

Abstract
The MDM2 oncogene is overexpressed in various human cancers. Its expression correlates with the phenotypes of high-grade, late-stage, and more resistant tumors. The auto-regulatory loop between MDM2 and the tumor suppressor p53 has long been considered the epitome of a rational target for cancer therapy. As such, many novel agents have been generated to interfere with the interaction of the two proteins, which results in the activation of p53. Among these agents are several small molecule inhibitors synthesized based upon the crystal structures of the MDM2-p53 complex. With use of high-throughput screening, several specific and effective agents for inhibition of the protein-protein interaction were discovered. Recent investigations, however, have demonstrated that many proteins regulate the MDM2-p53 interaction, and that MDM2 may have p53-independent oncogenic functions. In order for novel MDM2 inhibitors to be translated to the clinic, it is necessary to obtain a better understanding of the regulation of MDM2 and of the MDM2-p53 interaction. In particular, the implications of various interactions between certain regulator(s) and MDM2/p53 under different circumstances need to be elucidated to determine which pathway(s) represent the best targets for therapy. Targeting both MDM2 itself and regulators of MDM2 and the MDM2-p53 interaction, or use of MDM2 inhibitors in combination with conventional treatments, may improve prospects for tumor eradication.
Keywords: MDM2, p53, oncogene, regulation, protein-protein interaction, apoptosis, cell cycle arrest
1. INTRODUCTION
1.1. Oncogene Addiction is a Key Feature of Human Cancers
Oncogenes and tumor suppressor genes are involved in carcinogenesis and tumor progression. Although loss of a tumor suppressor can lead to oncogenesis, many human tumors possess at least one activated oncogene. The activation of one or more oncogenes, along with environmental stress or another cellular “hit,” is sufficient to initiate carcinogenesis [1, 2]. There is a long and still-growing list of gene products implicated in initiating and stimulating carcinogenesis and cancer progression [2–6]. These oncoproteins can be grouped into several categories: growth factors (e.g., EGFR), transcription factors (c-Myc), tyrosine kinases (Her2/neu), serine/threonine kinases (Raf), other intermediate signaling molecules (PI3K), and cell-cycle or apoptosis-related proteins (Bcl-2).
Continuous expression of at least one oncogene is often necessary and sufficient for cells to attain unchecked growth and proliferation under conditions normally incompatible with survival. Numerous oncogenes have been examined as targets for therapy, and inhibiting their expression or activity is often sufficient to inhibit tumor growth, demonstrating that breaking the addiction can end cancer development and progression. Overcoming the addiction of cells to these oncogenic proteins results in a reversion to a normal phenotype or, more frequently, to apoptosis of transformed cells [2, 7].
1.2. Targeting Oncogenes for Cancer Prevention and Therapy
Given the extent of oncogene addiction and the number of oncogenes that have been identified, developing strategies to inhibit their expression or activity in the clinic has been a major research priority for more than a decade. Considerable progress has been made, especially for oncogenes within the tyrosine kinase category. Several agents targeting tyrosine kinases are currently being used to treat human cancer, including Herceptin® against Her2/neu expressing cells, Gleevec® against cells expressing bcr/abl, and Iressa® against cells over-expressing EGFR. Development of these agents was a major breakthrough, and they have served as a proof-of-principle that targeted therapy is a novel and effective anti-cancer strategy. This is especially true in the case of Gleevec, which significantly improved the survival of CML (chronic myelogenous leukemia) patients [8]. Other categories of oncogenes, including the cell cycle-related and apoptosis-related oncogenes, have proved more difficult to target with small molecules, but studies are progressing.
In fact, a variety of agents, ranging from antisense oligonucleotides to antibodies to rationally targeted small molecules, are being investigated for their potential to inhibit oncogene expression. Other cancer prevention strategies make use of natural products or synthetic agents with low toxicity profiles to inhibit the expression of oncogenes in the hope of preventing oncogenesis and cancer progression. In particular, genistein, a soy isoflavone, inhibits tyrosine kinase and decreases the expression of various growth factors and their receptors [9]. Other anti-oncogenic functions of genistein have been discovered [10], and other natural products have demonstrated similar effects [11]. There is hope that these agents, which have little or no apparent toxicity, can be used to prevent cancer initiation or to inhibit tumor growth in early stages.
1.3. The MDM2 Oncogene
History
The MDM2 (mouse double-minute 2, the human homologue is also called HDM2) oncogene was first identified in the early 1990’s following the observation that its overexpression in 3T3DM cells was sufficient to confer tumorigenicity in mice [12]. MDM2 is overexpressed in most human cancers, including 56% of breast cancers, 65% of colon cancers, 41% of prostate cancers, and 39% of lung cancers, as well as 80% of lymphomas and 70% of leukemias [13]. MDM2 overexpression is associated with tumors that are more invasive, in higher/later stages, with greater metastatic potential, and with resistance to chemotherapeutic agents and radiation [13]. These factors implicate MDM2 as an oncogene, and indicate that it represents a target for therapy.
The human MDM2 gene is located on chromosome 12q13–15 [14]. The protein contains 491 amino acids, and migrates as a 90–95 kD band when separated by SDS-denaturing gel electrophoresis. MDM2 has a nuclear localization signal (NLS), a nuclear export signal (NES), a central acidic domain, a C-terminal zinc-finger domain, and a RING finger domain possessing E3 ligase activity [15, 16]. Under normal conditions, MDM2 is expressed in the nucleus, but it translocates to the cytoplasm to allow the degradation of some of its targets by the proteasome [17–19].
The MDM2-p53 Interaction
Not long after discovering the oncogenic potential of MDM2, the oncoprotein was established as a negative regulator of the tumor suppressor p53 [17]. The p53 protein maintains genomic stability and, under normal conditions, is present at low levels. In response of stresses, p53 is activated and induces cell cycle arrest, apoptosis, senescence, and cell differentiation [20, 21]. MDM2 binds to p53, masking its transactivation domain, and makes use of its intrinsic E3 ligase activity to ubiquitinate p53 and target it to the proteasome for degradation [22, 23]. Completing an elegant regulatory system, p53 in turn regulates MDM2 by promoting its transcription [15]. MDM2 has an intronic p53-dependent promoter (P2), which is highly conserved between humans and mice. The P2 structure has two tandem p53-binding elements, and promoter activation requires simultaneous binding of p53 to both [14]. Thus, the tumor suppressor and oncogene participate in an auto-regulatory feedback loop. Under normal homeostatic conditions, the two proteins regulate each other and prevent both extensive apoptosis and unnecessary cell growth. On the other hand, when MDM2 is overexpressed, the activity of p53 is inhibited. Thus, the extent of activation of p53 in response to DNA-damaging chemotherapeutic agents or radiation is compromised when MDM2 is overexpressed, allowing cancer cells to escape treatment-related arrest or death [24].
Based on its role in p53 regulation and its effects on the response to cancer therapies, MDM2 has been considered as a target for therapy for the past decade. Major research efforts have been directed toward the MDM2-p53 interaction and toward inhibition of the interaction. Although there are extensive new findings about MDM2, p53, and their interaction, this review is focused on the cofactors and co-regulators involved in regulating the proteins and their interaction and on agents developed to target the interaction. Although other topics related to MDM2 and p53 regulation are not covered, interested readers can refer to the several excellent reviews published in the past few years [25–30].
2. REGULATION OF THE MDM2-p53 INTERACTION
MDM2 and p53 are components of an elegant auto-regulatory feedback loop. The interaction involves a large number of factors, and the extent of regulation of the expression and activity of MDM2 and p53 is more extensive than was originally thought. Accumulating evidence indicates that, in order to achieve the subtle differences in activity that are necessary to cause the various changes in cell cycle progression and arrest, apoptosis, angiogenesis, and other processes (under both homeostatic and stressed conditions and in different cell types), other molecules must regulate MDM2, p53, and their interaction.
In support of this concept, there is a diverse and growing group of proteins known to regulate the MDM2-p53 interaction and the activities of the two proteins. These co-factors alter MDM2 or p53 conformation, binding, localization, and expression, and modulate the E3 ligase activity of MDM2 toward itself, p53, and other substrates. The MDM2-p53 interaction regulates a variety of different processes, with some overlap between the different molecule in their activity. The following describes some of the more recent discoveries of how the interaction is regulated. Table 1 presents many of the MDM2-binding proteins and the outcome of their interactions with MDM2 and p53.
Table 1.
MDM2-Interactive Proteins and their Effects
| Protein (Alternate Name(s)) | Binding Site(s) on the Protein | Binding Site on MDM2 | Binding Site on p53 | Effect of Interaction with the Protein on MDM2 | Effect of Interaction with the Protein on p53 | Effect of MDM2 and/or p53 on the Protein | Ref. |
|---|---|---|---|---|---|---|---|
| 14-3-3-σ | aa153–248 (to both MDM2 and p53) | C-ter | C-ter | Decreased stability, translocation to cytoplasm | Increased stability, increased activity | p53 increases its activity following DNA damage | [33] |
| P14 (ARF) | aa1–14 (MDM2) | aa235–264, 270–289 | n/a (does not bind) | Decreased activity, nucleolar localization | Increased stability | MDM2 decreases its transcriptional activity and increases degradation | [36, 38] |
| Nucleophosmin (NPM, B23) | aa187–295 (p53) | N-ter (1–110) and C-ter (280–491) | N-ter | Decreased activity toward p53 (inhibited binding) | Increased stability (controversial) | Effect unknown | [44] |
| C5NF (COP9, COPS5,Jab1) | aa1–110 (p53) | Binding site is unknown | N-ter (82–105) | Decreased auto-ubiquitination, increased stability | Increased phosphorylation and degradation, nuclear export | Effect unknown | [50, 52] |
| Gankyrin (PSM10) | Full-length necessary for binding | aa412–437 | Unknown, may be indirect | Enhanced E3 ligase activity (auto- and substrate ubiquitination) | Enhanced ubiquitination and degradation | Effect unknown | [56] |
| HAUSP/USP7 | aa53–208 | aa1–110 and aa276–491 | aa357–382 | Increased stability due to de-ubiquitination | Decreased stability due to increased MDM2-mediated ubiquitination (HAUSP can also de-ubiquitinatep53) | Effect unknown | [59, 60] |
| JMY | Binding site is unknown | RING finger domain | Unknown, may be indirect | Effect unknown | Decreased transcriptional activity | MDM2 increases proteasomal degradation | [65] |
| KAP1 (TRIM28) | aa239–419 | aa150–230, with 200–230 critical | Unknown, may be indirect | Effect unknown | Inactivation | Effect unknown | [67] |
| MDM4/MDMX | RING finger domain | aa276–491(zinc finger and RING finger domain) | N-terminal | Increased stability, increased activity (in some circumstances) | Increased stability and activity; NEDDylation | MDM2 increases its ubiquitination | [69] |
| MTBP | aa515–894 | aa167–304 (200–304critical region) | aa293–393 | Increased stability | Increased ubiquitination and degradation | None/unknown | [76, 77] |
| Nucleolin | Binding site is unknown | Binding site is unknown | aa363–393 | Inhibits auto-and p53 ubiquitination; decreases stability | Increased stability, transcriptional activity | Unknown | [81, 83] |
| NUMB | aa92–165 or aa352–592 (2 binding domains to MDM2) | aa1–134 | aa537–551 | No known effect | Enhanced activity | MDM2 increases its degradation | [84, 87] |
| P21 | Binding site is unknown | aa180–298 | p21 is atranscriptional target of p53 | No known effect | Decreased expression (decreased transcription) | MDM2 increases its proteasomal degradation | [89, 90] |
| P73 | aa1–70 | aa1–150 | DNA binding domain (aa100–300) | Effect unknown | Increased stability and transcription; NEDDylation | Interaction with MDM2 decreases its transcriptional activity | [94, 95] |
| PA28γ(REGγ, PSME3, Ki antigen) | 96–103 (MDM2), 86–96 (p53) | Binding site is unknown | Binding site is unknown | Increased stability | Increased ubiquitination | Effect unknown | [99] |
| PCAF | aa1–493 | aa1–150 and aa 294–384 | aa 300–369 or aa 358–393 | Inhibits binding to p53; stimulates auto-ubiquitination | Decreased acetylation; stabilization and activation | MDM2 increases its proteasomal degradation | [101] |
| PML | aa300–633 | aa202–304 (aa222–272 critical region) | Unknown, may be indirect | Nuclear localization | CK1-mediated phosphorylation; decreased ubiquitination; increased stabilization and activation | Interaction with MDM2 leads to cytoplasmic localization | [102, 104] |
| pRb | aa792–928 (C-pocket) | aa273–321(?), aa254–264 (central acidic domain) | Indirect interaction | Decreased E3 ligase activity toward p53 | Increases p53-mediated apoptosis | MDM2 binding decreases activity | [105, 107] |
| L5 | Binding site is unknown | aa221–274 | Unknown, may be indirect | Decreased activity | Increased stability and activity/decreased MDM2 -mediated □ biquityination | Effect unknown | [108, 110] |
| L11 | Central domain (aa51–108)(to MDM2) | aa284–374 | Unknown, may be indirect | Increased stabilization | Increased activity | Increased stability | [108, 109] |
| L23 | aa43–70 | aa150–301 and 384–425 | Unknown, may be indirect | Decreased ubiquitination of p53 | Increased stability and activity | Effect unknown | [108, 110] |
| L26 | Binding site is unknown | Binding site is unknown | Unknown, may be indirect interaction with protein (directly interacts with p53 mRNA) | Decreased ubiquitination of p53 | Increased synthesis and protection form MDM2-mediated ubiquitination | MDM2 increases its ubiquitination | [112] |
| S7 | aa59–134 | aa180–298 | Unknown, may be indirect | Effect unknown | Increased stability and activity | Effect unknown | [113] |
| Tip60 | aa258–364 | Binding site is unknown | DNA binding domain | Localization to PML bodies | Decreased MDM2-mediated NEDDylation; acetylation | MDM2 increases its proteasomal degradation | [116, 117] |
| TSG101 | Ubiquitin-conjugating E2 variant (UEV) domain | Binding site is unknown | Binding site is unknown, likely indirect | Inhibits ubiquitination; increased stabilization | Decreased activity | MDM2 increases its proteolysis | [120, 122] |
| β-arrestins | Binding site is unknown | aa161–400 (β-arrestin 2, P-arrestin 1 is unknown) | Binding site is unknown, likely indirect | Cytoplasmic localization, decreased E3 ligase activity (β-A2) | Increased protein stability and activity (β-arrestin 2) | Effect unknown (β-arrestin 2), increased ubiquitination (β-arrestin 1) | [123, 124] |
| HIF-1 | aa1–245 | p53 binding site (aa19–220) | DNA binding site | Increased activity toward p53 (controversial) | Decreased stability (controversial) | MDM2 may increase its stability (controversial) | [127, 130] |
| IGFR | Binding site is unknown | Binding site is unknown | Binding site is unknown, likely indirect | Effect unknown | Increased stability | MDM2 increases its proteasomal degradation | [138, 139] |
| Estrogen Receptor (α) | Ligand binding domain | Binding site is unknown | N-terminal end (aa1–103) | Increased transcription following estrogen ligation by the ER | Increased stability and activity | Interaction with MDM2 decreases its stability; it also increases its activity when p53 is absent | [146] |
| Androgen Receptor | Binding site is unknown | Binding site is unknown | Binding site is unknown, may be indirect | Effect unknown | Effect unknown | MDM2 increases its ubiquitination and proteolysis. MDM2 also decreases its transcriptional activity | [118] |
| Glucocorticoid Receptor | Binding site is unknown | Binding site is unknown | aa76–320 | Effect unknown | Increased ubiquitination (especially upon GR agonism) | MDM2 increases its ubiquitination | [144, 145] |
| TGFβ | Binding site is unknown | C-terminal half of p53-binding site, Zn coordination residues of RING finger domain, NLS (aa 177–192) | Unknown whether there is direct binding | Effect unknown | No significant effect | Effect unknown, MDM2 may mediate TGF-β resistance | [152] |
| E2F1 | aa359–407 (activation domain) | aa1–220 | DNA-binding domain | Effect unknown | Effect unknown | p53 represses its activation/function; MDM2 increases its stability and stimulates activity | [53] |
2.1. Regulation of the MDM2-p53 Interaction with Respect to the Cell Cycle and Apoptosis
14-3-3-σ
The 14-3-3-σ protein is a downstream target of p53 that is expressed following cell stress, such as exposure to radiation [31]. 14-3-3-σ negatively regulates cell cycle progression via interactions with CDK2/4 and CDC2, preventing the cyclin-CDK/CDC interaction and arresting cells in the G2 phase [32, 33]. The protein has further effects on the cell cycle by decreasing p53 degradation via an increase in MDM2 auto-ubiquitination and degradation, as well as by causing the translocation of MDM2 to the cytoplasm [34]. Moreover, 14-3-3-σ inhibits MDM2-mediated degradation of Rb and NEDDylation of p53 [34]. 14-3-3-σ is down-regulated in breast and other cancers, possibly as a result of decreased p53 transcriptional activity [35]. These findings suggest that 14-3-3-σ could represent a target for gene therapy or that a 14-3-3-σ mimetic might have anti-cancer effects.
ARF(p14/p19) and Nucleophosmin (B23)
p14ARF (p19 in mice) forms a ternary complex with MDM2 and p53, leading to antagonism of the MDM2-mediated ubiquitination of p53 [36]. The protein also changes the conformation of MDM2, leading to its association with nucleoli [37, 38]. ARF may inhibit the E3 ligase activity of MDM2, although this has not been demonstrated in vivo [39]. Regardless of which mechanism(s) it uses, overexpression of p14 by an adenoviral vector leads to anti-tumor effects and potentiates the effects of chemotherapy in tumor-bearing mice [40]. Moreover, mice with ARF knocked out demonstrate increased susceptibility to cancer [41]. However, ARF has other, p53-independent activities, suggesting that the effects of knockdown may not be exclusively due to its effects on p53 [42].
Another MDM2-interactive protein, nucleophosmin (NPM, B23, NO38, numatrin), is involved in the MDM2-p14ARF interaction [43], stabilizing ARF and increasing its concentration in the nucleolus. NPM also apparently competes for binding of MDM2 with p53, resulting in decreased p53 ubiquitination, because both proteins (NPM and p53) bind to the same region of MDM2 [44]. NPM mutations and overexpression have been observed in human cancers [45]. Nevertheless, it appears that wild-type NPM decreases tumor initiation, likely via a p53-independent role in preserving genomic integrity. Moreover, the protein may also interact with p53 to activate its transcriptional activity [46]. Thus, there may be several layers of regulation to the MDM2-ARF-NPM-p53 pathway. Thus, it seems likely that still another regulator is involved in the process that serves to release ARF from NPM.
Targeting NPM for cancer therapy is controversial since eliminating its expression leads to genomic instability. More information is needed to determine whether the protein can serve as a therapeutic target. While overexpression of ARF via an adenovirus leads to anti-tumor effects [40], the effects of ARF on the MDM2-p53 interaction are not its only activities. It appears that ARF also regulates cell growth via additional pathways and interacts with numerous other proteins [47]. Therefore, if ARF and/or nucleophosmin are targeted, care must be taken so that these pathways are not affected.
CSN5
CSN5 (Jab1, COPS5, COP9), a component of the COP9 signalosome, may regulate a variety of processes, including cell cycle progression. It was found by yeast-2-hybrid assays to interact with p27, which it translocates from the nucleus to the cytoplasm [48]. The protein is also involved in regulating SCF ubiquitin ligases, possibly by decreasing their NEDDylation [49]. Since many cell cycle regulating proteins are subject to ubiquitination by SCF li-gases, it is likely that CSN5 contributes to their regulation, adding another layer by which CSN5 regulates the cell cycle.
Moreover, CSN5 promotes p53 phosphorylation and increases its proteasomal degradation [50]; it also promotes p53 nuclear export [51]. The protein also decreases MDM2 degradation by decreasing MDM2 auto-ubiquitination [52]. While no p53-independent/MDM2-dependent functions are known for CSN5, the CSN complex is involved in the degradation of a variety of other cell cycle regulating proteins [52], and their degradation may also be dependent upon MDM2. In particular, it seems likely that the protein may function in MDM2-mediated regulation of E2F1, which is accomplished via interruption of E2F1’s interaction with SCF [53]. More research is needed into the MDM2-CSN5-p53 interaction before it can be considered a target, but the present data indicate that the protein is associated with regulation of the cell cycle.
Gankyrin
Gankyrin, also known as PSM10, is a protein commonly over-expressed in early hepatocarcinogenesis and in hepatocellular carcinomas [54, 55]. This seven-repeat protein is associated with the 19S regulatory complex of the 26S proteasome. Data derived in vitro and in vivo suggest that gankyrin facilitates the p53-MDM2 interaction by binding to MDM2 [56]. This association results in increased ubiquitination of p53 and increases its degradation, possibly through both the greater extent of ubiquitination and enhanced interaction with the 26S proteasome [56]. Even in the absence of p53, however, the protein enhances the auto-ubiquitination of MDM2. Gankyrin also forms complexes with other proteins, such as CDK4 and pRb [57, 58], supporting the idea that it functions in cell cycle regulation. Since inhibition of gankyrin leads to apoptosis in cancer cells, it is a possible target for cancer therapy [56].
HAUSP
HAUSP (herpes virus-associated ubiquitin-specific protease, also known as USP7- ubiquitin specific protease 7) was initially identified as a p53-interactive protein that stabilizes p53 [59]. HAUSP was later found to bind to MDM2 as well [60]. Binding of HAUSP to either p53 or MDM2 leads to their de-ubiquitination, and binding to MDM2 leads to p53 destabilization due to increased MDM2 stability [60]. Exactly how these apparently divergent activities of HAUSP are accomplished is unknown, but HAUSP may act differently under different conditions (e.g., its activity under homeostatic conditions may differ from its activity following DNA damage). Such a premise gained support following mathematical modeling, which led to the suggestion that HAUSP acts as a switch to change the specificity of MDM2’s E3 ligase from auto-ubiquitination to predominantly p53 ubiquitination [61].
In support of this, recent studies have identified Daxx as another interactive protein involved in regulating the effects of HAUSP on MDM2 and p53. Binding of Daxx apparently decreases the auto-ubiquitination of MDM2, leading to increased p53 ubiquitination [62]. Other molecules, including ATM kinase and MDMX, are also involved in this protein complex and are likely regulate MDM2 and p53 [63]. Further investigations are needed to elucidate the identities of the proteins involved in the HAUSP-MDM2-p53 complex and to determine their roles in cancer initiation and progression. Nevertheless, targeting of HAUSP and reducing its activity may result in anti-cancer effects.
JMY
DNA damage increases the accumulation of JMY, a p53 co-transcription factor responsible for augmentation of the p53 response to damage [64]. Consistent with its regulation of the p53-MDM2 regulatory loop, the MDM2 RING domain is required for the ubiquitin-dependent degradation of JMY [65]. Complicating interpretation of the interaction, however, are the facts that JMY functions as a transcriptional target of E2F1, and over-expression of both wild-type p53 and E2F1 increases apoptosis [66]. This may explain the apparently dual roles of E2F1 in its interactions with MDM2 versus p53 (cell growth versus apoptosis). More investigations are needed to evaluate the potential of overexpressing or knocking down JMY for therapy, but it appears to be involved in the p53-MDM2-E2F1 interaction.
KAP1
KAP1, also known as TRIM28, mediates transcriptional control by associating with specific regions of chromatin. KAP1 cooperates with MDM2 in the ubiquitination of p53 by binding to the central acidic domain of MDM2 [67]. This interaction stimulates the formation of the p53-HDAC complex and inhibits the protective acetylation of p53. RNA interference (RNAi)-mediated inhibition of KAP1 increased p53 transcriptional activity and apoptosis in HT1080 (wt p53) cells. Following exposure of MCF-7 cells to actinomycin D or gamma-radiation, there is a reduction in KAP1 that correlates with an induction of p21, a p53 gene product [68]. This decrease in KAP1 is also associated with cell cycle arrest and inhibition of clonal cell growth. Thus, it appears that KAP1 helps regulate the apoptotic response following DNA damage as well as cell cycle progression under both normal and stressed conditions. More work is needed to determine the exact role of KAP1 in the MDM2-p53 (and possibly other proteins) interaction.
MDMX/MDM4
Since the discovery of MDMX (also called MDM4) in 1996 [69], major advances have been made in understanding its structural and functional relevance and its interaction with MDM2. These proteins form hetero-complexes and regulate each other’s stability and activity. The interaction of MDMX with MDM2 may be required for the effects of MDM2 on p53. Recent studies in vitro suggest that wild-type MDMX regulates p53 abundance by modulating the levels of “free” MDM2 [70]; thus, the degree of p53 degradation is determined by the balance between MDM2 and MDMX. Although MDM2 regulates the stability of p53, MDMX may regulate its activity. In cell culture studies, MDMX overexpression prevents p53 activation and cell cycle arrest; further, decreased tumor formation is observed in MDMX-knockout xenograft models [71]. Over-expression of MDMX in vivo decreases sensitivity to 5-FU but does not accelerate tumor growth [70]. Similar to MDM2, MDMX is overexpressed in many cancers, including breast carcinoma, soft tissue sarcoma, lung carcinoma, and retinoblastoma [72]. However, despite their high level of structural conservation, the homologues have distinct functions.
For example, the C-terminal of the RING finger domain is essential for the E3 ligase activity of MDM2. Complete deletion of the C-terminal tail of MDM2 results in loss of E3 activity as well as the capacity to oligomerize with itself or MDMX [73]. Although MDMX does not possess any intrinsic E3 ligase activity, the MDM2-MDMX heterodimer is more stable, and has greater E3 ligase activity than the MDM2-homodimer, suggesting that MDMX is involved in the ubiquitination of p53 and other substrates, likely by modulating the stability and activity of MDM2.
Another study examined both the expression of MDMX and a splice variant, MDMX-S (HDMX-S). This transcript contains an internal deletion of 68 base pairs, which causes a frame shift and ultimately results in a premature stop codon. The truncated protein appears to bind and inactivate p53 better than MDM2 or full-length MDMX [74]. MDMX-S contains 13 novel C-terminal amino acids that are responsible for its high affinity to p53, and this spice variant is targeted more efficiently to the nucleus than MDMX [75]. When these amino acids are deleted, MDMX-S does not bind to and inactivate p53. New studies are now examining specific ways to target MDMX, its variants, or both MDMX and MDM2 simultaneously [71].
MTBP
MDM2-binding protein (MTBP) was identified via a yeast two-hybrid screen [76]. Data derived in vitro suggest that MTBP differentially regulates both MDM2 auto-ubiquitination and ubiquitination of p53 [77]. Knockdown of MTBP in non-stressed, p53-transfected H1299 cells reveals an increase in p53 transcriptional activity, demonstrating that MTBP is a necessary cofactor for MDM2-mediated p53 regulation. Although gamma irradiation results in MTBP-mediated stabilization of MDM2, exposure to UV radiation down-regulates the expression of both MDM2 and MTBP. This suggests that, like HAUSP, the proteins have different functions under different conditions [77]. The protein also induces G1 cell cycle arrest in a p53-independent manner, but the arrest is blocked by MDM2 overexpression [76]. Further, homozygous disruption of MTBP results in early embryonic lethality that can not be rescued by additional knockout of p53 [78]. Taken together, these data support the role of MTBP as a cofactor for MDM2 stabilization and MDM2-mediated p53 degradation; they also indicate that the protein has p53-independent effects on the cell cycle [78]. Its effects under different conditions (homeostasis vs. stress) warrant further investigation.
Nucleolin
Nucleolin, a c-myc gene product [79], contributes to the inhibition of chromosomal DNA replication following cellular stress and is involved in ribosome assembly [80]. An interaction between p53 and nucleolin was established in 2002 [81]. It has been suggested that nucleolin inhibits p53 translation [82], although this has not been confirmed. In contrast, there is an interaction between MDM2 and nucleolin, and data derived in vitro and in vivo suggest that nucleolin stimulates p53 activity by inhibiting its MDM2-mediated ubiquitination [83]. As would be expected, knockdown of nucleolin results in reduced levels of p53 [83]. Like several other proteins, the MDM2-nucleolin interaction also diminishes MDM2 expression (in addition to p53). This effect is observed both in the presence and absence of p53 [83].
NUMB
NUMB functions in the determination of cell fate during development. The interaction between the N-terminus of MDM2 and NUMB was discovered during a screen for MDM2-interactive partners [84], and MDM2 was subsequently demonstrated to regulate NUMB by ubiquitinating it [85]. Decreased p53 levels and increased chemoresistance in breast cancer may be due to the frequent loss of NUMB expression [86]. Loss of NUMB in human breast tumors correlates with a poorer prognosis [87].
Although MDM2 is responsible for the ubiquitination and degradation of NUMB [85], when NUMB simultaneously binds p53 and MDM2, p53 degradation is inhibited [87]. Furthermore, knockdown of NUMB in MCF10A cells enhances p53 ubiquitination, confirming the significance of NUMB in MDM2-mediated regulation of p53. In addition, MDM2 interacts with all four isoforms of NUMB [87]. Given its strong interaction with these isoforms and the association of NUMB expression with prognosis, it appears that the MDM2-NUMB complex represents a new target for therapy.
p21
While p21 has long been used as a marker of p53 (and therefore MDM2) activity, it was discovered a few years ago that MDM2 directly interacts with p21 and enhances its degradation in a p53-independent manner [88, 89]. More recent studies indicate that interaction with MDM2 is required for p21 to exert its effects on Cdk2 [90]. Further, p21 may add an additional layer of regulation to the MDM2-p53 interaction by decreasing the transcription of p53 [91]. These various activities make p21 a molecular target, the inhibition of which would be particularly effective when combined with therapies targeting MDM2 or the MDM2-p53 interaction.
p73
p73, a homologue of the tumor suppressor gene p53, is involved in tumorigenesis [92]. Although p53 and p73 have similar structures and sequences, p73 is rarely mutated in human cancers [93]. Moreover, the interaction between p73 and MDM2 does not promote the ubiquitination and proteolytic degradation of p73 [94].
The p73 gene has distinct promoters allowing the translation of several protein isoforms, two of which are opposite in effect: the full-length transactivating (TA) p73, which is pro-apoptotic, and DeltaNp73 (lacking the N-terminal transactivating domain), which is pro-survival. The binding of TAp73 to the MDM2 promoter prevents p53 from increasing MDM2 transcription, allowing increased p53 stability and activity [95]. Knocking down p73 expression by use of small interfering RNA (siRNA) decreased p53 activity, suggesting that the TAp73 activity predominates [95]. Perhaps the loss of p73-MDM2 binding allows MDM2 to interact with p53 and to promote its degradation. In agreement with this, loss of p73 expression increases malignancy in vivo, and increases resistance to ionizing radiation in a clonal model of mouse epidermal carcinogenesis [96]. However, MDM2 mediates the NEDDylation of TAp73, a process that promotes the translocation of the p73 protein to the cytoplasm and attenuates its p53-transactivation function [97]. These discrepancies warrant further examination of the p73-MDM2 interaction and its biological consequences.
PA28γ
Although PA28γ(REGγ, PSME3, Ki antigen) affects proliferation and cell cycle progression, its mechanism is not known [98]. Adding another layer to the MDM2-mediated proteasomal degradation of p53, the proteasome activator PA28γ regulates the MDM2-p53 interaction (independent of its proteasome-activator function) and serves as a necessary co-factor for p53 degradation [99]. The PA28γ protein binds to MDM2 via its unique homologue-specific insert region, which differentiates it from other PA28 family members. Moreover, in addition to specifically binding and regulating the MDM2-p53 interaction, PA28γ knockdown enhances the sensitivity of cells to MDM2 knockdown, suggesting a potential new combination therapy [99]. Further, PA28γ binds p21 to regulate its degradation in an ubiquitin-independent manner; it also binds p14/p19ARF and p16(INK4A) [100]. Since PA28γ, p21, and p14ARF are MDM2-interactive proteins, they may form a complex to enhance proteasomal degradation of the various proteins.
PCAF
MDM2 mediates proteasomal degradation of PCAF (p300/CREB-binding protein associated factor), and PCAF is a factor in the regulation of MDM2 [101]. Newly published data suggest that PCAF is involved in the reversible acetylation of p53. This acetylation stabilizes the p53 protein and prevents p53-MDM2 binding by changing the conformation of p53 [101]. In addition to having his-tone acetyltransferase activity, PCAF also serves as a ubiquitination factor with intrinsic E3 activity toward MDM2. PCAF knockdown (in HeLa and U20S cells) induces stabilization of MDM2. Thus, the three proteins may constitute a large, auto-regulatory feedback loop.
PML
PML (promyelocytic leukemia protein) is responsible for the localization of proteins to the nucleus. PML protects p53 from MDM2-mediated ubiquitination [102]; this occurs because PML sequesters MDM2 in the nucleus [103]. The mechanism responsible for this inhibitory effect is now known. CK1 (casein kinase 1) phosphorylates p53 at Thr18. In response to stress and DNA damage, CK1 accumulates in the cell and, along with p53, localizes to the PML nuclear bodies [104]. This localization induces CK-1 mediated phosphorylation, which alters the conformation of p53, thus protecting it from MDM2-mediated degradation.
pRb
The MDM2-pRb interaction modulates the inhibitory effects of the Rb protein on cell growth, preventing pRb-mediated G1 arrest. MDM2 suppresses Rb function by binding to the C-pocket of Rb via its central acidic domain and inhibits the formation of Rb-E2FDNA complexes. Rb then modulates the functions of MDM2 by forming an complex with it, altering its binding capabilities [105]. A ternary complex made up of MDM2, pRb, and p53 inhibits the degradation of p53 and consequently rescues its pro-apoptotic functions [106]. Binding of pRb with MDM2 also regulates the activity of transcription factor Sp1 [107], which has implications for muscle function. Reports have indicated that pRb and MDM2 interact via amino acid residues 273–321 [107] or that residues 254–264 are essential for this interaction [105]. MDM2 deletion mutants are unable to interact with and suppress Rb function, whereas wild-type MDM2 stimulates E2F transactivation activity and represses the growth inhibitory functions of Rb [105].
Ribosomal Proteins
Proteins from the large subunit of the ribosome, L5, L11, and L23, are established MDM2-interacting proteins [108–110]. However, unlike L5 and L23, L11 differentially regulates the levels of ubiquitinated p53 and MDM2 [109]. This L11-mediated post-ubiquitination mechanism also inhibits 26S proteasome degradation of MDM2 and extends the half-life of L11 in cells. Mycophenolic acid, an immunosuppressive agent that activates p53, requires L5 and L11 in order to exert its effect on p53 [111]. Another ribosomal protein, L26, also increases p53 synthesis by binding to p53 mRNA and protects p53 by binding to MDM2. In turn, MDM2 ubiquitinates L26 and decreases its capacity to increase p53 synthesis [112].
Our group recently discovered another novel MDM2-interacting ribosomal protein, S7. This protein, from the small subunit, was identified using yeast two-hybrid screening, and was shown to bind MDM2 in vitro and in vivo [113]. The interaction induces p53 stabilization by forming a ternary complex of MDM2-p53-S7, thus preventing the ubiquitination of p53 by MDM2. Additionally, over-expression of S7 inhibits cell proliferation, induces apoptosis, and increases p53 transactivation activity. S7 re-localizes MDM2 to the nucleolus and inhibits MDM2 auto-ubiquitination in addition to p53 ubiquitination. S7 may itself serve as an MDM2 substrate, completing the regulatory loop [114].
Tip60
HIV-1 Tat-interacting 60kDa protein, known as Tip60, is involved in DNA repair and apoptosis and is believed to participate in signal transduction [115]. MDM2 interacts with Tip60 and promotes its proteasomal degradation [116]. Whereas the MDM2 antagonist, p14ARF, selectively blocks all MDM2-mediated ubiquitin conjugation, Tip60 preferentially inhibits the MDM2-mediated conjugation of NEDD8 to p53 [117]. Upon binding, Tip60 and MDM2 re-localize to the polymorphonuclear leukocyte (PMNL) bodies, nuclear compartments that contribute to the phosphorylation and acetylation of p53. However, Tip60 mutants do not re-localize, despite the fact that they retain the ability to bind MDM2 [117]. Tip60 also apparently interacts with MDM2 and HDAC to regulate the activity and stability of the androgen receptor (AR) [118]. Further studies are needed to fully elucidate the biological consequences of this interaction.
TSG101
Tumor susceptibility gene 101, initially identified in a genetic screen of mouse fibroblasts undergoing neoplastic transformation, affects genomic stability and cell cycle regulation. This protein is tightly regulated, as both TSG101 deficiency and TSG101 overexpression can result in neoplastic transformation and the formation of metastatic tumors in nude mice [119]. TSG101 binds to MDM2 via the ubiquitin enzyme variant (UEV) domain [120]. This interaction prevents MDM2 auto-ubiquitination, thus prolonging its half-life and facilitating MDM2-mediated p53 inhibition [120]. Moreover, MDM2 elevation may correlate with increased proteolysis of both TSG101 and p53. However, this activity is controversial, as another group found no role for TSG101 in stabilizing MDM2 [121]. Nevertheless, TSG101 stabilizes p75MDM2 and p90MDM2, the two predominant MDM2 isoforms in both human and murine cells [122]. Although TSG101 prevents the degradation of both isoforms, only p90MDM2 promotes the proteolysis of TSG101 and p53. More information is needed about the TSG101-MDM2-p53 interaction and its relevance for the proteins involved.
2.2. Regulation of the MDM2-p53 Interaction with Respect to Cell Growth and Differentiation
β-Arrestins
β-Arrestins act as adapters for G-protein coupled receptors (GPCRs). GPCR signaling leads to binding between β-arrestin 2 and MDM2. This results in translocation of MDM2 to the cytoplasm, decreasing both MDM2 auto-ubiquitination and its ubiquitination of p53 [123]. Further, overexpression of β-arrestin 2 leads to increased p53 activity, and knockdown of the protein with siRNA decreases p53 protein and activity levels [123]. It appears that another β-arrestin (β-arrestin 1) also brings together MDM2 in a ternary complex with IGF-1R and that IGF-1 stimulation leads to the ubiquitination of both IGF-R1 and β-arrestin 1 by MDM2’s E3 ligase [124, 125]. While these interactions (with IGFR) affect p53, they would still be operative even in p53 null/mutant cells, implicating MDM2 in another growth pathway, and possibly presenting a new area for therapeutic intervention.
HIF-1
MDM2 has been implicated in the regulation of hypoxia inducible factor-1 (HIF1) by inducing its ubiquitination and degradation [126]. However, in studies with HCT116 cells, MDM2 overexpression results in an increase in HIF-1 expression levels and a subsequent increase in VEGF [127]. Similarly, studies conducted in renal carcinoma cells reveal that HIF-1 expression levels decrease following HDM2 knockdown via siRNA [128]. Following this knockdown, there is an increase in the levels of VEGF and PAI-1 [128]. In addition, decreased p53 expression (which frequently occurs when MDM2 is overexpressed) results in increased HIF-1 and an increase in VEGF transcription. Another study, involving “normal” astrocytes, indicated that MDM2 and p53 do not regulate HIF-1, even in response to DNA damage [129]. Further, the phosphorylation state of HIF-1 may affect MDM2 and p53, with the de-phosphorylated form of the protein leading to greater MDM2-mediated p53 degradation [130]. These findings suggest that the effects of the HIF-1-MDM2-p53 interaction may be cell-type- and cell-condition-specific. Further investigation is needed to determine their role in cancer and other diseases.
Heat Shock Proteins
Given the variety of co-factors and binding proteins with which it interacts, as well as its translocation between different cellular compartments, it is not surprising that MDM2 binds to heat shock proteins (Hsps). In fact, mutant p53 leads to the formation of a complex between MDM2-p53 and Hsp90. This interaction may inhibit the E3 ligase activity of MDM2, decreasing both MDM2 auto-ubiquitination and p53 ubiquitination [131]. The interaction, which seems to prefer mutant p53, shifts the conformation of wild-type p53 toward a more mutant-like conformation to prevent the ubiquitination of p53 [132]. In the absence of Hsp90, p53 instead interacts with Hsp70 and CHIP (carboxy terminus of Hsp70-interacting protein) to achieve similar effects [133]. Additionally, it appears that MDM2 itself may act as a type of chaperone for p53, altering its conformation, thus leading to changes in its stability [132] and activity (mediated by MDM2’s ATP binding region) [134]. Moreover, the Hsps also interact with a variety of MDM2-interactive/regulatory proteins, including the AR [135], PA28γ [136], and HIF-1α [137].
IGFR
As mentioned in the β-arrestin section above, MDM2 is involved in regulation of IGFR [138]. Decreasing expression of either p53 or MDM2 in p53 wild-type cells leads to a decrease in IGFR, but decreasing both simultaneously abrogates the effect. This suggests that wt p53 prevents MDM2 from binding to the IGFR, resulting in decreased IGFR ubiquitination and degradation [138]. β-Arrestin is apparently also involved in this process [124, 125]. Moreover, a new study demonstrates that IGF signaling likely regulates p53 transcriptional activity [139]. These findings suggest that the IGFR-MDM2 interaction is associated with p53 regulation.
Nuclear Receptors (AR, ER, GR)
Interactions between MDM2 and the androgen, estrogen, and glucocorticoid receptors have been suggested [140, 141]. For example, MDM2 is necessary for efficient ubiquitination of the AR; it also acts in opposition to the Tip60 co-factor to decrease the transcription factor activity of the AR [118, 142]. Further, the expression level of p53 also regulates AR stability and sensitivity, with decreases in p53 leading to decreases in the AR [143]. Similarly, the glucocorticoid receptor (GR), p53, and MDM2 form a ternary complex that leads to the ubiquitination of the GR and p53 [144]. Despite their homology, p73 does not cause a similar effect [92]. However, interactions between the GR, the estrogen receptor (ER), and MDM2 lead to an increase in GR ubiquitination [145].
MDM2 forms a ternary complex with ERα and p53, and estrogen signaling impacts p53 stability and activity [146]. Apparently, the interaction with the ER contributes to decreased MDM2-mediated p53 ubiquitination, and MDM2-ER binding leads to the degradation of the ER, independent of estrogen exposure [147]. Moreover, p53 induction by DNA damage leads to a decrease in MDM2-mediated ER turnover, suggesting that p53 enhances the effects of MDM2 on the ER. The various nuclear receptor complexes and their interactions with MDM2 and p53 need to be investigated further. These could present novel targets for cancer therapy, especially for breast and prostate cancers.
TGF-β Signaling
The regulatory role of transforming growth factor beta (TGF-β) on apoptosis (via the SMAD or Daxx pathways), cell cycle progression, proliferation, and cell differentiation processes is well-established [148]. MDM2 may confer TGF-β resistance (overcoming G1 arrest) by directly interacting with the cellular machinery involved in cell cycle arrest. This resistance is associated with dys-regulated cell growth and tumor progression [149, 150]. Nevertheless, other studies suggest that the regulation of TGF-β signaling by MDM2 may be cell-specific [151]. Recent functional studies in mink lung epithelial and human mammary cells have identified three structural elements that are essential for MDM2-mediated TGF-β resistance: the C-terminal end of the p53-binding domain, the NLS, and the RING finger domain of MDM2 [152]. These regions of MDM2, while not yet confirmed as having a functional interaction with TGF-β, may represent potential targets for inhibiting resistance.
Other Interactive Proteins
Numerous other proteins also interact with p53 and/or MDM2 to affect their interaction, stability, and activities. For example, among the proteins that increase the stability or activity of MDM2 are AAA ATPase p97 (VCP, CDC48, UBX-domain-containing adaptor) [153], G3BP2 (Ras-GTPase activating protein-SH3-domain-binding protein 2) [154], Nbs1 [155], nucleostemin [156], Plk1 [157], USP2a (ubiquitin-specific protease 2a) [158], Wip1 phosphatase (PPM1D) [159], and YY1 [160]. Similarly, PACT (p53-associated cellular protein-testes derived, P2P-R, RBBP6) interacts with both p53 and MDM2 to cause an increase in their binding affinity (and subsequent degradation of p53) [161]. Other proteins, including IFIXalpha1 hematopoeitic interferon (IFN)-inducible nuclear protein [162], Merlin [163], and p300 [164] decrease MDM2 activity and increase p53. (Fig. 1) depicts various proteins that regulate MDM2 and the MDM2-p53 interaction.
Fig. (1).

MDM2 interactive proteins and their localization.
3. TARGETING THE MDM2-p53 INTERACTION FOR THERAPY
In view of its numerous activities, MDM2 has been implicated in a variety of other diseases in addition to cancer. Among these are diabetes, cardiovascular disease, and various infectious diseases [165–167]. As would be expected, the MDM2-p53 interaction is a target for therapeutic intervention, particularly for cancer. Since increasing the expression and activity of wild-type p53 is the ultimate goal, a major push toward p53 gene therapy has been underway for many years [168, 169]. Although the overall enthusiasm for gene therapy has subsided following several highly publicized failures, it still offers a way to induce long-lasting expression of p53. Other investigators have proposed the use of vaccines against mutant p53 [170, 171] in the hope that any remaining wild-type protein will have enough function to prevent tumor formation and sensitize tumors to chemotherapy and radiation. Still others have generated small molecules or antibodies that bind to mutant p53 to restore normal conformation and/or activity (e.g., ellipticine) [172, 173].
Although the agents aimed at increasing wild-type p53 expression or restoring p53 function to mutants have demonstrated utility in pre-clinical and clinical trials, none has addressed the root problem associated with many cancers and other diseases: MDM2 over-expression. Even if cells express wild-type p53 or have functional p53 restored, if MDM2 is overexpressed, it will outweigh the benefits of such therapies. For these reasons, numerous other molecules have been developed to target either MDM2 itself or the MDM2-p53 interaction.
3.1. Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) were among the first compounds used to inhibit MDM2 [174]. The antisense strategy has a fairly long history, and ASOs have been used to target a variety of cancer-related molecules, including oncogenes [175, 176]. Pre-clinical studies of ASOs have demonstrated that they inhibit the expression of a variety of gene products and produce anti-tumor effects in cancer models. There have been numerous clinical trials of ASOs, several involving advanced (phase II/III) trials [177].
The ASOs are theorized to work via several mechanisms [178, 179], but most of the anti-MDM2 ASOs likely exert their effects via translation arrest (interfering with protein synthesis from mRNA) or RNase-mediated mRNA degradation. In brief, cells are transfected with the nucleotides along with a cationic lipid, or the ASOs are administered to animals (typically “naked”), where they enter the cytoplasm of cells and interact with complementary MDM2 mRNA [178, 179]. This interaction prevents the formation of the protein product, decreasing expression of MDM2.
Decreasing MDM2 expression by exposure of cultured cells to ASOs leads to decreased cell survival and proliferation, increased apoptosis, and cell cycle arrest [140, 180, 181]. Moreover, it also sensitizes cells to treatment with a variety of conventional anti-cancer agents, including hormones, radiation, and chemotherapeutics [182, 183]. Accompanying the decrease in MDM2 expression is an increase and/or stabilization of wild-type p53 protein, increased Bax and p21, and decreased E2F1 and Bcl-2 expression [140, 183].
Similar effects are noted when MDM2 is knocked down by ASOs in tumor models in vivo. In animal cancer models, administration of anti-MDM2 ASOs leads to decreased tumor growth, increased animal survival, increased sensitivity to conventional cancer therapies (radiation and various chemotherapeutic agents) and a reversal, to some extent, of the androgen-independent phenotype [183]. The changes in protein expression are also found in vivo [184]. These results suggest that the anti-MDM2 ASOs represent an effective, novel anti-cancer strategy. However, given the relatively poor performance of ASOs in the clinic (only a single ASO has been approved by the FDA for clinical use), research into these molecules is barely progressing. No anti-MDM2 ASOs have been examined in the clinic, but it is possible that, if better ways to evaluate ASOs clinically are determined or if there is an increased interest in ASO-based therapies, these molecules may undergo further development.
3.2. Other Gene Silencing Strategies
Other nucleic-acid-based therapies, including use of ribozymes and RNAi, are also being explored. Ribozymes are catalytic molecules based on intron-splicing enzymes of Tetrahymena [185]. The enzymes have been modified to splice exogenous sequences, and, like ASOs, have been used to decrease expression of a variety of oncogenes and other targets [186, 187]. Ribozymes act by binding and catalytically degrading complementary mRNA, preventing expression of the target protein. Therefore, while they act by different mechanisms, ribozymes and ASOs produce the same end result.
An anti-MDM2 ribozyme led to decreased proliferation and increased apoptosis of cells, although this agent was apparently not evaluated in animals [188]. Given their catalytic nature, ribozymes are more difficult to design and synthesize than ASOs, which is perhaps responsible for the limited number of studies. Additionally, like ASOs, ribozymes have largely fallen out of favor as therapeutic agents. Although there were a few recent clinical studies of ribozymes, none are currently active [177].
The newer gene silencing strategy, RNAi, is being used for similar purposes as the ASOs and ribozymes. Like these strategies, RNAi makes use of short sequences of nucleotides to bring about enzymatic destruction of the target mRNA sequence. However, in the case of RNAi (small interfering RNA-siRNA, short hairpin RNA-shRNA or micro RNA-miRNA), double-stranded sequences of RNA enter the RISC (RNA-induced silencing complex) enzyme complex, where the RNA binds to complementary mRNA, leading to the destruction of the target.
Studies have primarily made use of RNAi-mediated knockdown of MDM2 to examine its activities and to evaluate novel regulatory mechanisms [189, 190]. However, exposure to RNAi leads to similar effects on proliferation, survival, apoptosis, and cell cycle progression as were observed following treatment with ASOs or ribozymes targeting MDM2 [191, 192].
Each of the gene silencing strategies has its advantages and disadvantages [187], but none has produced an MDM2-targeting agent for clinical trials. Nevertheless, numerous agents from each class have been examined, and several, targeting other molecules, are in current clinical trials. As the information about MDM2 becomes clearer, there may eventually be a gene silencing strategy aimed toward reduction of MDM2 in humans.
3.3. Small Molecule Inhibitors
Numerous other rationally designed agents specifically targeting MDM2’s interaction with p53 or its capacity to mediate p53 degradation are in development. While the gene silencing strategies have demonstrated some efficacy, their development is essentially static. The various small molecule inhibitors promise better specificity, greater efficacy, and fewer off-target effects than the gene silencing strategies or p53-increasing strategies. Nevertheless, the rationally designed small molecules described below have had varying degrees of success, leaving room for improvement.
Nutlins
Perhaps the best-known small-molecule MDM2 inhibitors are the nutlins, which were developed by Roche. The compounds were synthesized following extensive analysis of the MDM2 crystal structure, which indicated the presence of a hydrophobic pocket, which binds a peptide simulating the transactivational domain of p53 [193]. A library screening yielded hits with nutlins, which are cis-imidazole derivatives. Nutlins exert their activity by binding to MDM2, releasing p53, and preventing the binding between the two proteins, resulting in decreased degradation of p53.
The nutlins (particularly nutlin-3) have potent activities against cancer cells. Not surprisingly, given the frequency of MDM2 over-expression, a variety of cancer types are sensitive to the nutlins. For example, exposure to nutlin-3 led to an increase in p53 and subsequent apoptosis in acute myelogenous leukemia cells and increased their sensitivity to chemotherapy [194]. Similar effects were noted in multiple myeloma cells [195]. Nutlin-3 also leads to enhanced sensitivity to radiation [196]. Nutlin-3 causes apoptosis of cancer cells, but it does not have the same effect in normal hematopoeitic progenitor cells [194]. Although they exert some activity in cells lacking p53 or with mutant p53, the nutlins are most effective in p53 wild-type cells. Although nutlin-3 is not being developed clinically due to its poor “drug-like” properties, its activity demonstrated proof-of-principle that protein-protein interactions, including that between MDM2 and p53, can be inhibited, at least in vitro.
Other Small Molecule Inhibitors
To prevent the binding of p53 and MDM2, a variety of other peptide-based and non-peptidic agents have been designed, again based upon crystal structures and computer modeling of the interaction. With their physical properties and the associated difficulties in delivering them to target cells, peptide-based mimics are relatively poor drug candidates, even though they demonstrate anti-proliferative and pro-apoptotic effects in human cancer cell lines [197, 198]. Better delivery methods (e.g., encapsulation in nanoparticles) might improve the outlook for such molecules.
Following library screens or structural chemistry studies, numerous non-peptide compounds have been generated to target the MDM2-p53 interaction. For example, compounds based on a terphenyl scaffold were synthesized following computational modeling to mimic the face of an α-helical peptide [199]. These compounds inhibit MDM2-p53 binding and increase p53 expression and activity in cancer cells [200]. Similarly, a series of 2-alkyl-3-aryl-3-alkoxyisoindolinone-based compounds increases p53 transcriptional activity in SJSA cells, as evidenced by increased MDM2 and p21 [201]. A spiro-oxindole (MI-63) decreases the growth of LNCaP cells and leads to increases in p53, MDM2, and p21 protein [202]. Yet another class of compounds, benzodiazepine diones (specifically TDP521252 and TDP665759), increase p53 activity, and decrease the growth of both cancer cells in vitro and xenograft tumors [203, 204].
While most of the compounds mentioned above function by mimicking Phe19, Trp23, and Leu26 of p53 in order to compete for MDM2 binding, another set of compounds (HLI98L-E) was designed to target the RING finger domain of MDM2 to prevent its E3 ligase activity [205]. The HLI98 compounds increase expression of both p53 and MDM2 in fibroblasts, but demonstrate off-target/non-specific effects, decreasing interest in their further development due to predicted high toxicity to host cells [205]. A screen of more than 100,000 natural product extracts identified sempervirine as another inhibitor of the E3 ligase of MDM2. In p53 wild-type cells, the compound leads to inhibition of the ubiquitination of p53 and MDM2, accumulation of p53, and apoptosis, apparently with better specificity for MDM2 than the HLI98 compounds [206]. Despite their promising activity, none of these small molecule compounds have been examined in human clinical trials, and many have not progressed even to animal studies. This is due in part to their non-specific cytotoxicity and the lack of successful delivery of the compounds to target tissues in animals. More work is needed to improve the solubility, stability, delivery, bioavailability, and safety of molecules that inhibit the MDM2-p53 interaction.
Natural Product Inhibitors of MDM2
A variety of compounds with anti-cancer activity (with previously uncharacterized mechanisms) may act via inhibition of MDM2. In particular, natural compounds may be a source of novel MDM2 inhibitors (as they were for sempervirine). For example, another series of compounds developed by Roche, the chalcones (1,3-diphenyl-2-propen-1-ones), derived from compounds found in licorice, also interfere with the interaction between MDM2 and p53 [207]. While the original chalcones are too toxic to be considered for further development and have relatively weak binding to MDM2, boronic chalcone derivatives exhibit potent activity against human breast cancer cells with lower toxicity to normal human breast epithelial cells [208]. These agents may exert their effects through non-MDM2 mechanisms of action, including inhibition of the proteasome [209]. While an agent having more than one type of anti-cancer activity may be attractive, a well-defined (and solitary) mechanism of action is generally necessary for an agent to move into clinical trials.
Chlorofusin, a natural product derived from a fungus (fusarium species), also inhibits the MDM2-p53 interaction due to direct binding to the MDM2 N-terminal [210, 211]. Synthetic portions of the compound are not effective inhibitors of MDM2 [212, 213], but there is contention about the stereochemistry of the molecule. Further studies are needed to determine the activity of the compound and its derivatives. Still another natural compound, apigenin, also inhibits MDM2 expression, and this activity (along with inhibition of HIF1 and other molecules) is postulated to be responsible for the decrease in angiogenesis following its administration [214].
In our laboratory, we have seen that several chemopreventive and therapeutic natural products, including genistein, curcumin, and ginsenosides [10, 11, 215, 216] inhibit MDM2 expression at the mRNA and/or protein level. While the mechanisms by which the downregulation occurs differ, with genistein affecting the NFAT transcription site on the MDM2 promoter [10], curcumin acting on MDM2 transcription via the PI3K/mTOR/ETS2 pathway [11], and the ginsenosides acting by yet-uncharacterized mechanisms [215, 216], all lead to significant decreases in MDM2 expression, increased cellular apoptosis, and decreased proliferation. All are also effective in vivo, inhibiting tumor growth. It is possible, however, that these compounds exert their effects via other mechanisms of action (e.g., inhibition of tyrosine kinases in the case of genistein and downregulation of the AR in the case of ginsenosides) [215]. Thus, further investigations are needed to determine the efficacy, safety, and primary mechanisms of action of the compounds for treating MDM2-related cancer and other diseases. Table 2 shows the structures of various small molecule inhibitors of MDM2.
Table 2.
Structures of the Small-Molecule Inhibitors of MDM2 and the MDM2-p53 Interaction
| Compound | Structure | Target | Ref. |
|---|---|---|---|
| Nutlin-3 | 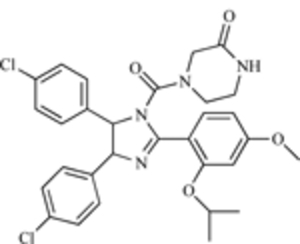 |
MDM2-p53 Interaction | [193] |
| Terphenyl helix mimetic (core structure) |  |
MDM2-p53 Interaction | [199] |
| Isoindolinone (core structure) |  |
MDM2-p53 nteraction | [201] |
| Spiro-oxindole (MI-63) |  |
MDM2-p53 Interaction | [202] |
| 1,4-benzodiazepin −2,5-dione | 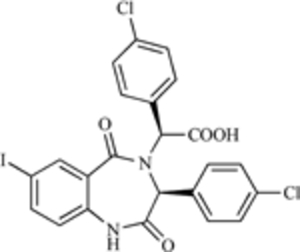 |
MDM2-p53 Interaction | [203] |
| HLI98C | 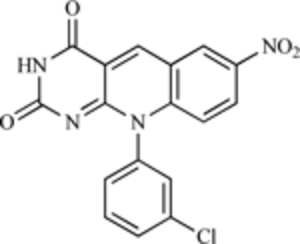 |
E3 Ubiquitin Ligase | [205] |
| Sempervirine | 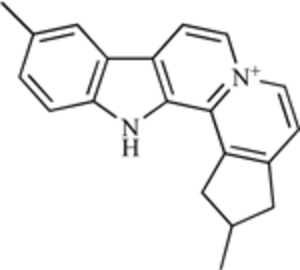 |
E3 Ubiquitin Ligase | [206] |
| Chalcone (generic core structure) | 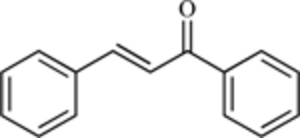 |
MDM2-p53 Interaction | [207] |
| Cyclic peptide derived from chlorofusin | 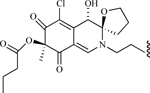 |
MDM2-p53 Interaction | [213] |
| Apigenin | 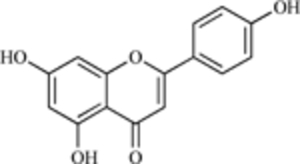 |
MDM2 Expression | [214] |
| Genistein | 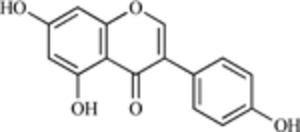 |
MDM2 expression | [10] |
| Curcumin | 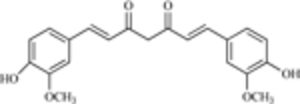 |
MDM2 expression | [11] |
| 25-OCH3-PPD (a ginsenoside) | 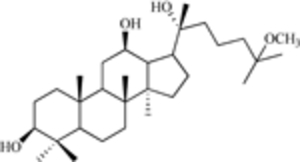 |
MDM2 expression | [15] |
| 25-OH-PPD (a ginsenoside) | 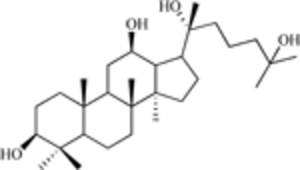 |
MDM2 expression | [16] |
4. FURTHER CHARACTERIZATION OF MDM2
4.1. Evidence of p53-Independent Effects of MDM2
Increasing evidence suggests that MDM2 has p53-independent activities, including p53-independent oncogenic effects. For example, when MDM2 is specifically targeted (via a K14 promoter) to the epidermis in both normal and p53-null mice, they develop lesions that frequently progress to squamous cell carcinomas, indicating that the MDM2-mediated development of cancer occurs in a p53-independent manner [217]. This theory is supported by the existence of numerous naturally occurring MDM2 variants, some of which lack the p53-binding domain but still transform NIH3T3 cells [218]. MDM2 regulates muscle differentiation via interactions with Sp1 and Rb in a p53-independent manner [107]. In addition, numerous other proteins, including several discussed above for their p53-dependent effects, interact with MDM2 independently of p53 to achieve oncogenic effects.
4.2. p53-Independent Protein-Protein Interactions with MDM2
Our laboratory has previously reviewed numerous proteins that interact with MDM2 in the absence of p53 [140]. These include many of those described above (which have additional activities in the absence of p53), as well as several not discussed in this review, including DNA polymerase ε, NF-κB, PSD-95, and the β-adrenergic receptor [140].
Recent investigations have brought to light numerous p53-independent functions and interactions for several other MDM2 interactive proteins. For example, while the effects of the 14-3-3-σ-MDM2 interaction are important for p53, it appears that this interaction also determines the stability or activity of other proteins, including Rb. MDM2 also regulates JMY independent of its p53-binding domain and the p53 status of cells [65]. This is in contrast to previous studies suggesting that the major function of JMY is to regulate the p53 response.
The E2F transcription factor 1 (E2F1) regulates cell cycle progression, DNA synthesis, and the activity of tumor suppressor proteins. As mentioned under CSN5 in the MDM2-p53 regulation section above, MDM2 stabilizes E2F1 by directly binding to the protein and preventing E2F1-mediated cell cycle arrest and apoptosis [53]. This occurs because MDM2 prevents the interaction between E2F1 and SCF (SKP2), the E2F1 E3 ligase necessary for E2F1 ubiquitination [53, 219]. Although the MDM2 NLS overlaps the binding domain of E2F1, deletion of the MDM2 NLS does not reduce E2F1 binding and stabilization.
As demonstrated in other studies, MDM2 promotes cell transformation and inhibits pRb activity via ubiquitination in a p53-independent manner [220]. Furthermore, small-cell lung cancers that overexpress MDM2 exhibit a correlative decrease in pRb expression, regardless of their p53 status.
Co-immunoprecipitation of endogenous HIF-1 and MDM2 in lysates of hypoxic HCT116 cells (p53WT and p53 −/−) suggest that the association between MDM2 and HIF-1 is also p53-independent. More studies are needed to clarify the underlying mechanism and physiological significance of the MDM2-HIF-1 interaction, but since hypoxia is a frequent condition within tumors, particularly large tumors, the interaction will likely have implications for cancer therapy.
While the ribosomal proteins may function as sensors of biogenic stress that activate p53, the L11 ribosomal protein is believed to promote the accumulation of ubiquitinated and native MDM2 in a p53-independent manner [109]. p14 also interacts with MDM2 independent of p53 to allow a response to DNA damage [221]. These findings indicate that, in the absence of functional p53, cellular stress still elicits a response that affects cell cycle progression.
It is likely that many proteins that interact with MDM2 and/or regulate the MDM2-p53 interaction are yet to be discovered. Both MDM2 and p53 are central to diverse biological processes, including embryonic development and the response to cytotoxic stresses. (Fig. 2) provides a summary of the numerous MDM2-interactive proteins (p53-dependent and –independent) and their primary activities. The interactions of these proteins shed new light on how MDM2 is regulated, how MDM2 functions in various processes under different conditions, and provide information relative to the tumorigenesis process. However, this multitude of proteins also adds to the confusion surrounding the regulation of MDM2 and p53 and indicates that extensive work that must be accomplished to ascertain their functional relevance.
Fig. (2).

The numerous p53-dependent and –independent MDM2 interactive proteins and their general functions.
4.3. p53-Independent Regulation of MDM2
MDM2 Transcription
Although MDM2 regulates the activity and stability of a variety of proteins (and is regulated by a variety of proteins) independent of its interaction with p53, the transcription of MDM2 can be p53-independent. For example, PTEN both decreases MDM2 stability and decreases its transcription [222]. MDM2 has two promoters (P1 and P2) that regulate its transcription. While p53 activates MDM2 transcription by binding to P2, leading to transcription of a short MDM2 lacking exon 1 (p75), activation of P1 activates transcription of a longer p90 MDM2 transcript, which lacks exon 2, in [223]. PTEN disrupts activation of the P1 promoter, thereby decreasing MDM2 expression, independent of p53 [222]. In contrast, the p72 RNA helicase cooperates with PCAF and CBP/p300 to enhance both p53-dependent and –independent transcription of MDM2 [224].
To increase MDM2 expression, the ER increases p53-dependent transcription of MDM2 and binds to the MDM2 promoter itself in a p53-independent manner [145]. MDM2 is also a target of MYCN, and its induction of MDM2 transcription leads to down-regulation of p53 [225]. The Fli-1 Ets transcription factor acts in a similar manner, increasing its transcription and further decreasing p53 stability and activity [226]. The Ras-Raf-MEK-MAPK pathway also increases MDM2 transcription [227], and MEK-mediated phosphorylation of MDM2 enhances its export from the nucleus [228].
Polymorphisms in the MDM2 Promoter
There are also MDM2 mutations that affect its expression, but a complete discussion of this topic warrants a full-length review. However, in brief, several SNPs (single nucleotide polymorphisms) of MDM2 have been identified, the most important being SNP309/G2580T [229]. This SNP increases MDM2 transcription by Sp1, which subsequently leads to increased degradation of p53 by MDM2 [229]. While some studies have suggested that SNP309 increases the risk of developing cancer and for developing more aggressive cancer [230–232], its impact is still controversial, with other studies finding no effect of SNP309 on cancer risk or aggressiveness [233, 234]. It is possible that the effects are cell/tissue-specific and may depend on additional factors, such as hormone status [231] or mutations to other genes [232]. More intensive studies of the different mutations and their effects on different cells/tissues under diverse conditions are needed.
MDM2 Post-Translational Modifications
Again, a full description of the number and functions of the various known MDM2 post-translational modifications would constitute a review in itself. However, it bears mentioning that there are numerous MDM2 post-translational modifications that alter its stability, activity, localization, and interactions with other proteins. These modifications, including ubiquitination, phosphorylation, acetylation, and SUMOylation, are mediated by diverse proteins [30]. The effects of MDM2 ubiquitination and the resulting activity against p53 are known [235]; other modifications affect MDM2 stability and activity. For example, cyclin G interacts with both PP2A and MDM2, leading to de-phosphorylation of MDM2 on T216/S166 (murine/human), increasing MDM2’s binding with p53 (and subsequently its ubiquitination of p53) [236]. In fact, it appears that MDM2 must be hyper-phosphorylated in order to down-regulate p53 expression efficiently. Regarding the activity of various mutants of MDM2 (Ser-Ala), the non-phosphorylated proteins retain E3 ligase activity but do not lead to efficient p53 degradation [237].
Further indicating that MDM2 phosphorylation controls its regulation of p53, irradiation leads to lower levels of hyper-phosphorylated MDM2 and greater p53 stability [237]. In contrast, phosphorylation of MDM2 by Akt inhibits HAUSP-mediated MDM2 de-ubiquitination. This results in a decrease in MDM2 stability and a subsequent increase in p53 stability [238]. Other enzymes, such as CK2, PI3K/Akt, and DNA PK, as well as members of the Ras-Raf-MEK-MAPK pathway, also regulate MDM2 phosphorylation [30]. Thus, the affected MDM2 residues and the localization of the protein when it is phosphorylated/de-phosphorylated determine its activity and stability.
MDM2 can be SUMOylated within its RING finger domain. This leads to a decrease in MDM2 auto-ubiquitination but to an increase in the ubiquitination of p53 [239]. MDM2 is SUMOylated within its NLS via RanBP2 and PIASxβ/PIAS1 [240]. The ARF (p14/p19) protein also leads to increased SUMOylation of MDM2, independent of p53 [241]. While the SUMOylation of MDM2 by ARF does not appear to affect the p53-MDM2 loop, it may affect the p53-independent activities of MDM2 [241].
Acetylation of MDM2 also occurs. Both p300 and CEBP lead to acetylation of MDM2’s RING finger domain, decreasing its activity against both p53 and itself [242]. It is likely that there are also other acetylation sites and that acetylation of p53 is necessary for its interaction with MDM2 [243]. Targeting these regulators of MDM2 may be useful for restoring p53, decreasing the oncogenic activity of MDM2 in p53 null/mutant cells, or for modifying the activities of the various proteins to accomplish desired cellular effects.
5. THE NEXT CHAPTER: TARGETING THE p53-INDEPENDENT ACTIVITIES OF MDM2
Although none of the existing anti-MDM2 or anti-MDM2/p53 agents were developed to target specifically the p53-independent activities of MDM2, several of these would be effective for this purpose. In fact, ASOs and RNAi were used to discover many of the p53-independent interactions [e.g., see 53, 89].
The gene silencing strategies (ASOs, RNAi, and ribozymes) can be used to target MDM2 specifically, regardless of the cell type or status. Silencing of MDM2 expression leads to an increase in p53 expression and activity and decreases tumor growth in p53-null models [184].
Although the nutlins were rationally designed to target the MDM2-p53 interaction, their effects (even in p53 wild-type cells) may depend on other cellular factors. For example, the outcome of treatment with nutlin-3 (cell cycle arrest or apoptosis) depends on expression of Rb [244]. Since MDM2 interacts with both E2F1 and Rb regardless of the p53 status of cells, this reflects anti-tumor effects of an interaction with MDM2 itself. Nutlins may also disrupt the interaction of MDM2 with other molecules that bind within or near the p53-binding domain.
The natural product inhibitors of MDM2 could target the p53-independent activities of MDM2. Their mechanisms need to be more clearly defined before conclusions are made, but the compounds (sempervirine, apigenin, genistein, curcumin, ginsenosides) appear to decrease MDM2 expression and/or activity in animal models, suggesting that they could be effective therapeutic or preventive agents.
It is likely that targeting individual interactive molecules or the interaction of MDM2 with specific co-factors or regulators (dependent or independent of p53) will be an effective strategy. However, more research is needed to elucidate the role of each of these interactions, and to define the circumstances under which the interaction can be successfully targeted.
6. MOVING MDM2 INHIBITION TO THE CLINIC
6.1. Problems with the Current Generation of MDM2 Inhibitors
Although there are numerous agents in pre-clinical development, no anti-MDM2 compounds are being examined in clinical trials. There have been several trials of agents targeting p53, and many p53-targeting strategies are being clinically investigated, including an anti-p53 ASO and an adenoviral vector for delivery of wild-type p53 [168, 177, 245]. The lack of MDM2-targeting agents in clinical trials can be partially explained by the pace of research. The anti-MDM2 ASOs were developed in the mid-late 1990’s; most of the small molecule inhibitors and other anti-MDM2 molecules are more recent discoveries. It is the authors’ opinion that MDM2 is a valid target for therapy and that an agent targeting MDM2 will soon be evaluated in a clinical trial. Table 3 summarizes the current approaches being used to target MDM2 and the MDM2-p53 interaction and suggests areas that could be used for future investigations.
Table 3.
Summary of Existing Approaches Targeting MDM2 or the MDM2-p53 Interaction, and Potential Areas for Future Development
| Target | Existing Agents | Current approach | Potential approaches | References |
|---|---|---|---|---|
| MDM2-p53 interaction | Nutlins, terphenyl helix mimetics, isoindolinones, spiro-oxindoles, benzodiazepine diones, chalcones, chlorofusin | Small molecule inhibitors, natural products | Other small molecules (different specificity, lower toxicity to normal cells) | [193–196, 199–204, 207–213] |
| MDM2 E3 ligase | HLI98 compounds, sempervirine | Small molecule inhibitors, natural products | Novel small molecules, peptide mimetics (eg: Arf or HAUSP mimetics) or agents that alter the expression/activity of MDMX | [205, 206] |
| MDM2 expression | ASOs, Ribozymes, siRNA, apigenin, genistein, curcumin, ginsenosides | Gene silencing, natural products | New natural products or gene silencing agents, aptamers | [10, 11, 140, 180–184, 188–192, 214–216] |
| MDM2 splicing variants | MDM2 ASOs (maybe), siRNA | Gene silencing, possibly natural products (this has not been evaluated) | Variant-specific gene silencing, variant-specific natural products, gene therapy to “repair” the variant (eg: a trans-splicing ribozyme) | [140, 180–184, 189–192] |
| MDM2 SNPs | MDM2 ASOs (maybe), siRNA | Gene silencing, possibly natural products (this has not been evaluated) | SNP-specific gene silencing, SNP-specific natural products, gene therapy | [140, 180–184, 189–192] |
| MDM2 post-translational modifications | No current agents | No current approaches | Kinase inhibitors (eg: Deguelin, PD98059), enzyme inhibitors (for other processes-ubiquitination, acetylation, SUMOylation), peptide mimetics to act as substrates | n/a |
| Other MDM2-interactive proteins | ASOs, siRNA | Gene silencing | Small molecule inhibitors, gene silencing, peptide mimetics | [140, 180–184, 189–192] |
In order for an MDM2-targeting agent to enter human trials, several obstacles must be overcome. First, as discussed above, almost all of the existing agents specifically target the MDM2-p53 interaction. While the interaction is a valid target for cancer therapy, p53 is mutated or lacking in approximately half of human cancers [246], suggesting that these molecules may not be effective for tumors lacking p53 or with mutant p53. Moreover, a recent study indicated that the MDM2-p53 interaction is less important than its E3 ligase activity toward p53 (at least for development), indicating that the development of the complex protein-protein interaction inhibitors may be unnecessary [247]
For the various molecules targeting the MDM2-p53 interaction, there are a number of problems, including non-specific cytotoxicity (no difference in the toxicity to cancer versus normal cells). Modifications to the chemical structures, particularly the boron derivatization of the chalcones, have decreased the toxicity of some of the agents but improvements are still needed [248, 208]. Moreover, long-term toxicity studies of MDM2 inhibition in animal models (longer than a few weeks) are yet to be accomplished. Since complete loss of MDM2 in transgenic mice results in embryonic death in p53wt animals [249, 250], the effects of MDM2 inhibition on animal survival, cellular repair/recovery, and reproduction need to be assessed. In particular, the effects of the various molecules on MDM2 and p53-related proteins (MDMX, p63, p73, other E3 ligases) as well as the other MDM2-interactive proteins (p53-related and –independent) need to be examined.
Another factor is that MDM2 shuttles between the nucleus and the cytoplasm, and apparently has functions in both locations. Thus, delivery to appropriate sites is a concern for successful use of novel agents. Depending on which function or interaction is being targeted, it may be necessary to ensure nuclear delivery of the agent or to specifically target the cytoplasmic protein or modifications made to MDM2 in the cytoplasm. The gene silencing strategies would be useful for inhibiting the activities of MDM2 in both locations, but small molecules that cannot penetrate the nucleus will be effective for inhibiting only the cytoplasmic MDM2.
The successful translation of MDM2-targeting agents to human clinical trials will face the same problems as most new agents specificity, toxicity, delivery, and activity. Pre-clinical studies in animal cancer models have demonstrated that many of the agents are safe and active and that they can be delivered to tumors. However, due to the wide array of p53-independent activities of MDM2, more work is needed to develop a successful MDM2 inhibitor for use against human cancers. Nevertheless, other specific and rationally targeted drugs (Herceptin®, Gleevec®) have been successful; the future for MDM2 could be equally bright.
6.2. Combination Therapy with Agents Targeting MDM2
Although animal studies suggest that agents targeting MDM2 may be effective when used as monotherapy, combining MDM2-targeted agents with other anti-cancer agents or approaches may be a better course of action. In our studies with anti-MDM2 ASOs, there were increases in anti-cancer activity when the ASOs were used in combination with various chemotherapeutic agents or radiation therapy in animal models of cancer [182, 183]. Other agents (targeting either MDM2 alone or targeting the MDM2-p53 protein-protein interaction) have similar effects [208, 251, 252]. It is noteworthy that combination therapy is used in the clinic to overcome intrinsic and acquired resistance of tumors to single agents.
As mentioned above, with the large number of MDM2-interactive proteins being discovered, it may be beneficial to target specific MDM2-interactive proteins or to target both the MDM2-p53 interaction and another MDM2-protein interaction. Targeting two or more molecules or protein-protein interactions simultaneously could be used to tailor therapy to a specific cancer or to ensure more complete inhibition of a particular part of the p53 pathway. More work is needed to define the optimal combinations. The number of MDM2-p53 interactive proteins will likely be large. Studies should now focus on determining the core proteins involved in regulating the MDM2-p53 interaction. Preliminary studies indicate that dual targeting of MDM2 and known MDM2-interactive proteins improves the therapeutic outcome. For example, targeting both MDM2 and PA28γ (a regulator of the MDM2-p53 interaction) by an ASO and siRNA, respectively, increases the destruction of cancer cells and their sensitivity to radiation [99]. Similarly, targeting both the p53-dependent and p53-independent effects of MDM2 should increase the therapeutic response by affecting cells with both wild-type and mutant/null p53 phenotypes.
7. FUTURE DIRECTIONS
7.1. Areas that Need Further Investigation
Despite the many findings reviewed here, it is apparent that our current understanding of MDM2 is not sufficient. In addition to the large number of interactive proteins, links between them are also being established. While early investigations identifed proteins that bound MDM2, their complex interactions have not been sufficiently investigated. As an example, although E3 ligase activity of MDM2 was established early in the 1990’s, it was not known until 2004 that it could also mediate ubiquitin-independent proteasomal degradation of proteins [89]. Numerous interactive proteins that link MDM2 to the proteasome have now been identified; these include p53, gankyrin, MDMX, 14-3-3-σ, Rb, p21, PA28-γ, β-arrestins (1 & 2), and the IGFR. The interactions go beyond ubiquitination of proteins to target them for degradation. Links between some of these proteins have been established, but more research is needed to determine how and under what circumstances these pathways are involved.
Further investigation of MDM2 splicing variants is needed. More than forty alternative or aberrant splicing variants of MDM2 have been identified; most of these are localized in the nucleus [253]. The activities of the different variants are being investigated; several, including MDM2-B, are hypothesized to exert oncogenic effects [254]. Since most of the variants lack the p53-binding domain, their oncogenic effects are apparently p53-independent. Despite some reports of oncogenic activity, the functions of the variants (particularly MDM2-B) remain controversial [255]. Like other activities of MDM2, the effects of the variants may be cell-type dependent. Nevertheless, the expression of variants that may or may not be affected by MDM2-targeting therapies should be taken into account.
Perhaps the most important investigations will be those that determine the effects of various challenges and stressors on MDM2 expression and those that determine how different cell types regulate this process. With more advanced molecular biology techniques and more powerful computers for data analysis, it should eventually be possible to model particular cells, organs, and organisms in order to predict the effects of various conditions. These results can then be used to achieve specific therapeutic outcomes, for example, to protect patients’ normal cells from the harmful effects of radiation, while enhancing the effects on tumors. These large-scale studies in silico are not yet possible, making it difficult to determine the number of p53-MDM2 interactive complexes. Research should now focus on determining which molecules are most important under different circumstances (such as development, carcinogenesis, and metastasis) and on evaluating these molecules as targets for therapy.
7.2. Other Properties of MDM2 that can be Targeted
MDM2 has several domains that can be targets for therapy, including its p53-binding domain, its NLS, its NES, and its C-terminal RING finger domain. The post-translational modifications and splicing variants of MDM2 may also present novel objectives for inhibiting MDM2. The p53-binding domain has been extensively targeted, and a few agents have focused on the E3 ligase activity of the RING finger domain. Other regions of MDM2, including the binding domains for other proteins as well as its post-translational modifications and its variants, and have not been considered. Further, the numerous MDM2-interactive (p53-dependent and –independent) proteins can also be targeted to improve the therapeutic response.
Although much of the focus on cancer is directed toward designing new therapeutic agents and improving the response to conventional agents, the MDM2 oncogene and its myriad interactive proteins also represent targets for cancer prevention. Since some of the small molecules and gene silencing techniques may be too toxic for long-term use, there may be a few agents (e.g., natural products such as curcumin or the ginsenosides) that can be used long-term without any adverse effects. Studies examining long-term down-regulation of MDM2 should be accomplished to determine the effects on oncogenesis and normal cellular processes.
8. CONCLUSIONS
In summary, the MDM2 protein and the MDM2-p53 auto-regulatory loop are part of a complex network that appears more complicated with each new discovery. However, the proteins are central to both apoptosis and cell cycle progression, two of the most important cellular processes. Considerable research is still needed, but the MDM2-p53-“other protein” interactions represent novel targets for therapy and present the opportunity to individualize treatments based upon a patient’s particular cancer type and expression status.
ACKNOWLEDGEMENTS
We apologize to our colleagues for omitting some interesting and important work about MDM2 and the regulation of the MDM2-p53 interaction, primarily related to its transcriptional and post-translational regulation. Given the constraints of this review, we had to condense the information and/or omit discussions about several molecules, but we believe that we have presented an extensive and accurate picture of the diversity of MDM2’s functions and have included the most up-to-date research published about the topics. We would like to thank past and current members of the Zhang laboratory who have worked diligently to help elucidate the activity, relevance, and potential of MDM2 and MDM2-interactive proteins as cancer targets. In particular, we thank Dr. Zhuo Zhang for critical review of this manuscript and for his many helpful suggestions, and Drs. Feng Wang and Gu Jing for their assistance in preparing better and more attractive figures. RZ was supported in part by NIH/NCI grants R01 CA112029 and R01 CA121211, and ER was supported in part by the USA Department of Defense Prostate Cancer Research Program (grant number W81XWH-06-1-0063) and a T32 fellowship (CA075930) from the NIH/UAB Gene Therapy Center.
Biography

Author Profile: Ruiwen Zhang, MD, PhD, DABT, is Professor of Pharmacology and Toxicology, Director of the Cancer Pharmacology Laboratory, Senior Scientist in the Comprehensive Cancer Center, Center for AIDS Research, and Gene Therapy Center at the University of Alabama at Birmingham. He has extensive experience in clinical pharmacology, molecular therapeutics, and toxicology.
REFERENCES
- [1].Croce CM N. Engl. J. Med, 2008, 358, 502. [DOI] [PubMed] [Google Scholar]
- [2].Letai AG Nat. Rev. Cancer, 2008, 8, 121. [DOI] [PubMed] [Google Scholar]
- [3].Zhang Z; Li M; Rayburn ER; Hill DL; Zhang R; Wang H Am. J. Pharmacogenomics, 2005, 5, 173. [DOI] [PubMed] [Google Scholar]
- [4].Zhang Z; Li M; Rayburn ER; Hill DL; Zhang R; Wang H Am. J. Pharmacogenomics, 2005, 5, 247. [DOI] [PubMed] [Google Scholar]
- [5].Zhang Z; Li M; Rayburn ER; Hill DL; Zhang R; Wang H Am. J. Pharmacogenomics, 2005, 5, 327. [DOI] [PubMed] [Google Scholar]
- [6].Zhang Z; Li M; Rayburn ER; Hill DL; Zhang R; Wang H Am. J. Pharmacogenomics, 2005, 5, 397. [DOI] [PubMed] [Google Scholar]
- [7].Weinstein IB; Joe AK Nat. Clin. Pract. Oncol, 2006, 3, 448. [DOI] [PubMed] [Google Scholar]
- [8].Carella AM; Lerma E Curr. Stem Cell Res. Ther, 2007, 2, 249. [DOI] [PubMed] [Google Scholar]
- [9].Wang J; Meyering-Vos M; Hoffmann KH Mol. Cell Endocrinol, 2004, 227, 41. [DOI] [PubMed] [Google Scholar]
- [10].Li M; Zhang Z; Hill DL; Chen X; Wang H; Zhang R Cancer Res, 2005, 65, 8200. [DOI] [PubMed] [Google Scholar]
- [11].Li M; Zhang Z; Hill DL; Wang H; Zhang R Cancer Res., 2007, 67, 1988. [DOI] [PubMed] [Google Scholar]
- [12].Fakharzadeh SS; Trusko SP; George DL EMBO J., 1991, 10, 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rayburn E; Zhang R; He J; Wang H Curr. Cancer Drug Targets, 2005, 5, 27. [DOI] [PubMed] [Google Scholar]
- [14].Oliner JD; Kinzler KW; Meltzer PS; George DL; Vogelstein B Nature, 1992, 358, 80. [DOI] [PubMed] [Google Scholar]
- [15].Iwakuma T; Lozano G Mol. Cancer Res, 2003, 1, 993. [PubMed] [Google Scholar]
- [16].Honda R; Tanaka H; Yasuda H FEBS Lett., 1997, 420, 25. [DOI] [PubMed] [Google Scholar]
- [17].Momand J; Zambetti GP; Olson DC; George D; Levine AJ Cell, 1992, 69, 1237. [DOI] [PubMed] [Google Scholar]
- [18].Bottger A; Bottger V; Garcia-Echeverria C; Chene P; Hochkeppel HK; Sampson W; Ang K; Howard SF; Picksley SM; Lane DP J. Mol. Biol, 1997, 269, 744. [DOI] [PubMed] [Google Scholar]
- [19].Wsierska-Gadek J; Horky M Ann. N. Y. Acad. Sci, 2003, 1010, 266. [DOI] [PubMed] [Google Scholar]
- [20].Helton ES; Chen XJ Cell Biochem., 2007, 100, 883. [DOI] [PubMed] [Google Scholar]
- [21].Harris SL; Levine AJ Oncogene, 2005, 24, 2899. [DOI] [PubMed] [Google Scholar]
- [22].Kubbutat MH; Jones SN; Vousden KH Nature, 1997, 387, 299. [DOI] [PubMed] [Google Scholar]
- [23].Fuchs SY; Adler V; Buschmann T; Wu X; Ronai Z Onco-gene, 1998, 17, 2543. [DOI] [PubMed] [Google Scholar]
- [24].Kastan MB; Onyekwere O; Sidransky D; Vogelstein B; Craig RW Cancer Res., 1991, 51, 6304. [PubMed] [Google Scholar]
- [25].Bond GL; Hu W; Levine AJ Curr. Cancer Drug Targets, 2005, 5, 3. [DOI] [PubMed] [Google Scholar]
- [26].Vousden KH; Lane DP Nat. Rev. Mol. Cell Biol, 2007, 8, 275. [DOI] [PubMed] [Google Scholar]
- [27].Laptenko O; Prives C Cell Death Differ, 2006, 13, 951. [DOI] [PubMed] [Google Scholar]
- [28].Raver-Shapira N; Oren M Cell Cycle, 2007, 6, 2656. [DOI] [PubMed] [Google Scholar]
- [29].Olsson A; Manzl C; Strasser A; Villunger A Cell Death Differ., 2007, 14, 1561. [DOI] [PubMed] [Google Scholar]
- [30].Meek DW; Knippschild U Mol. Cancer Res, 2003, 1, 1017. [PubMed] [Google Scholar]
- [31].Hermeking H; Lengauer C; Polyak K; He TC; Zhang L; Thiagalingam S; Kinzler KW; Vogelstein B Mol. Cell, 1997, 1, 3. [DOI] [PubMed] [Google Scholar]
- [32].Laronga C; Yang HY; Neal C; Lee MH J. Biol. Chem, 2000, 275, 23106. [DOI] [PubMed] [Google Scholar]
- [33].Yang HY; Wen YY; Chen CH; Lozano G; Lee MH Mol. Cell Biol, 2003, 23, 7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yang HY; Wen YY; Lin YI; Pham L; Su CH; Yang H; Chen J; Lee MH Oncogene, 2007, 26, 7355. [DOI] [PubMed] [Google Scholar]
- [35].Lee MH; Lozano G Semin. Cancer Biol, 2006, 16, 225. [DOI] [PubMed] [Google Scholar]
- [36].Stott FJ; Bates S; James MC; McConnell BB; Starborg M; Brookes S; Palmero I; Ryan K; Hara E; Vousden KH; Peters G EMBO J., 1998, 17, 5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bothner B; Lewis WS; DiGiammarino EL; Weber JD; Bothner SJ; Kriwacki RW J. Mol. Biol, 2001, 314, 263. [DOI] [PubMed] [Google Scholar]
- [38].Weber JD; Kuo ML; Bothner B; DiGiammarino EL; Kriwacki RW; Roussel MF; Sherr CJ Mol. Cell Biol, 2000, 20, 2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Honda R; Yasuda H EMBO J., 1999, 18, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Deng X; Kim M; Vandier D; Jung YJ; Rikiyama T; Sgagias MK; Goldsmith M; Cowan KH Biochem. Biophys. Res. Commun, 2002, 296, 792. [DOI] [PubMed] [Google Scholar]
- [41].Lowe SW; Sherr CJ Curr. Opin. Genet. Dev, 2003, 13, 77. [DOI] [PubMed] [Google Scholar]
- [42].Widlund HR; Fisher DE Pigment Cell Res., 2007, 20, 339. [DOI] [PubMed] [Google Scholar]
- [43].Lindstrom MS; Zhang Y Cell Biochem. Biophys, 2006, 46, 79. [DOI] [PubMed] [Google Scholar]
- [44].Kurki S; Peltonen K; Laiho M Cell Cycle, 2004, 3, 976. [PubMed] [Google Scholar]
- [45].Gjerset RA J. Mol. Histol, 2006, 37, 239. [DOI] [PubMed] [Google Scholar]
- [46].Colombo E; Marine JC; Danovi D; Falini B; Pelicci PG Nat. Cell Biol, 2002, 4, 529. [DOI] [PubMed] [Google Scholar]
- [47].Gallagher SJ; Kefford RF; Rizos H Int. J. Biochem. Cell Biol, 2006, 38, 1637. [DOI] [PubMed] [Google Scholar]
- [48].Tomoda K; Kubota Y; Arata Y; Mori S; Maeda M; Tanaka T; Yoshida M; Yoneda-Kato N; Kato JY J. Biol. Chem, 2002, 277, 2302. [DOI] [PubMed] [Google Scholar]
- [49].Cope GA; Deshaies RJ Cell, 2003, 114, 663. [DOI] [PubMed] [Google Scholar]
- [50].Bech-Otschir D; Kraft R; Huang X; Henklein P; Kapelari B; Pollmann C; Dubiel W EMBO J., 2001, 20, 1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lee EW; Oh W; Song J Mol. Cell, 2006, 22, 133. [PubMed] [Google Scholar]
- [52].Zhang XC; Chen J; Su CH; Yang HY; Lee MH J. Cell Biochem, 2008, 103, 1219. [DOI] [PubMed] [Google Scholar]
- [53].Zhang Z; Wang H; Li M; Rayburn ER; Agrawal S; Zhang R Oncogene, 2005, 24, 7238. [DOI] [PubMed] [Google Scholar]
- [54].Dawson S; Apcher S; Mee M; Higashitsuji H; Baker R; Uhle S; Dubiel W; Fujita J; Mayer RJ J. Biol. Chem, 2002, 277, 10893. [DOI] [PubMed] [Google Scholar]
- [55].Umemura A; Itoh Y; Itoh K; Yamaguchi K; Nakajima T; Higashitsuji H; Onoue H; Fukumoto M; Okanoue T; Fujita J Hepatology, 2008, 47, 493. [DOI] [PubMed] [Google Scholar]
- [56].Higashitsuji H; Higashitsuji H; Itoh K; Sakurai T; Nagao T; Sumitomo Y; Masuda T; Dawson S; Shimada Y; Mayer RJ; Fujita J Cancer Cell, 2005, 8, 75. [DOI] [PubMed] [Google Scholar]
- [57].Dawson S; Higashitsuji H; Wilkinson AJ; Fujita J; Mayer RJ Trends Cell Biol., 2006, 16, 229. [DOI] [PubMed] [Google Scholar]
- [58].Mayer RJ; Fujita J Biochem. Soc. Trans, 2006, 34, 746. [DOI] [PubMed] [Google Scholar]
- [59].Li M; Chen D; Shiloh A; Luo J; Nikolaev AY; Qin J; Gu W Nature, 2002, 416, 648. [DOI] [PubMed] [Google Scholar]
- [60].Cummins JM; Vogelstein B Cell Cycle, 2004, 3, 689. [PubMed] [Google Scholar]
- [61].Brazhnik P; Kohn KW Math Biosci., 2007, 210, 60. [DOI] [PubMed] [Google Scholar]
- [62].Tang J; Qu LK; Zhang J; Wang W; Michaelson JS; Degenhardt YY; El-Deiry WS; Yang X Nat. Cell Biol, 2006, 8, 855. [DOI] [PubMed] [Google Scholar]
- [63].Ronai Z Nat. Cell Biol, 2006, 8, 790. [DOI] [PubMed] [Google Scholar]
- [64].Shikama N; Lee CW; France S; Delavaine L; Lyon J; Krstic-Demonacos M; La Thangue NB Mol. Cell, 1999, 4, 365. [DOI] [PubMed] [Google Scholar]
- [65].Coutts AS; Boulahbel H; Graham A; La Thangue NB EMBO Rep., 2007, 8, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hershko T; Chaussepied M; Oren M; Ginsberg D Cell Death Differ., 2005, 12, 377. [DOI] [PubMed] [Google Scholar]
- [67].Wang C; Ivanov A; Chen L; Fredericks WJ; Seto E; Rauscher FJ 3rd; Chen J EMBO J., 2005, 24, 3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Okamoto K; Li H; Jensen MR; Zhang T; Taya Y; Thorgeirsson SS; Prives C Mol. Cell, 2002, 9, 761. [DOI] [PubMed] [Google Scholar]
- [69].Shvarts A; Steegenga WT; Riteco N; van Laar T; Dekker P; Bazuine M; van Ham RC; van der Houven van Oordt W; Hateboer G; van der Eb AJ; Jochemsen AG EMBO J., 1996, 15, 5349. [PMC free article] [PubMed] [Google Scholar]
- [70].Gilkes DM; Chen J Cell Cycle, 2007, 6, 151. [DOI] [PubMed] [Google Scholar]
- [71].Hu B; Gilkes DM; Chen J Cancer Res., 2007, 67, 8810. [DOI] [PubMed] [Google Scholar]
- [72].Marine JC; Dyer MA; Jochemsen AG J. Cell Sci, 2007, 120, 371. [DOI] [PubMed] [Google Scholar]
- [73].Uldrijan S; Pannekoek WJ; Vousden KH EMBO J., 2007, 26, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Rallapalli R; Strachan G; Cho B; Mercer WE; Hall DJ J. Biol. Chem, 1999, 274, 8299. [DOI] [PubMed] [Google Scholar]
- [75].Rallapalli R; Strachan G; Tuan RS; Hall DJ J. Cell Biochem, 2003, 89, 563. [DOI] [PubMed] [Google Scholar]
- [76].Boyd MT; Vlatkovic N; Haines DS J. Biol. Chem, 2000, 275, 31883. [DOI] [PubMed] [Google Scholar]
- [77].Brady M; Vlatkovic N; Boyd MT Mol. Cell Biol, 2005, 25, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Iwakuma T; Tochigi Y; Van Pelt CS; Caldwell LC; Terzian T; Parant JM; Chau GP; Koch JG; Eischen CM; Lozano G Oncogene, 2007, 2177. [DOI] [PubMed] [Google Scholar]
- [79].Greasley PJ; Bonnard C; Amati B Nucleic Acids Res., 2000, 28, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kondo K; Inouye MJ Biol. Chem, 1992, 267, 16252. [PubMed] [Google Scholar]
- [81].Daniely Y; Dimitrova DD; Borowiec JA Mol. Cell Biol, 2002, 22, 6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Takagi M; Absalon MJ; McLure KG; Kastan MB Cell, 2005, 123, 49. [DOI] [PubMed] [Google Scholar]
- [83].Saxena A; Rorie CJ; Dimitrova D; Daniely Y; Borowiec JA Oncogene, 2006, 25, 7274. [DOI] [PubMed] [Google Scholar]
- [84].Juven-Gershon T; Shifman O; Unger T; Elkeles A; Haupt Y; Oren M Mol. Cell Biol, 1998, 18, 3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Yogosawa S; Miyauchi Y; Honda R; Tanaka H; Yasuda H Biochem. Biophys. Res. Commun, 2003, 302, 869. [DOI] [PubMed] [Google Scholar]
- [86].Pece S; Serresi M; Santolini E; Capra M; Hulleman E; Galimberti V; Zurrida S; Maisonneuve P; Viale G; Di Fiore PP J. Cell Biol, 2004, 167, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Colaluca IN; Tosoni D; Nuciforo P; Senic-Matuglia F; Galimberti V; Viale G; Pece S; Di Fiore PP Nature, 2008, 451, 76. [DOI] [PubMed] [Google Scholar]
- [88].Jin Y; Lee H; Zeng SX; Dai MS; Lu H EMBO J., 2003, 22, 6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zhang Z; Wang H; Li M; Agrawal S; Chen X; Zhang RJ Biol. Chem, 2004, 279, 16000. [DOI] [PubMed] [Google Scholar]
- [90].Giono LE; Manfredi JJ Mol. Cell Biol, 2007, 27, 4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Broude EV; Demidenko ZN; Vivo C; Swift ME; Davis BM; Blagosklonny MV; Roninson IB Cell Cycle, 2007, 6, 1468. [PubMed] [Google Scholar]
- [92].Zhang L; Nie L; Maki CG Mol. Cancer, 2006, 5, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Oberst A; Rossi M; Salomoni P; Pandolfi PP; Oren M; Melino G; Bernassola F Biochem. Biophys. Res. Commun, 2005, 331, 707. [DOI] [PubMed] [Google Scholar]
- [94].Ozaki T; Hosoda M; Miyazaki K; Hayashi S; Watanabe K; Nakagawa T; Nakagawara A Cancer Lett., 2005, 228, 29. [DOI] [PubMed] [Google Scholar]
- [95].Malaguarnera R; Vella V; Pandini G; Sanfilippo M; Pezzino V; Vigneri R; Frasca F Mol. Cancer Res, 2008, 6, 64. [DOI] [PubMed] [Google Scholar]
- [96].Johnson J; Lagowski J; Lawson S; Liu Y; Kulesz-Martin M Oncogene, 2008, 27, 2780. [DOI] [PubMed] [Google Scholar]
- [97].Watson IR; Blanch A; Lin DC; Ohh M; Irwin MS J. Biol. Chem, 2006, 281, 34096. [DOI] [PubMed] [Google Scholar]
- [98].Rechsteiner M; Hill CP Trends Cell Biol., 2005, 15, 27. [DOI] [PubMed] [Google Scholar]
- [99].Zhang Z; Zhang R EMBO J., 2008, 27, 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Chen X; Barton LF; Chi Y; Clurman BE; Roberts JM Mol. Cell, 2007, 26, 843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Linares LK; Kiernan R; Triboulet R; Chable-Bessia C; Latreille D; Cuvier O; Lacroix M; Le Cam L; Coux O; Benkirane M Nat. Cell Biol, 2007, 9, 331. [DOI] [PubMed] [Google Scholar]
- [102].Louria-Hayon I; Grossman T; Sionov RV; Alsheich O; Pandolfi PP; Haupt YJ Biol. Chem, 2003, 278, 33134. [DOI] [PubMed] [Google Scholar]
- [103].Bernardi R; Scaglioni PP; Bergmann S; Horn HF; Vousden KH; Pandolfi PP Nat. Cell Biol, 2004, 6, 665. [DOI] [PubMed] [Google Scholar]
- [104].Alsheich-Bartok O; Haupt S; Alkalay-Snir I; Saito S; Appella E; Haupt Y Oncogene, 2008, 27, 3653. [DOI] [PubMed] [Google Scholar]
- [105].Sdek P; Ying H; Zheng H; Margulis A; Tang X; Tian K; Xiao ZX J. Biol. Chem, 2004, 279, 53317. [DOI] [PubMed] [Google Scholar]
- [106].Hsieh JK; Chan FS; O’Connor DJ; Mittnacht S; Zhong S; Lu X Mol. Cell, 1999, 3, 181. [DOI] [PubMed] [Google Scholar]
- [107].Guo CS; Degnin C; Fiddler TA; Stauffer D; Thayer MJ J. Biol. Chem, 2003, 278, 22615. [DOI] [PubMed] [Google Scholar]
- [108].Dai MS; Lu HJ Biol. Chem, 2004, 279, 44475. [DOI] [PubMed] [Google Scholar]
- [109].Dai MS; Shi D; Jin Y; Sun XX; Zhang Y; Grossman SR; Lu HJ Biol. Chem, 2006, 281, 24304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Dai MS; Zeng SX; Jin Y; Sun XX; David L; Lu H Mol. Cell Biol, 2004, 24, 7654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Sun XX; Dai MS; Lu HJ Biol. Chem, 2008, 283, 12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Ofir-Rosenfeld YBK; Michael D; Kastan M; Oren M In International MDM2 Workshop IV, Woods Hole, MA, Abstract #73, 2008. [Google Scholar]
- [113].Chen D; Zhang Z; Li M; Wang W; Li Y; Rayburn ER; Hill DL; Wang H; Zhang R Oncogene, 2007, 26, 5029. [DOI] [PubMed] [Google Scholar]
- [114].Zhu YPM, Jacq X; Stahl J; Prives C, In International MDM2 Workshop IV, Woods Hole, MA, Abstract #96, 2008. [Google Scholar]
- [115].Sapountzi V; Logan IR; Robson CN Int. J. Biochem. Cell Biol, 2006, 38, 1496. [DOI] [PubMed] [Google Scholar]
- [116].Legube G; Linares LK; Lemercier C; Scheffner M; Khochbin S; Trouche D EMBO J., 2002, 21, 1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Dohmesen C; Koeppel M; Dobbelstein M Cell Cycle, 2008, 7, 222. [DOI] [PubMed] [Google Scholar]
- [118].Gaughan L; Logan IR; Neal DE; Robson CN Nucleic Acids Res., 2005, 33, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Li L; Cohen SN Cell, 1996, 85, 319. [DOI] [PubMed] [Google Scholar]
- [120].Li L; Liao J; Ruland J; Mak TW; Cohen SN Proc. Natl. Acad. Sci. U.S.A, 2001, 98, 1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Carstens MJ; Krempler A; Triplett AA; Van Lohuizen M; Wagner KU J. Biol. Chem, 2004, 279, 35984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Cheng TH; Cohen SN Mol. Cell Biol, 2007, 27, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Wang P; Gao H; Ni Y; Wang B; Wu Y; Ji L; Qin L; Ma L; Pei GJ Biol. Chem, 2003, 278, 6363. [DOI] [PubMed] [Google Scholar]
- [124].Girnita L; Shenoy SK; Sehat B; Vasilcanu R; Girnita A; Lefkowitz RJ; Larsson OJ Biol. Chem, 2005, 280, 24412. [DOI] [PubMed] [Google Scholar]
- [125].Girnita L; Shenoy SK; Sehat B; Vasilcanu R; Vasilcanu D; Girnita A; Lefkowitz RJ; Larsson OJ Biol. Chem, 2007, 282, 11329. [DOI] [PubMed] [Google Scholar]
- [126].Ravi R; Mookerjee B; Bhujwalla ZM; Sutter CH; Artemov D; Zeng Q; Dillehay LE; Madan A; Semenza GL; Bedi A Genes Dev., 2000, 14, 34. [PMC free article] [PubMed] [Google Scholar]
- [127].Nieminen AL; Qanungo S; Schneider EA; Jiang BH; Agani FH J. Cell Physiol, 2005, 204, 364. [DOI] [PubMed] [Google Scholar]
- [128].Carroll VA; Ashcroft M Cancer Res., 2008, 68, 545. [DOI] [PubMed] [Google Scholar]
- [129].Rempe DA; Lelli KM; Vangeison G; Johnson RS; Federoff HJ J. Biol. Chem, 2007, 282, 16187. [DOI] [PubMed] [Google Scholar]
- [130].Suzuki H; Tomida A; Tsuruo T Oncogene, 2001, 20, 5779. [DOI] [PubMed] [Google Scholar]
- [131].Peng Y; Chen L; Li C; Lu W; Chen JJ Biol. Chem, 2001, 276, 40583. [DOI] [PubMed] [Google Scholar]
- [132].Sasaki M; Nie L; Maki CG J. Biol. Chem, 2007, 282, 14626. [DOI] [PubMed] [Google Scholar]
- [133].Muller P; Hrstka R; Coomber D; Lane DP; Vojtesek B Oncogene, 2008. [DOI] [PubMed] [Google Scholar]
- [134].Wawrzynow B; Zylicz A; Wallace M; Hupp T; Zylicz MJ Biol. Chem, 2007, 282, 32603. [DOI] [PubMed] [Google Scholar]
- [135].Zoubeidi A; Zardan A; Beraldi E; Fazli L; Sowery R; Rennie P; Nelson C; Gleave M Cancer Res., 2007, 67, 10455. [DOI] [PubMed] [Google Scholar]
- [136].Minami M; Shinozaki F; Suzuki M; Yoshimatsu K; Ichikawa Y; Minami Y Biochem. Biophys. Res. Commun, 2006, 344, 1315. [DOI] [PubMed] [Google Scholar]
- [137].Minet E; Mottet D; Michel G; Roland I; Raes M; Remacle J; Michiels C FEBS Lett., 1999, 460, 251. [DOI] [PubMed] [Google Scholar]
- [138].Girnita L; Girnita A; Larsson O Proc. Natl. Acad. Sci. U.S.A, 2003, 100, 8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Xiong L; Kou F; Yang Y; Wu JJ Cell Biol., 2007, 178, 995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Zhang Z; Zhang R Curr. Cancer Drug Targets, 2005, 5, 9. [DOI] [PubMed] [Google Scholar]
- [141].Sengupta S; Wasylyk B Ann. N.Y. Acad. Sci, 2004, 1024, 54. [DOI] [PubMed] [Google Scholar]
- [142].Lin HK; Wang L; Hu YC; Altuwaijri S; Chang C EMBO J., 2002, 21, 4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Cronauer MV; Schulz WA; Burchardt T; Ackermann R; Burchardt M Oncogene, 2004, 23, 3541. [DOI] [PubMed] [Google Scholar]
- [144].Sengupta S; Wasylyk B Genes Dev., 2001, 15, 2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Kinyamu HK; Archer TK Mol. Cell Biol, 2003, 23, 5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Saji S; Nakashima S; Hayashi S; Toi M; Saji S; Nozawa Y Jpn. J. Cancer Res, 1999, 90, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Duong V; Boulle N; Daujat S; Chauvet J; Bonnet S; Neel H; Cavailles V Cancer Res., 2007, 67, 5513. [DOI] [PubMed] [Google Scholar]
- [148].Ellenrieder V; Buck A; Gress TM Int. J. Gastrointest. Cancer, 2002, 31, 61. [DOI] [PubMed] [Google Scholar]
- [149].Sun P; Dong P; Dai K; Hannon GJ; Beach D Science, 1998, 282, 2270. [DOI] [PubMed] [Google Scholar]
- [150].Yam CH; Siu WY; Arooz T; Chiu CH; Lau A; Wang XQ; Poon RY Cancer Res., 1999, 59, 5075. [PubMed] [Google Scholar]
- [151].Blain SW; Massague JJ Biol. Chem, 2000, 275, 32066. [DOI] [PubMed] [Google Scholar]
- [152].Kannemeier C; Liao R; Sun P Mol. Biol. Cell, 2007, 18, 2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Takemoto S; Trovato R; Cereseto A; Nicot C; Kislyakova T; Casareto L; Waldmann T; Torelli G; Franchini G Blood, 2000, 95, 3939. [PubMed] [Google Scholar]
- [154].Kim MM; Wiederschain D; Kennedy D; Hansen E; Yuan ZM Oncogene, 2007, 26, 4209. [DOI] [PubMed] [Google Scholar]
- [155].Alt JR; Bouska A; Fernandez MR; Cerny RL; Xiao H; Eischen CM J. Biol. Chem, 2005, 280, 18771. [DOI] [PubMed] [Google Scholar]
- [156].Ma H; Pederson T Mol. Biol. Cell, 2007, 18, 2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Ando K; Ozaki T; Yamamoto H; Furuya K; Hosoda M; Hayashi S; Fukuzawa M; Nakagawara AJ Biol. Chem, 2004, 279, 25549. [DOI] [PubMed] [Google Scholar]
- [158].Stevenson LF; Sparks A; Allende-Vega N; Xirodimas DP; Lane DP; Saville MK EMBO J., 2007, 26, 976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Lu X; Nguyen TA; Donehower LA Cell Cycle, 2008, 7, 164. [DOI] [PubMed] [Google Scholar]
- [160].Sui G; Affar el B; Shi Y; Brignone C; Wall NR; Yin P; Donohoe M; Luke MP; Calvo D; Grossman SR; Shi Y Cell, 2004, 117, 859. [DOI] [PubMed] [Google Scholar]
- [161].Li L; Deng B; Xing G; Teng Y; Tian C; Cheng X; Yin X; Yang J; Gao X; Zhu Y; Sun Q; Zhang L; Yang X; He F Proc. Natl. Acad. Sci. U.S.A, 2007, 104, 7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Ding Y; Lee JF; Lu H; Lee MH; Yan DH Mol. Cell Biol, 2006, 26, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Kim H; Kwak NJ; Lee JY; Choi BH; Lim Y; Ko YJ; Kim YH; Huh PW; Lee KH; Rha HK; Wang YP J. Biol. Chem, 2004, 279, 7812. [DOI] [PubMed] [Google Scholar]
- [164].Thomas A; White E Genes Dev., 1998, 12, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Cozar-Castellano I; Fiaschi-Taesch N; Bigatel TA; Takane KK; Garcia-Ocana A; Vasavada R; Stewart AF Endocr. Rev, 2006, 27, 356. [DOI] [PubMed] [Google Scholar]
- [166].Toth A; Nickson P; Qin LL; Erhardt PJ Biol. Chem, 2006, 281, 3679. [DOI] [PubMed] [Google Scholar]
- [167].Arany I; Yen A; Tyring SK Anticancer Res., 1997, 17, 1281. [PubMed] [Google Scholar]
- [168].Bossi G; Sacchi A Head Neck, 2007, 29, 272. [DOI] [PubMed] [Google Scholar]
- [169].Hupp TR; Lane DP; Ball KL Biochem. J, 2000, 352 Pt 1, 1. [PMC free article] [PubMed] [Google Scholar]
- [170].Daftarian P; Song GY; Ali S; Faynsod M; Longmate J; Diamond DJ; Ellenhorn JD Cancer Res, 2004, 64, 5407. [DOI] [PubMed] [Google Scholar]
- [171].Lomas M; Liauw W; Packham D; Williams K; Kelleher A; Zaunders J; Ward R Ann. Oncol, 2004, 15, 324. [DOI] [PubMed] [Google Scholar]
- [172].Wiman KG Cell Death Differ., 2006, 13, 921. [DOI] [PubMed] [Google Scholar]
- [173].Issaeva N; Bozko P; Enge M; Protopopova M; Verhoef LG; Masucci M; Pramanik A; Selivanova G Nat. Med, 2004, 10, 1321. [DOI] [PubMed] [Google Scholar]
- [174].Kondo S; Barnett GH; Hara H; Morimura T; Takeuchi J Oncogene, 1995, 10, 2001. [PubMed] [Google Scholar]
- [175].Leonetti C; Zupi G Curr. Pharm. Des, 2007, 13, 463. [DOI] [PubMed] [Google Scholar]
- [176].Zhang C; Pei J; Kumar D; Sakabe I; Boudreau HE; Gokhale PC; Kasid UN Methods Mol. Biol, 2007, 361, 163. [DOI] [PubMed] [Google Scholar]
- [177].Rayburn E; Wang W; Zhang R; Wang H Prog. Drug. Res, 2005, 63, 227. [DOI] [PubMed] [Google Scholar]
- [178].Rayburn ER; Wang H; Zhang R Expert Opin. Emerg. Drugs, 2006, 11, 337. [DOI] [PubMed] [Google Scholar]
- [179].www.clinicaltrials.gov, Accessed (March 07, 2008)
- [180].Bianco R; Ciardiello F; Tortora G Curr. Cancer Drug Targets, 2005, 5, 51. [DOI] [PubMed] [Google Scholar]
- [181].Bartel F; Harris LC; Wurl P; Taubert H Mol. Cancer Res, 2004, 2, 29. [PubMed] [Google Scholar]
- [182].Mu Z; Hachem P; Hensley H; Stoyanova R; Kwon HW; Hanlon AL; Agrawal S; Pollack A Prostate, 2008, 68, 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Wang H; Oliver P; Zhang Z; Agrawal S; Zhang R Ann. N.Y. Acad. Sci, 2003, 1002, 217. [DOI] [PubMed] [Google Scholar]
- [184].Zhang Z; Li M; Wang H; Agrawal S; Zhang R Proc. Natl. Acad. Sci. U.S.A, 2003, 100, 11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Sullenger BA; Cech TR Nature, 1994, 371, 619. [DOI] [PubMed] [Google Scholar]
- [186].Sullenger BA N. Engl. J. Med, 2004, 351, 2759. [DOI] [PubMed] [Google Scholar]
- [187].Rayburn E; Wang H; He J; Zhang R, Lett. Drug Des. Disc, 2005, 2(1), 1–18. [Google Scholar]
- [188].Castanotto D; Li JR; Michienzi A; Langlois MA; Lee NS; Puymirat J; Rossi JJ Biochem. Soc. Trans, 2002, 30, 1140. [DOI] [PubMed] [Google Scholar]
- [189].Brummelkamp TR; Fabius AW; Mullenders J; Madiredjo M; Velds A; Kerkhoven RM; Bernards R; Beijersbergen RL Nat. Chem. Biol, 2006, 2, 202. [DOI] [PubMed] [Google Scholar]
- [190].Zhang Z; Wang H; Li M; Rayburn E; Agrawal S; Zhang R Ann. N.Y. Acad. Sci, 2005, 1058, 205. [DOI] [PubMed] [Google Scholar]
- [191].Zhang C; Tang N; Liu X; Liang W; Xu W; Torchilin VP J. Control Release, 2006, 112, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Kaeser MD; Pebernard S; Iggo RD J. Biol. Chem, 2004, 279, 7598. [DOI] [PubMed] [Google Scholar]
- [193].Vassilev LT; Vu BT; Graves B; Carvajal D; Podlaski F; Filipovic Z; Kong N; Kammlott U; Lukacs C; Klein C; Fotouhi N; Liu EA Science, 2004, 303, 844. [DOI] [PubMed] [Google Scholar]
- [194].Kojima K; Konopleva M; Samudio IJ; Shikami M; Cabreira-Hansen M; McQueen T; Ruvolo V; Tsao T; Zeng Z; Vassilev LT; Andreeff M Blood, 2005, 106, 3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Stuhmer T; Bargou RC Cell Cycle, 2006, 5, 39. [DOI] [PubMed] [Google Scholar]
- [196].Cao C; Shinohara ET; Subhawong TK; Geng L; Woon Kim K; Albert JM; Hallahan DE; Lu B Mol. Cancer Ther, 2006, 5, 411. [DOI] [PubMed] [Google Scholar]
- [197].Bottger A; Bottger V; Sparks A; Liu WL; Howard SF; Lane DP Curr. Biol, 1997, 7, 860. [DOI] [PubMed] [Google Scholar]
- [198].Wasylyk C; Salvi R; Argentini M; Dureuil C; Delumeau I; Abecassis J; Debussche L; Wasylyk B Oncogene, 1999, 18, 1921. [DOI] [PubMed] [Google Scholar]
- [199].Yin H; Lee GI; Park HS; Payne GA; Rodriguez JM; Sebti SM; Hamilton AD Angew. Chem. Int. Ed. Engl, 2005, 44, 2704. [DOI] [PubMed] [Google Scholar]
- [200].Chen L; Yin H; Farooqi B; Sebti S; Hamilton AD; Chen J Mol. Cancer Ther, 2005, 4, 1019. [DOI] [PubMed] [Google Scholar]
- [201].Hardcastle IR; Ahmed SU; Atkins H; Farnie G; Golding BT; Griffin RJ; Guyenne S; Hutton C; Kallblad P; Kemp SJ; Kitching MS; Newell DR; Norbedo S; Northen JS; Reid RJ; Saravanan K; Willems HM; Lunec JJ Med. Chem, 2006, 49, 6209. [DOI] [PubMed] [Google Scholar]
- [202].Ding K; Lu Y; Nikolovska-Coleska Z; Wang G; Qiu S; Shangary S; Gao W; Qin D; Stuckey J; Krajewski K; Roller PP; Wang SJ Med. Chem, 2006, 49, 3432. [DOI] [PubMed] [Google Scholar]
- [203].Grasberger BL; Lu T; Schubert C; Parks DJ; Carver TE; Koblish HK; Cummings MD; LaFrance LV; Milkiewicz KL; Calvo RR; Maguire D; Lattanze J; Franks CF; Zhao S; Ramachandren K; Bylebyl GR; Zhang M; Manthey CL; Petrella EC; Pantoliano MW; Deckman IC; Spurlino JC; Maroney AC; Tomczuk BE; Molloy CJ; Bone RF J. Med. Chem, 2005, 48, 909. [DOI] [PubMed] [Google Scholar]
- [204].Koblish HK; Zhao S; Franks CF; Donatelli RR; Tominovich RM; LaFrance LV; Leonard KA; Gushue JM; Parks DJ; Calvo RR; Milkiewicz KL; Marugan JJ; Raboisson P; Cummings MD; Grasberger BL; Johnson DL; Lu T; Molloy CJ; Maroney AC Mol. Cancer Ther, 2006, 5, 160. [DOI] [PubMed] [Google Scholar]
- [205].Yang Y; Ludwig RL; Jensen JP; Pierre SA; Medaglia MV; Davydov IV; Safiran YJ; Oberoi P; Kenten JH; Phillips AC; Weissman AM; Vousden KH Cancer Cell, 2005, 7, 547. [DOI] [PubMed] [Google Scholar]
- [206].Sasiela CA; Stewart D; Kitagaki J; Safiran YJ; Yang Y; Weissman AM; Oberoi P; Davydov IV; Goncharova E; Beutler JA; McMahon JB; O’Keefe BR J. Biomol. Screen, 2008, 13, 229. [DOI] [PubMed] [Google Scholar]
- [207].Stoll R; Renner C; Hansen S; Palme S; Klein C; Belling A; Zeslawski W; Kamionka M; Rehm T; Muhlhahn P; Schumacher R; Hesse F; Kaluza B; Voelter W; Engh RA; Holak TA Biochemistry, 2001, 40, 336. [DOI] [PubMed] [Google Scholar]
- [208].Kumar SK; Hager E; Pettit C; Gurulingappa H; Davidson NE; Khan SR J. Med. Chem, 2003, 46, 2813. [DOI] [PubMed] [Google Scholar]
- [209].Achanta G; Modzelewska A; Feng L; Khan SR; Huang P Mol. Pharmacol, 2006, 70, 426. [DOI] [PubMed] [Google Scholar]
- [210].Duncan SJ; Gruschow S; Williams DH; McNicholas C; Purewal R; Hajek M; Gerlitz M; Martin S; Wrigley SK; Moore MJ Am. Chem. Soc, 2001, 123, 554. [DOI] [PubMed] [Google Scholar]
- [211].Duncan SJ; Cooper MA; Williams DH Chem. Commun. (Camb), 2003, 3, 316. [DOI] [PubMed] [Google Scholar]
- [212].Woon EC; Arcieri M; Wilderspin AF; Malkinson JP; Searcey MJ Org. Chem, 2007, 72, 5146. [DOI] [PubMed] [Google Scholar]
- [213].Malkinson JP; Zloh M; Kadom M; Errington R; Smith PJ; Searcey M Org. Lett, 2003, 5, 5051. [DOI] [PubMed] [Google Scholar]
- [214].Fang J; Xia C; Cao Z; Zheng JZ; Reed E; Jiang BH FASEB J., 2005, 19, 342. [DOI] [PubMed] [Google Scholar]
- [215].Wang W; Wang H; Rayburn ER; Zhao Y; Hill DL; Zhang R Br. J. Cancer, 2008, 98, 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Wang W; Rayburn ER; Hao M; Zhao Y; Hill DL; Zhang R; Wang H Prostate, 2008, 68, 809. [DOI] [PubMed] [Google Scholar]
- [217].Ganguli G; Abecassis J; Wasylyk B EMBO J., 2000, 19, 5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Liang H; Atkins H; Abdel-Fattah R; Jones SN; Lunec J Gene, 2004, 338, 217. [DOI] [PubMed] [Google Scholar]
- [219].Marti A; Wirbelauer C; Scheffner M; Krek W Nat. Cell Biol, 1999, 1, 14. [DOI] [PubMed] [Google Scholar]
- [220].Miwa S; Uchida C; Kitagawa K; Hattori T; Oda T; Sugimura H; Yasuda H; Nakamura H; Chida K; Kitagawa M Biochem. Biophys. Res. Commun, 2006, 340, 54. [DOI] [PubMed] [Google Scholar]
- [221].Tompkins VSHJ; Lushnikova T; Eischen CM; Quelle DE In International MDM2 Workshop IV, Woods Hole, MA, Abstract #15, 2008. [Google Scholar]
- [222].Chang CJ; Freeman DJ; Wu HJ Biol. Chem, 2004, 279, 29841. [DOI] [PubMed] [Google Scholar]
- [223].Barak Y; Gottlieb E; Juven-Gershon T; Oren M Genes Dev., 1994, 8, 1739. [DOI] [PubMed] [Google Scholar]
- [224].Shin S; Janknecht RJ Cell Biochem., 2007, 101, 1252. [DOI] [PubMed] [Google Scholar]
- [225].Slack A; Chen Z; Tonelli R; Pule M; Hunt L; Pession A; Shohet JM Proc. Natl. Acad. Sci. U.S.A, 2005, 102, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Truong AH; Cervi D; Lee J; Ben-David Y Oncogene, 2005, 24, 962. [DOI] [PubMed] [Google Scholar]
- [227].Ries S; Biederer C; Woods D; Shifman O; Shirasawa S; Sasazuki T; McMahon M; Oren M; McCormick F Cell, 2000, 103, 321. [DOI] [PubMed] [Google Scholar]
- [228].Phelps M; Phillips A; Darley M; Blaydes JP J. Biol. Chem, 2005, 280, 16651. [DOI] [PubMed] [Google Scholar]
- [229].Bond GL; Hu W; Bond EE; Robins H; Lutzker SG; Arva NC; Bargonetti J; Bartel F; Taubert H; Wuerl P; Onel K; Yip L; Hwang SJ; Strong LC; Lozano G; Levine AJ Cell, 2004, 119, 591. [DOI] [PubMed] [Google Scholar]
- [230].Lind H; Zienolddiny S; Ekstrom PO; Skaug V; Haugen A Int. J. Cancer, 2006, 119, 718. [DOI] [PubMed] [Google Scholar]
- [231].Bond GL; Hirshfield KM; Kirchhoff T; Alexe G; Bond EE; Robins H; Bartel F; Taubert H; Wuerl P; Hait W; Toppmeyer D; Offit K; Levine AJ Cancer Res., 2006, 66, 5104. [DOI] [PubMed] [Google Scholar]
- [232].Boersma BJ; Howe TM; Goodman JE; Yfantis HG; Lee DH; Chanock SJ; Ambs SJ Natl. Cancer Inst, 2006, 98, 911. [DOI] [PubMed] [Google Scholar]
- [233].Bond GL; Levine AJ Oncogene, 2007, 26, 1317. [DOI] [PubMed] [Google Scholar]
- [234].Petenkaya A; Bozkurt B; Akilli-Ozturk O; Kaya HS; Gur-Dedeoglu B; Yulug IG Anticancer Res., 2006, 26, 4975. [PubMed] [Google Scholar]
- [235].Wu X; Bayle JH; Olson D; Levine AJ Genes Dev., 1993, 7, 1126. [DOI] [PubMed] [Google Scholar]
- [236].Okamoto K; Kitabayashi I; Taya Y Biochem. Biophys. Res. Commun, 2006, 351, 216. [DOI] [PubMed] [Google Scholar]
- [237].Blattner C; Hay T; Meek DW; Lane DP Mol. Cell Biol, 2002, 22, 6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Meulmeester E; Pereg Y; Shiloh Y; Jochemsen AG Cell Cycle, 2005, 4, 1166. [DOI] [PubMed] [Google Scholar]
- [239].Buschmann T; Fuchs SY; Lee CG; Pan ZQ; Ronai Z Cell, 2000, 101, 753. [DOI] [PubMed] [Google Scholar]
- [240].Miyauchi Y; Yogosawa S; Honda R; Nishida T; Yasuda HJ Biol. Chem, 2002, 277, 50131. [DOI] [PubMed] [Google Scholar]
- [241].Tago K; Chiocca S; Sherr CJ Proc. Natl. Acad. Sci. U.S.A, 2005, 102, 7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Wang X; Taplick J; Geva N; Oren M FEBS Lett., 2004, 561, 195. [DOI] [PubMed] [Google Scholar]
- [243].Lavin MF; Gueven N Cell Death Differ, 2006, 13, 941. [DOI] [PubMed] [Google Scholar]
- [244].Du WX, Z-X J In International MDM2 Workshop IV, Woods Hole, MA, Abstract #15, 2008. [Google Scholar]
- [245].Bouchet BP; de Fromentel CC; Puisieux A; Galmarini CM Crit. Rev. Oncol. Hematol, 2006, 58, 190. [DOI] [PubMed] [Google Scholar]
- [246].Bartek J; Bartkova J; Vojtesek B; Staskova Z; Lukas J; Rejthar A; Kovarik J; Midgley CA; Gannon JV; Lane DP Oncogene, 1991, 6, 1699. [PubMed] [Google Scholar]
- [247].Itahana K; Mao H; Jin A; Itahana Y; Clegg HV; Lindström MS; Bhat KP; Godfrey VL; Evan GI; Zhang Y Cancer Cell, 2007, 12, 355. [DOI] [PubMed] [Google Scholar]
- [248].Go ML; Wu X; Liu XL Curr. Med. Chem, 2005, 12, 481. [DOI] [PubMed] [Google Scholar]
- [249].Montes de Oca Luna R; Wagner DS; Lozano G Nature, 1995, 378, 203. [DOI] [PubMed] [Google Scholar]
- [250].Jones SN; Roe AE; Donehower LA; Bradley A Nature, 1995, 378, 206. [DOI] [PubMed] [Google Scholar]
- [251].Laurie NA; Schin-Shih C; Dyer MA Curr. Cancer Drug Targets, 2007, 7, 689. [DOI] [PubMed] [Google Scholar]
- [252].Drakos E; Thomaides A; Medeiros LJ; Li J; Leventaki V; Konopleva M; Andreeff M; Rassidakis GZ Clin. Cancer Res, 2007, 13, 3380. [DOI] [PubMed] [Google Scholar]
- [253].Schuster K; Fan L; Harris LC Mol. Cancer Res, 2007, 5, 403. [DOI] [PubMed] [Google Scholar]
- [254].Steinman HA; Burstein E; Lengner C; Gosselin J; Pihan G; Duckett CS; Jones SN J. Biol. Chem, 2004, 279, 4877. [DOI] [PubMed] [Google Scholar]
- [255].Fridman JS; Hernando E; Hemann MT; de Stanchina E; Cordon-Cardo C; Lowe SW Cancer Res., 2003, 63, 5703. [PubMed] [Google Scholar]