]
- Chaitidis P, Adel S, Anton M, Heydeck D, Kuhn H and Horn T, 2013. Lipoxygenase pathways in Homo neanderthalensis: functional comparison with Homo sapiens isoforms. J Lipid Res 54, 1397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaitidis P, O’Donnell V, Kuban RJ, Bermudez-Fajardo A, Ungethuem U and Kuhn H, 2005. Gene expression alterations of human peripheral blood monocytes induced by medium-term treatment with the TH2-cytokines interleukin-4 and −13. Cytokine 30, 366–77. [DOI] [PubMed] [Google Scholar]
- Chakrabarti SK, Wen Y, Dobrian AD, Cole BK, Ma Q, Pei H, Williams MD, Bevard MH, Vandenhoff GE, Keller SR, Gu J and Nadler JL, 2011. Evidence for activation of inflammatory lipoxygenase pathways in visceral adipose tissue of obese Zucker rats. Am J Physiol Endocrinol Metab 300, E175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH and Fishman RA, 1978. Brain edema: induction in cortical slices by polyunsaturated fatty acids. Science 201, 358–60. [DOI] [PubMed] [Google Scholar]
- Chan PH, Fishman RA, Caronna J, Schmidley JW, Prioleau G and Lee J, 1983. Induction of brain edema following intracerebral injection of arachidonic acid. Ann Neurol 13, 625–32. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay R, Dyukova E, Singh NK, Ohba M, Mobley JA and Rao GN, 2014. Vascular endothelial tight junctions and barrier function are disrupted by 15(S)-hydroxyeicosatetraenoic acid partly via protein kinase C epsilon-mediated zona occludens-1 phosphorylation at threonine 770/772. J Biol Chem 289, 3148–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay R, Tinnikov A, Dyukova E, Singh NK, Kotla S, Mobley JA and Rao GN, 2015. 12/15-Lipoxygenase-dependent ROS production is required for diet-induced endothelial barrier dysfunction. J Lipid Res 56, 562–77. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chawengsub Y, Gauthier KM and Campbell WB, 2009. Role of arachidonic acid lipoxygenase metabolites in the regulation of vascular tone. Am J Physiol Heart Circ Physiol 297, H495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Tsui S, Boeglin WE, Douglas RS, Brash AR and Smith TJ, 2006. Interleukin-4 induces 15-lipoxygenase-1 expression in human orbital fibroblasts from patients with Graves disease. Evidence for anatomic site-selective actions of Th2 cytokines. J Biol Chem 281, 18296–306. [DOI] [PubMed] [Google Scholar]
- Chen M, Yang ZD, Smith KM, Carter JD and Nadler JL, 2005. Activation of 12-lipoxygenase in proinflammatory cytokine-mediated beta cell toxicity. Diabetologia 48, 486–95. [DOI] [PubMed] [Google Scholar]
- Chisolm GM and Steinberg D, 2000. The oxidative modification hypothesis of atherogenesis: an overview. Free Radic Biol Med 28, 1815–26. [DOI] [PubMed] [Google Scholar]
- Chiu J and Dawes IW, 2012. Redox control of cell proliferation. Trends Cell Biol 22, 592–601. [DOI] [PubMed] [Google Scholar]
- Choi J, Chon JK, Kim S and Shin W, 2008. Conformational flexibility in mammalian 15S-lipoxygenase: Reinterpretation of the crystallographic data. Proteins 70, 1023–32. [DOI] [PubMed] [Google Scholar]
- Chu K, Vojtchovsky J, McMahon BH, Sweet RM, Berendzen J and Schlichting I, 2000. Structure of a ligand-binding intermediate in wild-type carbonmonoxy myoglobin. Nature 403, 921–3. [DOI] [PubMed] [Google Scholar]
- Cimen I, Astarci E and Banerjee S, 2011. 15-lipoxygenase-1 exerts its tumor suppressive role by inhibiting nuclear factor-kappa B via activation of PPAR gamma. J Cell Biochem 112, 2490–501. [DOI] [PubMed] [Google Scholar]
- Cimen I, Tuncay S and Banerjee S, 2009. 15-Lipoxygenase-1 expression suppresses the invasive properties of colorectal carcinoma cell lines HCT-116 and HT-29. Cancer Sci 100, 2283–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson HE, 2009. On the biosynthesis and biological role of eoxins and 15-lipoxygenase-1 in airway inflammation and Hodgkin lymphoma. Prostaglandins Other Lipid Mediat 89, 120–5. [DOI] [PubMed] [Google Scholar]
- Coffa G and Brash AR, 2004. A single active site residue directs oxygenation stereospecificity in lipoxygenases: stereocontrol is linked to the position of oxygenation. Proc Natl Acad Sci U S A 101, 15579–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffa G and Hill EM, 2000. Discovery of an 11 (R)- and 12(S)-lipoxygenase activity in ovaries of the mussel Mytilus edulis. Lipids 35, 1195–204. [DOI] [PubMed] [Google Scholar]
- Coffa G, Imber AN, Maguire BC, Laxmikanthan G, Schneider C, Gaffney BJ and Brash AR, 2005a. On the relationships of substrate orientation, hydrogen abstraction, and product stereochemistry in single and double dioxygenations by soybean lipoxygenase-1 and its Ala542Gly mutant. J Biol Chem 280, 38756–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffa G, Schneider C and Brash AR, 2005b. A comprehensive model of positional and stereo control in lipoxygenases. Biochem Biophys Res Commun 338, 87–92. [DOI] [PubMed] [Google Scholar]
- Cohen J, Arkhipov A, Braun R and Schulten K, 2006. Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophys J 91, 1844–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole BK, Lieb DC, Dobrian AD and Nadler JL, 2013. 12- and 15-lipoxygenases in adipose tissue inflammation. Prostaglandins Other Lipid Mediat 104–105, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole BK, Morris MA, Grzesik WJ, Leone KA and Nadler JL, 2012. Adipose tissue-specific deletion of 12/15-lipoxygenase protects mice from the consequences of a high-fat diet. Mediators Inflamm 2012, 851798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DJ, Kuhn H, Mulkins M, Highland E and Sigal E, 1992. Specific inflammatory cytokines regulate the expression of human monocyte 15-lipoxygenase. Proc Natl Acad Sci U S A 89, 217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DJ and Lu M, 2000. Regulation of human 12/15-lipoxygenase by Stat6-dependent transcription. Am J Respir Cell Mol Biol 22, 226–34. [DOI] [PubMed] [Google Scholar]
- Conte D, Falaschi P, Proietti A, D’Urso R, Citarella F, Nordio M, Romanelli F, Maggi R, Motta M and Isidori A, 1986. Role of arachidonate metabolism on the in vitro release of luteinizing hormone and prolactin from the anterior pituitary gland: possible involvement of lipoxygenase pathway. Neuroendocrinology 43, 428–34. [DOI] [PubMed] [Google Scholar]
- Cooke BA, Dix CJ, Habberfield AD and Sullivan MH, 1984. Control of steroidogenesis in Leydig cells: roles of Ca2+ and lipoxygenase products in LH and LHRH agonist action. Ann N Y Acad Sci 438, 269–82. [DOI] [PubMed] [Google Scholar]
- Cretu E, Trifan A, Aprotosoaie AC and Miron A, 2013. 15-lipoxygenase inhibition, superoxide and hydroxyl radicals scavenging activities of Cedrus brevifolia bark extracts. Rev Med Chir Soc Med Nat Iasi 117, 250–6. [PubMed] [Google Scholar]
- Cutuli L, Pirillo A, Uboldi P, Kuehn H and Catapano AL, 2014. 15-lipoxygenase-mediated modification of HDL3 impairs eNOS activation in human endothelial cells. Lipids 49, 317–26. [DOI] [PubMed] [Google Scholar]
- Cyrus T, Pratico D, Zhao L, Witztum JL, Rader DJ, Rokach J, FitzGerald GA and Funk CD, 2001. Absence of 12/15-lipoxygenase expression decreases lipid peroxidation and atherogenesis in apolipoprotein e-deficient mice. Circulation 103, 2277–82. [DOI] [PubMed] [Google Scholar]
- Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF and Funk CD, 1999. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J Clin Invest 103, 1597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dainese E, Sabatucci A, van Zadelhoff G, Angelucci CB, Vachette P, Veldink GA, Agro AF and Maccarrone M, 2005. Structural stability of soybean lipoxygenase-1 in solution as probed by small angle X-ray scattering. J Mol Biol 349, 143–52. [DOI] [PubMed] [Google Scholar]
- Dalli J, Colas RA and Serhan CN, 2013. Novel n-3 immunoresolvents: structures and actions. Sci Rep 3, 1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot JJ, Garssen GJ, Vliegenthart JF and Boldingh J, 1973. The detection of linoleic acid radicals in the anaerobic reaction of lipoxygenase. Biochim Biophys Acta 326, 279–84. [DOI] [PubMed] [Google Scholar]
- de La Houssaye BA, Mikule K, Nikolic D and Pfenninger KH, 1999. Thrombin-induced growth cone collapse: involvement of phospholipase A(2) and eicosanoid generation. J Neurosci 19, 10843–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marzo N, Sloane DL, Dicharry S, Highland E and Sigal E, 1992. Cloning and expression of an airway epithelial 12-lipoxygenase. Am J Physiol 262, L198–207. [DOI] [PubMed] [Google Scholar]
- DeCostanzo AJ, Voloshyna I, Rosen ZB, Feinmark SJ and Siegelbaum SA, 2010. 12-Lipoxygenase regulates hippocampal long-term potentiation by modulating L-type Ca2+ channels. J Neurosci 30, 1822–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovanni S, Knoblach SM, Brandoli C, Aden SA, Hoffman EP and Faden AI, 2003. Gene profiling in spinal cord injury shows role of cell cycle in neuronal death. Ann Neurol 53, 454–68. [DOI] [PubMed] [Google Scholar]
- Di Venere A, Horn T, Stehling S, Mei G, Masgrau L, Gonzalez-Lafont A, Kuhn H and Ivanov I, 2013. Role of Arg403 for thermostability and catalytic activity of rabbit 12/15-lipoxygenase. Biochim Biophys Acta 1831, 1079–88. [DOI] [PubMed] [Google Scholar]
- Dix CJ, Habberfield AD, Sullivan MH and Cooke BA, 1984. Inhibition of steroid production in Leydig cells by non-steroidal anti-inflammatory and related compounds: evidence for the involvement of lipoxygenase products in steroidogenesis. Biochem J 219, 529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix CJ, Habberfield AD, Sullivan MH and Cooke BA, 1985. Evidence for the involvement of lipoxygenase products in steroidogenesis. Biochem Soc Trans 13, 60–3. [DOI] [PubMed] [Google Scholar]
- Dobrian AD, Lieb DC, Cole BK, Taylor-Fishwick DA, Chakrabarti SK and Nadler JL, 2011. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res 50, 115–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrian AD, Lieb DC, Ma Q, Lindsay JW, Cole BK, Ma K, Chakrabarti SK, Kuhn NS, Wohlgemuth SD, Fontana M and Nadler JL, 2010. Differential expression and localization of 12/15 lipoxygenases in adipose tissue in human obese subjects. Biochem Biophys Res Commun 403, 485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey BR, Mootoo JE and Doyle SE, 1998. A role for lipoxygenase metabolites of arachidonic acid in porcine ovulation. Anim Reprod Sci 49, 269–79. [DOI] [PubMed] [Google Scholar]
- El Kebir D and Filep JG, 2013. Modulation of Neutrophil Apoptosis and the Resolution of Inflammation through beta2 Integrins. Front Immunol 4, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson MR and LeVine SM, 2004. Experimental allergic encephalomyelitis is exacerbated in mice deficient for 12/15-lipoxygenase or 5-lipoxygenase. Brain Res 1021, 140–5. [DOI] [PubMed] [Google Scholar]
- Epp N, Furstenberger G, Muller K, de Juanes S, Leitges M, Hausser I, Thieme F, Liebisch G, Schmitz G and Krieg P, 2007. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J Cell Biol 177, 173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ereso AQ, Cureton EL, Cripps MW, Sadjadi J, Dua MM, Curran B and Victorino GP, 2009. Lipoxin a(4) attenuates microvascular fluid leak during inflammation. J Surg Res 156, 183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezcurra D and Humaidan P, 2014. A review of luteinising hormone and human chorionic gonadotropin when used in assisted reproductive technology. Reprod Biol Endocrinol 12, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinmark SJ, Begum R, Tsvetkov E, Goussakov I, Funk CD, Siegelbaum SA and Bolshakov VY, 2003. 12-lipoxygenase metabolites of arachidonic acid mediate metabotropic glutamate receptor-dependent long-term depression at hippocampal CA3-CA1 synapses. J Neurosci 23, 11427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltenmark S, Gautam N, Brunnstrom A, Griffiths W, Backman L, Edenius C, Lindbom L, Bjorkholm M and Claesson HE, 2008. Eoxins are proinflammatory arachidonic acid metabolites produced via the 15-lipoxygenase-1 pathway in human eosinophils and mast cells. Proc Natl Acad Sci U S A 105, 680–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre N, Martinez-Clemente M, Lopez-Parra M, Gonzalez-Periz A, Horrillo R, Planaguma A, Camps J, Joven J, Tres A, Guardiola F, Bataller R, Arroyo V and Claria J, 2009. Increased susceptibility to exacerbated liver injury in hypercholesterolemic ApoE-deficient mice: potential involvement of oxysterols. Am J Physiol Gastrointest Liver Physiol 296, G553–62. [DOI] [PubMed] [Google Scholar]
- Fierro IM, Colgan SP, Bernasconi G, Petasis NA, Clish CB, Arita M and Serhan CN, 2003. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit human neutrophil migration: comparisons between synthetic 15 epimers in chemotaxis and transmigration with microvessel endothelial cells and epithelial cells. J Immunol 170, 2688–94. [DOI] [PubMed] [Google Scholar]
- Fischer KA, Van Leyen K, Lovercamp KW, Manandhar G, Sutovsky M, Feng D, Safranski T and Sutovsky P, 2005. 15-Lipoxygenase is a component of the mammalian sperm cytoplasmic droplet. Reproduction 130, 213–22. [DOI] [PubMed] [Google Scholar]
- Fleming J, Thiele BJ, Chester J, O’Prey J, Janetzki S, Aitken A, Anton IA, Rapoport SM and Harrison PR, 1989. The complete sequence of the rabbit erythroid cell-specific 15-lipoxygenase mRNA: comparison of the predicted amino acid sequence of the erythrocyte lipoxygenase with other lipoxygenases. Gene 79, 181–8. [DOI] [PubMed] [Google Scholar]
- Folcik VA, Nivar-Aristy RA, Krajewski LP and Cathcart MK, 1995. Lipoxygenase contributes to the oxidation of lipids in human atherosclerotic plaques. J Clin Invest 96, 504–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire MO and Van Dyke TE, 2013. Natural resolution of inflammation. Periodontol 2000 63, 149–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CD, Chen XS, Johnson EN and Zhao L, 2002. Lipoxygenase genes and their targeted disruption. Prostaglandins Other Lipid Mediat 68–69, 303–12. [DOI] [PubMed] [Google Scholar]
- Gan QF, Browner MF, Sloane DL and Sigal E, 1996. Defining the arachidonic acid binding site of human 15-lipoxygenase. Molecular modeling and mutagenesis. J Biol Chem 271, 25412–8. [DOI] [PubMed] [Google Scholar]
- Gan QF, Witkop GL, Sloane DL, Straub KM and Sigal E, 1995. Identification of a specific methionine in mammalian 15-lipoxygenase which is oxygenated by the enzyme product 13-HPODE: dissociation of sulfoxide formation from self-inactivation. Biochemistry 34, 7069–79. [DOI] [PubMed] [Google Scholar]
- Garreta A, Val-Moraes SP, Garcia-Fernandez Q, Busquets M, Juan C, Oliver A, Ortiz A, Gaffney BJ, Fita I, Manresa A and Carpena X, 2013. Structure and interaction with phospholipids of a prokaryotic lipoxygenase from Pseudomonas aeruginosa. FASEB J 27, 4811–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garscha U, Jernerén F, Chung D, Keller NP, Hamberg M and Oliw EH, 2007. Identification of dioxygenases required for Aspergillus development. Studies of products, stereochemistry, and the reaction mechanism. J Biol Chem 282, 34707–18. [DOI] [PubMed] [Google Scholar]
- Garssen GJ, Vliegenthart JF and Boldingh J, 1971. An anaerobic reaction between lipoxygenase, linoleic acid and its hydroperoxides. Biochem J 122, 327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garssen GJ, Vliegenthart JF and Boldingh J, 1972. The origin and structures of dimeric fatty acids from the anaerobic reaction between soya-bean lipoxygenase, linoleic acid and its hydroperoxide. Biochem J 130, 435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George J, Afek A, Shaish A, Levkovitz H, Bloom N, Cyrus T, Zhao L, Funk CD, Sigal E and Harats D, 2001. 12/15-Lipoxygenase gene disruption attenuates atherogenesis in LDL receptor-deficient mice. Circulation 104, 1646–50. [DOI] [PubMed] [Google Scholar]
- Gertow K, Nobili E, Folkersen L, Newman JW, Pedersen TL, Ekstrand J, Swedenborg J, Kühn H, Wheelock CE, Hansson GK, Hedin U, Haeggström JZ and Gabrielsen A, 2011. 12- and 15-lipoxygenases in human carotid atherosclerotic lesions: associations with cerebrovascular symptoms. Atherosclerosis 215, 411–6. [DOI] [PubMed] [Google Scholar]
- Gilbert NC, Rui Z, Neau DB, Waight MT, Bartlett SG, Boeglin WE, Brash AR and Newcomer ME, 2012. Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at Serine-663. FASEB J 26, 3222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillmor SA, Villasenor A, Fletterick R, Sigal E and Browner MF, 1997. The structure of mammalian 15-lipoxygenase reveals similarity to the lipases and the determinants of substrate specificity. Nat Struct Biol 4, 1003–9. [DOI] [PubMed] [Google Scholar]
- Glick D, Barth S and Macleod KF, 2010. Autophagy: cellular and molecular mechanisms. J Pathol 221, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami SK, 2013. Cellular redox, epigenetics and diseases. Subcell Biochem 61, 527–42. [DOI] [PubMed] [Google Scholar]
- Green-Mitchell SM, Tersey SA, Cole BK, Ma K, Kuhn NS, Cunningham TD, Maybee NA, Chakrabarti SK, McDuffie M, Taylor-Fishwick DA, Mirmira RG, Nadler JL and Morris MA, 2013. Deletion of 12/15-lipoxygenase alters macrophage and islet function in NOD-Alox15(null) mice, leading to protection against type 1 diabetes development. PLoS One 8, e56763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregus AM, Dumlao DS, Wei SC, Norris PC, Catella LC, Meyerstein FG, Buczynski MW, Steinauer JJ, Fitzsimmons BL, Yaksh TL and Dennis EA, 2013. Systematic analysis of rat 12/15-lipoxygenase enzymes reveals critical role for spinal eLOX3 hepoxilin synthase activity in inflammatory hyperalgesia. FASEB J 27, 1939–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths RE, Kupzig S, Cogan N, Mankelow TJ, Betin VM, Trakarnsanga K, Massey EJ, Parsons SF, Anstee DJ and Lane JD, 2012. The ins and outs of human reticulocyte maturation: autophagy and the endosome/exosome pathway. Autophagy 8, 1150–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronowicz G, Swift H and Steck TL, 1984. Maturation of the reticulocyte in vitro. J Cell Sci 71, 177–97. [DOI] [PubMed] [Google Scholar]
- Grossman S, Shahin I and Sredni B, 1979. Rat testis lipoxygenase-like enzyme. Characterization of products from linoleic acid. Biochim Biophys Acta 572, 293–7. [DOI] [PubMed] [Google Scholar]
- Grullich C, Duvoisin RM, Wiedmann M and van Leyen K, 2001. Inhibition of 15-lipoxygenase leads to delayed organelle degradation in the reticulocyte. FEBS Lett 489, 51–4. [DOI] [PubMed] [Google Scholar]
- Gustafson B, Hedjazifar S, Gogg S, Hammarstedt A and Smith U, 2015. Insulin resistance and impaired adipogenesis. Trends Endocrinol Metab 26, 193–200. [DOI] [PubMed] [Google Scholar]
- Haas U, Raschperger E, Hamberg M, Samuelsson B, Tryggvason K and Haeggstrom JZ, 2011. Targeted knock-down of a structurally atypical zebrafish 12S-lipoxygenase leads to severe impairment of embryonic development. Proc Natl Acad Sci U S A 108, 20479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hada T, Swift LL and Brash AR, 1997. Discovery of 5R-lipoxygenase activity in oocytes of the surf clam, Spisula solidissima. Biochim Biophys Acta 1346, 109–19. [DOI] [PubMed] [Google Scholar]
- Haeggstrom JZ and Funk CD, 2011. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev 111, 5866–98. [DOI] [PubMed] [Google Scholar]
- Hallenborg P, Jorgensen C, Petersen RK, Feddersen S, Araujo P, Markt P, Langer T, Furstenberger G, Krieg P, Koppen A, Kalkhoven E, Madsen L and Kristiansen K, 2010. Epidermis-type lipoxygenase 3 regulates adipocyte differentiation and peroxisome proliferator-activated receptor gamma activity. Mol Cell Biol 30, 4077–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamberg M and Samuelsson B, 1974. Prostaglandin endoperoxides. Novel transformations of arachidonic acid in human platelets. Proc Natl Acad Sci U S A 71, 3400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Walther M, Prassl R and Kuhn H, 2004. Structural flexibility of the N-terminal beta-barrel domain of 15-lipoxygenase-1 probed by small angle X-ray scattering. Functional consequences for activity regulation and membrane binding. J Mol Biol 343, 917–29. [DOI] [PubMed] [Google Scholar]
- Han H, Xu D, Liu C, Claesson HE, Bjorkholm M and Sjoberg J, 2014. Interleukin-4-mediated 15-lipoxygenase-1 trans-activation requires UTX recruitment and H3K27me3 demethylation at the promoter in A549 cells. PLoS One 9, e85085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Sun L, Xu Y, Liang H and Cheng Y, 2015. Activation of PPARgamma by 12/15-lipoxygenase during cerebral ischemia-reperfusion injury. Int J Mol Med 35, 195–201. [DOI] [PubMed] [Google Scholar]
- Hansen J, Garreta A, Benincasa M, Fuste MC, Busquets M and Manresa A, 2013. Bacterial lipoxygenases, a new subfamily of enzymes? A phylogenetic approach. Appl Microbiol Biotechnol 97, 4737–47. [DOI] [PubMed] [Google Scholar]
- Harats D, Ben-Shushan D, Cohen H, Gonen A, Barshack I, Goldberg I, Greenberger S, Hodish I, Harari A, Varda-Bloom N, Levanon K, Grossman E, Chaitidis P, Kuhn H and Shaish A, 2005. Inhibition of carcinogenesis in transgenic mouse models over-expressing 15-lipoxygenase in the vascular wall under the control of murine preproendothelin-1 promoter. Cancer Lett 229, 127–34. [DOI] [PubMed] [Google Scholar]
- Hartel B, Ludwig P, Schewe T and Rapoport SM, 1982. Self-inactivation by 13-hydroperoxylinoleic acid and lipohydroperoxidase activity of the reticulocyte lipoxygenase. Eur J Biochem 126, 353–7. [DOI] [PubMed] [Google Scholar]
- Hawkins DJ and Brash AR, 1987. Eggs of the sea urchin, Strongylocentrotus purpuratus, contain a prominent (11R) and (12R) lipoxygenase activity. J Biol Chem 262, 7629–34. [PubMed] [Google Scholar]
- Hawkins KE, DeMars KM, Singh J, Yang C, Cho HS, Frankowski JC, Dore S and Candelario-Jalil E, 2014. Neurovascular protection by post-ischemic intravenous injections of the lipoxin A4 receptor agonist, BML-111, in a rat model of ischemic stroke. J Neurochem 129, 130–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes RL and van Leyen K, 2013. 12/15-lipoxygenase expression is increased in oligodendrocytes and microglia of periventricular leukomalacia. Dev Neurosci 35, 140–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henricks PA, Engels F, van der Vliet H and Nijkamp FP, 1991. 9- and 13-hydroxy-linoleic acid possess chemotactic activity for bovine and human polymorphonuclear leukocytes. Prostaglandins 41, 21–7. [DOI] [PubMed] [Google Scholar]
- Hersberger M, 2010. Potential role of the lipoxygenase derived lipid mediators in atherosclerosis: leukotrienes, lipoxins and resolvins. Clin Chem Lab Med 48, 1063–73. [DOI] [PubMed] [Google Scholar]
- Heydeck D, Thomas L, Schnurr K, Trebus F, Thierfelder WE, Ihle JN and Kuhn H, 1998. Interleukin-4 and −13 induce upregulation of the murine macrophage 12/15-lipoxygenase activity: evidence for the involvement of transcription factor STAT6. Blood 92, 2503–10. [PubMed] [Google Scholar]
- Heydeck D, Upston JM, Viita H, Yla-Herttuala S and Stocker R, 2001. Oxidation of LDL by rabbit and human 15-lipoxygenase: prevalence of nonenzymatic reactions. J Lipid Res 42, 1082–8. [PubMed] [Google Scholar]
- Higgs GA, Salmon JA and Spayne JA, 1981. The inflammatory effects of hydroperoxy and hydroxy acid products of arachidonate lipoxygenase in rabbit skin. Br J Pharmacol 74, 429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann B and Steinhilber D, 2013. 5-Lipoxygenase inhibitors: a review of recent patents (2010–2012). Expert Opin Ther Pat 23, 895–909. [DOI] [PubMed] [Google Scholar]
- Hohne M, Bayer D, Prehn S, Schewe T and Rapoport SM, 1983. In vitro maturation of rabbit reticulocytes. III. Response of lipoxygenase. Biomed Biochim Acta 42, 1129–34. [PubMed] [Google Scholar]
- Horn T, Adel S, Schumann R, Sur S, Kakularam KR, Polamarasetty A, Redanna P, Kuhn H and Heydeck D, 2014. Evolutionary aspects of lipoxygenases and genetic diversity of human leukotriene signaling. Prog Lipid Res 57C, 13–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn T, Kakularam K. Reddy, Anton M, Richter C, Reddanna P and Kuhn H, 2013. Functional characterization of genetic enzyme variations in human lipoxygenases. Redox Biol 1, 566–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Mao-Ying QL, Wang J, Wang ZF, Mi WL, Wang XW, Jiang JW, Huang YL, Wu GC and Wang YQ, 2012. Lipoxins and aspirin-triggered lipoxin alleviate bone cancer pain in association with suppressing expression of spinal proinflammatory cytokines. J Neuroinflammation 9, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D and Glass CK, 1999. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 400, 378–82. [DOI] [PubMed] [Google Scholar]
- Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, Burcin T, Forlow SB, Stark MA, Smith DF, Clarke S, Srinivasan S, Hedrick CC, Pratico D, Witztum JL, Nadler JL, Funk CD and Ley K, 2004. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation 110, 2024–31. [DOI] [PubMed] [Google Scholar]
- Ikawa H, Yamamoto K, Takahashi Y, Ueda N, Hayashi Y, Yamamoto S, Ishimura K, Irahara M and Aono T, 1996. Arachidonate 12-lipoxygenase in porcine anterior pituitary cells: its localization and possible function in gonadotrophs. J Endocrinol 148, 33–41. [DOI] [PubMed] [Google Scholar]
- Ivanov I, Heydeck D, Hofheinz K, Roffeis J, O’Donnell VB, Kuhn H and Walther M, 2010. Molecular enzymology of lipoxygenases. Arch Biochem Biophys 503, 161–74. [DOI] [PubMed] [Google Scholar]
- Ivanov I, Romanov S, Ozdoba C, Holzhutter HG, Myagkova G and Kuhn H, 2004. Enantioselective substrate specificity of 15-lipoxygenase 1. Biochemistry 43, 15720–8. [DOI] [PubMed] [Google Scholar]
- Ivanov I, Saam J, Kuhn H and Holzhutter HG, 2005. Dual role of oxygen during lipoxygenase reactions. FEBS J 272, 2523–35. [DOI] [PubMed] [Google Scholar]
- Ivanov I, Schwarz K, Holzhutter HG, Myagkova G and Kuhn H, 1998. Omega-oxidation impairs oxidizability of polyenoic fatty acids by 15-lipoxygenases: consequences for substrate orientation at the active site. Biochem J 336 ( Pt 2), 345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I, Shang W, Toledo L, Masgrau L, Svergun DI, Stehling S, Gomez H, Di Venere A, Mei G, Lluch JM, Skrzypczak-Jankun E, Gonzalez-Lafont A and Kuhn H, 2012. Ligand-induced formation of transient dimers of mammalian 12/15-lipoxygenase: a key to allosteric behavior of this class of enzymes? Proteins 80, 703–12. [DOI] [PubMed] [Google Scholar]
- Janakiram NB, Mohammed A and Rao CV, 2011. Role of lipoxins, resolvins, and other bioactive lipids in colon and pancreatic cancer. Cancer Metastasis Rev 30, 507–23. [DOI] [PubMed] [Google Scholar]
- Janakiram NB and Rao CV, 2014. The role of inflammation in colon cancer. Adv Exp Med Biol 816, 25–52. [DOI] [PubMed] [Google Scholar]
- Jansen C, Hofheinz K, Vogel R, Roffeis J, Anton M, Reddanna P, Kuhn H and Walther M, 2011. Stereocontrol of arachidonic acid oxygenation by vertebrate lipoxygenases: newly cloned zebrafish lipoxygenase 1 does not follow the Ala-versus-Gly concept. J Biol Chem 286, 37804–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Arai K, Murata Y, Wang S, Stins MF, Lo EH and van Leyen K, 2008. Protecting against cerebrovascular injury: contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke 39, 2538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jisaka M, Kim RB, Boeglin WE and Brash AR, 2000. Identification of amino acid determinants of the positional specificity of mouse 8S-lipoxygenase and human 15S-lipoxygenase-2. J Biol Chem 275, 1287–93. [DOI] [PubMed] [Google Scholar]
- Johannesson M, Backman L, Claesson HE and Forsell PK, 2010. Cloning, purification and characterization of non-human primate 12/15-lipoxygenases. Prostaglandins Leukot Essent Fatty Acids 82, 121–9. [DOI] [PubMed] [Google Scholar]
- Keber R, Rozman D and Horvat S, 2013. Sterols in spermatogenesis and sperm maturation. J Lipid Res 54, 20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelavkar U, Wang S, Montero A, Murtagh J, Shah K and Badr K, 1998. Human 15-lipoxygenase gene promoter: analysis and identification of DNA binding sites for IL-13-induced regulatory factors in monocytes. Mol Biol Rep 25, 173–82. [DOI] [PubMed] [Google Scholar]
- Kelavkar UP, Wang S and Badr KF, 2000. Ku autoantigen (DNA helicase) is required for interleukins-13/−4-induction of 15-lipoxygenase-1 gene expression in human epithelial cells. Genes Immun 1, 237–50. [DOI] [PubMed] [Google Scholar]
- Kent G, Minick OT, Volini FI and Orfei E, 1966. Autophagic vacuoles in human red cells. Am J Pathol 48, 831–57. [PMC free article] [PubMed] [Google Scholar]
- Kenyon V, Rai G, Jadhav A, Schultz L, Armstrong M, Jameson JB 2nd, Perry S, Joshi N, Bougie JM, Leister W, Taylor-Fishwick DA, Nadler JL, Holinstat M, Simeonov A, Maloney DJ and Holman TR, 2011. Discovery of potent and selective inhibitors of human platelet-type 12-lipoxygenase. J Med Chem 54, 5485–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR and Sen CK, 2003. Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J Biol Chem 278, 43508–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Roy S, Slivka A, Craft TK, Chaki S, Rink C, Notestine MA, DeVries AC, Parinandi NL and Sen CK, 2005. Neuroprotective properties of the natural vitamin E alpha-tocotrienol. Stroke 36, 2258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesel L, Przylipiak A, Rabe T, Przylipiak M and Runnebaum B, 1987. Arachidonic acid and its lipoxygenase metabolites stimulate prolactin release in superfused pituitary cells. Hum Reprod 2, 281–5. [DOI] [PubMed] [Google Scholar]
- Kim GH, Ryan JJ and Archer SL, 2013. The role of redox signaling in epigenetics and cardiovascular disease. Antioxid Redox Signal 18, 1920–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klil-Drori AJ and Ariel A, 2013. 15-Lipoxygenases in cancer: a double-edged sword? Prostaglandins Other Lipid Mediat 106, 16–22. [DOI] [PubMed] [Google Scholar]
- Knapp MJ and Klinman JP, 2003. Kinetic studies of oxygen reactivity in soybean lipoxygenase-1. Biochemistry 42, 11466–75. [DOI] [PubMed] [Google Scholar]
- Knapp MJ, Seebeck FP and Klinman JP, 2001. Steric control of oxygenation regiochemistry in soybean lipoxygenase-1. J Am Chem Soc 123, 2931–2. [DOI] [PubMed] [Google Scholar]
- Krieg P, Rosenberger S, de Juanes S, Latzko S, Hou J, Dick A, Kloz U, van der Hoeven F, Hausser I, Esposito I, Rauh M and Schneider H, 2013. Aloxe3 knockout mice reveal a function of epidermal lipoxygenase-3 as hepoxilin synthase and its pivotal role in barrier formation. J Invest Dermatol 133, 172–80. [DOI] [PubMed] [Google Scholar]
- Kriska T, Cepura C, Magier D, Siangjong L, Gauthier KM and Campbell WB, 2012. Mice lacking macrophage 12/15-lipoxygenase are resistant to experimental hypertension. Am J Physiol Heart Circ Physiol 302, H2428–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke G, Katzenbeisser J, Uderhardt S, Zaiss MM, Scholtysek C, Schabbauer G, Zarbock A, Koenders MI, Axmann R, Zwerina J, Baenckler HW, van den Berg W, Voll RE, Kuhn H, Joosten LA and Schett G, 2009a. 12/15-lipoxygenase counteracts inflammation and tissue damage in arthritis. J Immunol 183, 3383–9. [DOI] [PubMed] [Google Scholar]
- Kronke G, Uderhardt S, Katzenbeisser J and Schett G, 2009b. The 12/15-lipoxygenase pathway promotes osteoclast development and differentiation. Autoimmunity 42, 383–5. [DOI] [PubMed] [Google Scholar]
- Kroschwald P, Kroschwald A, Kuhn H, Ludwig P, Thiele BJ, Hohne M, Schewe T and Rapoport SM, 1989. Occurrence of the erythroid cell specific arachidonate 15-lipoxygenase in human reticulocytes. Biochem Biophys Res Commun 160, 954–60. [DOI] [PubMed] [Google Scholar]
- Kuban RJ, Wiesner R, Rathman J, Veldink G, Nolting H, Solé VA and Kühn H, 1998. The iron ligand sphere geometry of mammalian 15-lipoxygenases. Biochem J 332 ( Pt 1), 237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H, 1996. Biosynthesis, metabolization and biological importance of the primary 15-lipoxygenase metabolites 15-hydro(pero)XY-5Z,8Z,11Z,13E-eicosatetraenoic acid and 13-hydro(pero)XY-9Z,11E-octadecadienoic acid. Prog Lipid Res 35, 203–26. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Banthiya S and van Leyen K, 2014. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn H, Barnett J, Grunberger D, Baecker P, Chow J, Nguyen B, Bursztyn-Pettegrew H, Chan H and Sigal E, 1993. Overexpression, purification and characterization of human recombinant 15-lipoxygenase. Biochim Biophys Acta 1169, 80–9. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Belkner J and Wiesner R, 1990a. Subcellular distribution of lipoxygenase products in rabbit reticulocyte membranes. Eur J Biochem 191, 221–7. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Belkner J, Wiesner R and Brash AR, 1990b. Oxygenation of biological membranes by the pure reticulocyte lipoxygenase. J Biol Chem 265, 18351–61. [PubMed] [Google Scholar]
- Kuhn H, Belkner J, Zaiss S, Fahrenklemper T and Wohlfeil S, 1994. Involvement of 15-lipoxygenase in early stages of atherogenesis. J Exp Med 179, 1903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H and Brash AR, 1990. Occurrence of lipoxygenase products in membranes of rabbit reticulocytes. Evidence for a role of the reticulocyte lipoxygenase in the maturation of red cells. J Biol Chem 265, 1454–8. [PubMed] [Google Scholar]
- Kuhn H, Heydeck D, Hugou I and Gniwotta C, 1997. In vivo action of 15-lipoxygenase in early stages of human atherogenesis. J Clin Invest 99, 888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H, Heydeck D and Sprecher H, 1991. On the mechanistic reasons for the dual positional specificity of the reticulocyte lipoxygenase. Biochim Biophys Acta 1081, 129–34. [DOI] [PubMed] [Google Scholar]
- Kuhn H and O’Donnell VB, 2006. Inflammation and immune regulation by 12/15-lipoxygenases. Prog Lipid Res 45, 334–56. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Romisch I and Belkner J, 2005a. The role of lipoxygenase-isoforms in atherogenesis. Mol Nutr Food Res 49, 1014–29. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Saam J, Eibach S, Holzhutter HG, Ivanov I and Walther M, 2005b. Structural biology of mammalian lipoxygenases: enzymatic consequences of targeted alterations of the protein structure. Biochem Biophys Res Commun 338, 93–101. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Salzmann-Reinhardt U, Ludwig P, Ponicke K, Schewe T and Rapoport S, 1986a. The stoichiometry of oxygen uptake and conjugated diene formation during the dioxygenation of linoleic acid by the pure reticulocyte lipoxygenase. Evidence for aerobic hydroperoxidase activity. Biochim Biophys Acta 876, 187–93. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Schewe T and Rapoport SM, 1986b. The stereochemistry of the reactions of lipoxygenases and their metabolites. Proposed nomenclature of lipoxygenases and related enzymes. Adv Enzymol Relat Areas Mol Biol 58, 273–311. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Sprecher H and Brash AR, 1990c. On singular or dual positional specificity of lipoxygenases. The number of chiral products varies with alignment of methylene groups at the active site of the enzyme. J Biol Chem 265, 16300–5. [PubMed] [Google Scholar]
- Kuhn H, Wiesner R, Stender H, Schewe T, Lankin VZ, Nekrasov A and Rapoport SM, 1986c. Requirement of monohydroperoxy fatty acids for the oxygenation of 15LS-HETE by reticulocyte lipoxygenase. FEBS Lett 203, 247–52. [DOI] [PubMed] [Google Scholar]
- Kumar P, Kalonia H and Kumar A, 2011. Role of LOX/COX pathways in 3-nitropropionic acid-induced Huntington’s disease-like symptoms in rats: protective effect of licofelone. Br J Pharmacol 164, 644–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundumani-Sridharan V, Dyukova E, Hansen DE 3rd and Rao GN, 2013. 12/15-Lipoxygenase mediates high-fat diet-induced endothelial tight junction disruption and monocyte transmigration: a new role for 15(S)-hydroxyeicosatetraenoic acid in endothelial cell dysfunction. J Biol Chem 288, 15830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankin VZ, Kuhn H, Hiebsch C, Schewe T, Rapoport SM, Tikhaze AK and Gordeeva NT, 1985. On the nature of the stimulation of the lipoxygenase from rabbit reticulocytes by biological membranes. Biomed Biochim Acta 44, 655–64. [PubMed] [Google Scholar]
- Lax Y, Grossman S, Rubinstein S, Magid N and Breitbart H, 1990. Role of lipoxygenase in the mechanism of acrosome reaction in mammalian spermatozoa. Biochim Biophys Acta 1043, 12–8. [DOI] [PubMed] [Google Scholar]
- Laybutt DR, Sharma A, Sgroi DC, Gaudet J, Bonner-Weir S and Weir GC, 2002. Genetic regulation of metabolic pathways in beta-cells disrupted by hyperglycemia. J Biol Chem 277, 10912–21. [DOI] [PubMed] [Google Scholar]
- Lebeau A, Esclaire F, Rostene W and Pelaprat D, 2001. Baicalein protects cortical neurons from beta-amyloid (25–35) induced toxicity. Neuroreport 12, 2199–202. [DOI] [PubMed] [Google Scholar]
- Lebeau A, Terro F, Rostene W and Pelaprat D, 2004. Blockade of 12-lipoxygenase expression protects cortical neurons from apoptosis induced by beta-amyloid peptide. Cell Death Differ 11, 875–84. [DOI] [PubMed] [Google Scholar]
- Lee HN and Surh YJ, 2012. Therapeutic potential of resolvins in the prevention and treatment of inflammatory disorders. Biochem Pharmacol 84, 1340–50. [DOI] [PubMed] [Google Scholar]
- Lee J, Kosaras B, Del Signore SJ, Cormier K, McKee A, Ratan RR, Kowall NW and Ryu H, 2011. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol 121, 487–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YW, Kuhn H, Kaiser S, Hennig B, Daugherty A and Toborek M, 2001. Interleukin 4 induces transcription of the 15-lipoxygenase I gene in human endothelial cells. J Lipid Res 42, 783–91. [PubMed] [Google Scholar]
- Lefterova MI, Haakonsson AK, Lazar MA and Mandrup S, 2014. PPARgamma and the global map of adipogenesis and beyond. Trends Endocrinol Metab 25, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnert N and Solomon EI, 2003. Density-functional investigation on the mechanism of H-atom abstraction by lipoxygenase. J Biol Inorg Chem 8, 294–305. [DOI] [PubMed] [Google Scholar]
- Lei ZM and Rao CV, 1992. The expression of 15-lipoxygenase gene and the presence of functional enzyme in cytoplasm and nuclei of pregnancy human myometria. Endocrinology 130, 861–70. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, 2014. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol 2, 749–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy BD, Romano M, Chapman HA, Reilly JJ, Drazen J and Serhan CN, 1993. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. J Clin Invest 92, 1572–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, 2012. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol 32, 2045–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb DC, Brotman JJ, Hatcher MA, Aye MS, Cole BK, Haynes BA, Wohlgemuth SD, Fontana MA, Beydoun H, Nadler JL and Dobrian AD, 2014. Adipose tissue 12/15 lipoxygenase pathway in human obesity and diabetes. J Clin Endocrinol Metab 99, E1713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liepelt A, Mossanen JC, Denecke B, Heymann F, De Santis R, Tacke F, Marx G, Ostareck DH and Ostareck-Lederer A, 2014. Translation control of TAK1 mRNA by hnRNP K modulates LPS-induced macrophage activation. RNA 20, 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limor R, Sharon O, Knoll E, Many A, Weisinger G and Stern N, 2008. Lipoxygenase-derived metabolites are regulators of peroxisome proliferator-activated receptor gamma-2 expression in human vascular smooth muscle cells. Am J Hypertens 21, 219–23. [DOI] [PubMed] [Google Scholar]
- Liu C, Schain F, Han H, Xu D, Andersson-Sand H, Forsell P, Claesson HE, Bjorkholm M and Sjoberg J, 2012. Epigenetic and transcriptional control of the 15-lipoxygenase-1 gene in a Hodgkin lymphoma cell line. Exp Cell Res 318, 169–76. [DOI] [PubMed] [Google Scholar]
- Liu M and Yokomizo T, 2015. The role of leukotrienes in allergic diseases. Allergol Int 64, 17–26. [DOI] [PubMed] [Google Scholar]
- Lovat PE, Oliverio S, Ranalli M, Corazzari M, Rodolfo C, Bernassola F, Aughton K, Maccarrone M, Hewson QD, Pearson AD, Melino G, Piacentini M and Redfern CP, 2002. GADD153 and 12-lipoxygenase mediate fenretinide-induced apoptosis of neuroblastoma. Cancer Res 62, 5158–67. [PubMed] [Google Scholar]
- Lovat PE, Ranalli M, Corazzari M, Raffaghello L, Pearson AD, Ponzoni M, Piacentini M, Melino G and Redfern CP, 2003. Mechanisms of free-radical induction in relation to fenretinide-induced apoptosis of neuroblastoma. J Cell Biochem 89, 698–708. [DOI] [PubMed] [Google Scholar]
- Lovercamp KW, Safranski TJ, Fischer KA, Manandhar G, Sutovsky M, Herring W and Sutovsky P, 2007. Arachidonate 15-lipoxygenase and ubiquitin as fertility markers in boars. Theriogenology 67, 704–18. [DOI] [PubMed] [Google Scholar]
- Lu X, Zhang J, Liu S, Zhang D, Xu Z, Wu J, Li J, Du G and Chen J, 2013. Overproduction, purification, and characterization of extracellular lipoxygenase of Pseudomonas aeruginosa in Escherichia coli. Appl Microbiol Biotechnol 97, 5793–800. [DOI] [PubMed] [Google Scholar]
- Ludwig P, Hohne M, Kuhn H, Schewe T and Rapoport SM, 1988. The biological dynamics of lipoxygenase in rabbit red cells in the course of an experimental bleeding anaemia. Unexpected effects of the calcium ionophore A 23187. Biomed Biochim Acta 47, 593–608. [PubMed] [Google Scholar]
- Ludwig P, Holzhutter HG, Colosimo A, Silvestrini MC, Schewe T and Rapoport SM, 1987. A kinetic model for lipoxygenases based on experimental data with the lipoxygenase of reticulocytes. Eur J Biochem 168, 325–37. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN and Bazan NG, 2005. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest 115, 2774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA and Nadler JL, 2010. 12-Lipoxygenase Products Reduce Insulin Secretion and {beta}-Cell Viability in Human Islets. J Clin Endocrinol Metab 95, 887–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas RL and Brash AR, 1983. Evidence for a lipoxygenase mechanism in the biosynthesis of epoxide and dihydroxy leukotrienes from 15(S)-hydroperoxyicosatetraenoic acid by human platelets and porcine leukocytes. Proc Natl Acad Sci U S A 80, 2884–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SR, Han HL, De Jonge J, Anderson RA and Zaneveld LJ, 1992. The human sperm acrosome reaction does not depend on arachidonic acid metabolism via the cyclooxygenase and lipoxygenase pathways. J Androl 13, 551–9. [PubMed] [Google Scholar]
- Madsen L, Petersen RK, Sorensen MB, Jorgensen C, Hallenborg P, Pridal L, Fleckner J, Amri EZ, Krieg P, Furstenberger G, Berge RK and Kristiansen K, 2003. Adipocyte differentiation of 3T3-L1 preadipocytes is dependent on lipoxygenase activity during the initial stages of the differentiation process. Biochem J 375, 539–49. [DOI] [PubMed] [Google Scholar]
- Mancini AD and Di Battista JA, 2011. The cardinal role of the phospholipase A(2)/cyclooxygenase-2/prostaglandin E synthase/prostaglandin E(2) (PCPP) axis in inflammostasis. Inflamm Res 60, 1083–92. [DOI] [PubMed] [Google Scholar]
- Mangino MJ, Brounts L, Harms B and Heise C, 2006. Lipoxin biosynthesis in inflammatory bowel disease. Prostaglandins Other Lipid Mediat 79, 84–92. [DOI] [PubMed] [Google Scholar]
- Martin H, 2010. Role of PPAR-gamma in inflammation. Prospects for therapeutic intervention by food components. Mutat Res 690, 57–63. [DOI] [PubMed] [Google Scholar]
- Martinez-Clemente M, Ferre N, Titos E, Horrillo R, Gonzalez-Periz A, Moran-Salvador E, Lopez-Vicario C, Miquel R, Arroyo V, Funk CD and Claria J, 2010. Disruption of the 12/15-lipoxygenase gene (Alox15) protects hyperlipidemic mice from nonalcoholic fatty liver disease. Hepatology 52, 1980–91. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Funk CD, Radmark O, Hoog JO, Jornvall H and Samuelsson B, 1988. Molecular cloning and amino acid sequence of human 5-lipoxygenase. Proc Natl Acad Sci U S A 85, 26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May C, Hohne M, Gnau P, Schwennesen K and Kindl H, 2000. The N-terminal beta-barrel structure of lipid body lipoxygenase mediates its binding to liposomes and lipid bodies. Eur J Biochem 267, 1100–9. [DOI] [PubMed] [Google Scholar]
- McDuffie M, Maybee NA, Keller SR, Stevens BK, Garmey JC, Morris MA, Kropf E, Rival C, Ma K, Carter JD, Tersey SA, Nunemaker CS and Nadler JL, 2008. Nonobese diabetic (NOD) mice congenic for a targeted deletion of 12/15-lipoxygenase are protected from autoimmune diabetes. Diabetes 57, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei G, Di Venere A, Nicolai E, Angelucci CB, Ivanov I, Sabatucci A, Dainese E, Kuhn H and Maccarrone M, 2008. Structural properties of plant and mammalian lipoxygenases. Temperature-dependent conformational alterations and membrane binding ability. Biochemistry 47, 9234–42. [DOI] [PubMed] [Google Scholar]
- Mele PG, Dada LA, Paz C, Neuman I, Cymeryng CB, Mendez CF, Finkielstein CV, Maciel F. Cornejo and Podesta EJ, 1997. Involvement of arachidonic acid and the lipoxygenase pathway in mediating luteinizing hormone-induced testosterone synthesis in rat Leydig cells. Endocr Res 23, 15–26. [DOI] [PubMed] [Google Scholar]
- Merched AJ, Ko K, Gotlinger KH, Serhan CN and Chan L, 2008. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J 22, 3595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messias AC, Harnisch C, Ostareck-Lederer A, Sattler M and Ostareck DH, 2006. The DICE-binding activity of KH domain 3 of hnRNP K is affected by c-Src-mediated tyrosine phosphorylation. J Mol Biol 361, 470–81. [DOI] [PubMed] [Google Scholar]
- Mhatre M, Floyd RA and Hensley K, 2004. Oxidative stress and neuroinflammation in Alzheimer’s disease and amyotrophic lateral sclerosis: common links and potential therapeutic targets. J Alzheimers Dis 6, 147–57. [DOI] [PubMed] [Google Scholar]
- Mikule K, Gatlin JC, de la Houssaye BA and Pfenninger KH, 2002. Growth cone collapse induced by semaphorin 3A requires 12/15-lipoxygenase. J Neurosci 22, 4932–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Steczko J, Stec B, Otwinowski Z, Bolin JT, Walter R and Axelrod B, 1996. Crystal structure of soybean lipoxygenase L-1 at 1.4 A resolution. Biochemistry 35, 10687–701. [DOI] [PubMed] [Google Scholar]
- Moin ST, Hofer TS, Sattar R and Ul-Haq Z, 2011. Molecular dynamics simulation of mammalian 15S-lipoxygenase with AMBER force field. Eur Biophys J 40, 715–26. [DOI] [PubMed] [Google Scholar]
- Moore K, Lovercamp K, Feng D, Antelman J, Sutovsky M, Manandhar G, van Leyen K, Safranski T and Sutovsky P, 2010. Altered epididymal sperm maturation and cytoplasmic droplet migration in subfertile male Alox15 mice. Cell Tissue Res 340, 569–81. [DOI] [PubMed] [Google Scholar]
- Morgan AH, Hammond VJ, Sakoh-Nakatogawa M, Ohsumi Y, Thomas CP, Blanchet F, Piguet V, Kiselyov K and O’Donnell VB, 2015. A novel role for 12/15-lipoxygenase in regulating autophagy. Redox Biol 4, 40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz B, Lilie H, Naarmann-de Vries IS, Urlaub H, Wahle E, Ostareck-Lederer A and Ostareck DH, 2014. Biophysical and biochemical analysis of hnRNP K: arginine methylation, reversible aggregation and combinatorial binding to nucleic acids. Biol Chem 395, 837–53. [DOI] [PubMed] [Google Scholar]
- Munger KA, Montero A, Fukunaga M, Uda S, Yura T, Imai E, Kaneda Y, Valdivielso JM and Badr KF, 1999. Transfection of rat kidney with human 15-lipoxygenase suppresses inflammation and preserves function in experimental glomerulonephritis. Proc Natl Acad Sci U S A 96, 13375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Garcia A, Thomas CP, Keeney DS, Zheng Y and Brash AR, 2014. The importance of the lipoxygenase-hepoxilin pathway in the mammalian epidermal barrier. Biochim Biophys Acta 1841, 401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naarmann IS, Harnisch C, Flach N, Kremmer E, Kuhn H, Ostareck DH and Ostareck-Lederer A, 2008. mRNA silencing in human erythroid cell maturation: heterogeneous nuclear ribonucleoprotein K controls the expression of its regulator c-Src. J Biol Chem 283, 18461–72. [DOI] [PubMed] [Google Scholar]
- Naarmann IS, Harnisch C, Muller-Newen G, Urlaub H, Ostareck-Lederer A and Ostareck DH, 2010. DDX6 recruits translational silenced human reticulocyte 15-lipoxygenase mRNA to RNP granules. RNA 16, 2189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadel JA, Conrad DJ, Ueki IF, Schuster A and Sigal E, 1991. Immunocytochemical localization of arachidonate 15-lipoxygenase in erythrocytes, leukocytes, and airway cells. J Clin Invest 87, 1139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelin MH, Srinivasan S, Lee J, Nadler JL and Hedrick CC, 2008. 12/15-Lipoxygenase activity increases the degradation of macrophage ATP-binding cassette transporter G1. Arterioscler Thromb Vasc Biol 28, 1811–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumiya S, Salmon JA, Flower RJ and Vane JR, 1982. Purification and properties of arachidonate-15-lipoxygenase from rabbit peritoneal polymorphonuclear leukocytes. Adv Prostaglandin Thromboxane Leukot Res 9, 77–82. [PubMed] [Google Scholar]
- Nasjletti A, 1998. Arthur C. Corcoran Memorial Lecture. The role of eicosanoids in angiotensin-dependent hypertension. Hypertension 31, 194–200. [DOI] [PubMed] [Google Scholar]
- Nassar GM, Morrow JD, Roberts LJ 2nd, Lakkis FG and Badr KF, 1994. Induction of 15-lipoxygenase by interleukin-13 in human blood monocytes. J Biol Chem 269, 27631–4. [PubMed] [Google Scholar]
- Natarajan R, Gu JL, Rossi J, Gonzales N, Lanting L, Xu L and Nadler J, 1993. Elevated glucose and angiotensin II increase 12-lipoxygenase activity and expression in porcine aortic smooth muscle cells. Proc Natl Acad Sci U S A 90, 4947–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neau DB, Bender G, Boeglin WE, Bartlett SG, Brash AR and Newcomer ME, 2014. Crystal structure of a lipoxygenase in complex with substrate: the arachidonic acid-binding site of 8R-lipoxygenase. J Biol Chem 289, 31905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomer ME and Brash AR, 2015. The structural basis for specificity in lipoxygenase catalysis. Protein Sci 24, 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigam S, Zafiriou MP, Deva R, Ciccoli R and der Merwe R. Roux-Van, 2007. Structure, biochemistry and biology of hepoxilins: an update. FEBS J 274, 3503–12. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hoshino A, Tsai L, Henley JR, Goshima Y, Tessier-Lavigne M, Poo MM and Hong K, 2003. Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature 423, 990–5. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Okamoto H, Watanabe T, Hori T, Hada T, Ueda N, Yamamoto S, Tsukamoto H, Watanabe K and Kirino T, 1992. Localization of arachidonate 12-lipoxygenase in canine brain tissues. J Neurochem 58, 1395–400. [DOI] [PubMed] [Google Scholar]
- Normandin M, Gagne J, Bernard J, Elie R, Miceli D, Baudry M and Massicotte G, 1996. Involvement of the 12-lipoxygenase pathway of arachidonic acid metabolism in homosynaptic long-term depression of the rat hippocampus. Brain Res 730, 40–6. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, Chen M, Pei H, Kimble SD, Keller SR, Carter JD, Yang Z, Smith KM, Wu R, Bevard MH, Garmey JC and Nadler JL, 2008. 12-Lipoxygenase-knockout mice are resistant to inflammatory effects of obesity induced by Western diet. Am J Physiol Endocrinol Metab 295, E1065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohira T, Arita M, Omori K, Recchiuti A, Van Dyke TE and Serhan CN, 2010. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J Biol Chem 285, 3451–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskolkova OV, Afonyushkin T, Preinerstorfer B, Bicker W, von Schlieffen E, Hainzl E, Demyanets S, Schabbauer G, Lindner W, Tselepis AD, Wojta J, Binder BR and Bochkov VN, 2010. Oxidized phospholipids are more potent antagonists of lipopolysaccharide than inducers of inflammation. J Immunol 185, 7706–12. [DOI] [PubMed] [Google Scholar]
- Ostareck-Lederer A, Ostareck DH, Standart N and Thiele BJ, 1994. Translation of 15-lipoxygenase mRNA is inhibited by a protein that binds to a repeated sequence in the 3’ untranslated region. EMBO J 13, 1476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostareck DH, Ostareck-Lederer A, Wilm M, Thiele BJ, Mann M and Hentze MW, 1997. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3’ end. Cell 89, 597–606. [DOI] [PubMed] [Google Scholar]
- Ostermann A, Waschipky R, Parak FG and Nienhaus GU, 2000. Ligand binding and conformational motions in myoglobin. Nature 404, 205–8. [DOI] [PubMed] [Google Scholar]
- Othman A, Ahmad S, Megyerdi S, Mussell R, Choksi K, Maddipati KR, Elmarakby A, Rizk N and Al-Shabrawey M, 2013. 12/15-Lipoxygenase-derived lipid metabolites induce retinal endothelial cell barrier dysfunction: contribution of NADPH oxidase. PLoS One 8, e57254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace-Asciak CR, 2009. The hepoxilins and some analogues: a review of their biology. Br J Pharmacol 158, 972–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace-Asciak CR, 2015. Pathophysiology of the hepoxilins. Biochim Biophys Acta 1851, 383–396. [DOI] [PubMed] [Google Scholar]
- Pallast S, Arai K, Pekcec A, Yigitkanli K, Yu Z, Wang X, Lo EH and van Leyen K, 2010. Increased nuclear apoptosis-inducing factor after transient focal ischemia: a 12/15-lipoxygenase-dependent organelle damage pathway. J Cereb Blood Flow Metab 30, 1157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallast S, Arai K, Wang X, Lo EH and van Leyen K, 2009. 12/15-Lipoxygenase targets neuronal mitochondria under oxidative stress. J Neurochem 111, 882–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekarova M, Kuhn H, Bezakova L, Ufer C and Heydeck D, 2015. Mutagenesis of triad determinants of rat Alox15 alters the specificity of fatty acid and phospholipid oxygenation. Arch Biochem Biophys 571, 50–57. [DOI] [PubMed] [Google Scholar]
- Pekcec A, Yigitkanli K, Jung JE, Pallast S, Xing C, Antipenko A, Minchenko M, Nikolov DB, Holman TR, Lo EH and van Leyen K, 2013. Following experimental stroke, the recovering brain is vulnerable to lipoxygenase-dependent semaphorin signaling. FASEB J 27, 437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotta I, 2013. Ultrastructural features of human atherosclerosis. Ultrastruct Pathol 37, 43–51. [DOI] [PubMed] [Google Scholar]
- Pfister SL and Campbell WB, 1992. Arachidonic acid- and acetylcholine-induced relaxations of rabbit aorta. Hypertension 20, 682–9. [DOI] [PubMed] [Google Scholar]
- Pfister SL, Spitzbarth N, Nithipatikom K, Edgemond WS, Falck JR and Campbell WB, 1998. Identification of the 11,14,15- and 11,12, 15-trihydroxyeicosatrienoic acids as endothelium-derived relaxing factors of rabbit aorta. J Biol Chem 273, 30879–87. [DOI] [PubMed] [Google Scholar]
- Pidgeon GP, Lysaght J, Krishnamoorthy S, Reynolds JV, O’Byrne K, Nie D and Honn KV, 2007. Lipoxygenase metabolism: roles in tumor progression and survival. Cancer Metastasis Rev 26, 503–24. [DOI] [PubMed] [Google Scholar]
- Piomelli D, 1991. Metabolism of arachidonic acid in nervous system of marine mollusk Aplysia californica. Am J Physiol 260, R844–8. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Shapiro E, Feinmark SJ and Schwartz JH, 1987a. Metabolites of arachidonic acid in the nervous system of Aplysia: possible mediators of synaptic modulation. J Neurosci 7, 3675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D, Volterra A, Dale N, Siegelbaum SA, Kandel ER, Schwartz JH and Belardetti F, 1987b. Lipoxygenase metabolites of arachidonic acid as second messengers for presynaptic inhibition of Aplysia sensory cells. Nature 328, 38–43. [DOI] [PubMed] [Google Scholar]
- Pirillo A, Uboldi P, Bolego C, Kuhn H and Catapano AL, 2008. The 15-lipoxygenase-modified high density lipoproteins 3 fail to inhibit the TNF-alpha-induced inflammatory response in human endothelial cells. J Immunol 181, 2821–30. [DOI] [PubMed] [Google Scholar]
- Pirillo A, Uboldi P, Ferri N, Corsini A, Kuhn H and Catapano AL, 2012. Upregulation of lectin-like oxidized low density lipoprotein receptor 1 (LOX-1) expression in human endothelial cells by modified high density lipoproteins. Biochem Biophys Res Commun 428, 230–3. [DOI] [PubMed] [Google Scholar]
- Pirillo A, Uboldi P, Kuhn H and Catapano AL, 2006. 15-Lipoxygenase-mediated modification of high-density lipoproteins impairs SR-BI- and ABCA1-dependent cholesterol efflux from macrophages. Biochim Biophys Acta 1761, 292–300. [DOI] [PubMed] [Google Scholar]
- Poeckel D, Berry KA, Murphy RC and Funk C.D. Zemski, 2009. Dual 12/15- and 5-lipoxygenase deficiency in macrophages alters arachidonic acid metabolism and attenuates peritonitis and atherosclerosis in ApoE knock-out mice. J Biol Chem 284, 21077–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontiki E and Hadjipavlou-Litina D, 2007. Synthesis of phenyl-substituted amides with antioxidant and anti-inflammatory activity as novel lipoxygenase inhibitors. Med Chem 3, 175–86. [DOI] [PubMed] [Google Scholar]
- Powell WS and Rokach J, 2015. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim Biophys Acta 1851, 340–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratico D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ and Lee VM, 2004. 12/15-lipoxygenase is increased in Alzheimer’s disease: possible involvement in brain oxidative stress. Am J Pathol 164, 1655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL and Tainer JA, 2007. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys 40, 191–285. [DOI] [PubMed] [Google Scholar]
- Rabier M, Chavis C, de Paulet A. Crastes and Damon M, 1987. Arachidonic acid metabolism in a cloned strain of rat pituitary tumor cells: correlation between 15 hydroxyeicosatetraenoic acid release and the prolactin secretory process. Prostaglandins Leukot Med 27, 27–42. [DOI] [PubMed] [Google Scholar]
- Rabier M, Chavis C, de Paulet A. Crastes and Damon M, 1988. 15-Lipoxygenase products stimulate prolactin secretion from a cloned strain of rat pituitary cells. Neuroendocrinology 47, 323–8. [DOI] [PubMed] [Google Scholar]
- Radmark O, Werz O, Steinhilber D and Samuelsson B, 2015. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim Biophys Acta 1851, 331–339. [DOI] [PubMed] [Google Scholar]
- Rai G, Joshi N, Jung JE, Liu Y, Schultz L, Yasgar A, Perry S, Diaz G, Zhang Q, Kenyon V, Jadhav A, Simeonov A, Lo EH, van Leyen K, Maloney DJ and Holman TR, 2014. Potent and selective inhibitors of human reticulocyte 12/15-lipoxygenase as anti-stroke therapies. J Med Chem 57, 4035–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai G, Kenyon V, Jadhav A, Schultz L, Armstrong M, Jameson JB, Hoobler E, Leister W, Simeonov A, Holman TR and Maloney DJ, 2010. Discovery of potent and selective inhibitors of human reticulocyte 15-lipoxygenase-1. J Med Chem 53, 7392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramis I, Catafau JR, Serra J, Bulbena O, Picado C and Gelpi E, 1991. In vivo release of 15-HETE and other arachidonic acid metabolites in nasal secretions during early allergic reactions. Prostaglandins 42, 411–20. [DOI] [PubMed] [Google Scholar]
- Rao JS, Rapoport SI and Kim HW, 2011. Altered neuroinflammatory, arachidonic acid cascade and synaptic markers in postmortem Alzheimer’s disease brain. Transl Psychiatry 1, e31. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rapoport S, Hartel B and Hausdorf G, 1984. Methionine sulfoxide formation: the cause of self-inactivation of reticulocyte lipoxygenase. Eur J Biochem 139, 573–6. [DOI] [PubMed] [Google Scholar]
- Rapoport SM and Schewe T, 1986. The maturational breakdown of mitochondria in reticulocytes. Biochim Biophys Acta 864, 471–95. [DOI] [PubMed] [Google Scholar]
- Rapoport SM, Schewe T, Wiesner R, Halangk W, Ludwig P, Janicke-Hohne M, Tannert C, Hiebsch C and Klatt D, 1979. The lipoxygenase of reticulocytes. Purification, characterization and biological dynamics of the lipoxygenase; its identity with the respiratory inhibitors of the reticulocyte. Eur J Biochem 96, 545–61. [DOI] [PubMed] [Google Scholar]
- Reimann I, Huth A, Thiele H and Thiele BJ, 2002. Suppression of 15-lipoxygenase synthesis by hnRNP E1 is dependent on repetitive nature of LOX mRNA 3’-UTR control element DICE. J Mol Biol 315, 965–74. [DOI] [PubMed] [Google Scholar]
- Richter C, Wendel A, Weser U and Azzi A, 1975. Inhibition by superoxide dismutase of linoleic acid peroxidation induced by lipoxidase. FEBS Lett 51, 300–3. [DOI] [PubMed] [Google Scholar]
- Rickert KW and Klinman JP, 1999. Nature of hydrogen transfer in soybean lipoxygenase 1: separation of primary and secondary isotope effects. Biochemistry 38, 12218–28. [DOI] [PubMed] [Google Scholar]
- Romano M, 2010. Lipoxin and aspirin-triggered lipoxins. ScientificWorldJournal 10, 1048–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanov S, Wiesner R, Myagkova G, Kuhn H and Ivanov I, 2006. Affinity labeling of the rabbit 12/15-lipoxygenase using azido derivatives of arachidonic acid. Biochemistry 45, 3554–62. [DOI] [PubMed] [Google Scholar]
- Rosolowsky M and Campbell WB, 1993. Role of PGI2 and epoxyeicosatrienoic acids in relaxation of bovine coronary arteries to arachidonic acid. Am J Physiol 264, H327–35. [DOI] [PubMed] [Google Scholar]
- Ross S, Essary B, de la Houssaye BA, Pan Z, Mikule K, Mubarak O and Pfenninger KH, 2000. Thrombin causes pseudopod detachment via a pathway involving cytosolic phospholipase A2 and 12/15-lipoxygenase products. Cell Growth Differ 11, 19–30. [PubMed] [Google Scholar]
- Roy B and Cathcart MK, 1998. Induction of 15-lipoxygenase expression by IL-13 requires tyrosine phosphorylation of Jak2 and Tyk2 in human monocytes. J Biol Chem 273, 32023–9. [DOI] [PubMed] [Google Scholar]
- Roy P, Sajan MP and Kulkarni AP, 1995. Lipoxygenase-mediated glutathione oxidation and superoxide generation. J Biochem Toxicol 10, 111–20. [DOI] [PubMed] [Google Scholar]
- Ryan A and Godson C, 2010. Lipoxins: regulators of resolution. Curr Opin Pharmacol 10, 166–72. [DOI] [PubMed] [Google Scholar]
- Saam J, Ivanov I, Walther M, Holzhutter HG and Kuhn H, 2007. Molecular dioxygen enters the active site of 12/15-lipoxygenase via dynamic oxygen access channels. Proc Natl Acad Sci U S A 104, 13319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs-Olsen C, Sanak M, Lang AM, Gielicz A, Mowinckel P, Carlsen K.C. Lodrup, Carlsen KH and Szczeklik A, 2010. Eoxins: a new inflammatory pathway in childhood asthma. J Allergy Clin Immunol 126, 859–867 e9. [DOI] [PubMed] [Google Scholar]
- Sala A, Folco G and Murphy RC, 2010. Transcellular biosynthesis of eicosanoids. Pharmacol Rep 62, 503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzmann U, Kuhn H, Schewe T and Rapoport SM, 1984. Pentane formation during the anaerobic reactions of reticulocyte lipoxygenase. Comparison with lipoxygenases from soybeans and green pea seeds. Biochim Biophys Acta 795, 535–42. [DOI] [PubMed] [Google Scholar]
- Sato H, Taketomi Y, Isogai Y, Miki Y, Yamamoto K, Masuda S, Hosono T, Arata S, Ishikawa Y, Ishii T, Kobayashi T, Nakanishi H, Ikeda K, Taguchi R, Hara S, Kudo I and Murakami M, 2010. Group III secreted phospholipase A2 regulates epididymal sperm maturation and fertility in mice. J Clin Invest 120, 1400–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savari S, Vinnakota K, Zhang Y and Sjolander A, 2014. Cysteinyl leukotrienes and their receptors: bridging inflammation and colorectal cancer. World J Gastroenterol 20, 968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schewe T, 2002. 15-lipoxygenase-1: a prooxidant enzyme. Biol Chem 383, 365–74. [DOI] [PubMed] [Google Scholar]
- Schewe T, Halangk W, Hiebsch C and Rapoport S, 1977. Degradation of mitochondria by cytosolic factors in reticulocytes. Acta Biol Med Ger 36, 563–72. [PubMed] [Google Scholar]
- Schewe T, Halangk W, Hiebsch C and Rapoport SM, 1975. A lipoxygenase in rabbit reticulocytes which attacks phospholipids and intact mitochondria. FEBS Lett 60, 149–52. [DOI] [PubMed] [Google Scholar]
- Schewe T, Kroschwald P, Kroschwald A, Ludwig P and Kuhn H, 1990. The erythroid arachidonate 15-lipoxygenase in rat reticulocytes. Biomed Biochim Acta 49, S42–6. [PubMed] [Google Scholar]
- Schewe T, Rapoport SM and Kuhn H, 1986. Enzymology and physiology of reticulocyte lipoxygenase: comparison with other lipoxygenases. Adv Enzymol Relat Areas Mol Biol 58, 191–272. [DOI] [PubMed] [Google Scholar]
- Schilstra MJ, Veldink GA and Vliegenthart JF, 1994. The dioxygenation rate in lipoxygenase catalysis is determined by the amount of iron (III) lipoxygenase in solution. Biochemistry 33, 3974–9. [DOI] [PubMed] [Google Scholar]
- Schneider C, Pratt DA, Porter NA and Brash AR, 2007. Control of oxygenation in lipoxygenase and cyclooxygenase catalysis. Chem Biol 14, 473–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Wortmann M, Mandal PK, Arpornchayanon W, Jannasch K, Alves F, Strieth S, Conrad M and Beck H, 2010. Absence of glutathione peroxidase 4 affects tumor angiogenesis through increased 12/15-lipoxygenase activity. Neoplasia 12, 254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnurr K, Belkner J, Ursini F, Schewe T and Kuhn H, 1996. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase controls the activity of the 15-lipoxygenase with complex substrates and preserves the specificity of the oxygenation products. J Biol Chem 271, 4653–8. [DOI] [PubMed] [Google Scholar]
- Schnurr K, Borchert A and Kuhn H, 1999. Inverse regulation of lipid-peroxidizing and hydroperoxyl lipid-reducing enzymes by interleukins 4 and 13. FASEB J 13, 143–54. [DOI] [PubMed] [Google Scholar]
- Schroder M, Hafner AK, Hofmann B, Radmark O, Tumulka F, Abele R, Dotsch V and Steinhilber D, 2014. Stabilisation and characterisation of the isolated regulatory domain of human 5-lipoxygenase. Biochim Biophys Acta 1842, 1538–47. [DOI] [PubMed] [Google Scholar]
- Schwarz K, Borngraber S, Anton M and Kuhn H, 1998. Probing the substrate alignment at the active site of 15-lipoxygenases by targeted substrate modification and site-directed mutagenesis. Evidence for an inverse substrate orientation. Biochemistry 37, 15327–35. [DOI] [PubMed] [Google Scholar]
- Schwenk U, Morita E, Engel R and Schroder JM, 1992. Identification of 5-oxo-15-hydroxy-6,8,11,13-eicosatetraenoic acid as a novel and potent human eosinophil chemotactic eicosanoid. J Biol Chem 267, 12482–8. [PubMed] [Google Scholar]
- Scott EE and Gibson QH, 1997. Ligand migration in sperm whale myoglobin. Biochemistry 36, 11909–17. [DOI] [PubMed] [Google Scholar]
- Sears DD, Miles PD, Chapman J, Ofrecio JM, Almazan F, Thapar D and Miller YI, 2009. 12/15-lipoxygenase is required for the early onset of high fat diet-induced adipose tissue inflammation and insulin resistance in mice. PLoS One 4, e7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segraves EN and Holman TR, 2003. Kinetic investigations of the rate-limiting step in human 12- and 15-lipoxygenase. Biochemistry 42, 5236–43. [DOI] [PubMed] [Google Scholar]
- Sendobry SM, Cornicelli JA, Welch K, Bocan T, Tait B, Trivedi BK, Colbry N, Dyer RD, Feinmark SJ and Daugherty A, 1997. Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties. Br J Pharmacol 120, 1199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Dalli J, Colas RA, Winkler JW and Chiang N, 2015. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim Biophys Acta 1851, 397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Dalli J, Karamnov S, Choi A, Park CK, Xu ZZ, Ji RR, Zhu M and Petasis NA, 2012. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J 26, 1755–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN and Petasis NA, 2011. Resolvins and protectins in inflammation resolution. Chem Rev 111, 5922–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahin I, Grossman S and Sredni B, 1978. Lipoxygenase-like enzyme in rat testis microsomes. Biochim Biophys Acta 529, 300–8. [DOI] [PubMed] [Google Scholar]
- Shang W, Ivanov I, Svergun DI, Borbulevych OY, Aleem AM, Stehling S, Jankun J, Kuhn H and Skrzypczak-Jankun E, 2011. Probing dimerization and structural flexibility of mammalian lipoxygenases by small-angle X-ray scattering. J Mol Biol 409, 654–68. [DOI] [PubMed] [Google Scholar]
- Shankaranarayanan P, Chaitidis P, Kuhn H and Nigam S, 2001. Acetylation by histone acetyltransferase CREB-binding protein/p300 of STAT6 is required for transcriptional activation of the 15-lipoxygenase-1 gene. J Biol Chem 276, 42753–60. [DOI] [PubMed] [Google Scholar]
- Shappell SB, Gupta RA, Manning S, Whitehead R, Boeglin WE, Schneider C, Case T, Price J, Jack GS, Wheeler TM, Matusik RJ, Brash AR and Dubois RN, 2001. 15S-Hydroxyeicosatetraenoic acid activates peroxisome proliferator-activated receptor gamma and inhibits proliferation in PC3 prostate carcinoma cells. Cancer Res 61, 497–503. [PubMed] [Google Scholar]
- Shen J, Herderick E, Cornhill JF, Zsigmond E, Kim HS, Kuhn H, Guevara NV and Chan L, 1996. Macrophage-mediated 15-lipoxygenase expression protects against atherosclerosis development. J Clin Invest 98, 2201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shillabeer G, Kumar V, Tibbo E and Lau DC, 1998. Arachidonic acid metabolites of the lipoxygenase as well as the cyclooxygenase pathway may be involved in regulating preadipocyte differentiation. Metabolism 47, 461–6. [DOI] [PubMed] [Google Scholar]
- Sloane DL, Browner MF, Dauter Z, Wilson K, Fletterick RJ and Sigal E, 1990. Purification and crystallization of 15-lipoxygenase from rabbit reticulocytes. Biochem Biophys Res Commun 173, 507–13. [DOI] [PubMed] [Google Scholar]
- Sloane DL, Dixon RA, Craik CS and Sigal E, 1991a. Expression of cloned human 15-lipoxygenase in eukaryotic and prokaryotic systems. Adv Prostaglandin Thromboxane Leukot Res 21A, 25–8. [PubMed] [Google Scholar]
- Sloane DL, Leung R, Barnett J, Craik CS and Sigal E, 1995. Conversion of human 15-lipoxygenase to an efficient 12-lipoxygenase: the side-chain geometry of amino acids 417 and 418 determine positional specificity. Protein Eng 8, 275–82. [DOI] [PubMed] [Google Scholar]
- Sloane DL, Leung R, Craik CS and Sigal E, 1991b. A primary determinant for lipoxygenase positional specificity. Nature 354, 149–52. [DOI] [PubMed] [Google Scholar]
- Sobrado M, Pereira MP, Ballesteros I, Hurtado O, Fernandez-Lopez D, Pradillo JM, Caso JR, Vivancos J, Nombela F, Serena J, Lizasoain I and Moro MA, 2009. Synthesis of lipoxin A4 by 5-lipoxygenase mediates PPARgamma-dependent, neuroprotective effects of rosiglitazone in experimental stroke. J Neurosci 29, 3875–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumya SJ, Binu S, Helen A, Reddanna P and Sudhakaran PR, 2013. 15(S)-HETE-induced angiogenesis in adipose tissue is mediated through activation of PI3K/Akt/mTOR signaling pathway. Biochem Cell Biol 91, 498–505. [DOI] [PubMed] [Google Scholar]
- Spanbroek R, Grabner R, Lotzer K, Hildner M, Urbach A, Ruhling K, Moos MP, Kaiser B, Cohnert TU, Wahlers T, Zieske A, Plenz G, Robenek H, Salbach P, Kuhn H, Radmark O, Samuelsson B and Habenicht AJ, 2003. Expanding expression of the 5-lipoxygenase pathway within the arterial wall during human atherogenesis. Proc Natl Acad Sci U S A 100, 1238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spite M, Claria J and Serhan CN, 2014. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab 19, 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg D, 2009. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res 50 Suppl, S376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streckert G and Stan HJ, 1975. Conversion of linoleic acid hydroperoxide by soybean lipoxygenase in the presence of guaiacol: identification of the reaction products. Lipids 10, 847–54. [DOI] [PubMed] [Google Scholar]
- Suardiaz R, Masgrau L, Lluch JM and Gonzalez-Lafont A, 2013. An insight into the regiospecificity of linoleic acid peroxidation catalyzed by mammalian 15-lipoxygenases. J Phys Chem B 117, 3747–54. [DOI] [PubMed] [Google Scholar]
- Suardiaz R, Masgrau L, Lluch JM and Gonzalez-Lafont A, 2014a. Introducing mutations to modify the C13/C9 ratio in linoleic acid oxygenations catalyzed by rabbit 15-lipoxygenase: a QM/MM and MD study. Chemphyschem 15, 4049–54. [DOI] [PubMed] [Google Scholar]
- Suardiaz R, Masgrau L, Lluch JM and Gonzalez-Lafont A, 2014b. Regio- and stereospecificity in the oxygenation of arachidonic acid catalyzed by Leu597 mutants of rabbit 15-lipoxygenase: a QM/MM study. Chemphyschem 15, 2303–10. [DOI] [PubMed] [Google Scholar]
- Sun D and Funk CD, 1996. Disruption of 12/15-lipoxygenase expression in peritoneal macrophages. Enhanced utilization of the 5-lipoxygenase pathway and diminished oxidation of low density lipoprotein. J Biol Chem 271, 24055–62. [PubMed] [Google Scholar]
- Sutovsky P, Van Leyen K, McCauley T, Day BN and Sutovsky M, 2004. Degradation of paternal mitochondria after fertilization: implications for heteroplasmy, assisted reproductive technologies and mtDNA inheritance. Reprod Biomed Online 8, 24–33. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Kishimoto K, Yoshimoto T, Yamamoto S, Kanai F, Ebina Y, Miyatake A and Tanabe T, 1994. Site-directed mutagenesis studies on the iron-binding domain and the determinant for the substrate oxygenation site of porcine leukocyte arachidonate 12-lipoxygenase. Biochim Biophys Acta 1210, 308–16. [DOI] [PubMed] [Google Scholar]
- Svensson-Ek M, Abramson J, Larsson G, Tornroth S, Brzezinski P and Iwata S, 2002. The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J Mol Biol 321, 329–39. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Glasgow WC, Suzuki H, Taketani Y, Yamamoto S, Anton M, Kuhn H and Brash AR, 1993. Investigation of the oxygenation of phospholipids by the porcine leukocyte and human platelet arachidonate 12-lipoxygenases. Eur J Biochem 218, 165–71. [DOI] [PubMed] [Google Scholar]
- Takayama H, Gimbrone MA Jr. and Schafer AI, 1987. Vascular lipoxygenase activity: synthesis of 15-hydroxyeicosatetraenoic acid from arachidonic acid by blood vessels and cultured vascular endothelial cells. Thromb Res 45, 803–16. [DOI] [PubMed] [Google Scholar]
- Thiele BJ, Andree H, Hohne M and Rapoport SM, 1982. Lipoxygenase mRNA in rabbit reticulocytes. Its isolation, characterization and translational repression. Eur J Biochem 129, 133–41. [DOI] [PubMed] [Google Scholar]
- Thiele BJ, Fleming J, Kasturi K, O’Prey J, Black E, Chester J, Rapoport SM and Harrison PR, 1987. Cloning of a rabbit erythroid-cell-specific lipoxygenase mRNA. Gene 57, 111–9. [DOI] [PubMed] [Google Scholar]
- Toledo L, Masgrau L, Lluch JM and Gonzalez-Lafont A, 2011. Substrate binding to mammalian 15-lipoxygenase. J Comput Aided Mol Des 25, 825–35. [DOI] [PubMed] [Google Scholar]
- Toledo L, Masgrau L, Maréchal JD, Lluch JM and González-Lafont A, 2010. Insights into the mechanism of binding of arachidonic acid to mammalian 15-lipoxygenases. J Phys Chem B 114, 7037–46. [DOI] [PubMed] [Google Scholar]
- Trebus F, Heydeck D, Schimke I, Gerth C and Kuhn H, 2002. Transient experimental anemia in cholesterol-fed rabbits induces systemic overexpression of the reticulocyte-type 15-lipoxygenase and protects from aortic lipid deposition. Prostaglandins Leukot Essent Fatty Acids 67, 419–28. [DOI] [PubMed] [Google Scholar]
- Tsao CH, Shiau MY, Chuang PH, Chang YH and Hwang J, 2014. Interleukin-4 regulates lipid metabolism by inhibiting adipogenesis and promoting lipolysis. J Lipid Res 55, 385–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda N, Yamamoto S, Oates JA and Brash AR, 1986. Stereoselective hydrogen abstraction in leukotriene A4 synthesis by purified 5-lipoxygenase of porcine leukocytes. Prostaglandins 32, 43–8. [DOI] [PubMed] [Google Scholar]
- Upston JM, Neuzil J and Stocker R, 1996. Oxidation of LDL by recombinant human 15-lipoxygenase: evidence for alpha-tocopherol-dependent oxidation of esterified core and surface lipids. J Lipid Res 37, 2650–61. [PubMed] [Google Scholar]
- Upston JM, Neuzil J, Witting PK, Alleva R and Stocker R, 1997. Oxidation of free fatty acids in low density lipoprotein by 15-lipoxygenase stimulates nonenzymic, alpha-tocopherol-mediated peroxidation of cholesteryl esters. J Biol Chem 272, 30067–74. [DOI] [PubMed] [Google Scholar]
- van Leyen K, 2013. Lipoxygenase: an emerging target for stroke therapy. CNS Neurol Disord Drug Targets 12, 191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leyen K, Arai K, Jin G, Kenyon V, Gerstner B, Rosenberg PA, Holman TR and Lo EH, 2008. Novel lipoxygenase inhibitors as neuroprotective reagents. J Neurosci Res 86, 904–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leyen K, Duvoisin RM, Engelhardt H and Wiedmann M, 1998. A function for lipoxygenase in programmed organelle degradation. Nature 395, 392–5. [DOI] [PubMed] [Google Scholar]
- van Leyen K, Kim HY, Lee SR, Jin G, Arai K and Lo EH, 2006. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke 37, 3014–8. [DOI] [PubMed] [Google Scholar]
- Van Os CP, Rijke-Schilder GP, Van Halbeek H, Verhagen J and Vliegenthart JF, 1981. Double dioxygenation of arachidonic acid by soybean lipoxygenase-1. Kinetics and regio-stereo specificities of the reaction steps. Biochim Biophys Acta 663, 177–93. [DOI] [PubMed] [Google Scholar]
- Vanderhoek JY and Bailey JM, 1984. Activation of a 15-lipoxygenase/leukotriene pathway in human polymorphonuclear leukocytes by the anti-inflammatory agent ibuprofen. J Biol Chem 259, 6752–6. [PubMed] [Google Scholar]
- Vogel R, Jansen C, Roffeis J, Reddanna P, Forsell P, Claesson HE, Kuhn H and Walther M, 2010. Applicability of the triad concept for the positional specificity of mammalian lipoxygenases. J Biol Chem 285, 5369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther M, Anton M, Wiedmann M, Fletterick R and Kuhn H, 2002. The N-terminal domain of the reticulocyte-type 15-lipoxygenase is not essential for enzymatic activity but contains determinants for membrane binding. J Biol Chem 277, 27360–6. [DOI] [PubMed] [Google Scholar]
- Walther M, Hofheinz K, Vogel R, Roffeis J and Kühn H, 2011. The N-terminal β-barrel domain of mammalian lipoxygenases including mouse 5-lipoxygenase is not essential for catalytic activity and membrane binding but exhibits regulatory functions. Archives of Biochemistry and Biophysics 516, 1–9. [DOI] [PubMed] [Google Scholar]
- Walther M, Ivanov I, Myagkova G and Kuhn H, 2001. Alterations of lipoxygenase specificity by targeted substrate modification and site-directed mutagenesis. Chem Biol 8, 779–90. [DOI] [PubMed] [Google Scholar]
- Walther M, Wiesner R and Kuhn H, 2004. Investigations into calcium-dependent membrane association of 15-lipoxygenase-1. Mechanistic roles of surface-exposed hydrophobic amino acids and calcium. J Biol Chem 279, 3717–25. [DOI] [PubMed] [Google Scholar]
- Wang H, Li J, Follett PL, Zhang Y, Cotanche DA, Jensen FE, Volpe JJ and Rosenberg PA, 2004. 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes. Eur J Neurosci 20, 2049–58. [DOI] [PubMed] [Google Scholar]
- Watanabe T and Haeggstrom JZ, 1993. Rat 12-lipoxygenase: mutations of amino acids implicated in the positional specificity of 15- and 12-lipoxygenases. Biochem Biophys Res Commun 192, 1023–9. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Medina JF, Haeggstrom JZ, Radmark O and Samuelsson B, 1993. Molecular cloning of a 12-lipoxygenase cDNA from rat brain. Eur J Biochem 212, 605–12. [DOI] [PubMed] [Google Scholar]
- Weibel GL, Joshi MR, Alexander ET, Zhu P, Blair IA and Rothblat GH, 2009. Overexpression of human 15(S)-lipoxygenase-1 in RAW macrophages leads to increased cholesterol mobilization and reverse cholesterol transport. Arterioscler Thromb Vasc Biol 29, 837–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel M and Heller AR, 2009. Anticancer actions of omega-3 fatty acids--current state and future perspectives. Anticancer Agents Med Chem 9, 457–70. [DOI] [PubMed] [Google Scholar]
- Wennman A, Karkehabadi S and Oliw EH, 2014. Kinetic investigation of the rate-limiting step of manganese- and iron-lipoxygenases. Arch Biochem Biophys 555–556, 9–15. [DOI] [PubMed] [Google Scholar]
- Wiesner R, Suzuki H, Walther M, Yamamoto S and Kuhn H, 2003. Suicidal inactivation of the rabbit 15-lipoxygenase by 15S-HpETE is paralleled by covalent modification of active site peptides. Free Radic Biol Med 34, 304–15. [DOI] [PubMed] [Google Scholar]
- Winkler FK, D’Arcy A and Hunziker W, 1990. Structure of human pancreatic lipase. Nature 343, 771–4. [DOI] [PubMed] [Google Scholar]
- Wittwer J and Hersberger M, 2007. The two faces of the 15-lipoxygenase in atherosclerosis. Prostaglandins Leukot Essent Fatty Acids 77, 67–77. [DOI] [PubMed] [Google Scholar]
- Witztum JL and Steinberg D, 1991. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest 88, 1785–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Bhattacharjee A, Roy B, Feldman GM and Cathcart MK, 2004. Role of protein kinase C isoforms in the regulation of interleukin-13-induced 15-lipoxygenase gene expression in human monocytes. J Biol Chem 279, 15954–60. [DOI] [PubMed] [Google Scholar]
- Xu B, Bhattacharjee A, Roy B, Xu HM, Anthony D, Frank DA, Feldman GM and Cathcart MK, 2003. Interleukin-13 induction of 15-lipoxygenase gene expression requires p38 mitogen-activated protein kinase-mediated serine 727 phosphorylation of Stat1 and Stat3. Mol Cell Biol 23, 3918–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhang Y, Xiao Y, Ma S, Liu Q, Dang S, Jin M, Shi Y, Wan B and Zhang Y, 2013. Inhibition of 12/15-lipoxygenase by baicalein induces microglia PPARbeta/delta: a potential therapeutic role for CNS autoimmune disease. Cell Death Dis 4, e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Zhuo JM, Chu J, Chinnici C and Pratico D, 2010. Amelioration of the Alzheimer’s disease phenotype by absence of 12/15-lipoxygenase. Biol Psychiatry 68, 922–9. [DOI] [PubMed] [Google Scholar]
- Yao Y, Clark CM, Trojanowski JQ, Lee VM and Pratico D, 2005. Elevation of 12/15 lipoxygenase products in AD and mild cognitive impairment. Ann Neurol 58, 623–6. [DOI] [PubMed] [Google Scholar]
- Yigitkanli K, Pekcec A, Karatas H, Pallast S, Mandeville E, Joshi N, Smirnova N, Gazaryan I, Ratan RR, Witztum JL, Montaner J, Holman TR, Lo EH and van Leyen K, 2013. Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke. Ann Neurol 73, 129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yla-Herttuala S, Rosenfeld ME, Parthasarathy S, Glass CK, Sigal E, Witztum JL and Steinberg D, 1990. Colocalization of 15-lipoxygenase mRNA and protein with epitopes of oxidized low density lipoprotein in macrophage-rich areas of atherosclerotic lesions. Proc Natl Acad Sci U S A 87, 6959–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Lim JY and Hwang SW, 2013. Resolvins: Endogenously-Generated Potent Painkilling Substances and their Therapeutic Perspectives. Curr Neuropharmacol 11, 664–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto T, Suzuki H, Yamamoto S, Takai T, Yokoyama C and Tanabe T, 1990. Cloning and sequence analysis of the cDNA for arachidonate 12-lipoxygenase of porcine leukocytes. Proc Natl Acad Sci U S A 87, 2142–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinaga M, Buchanan FG and DuBois RN, 2004. 15-LOX-1 inhibits p21 (Cip/WAF 1) expression by enhancing MEK-ERK ½ signaling in colon carcinoma cells. Prostaglandins Other Lipid Mediat 73, 111–22. [DOI] [PubMed] [Google Scholar]
- Zafiriou MP, Zelarayan LC, Noack C, Renger A, Nigam S and Siafaka-Kapadai A, 2011. Hepoxilin A(3) protects beta-cells from apoptosis in contrast to its precursor, 12-hydroperoxyeicosatetraenoic acid. Biochim Biophys Acta 1811, 361–9. [DOI] [PubMed] [Google Scholar]
- Zhang B, Jia H, Liu J, Yang Z, Jiang T, Tang K, Li D, Huang C, Ma J, Shen GX, Ye D and Huang B, 2010. Depletion of regulatory T cells facilitates growth of established tumors: a mechanism involving the regulation of myeloid-derived suppressor cells by lipoxin A4. J Immunol 185, 7199–206. [DOI] [PubMed] [Google Scholar]
- Zhang HJ, Sun CH, Kuang HY, Jiang XY, Liu HL, Hua WF, Liu ZJ, Zhou H, Sui H and Qi R, 2012. 12(S)-hydroxyeicosatetraenoic acid levels link to coronary artery disease in type 2 diabetic patients. J Endocrinol Invest In Press [DOI] [PubMed] [Google Scholar]
- Zhang HP, Jia CE, Lv Y, Gibson PG and Wang G, 2014. Montelukast for prevention and treatment of asthma exacerbations in adults: Systematic review and meta-analysis. Allergy Asthma Proc 35, 278–87. [DOI] [PubMed] [Google Scholar]
- Zhang J, 2013. Autophagy and Mitophagy in Cellular Damage Control. Redox Biol 1, 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li J, Jimenez DA, Levitan ES, Aizenman E and Rosenberg PA, 2004. Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J Neurosci 24, 10616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Pratico D, Rader DJ and Funk CD, 2005. 12/15-Lipoxygenase gene disruption and vitamin E administration diminish atherosclerosis and oxidative stress in apolipoprotein E deficient mice through a final common pathway. Prostaglandins Other Lipid Mediat 78, 185–93. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Bhattacharjee S, Jones BM, Hill J, Dua P and Lukiw WJ, 2013. Regulation of Neurotropic Signaling by the Inducible, NF-kB-Sensitive miRNA-125b in Alzheimer’s Disease (AD) and in Primary Human Neuronal-Glial (HNG) Cells. Mol Neurobiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y and Brash AR, 2010a. Dioxygenase activity of epidermal lipoxygenase-3 unveiled: typical and atypical features of its catalytic activity with natural and synthetic polyunsaturated fatty acids. J Biol Chem 285, 39866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y and Brash AR, 2010b. On the role of molecular oxygen in lipoxygenase activation: comparison and contrast of epidermal lipoxygenase-3 with soybean lipoxygenase-1. J Biol Chem 285, 39876–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D and Ran Y, 2012. Role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in hypoxia-induced pulmonary hypertension. J Physiol Sci 62, 163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X, Morris JS, Broaddus R and Shureiqi I, 2009. 15-LOX-1 transcription suppression through the NuRD complex in colon cancer cells. Oncogene 28, 1496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X, Shen L, Issa JP, Moy O, Morris JS, Lippman SM and Shureiqi I, 2008. 15-Lipoxygenase-1 transcriptional silencing by DNA methyltransferase-1 independently of DNA methylation. FASEB J 22, 1981–92. [DOI] [PMC free article] [PubMed] [Google Scholar]