

ABSTRACT
TAFRO syndrome is a novel disease concept characterized by Thrombocytopenia, Anasarca, myeloFibrosis, Renal dysfunction, Organomegaly, multiple lymphadenopathy and a histopathological pattern of atypical Castleman’s disease. A 58-year-old man was diagnosed as TAFRO syndrome by clinical and histopathological findings. After receiving intensive immunosuppressive therapy, his thrombocytopenia and anasarca had not improved. He developed complications such as methicillin-resistant Staphylococcus aureus sepsis, gastrointestinal bleeding, peritonitis caused by Stenotrophomonas maltophilia, gastrointestinal perforation, and disseminated candidiasis resulting in death. Autopsy revealed disseminated candidiasis and hemophagocytic lymphohistiocytosis, with no evidence of TAFRO syndrome. During treatment, we regarded his lasting thrombocytopenia and anasarca as insufficient control of TAFRO syndrome. However, the autopsy revealed that thrombocytopenia was caused by secondary hemophagocytic lymphohistiocytosis caused by over-immunosuppression. We reviewed the published literature to identify indicators of adequate treatment, which suggested improvement of platelet count and anasarca several weeks after initial therapy. This indicated that we could not depend on the platelet count and anasarca in acute medical care after initial treatment. We should treat TAFRO syndrome based on patients’ clinical status and obviate the risk of treatment-related complications caused by over-immunosuppression.
Key Words: TAFRO syndrome, immunosuppression, course of treatment, disseminated candidiasis, hemophagocytic lymphohistiocytosis
INTRODUCTION
TAFRO syndrome is a novel systemic inflammatory disorder characterized by Thrombocytopenia, Anasarca, myeloFibrosis, Renal dysfunction, Organomegaly, and multiple lymphadenopathy of mild degree, with histopathology of atypical Castleman’s disease.1,2 TAFRO syndrome was first reported in 2010,3 and newly proposed diagnostic criteria were published in 2016.4 The initial cases were reported from Japan,1 and there has been a recent increase in the number of reports worldwide.5,6 Some TAFRO syndrome patients have been successfully treated with early aggressive treatments, such as glucocorticoids and/or immunosuppressants including cyclosporine A, tocilizumab, and rituximab4,7; in contrast, some patients have died from the disease or complications.8-13 Here, we report the case of a Japanese man with TAFRO syndrome treated with glucocorticoid, tocilizumab, and intravenous immunoglobulin who subsequently died of disseminated candidiasis and secondary hemophagocytic lymphohistiocytosis because of over-immunosuppression. We were unable to determine whether the thrombocytopenia and anasarca were caused by insufficient treatment of TAFRO syndrome or over-immunosuppression. To identify clinical indicators of TAFRO syndrome, we reviewed past cases.
CASE REPORT
A 58-year-old Japanese man with lower abdominal pain and loss of appetite that had lasted for 2 weeks was admitted to our hospital. He underwent an upper gastrointestinal endoscopy without significant findings. His medical history showed aortic dissection (DeBakey IIIb) 8 years prior to admission, which was treated conservatively. Upon admission, his body temperature was 37.3°C. Physical examination revealed left abdominal tenderness with no swollen superficial lymph nodes and no leg edema. He had mild thrombocytopenia, hypoalbuminemia, and elevated C-reactive protein (CRP), alkaline phosphatase, γ-glutamyl transferase, creatinine, fibrinogen, and D-dimer levels. Serum soluble interleukin-2 receptor level and rheumatoid factor were also elevated. Antinuclear antibody index was 640 (speckled type) and anti-Sjögren’s-syndrome-related antigen A antibody index was 240. Anti-Sjögren’s-syndrome-related antigen B antibody and anti-platelet antibody were negative. Serum levels of β-D-glucan were normal (<6.0 pg/mL) and blood cultures were negative. Urine testing showed mild proteinuria without hematuria (Table 1). Enhanced whole-body computed tomography demonstrated small pleural effusion; however, there were no other abnormal findings without aortic dissection.
Table 1.
Laboratory data of the present case
| Complete blood count | Biochemistry | Virologic test | |||
| White blood cells | 9.000/µL | Total protein | 6.3 g/dL | HIV Ab | 0.1 s/co |
| Segmented | 86% | Albumin | 2.2 g/dL | HBs Ag | 0.00 IU/mL |
| Lymphocytes | 3% | BUN | 33.0 mg/dL | HCV Ab | 0.1 s/co |
| Monocytes | 8% | Creatinine | 1.89 mg/dL | IGRA | (-) |
| Atypical Lymphocytes | 3% | Uric acid | 6.4 mg/dL | HHV-8-DNA | (-) |
| Red blood cells | 3.95 × | Total bilirubin | 1.4 mg/dL | Immunologic test | |
| 106/µL | AST | 21 IU/L | IgG | 1,508 mg/dL | |
| Hemoglobin | 12.1 g/dL | ALT | 5 IU/L | IgA | 185 mg/dL |
| Hematocrit | 35.7% | LDH | 260 IU/L | IgM | 64 mg/dL |
| MCV | 90.4 fL | ALP | 851 IU/L | C3 | 98.9 mg/dL |
| MCH | 30.6 pg | γ-GTP | 206 IU/L | C4 | 20.8 mg/dL |
| MCHC | 33.9 g/dL | Na | 131 mEq/L | ANA | × 640 |
| Platelet | 86,000 /µL | K | 3.3 mEq/L | Speckled | |
| IPF | 21.8% | Cl | 92 mEq/L | RF | 37.1 IU/mL |
| Coagulation test | Ca | 7.6 mg/dL | Anti-SS-A | > 240 index | |
| Prothrombin time | 14.6 sec | CK | 123 IU/L | antibody | |
| APTT | 35.9 sec | CRP | 34.54 mg/dL | Anti-SS-B | 1.7 index |
| Fibrinogen | 877 mg/dL | Glucose | 81 mg/dL | antibody | |
| D-dimer | 3.17 µg/dL | Ferritin | 559 ng/mL | Anti-dsDNA | 0.7 IU/mL |
| Haptoglobin | 293 mg/dL | antibody | |||
| Urine test | Cytokines | PR-3 ANCA | < 1.0 U/mL | ||
| U-glucose | (-) | sIL-2R | 4,660 U/mL | MPO-ANCA | < 1.0 U/mL |
| U-protein | (1+) | IL-6 | 72.7 pg/mL | Anti-platelet | (-) |
| U-occult blood | (-) | (NR<4) | antibody | ||
| β-D-glucan | < 6.0 pg/ml | ||||
MCV: mean corpuscular volume, MCH: mean corpuscular hemoglobin, MCHC: mean corpuscular hemoglobin concentration, IPF: immature platelet fraction, APTT: activated partial thromboplastin, BUN: blood urea nitrogen, AST: aspartate aminotransferase, ALT: alanine aminotransferase, LDH: lactate dehydrogenase, ALP: alkaline phosphatase, γ-GTP: γ-glutamyl transferase, CK: creatine kinase, CRP: C-reactive protein, HIV Ab: human immunodeficiency virus antibody, HBs Ag: hepatitis B antigen, HCV Ab: hepatitis C virus antibody, IGRA: interferon gamma release assay, HHV-8: human herpesvirus-8, IgG, A, M: immunoglobulin G, A, M, C3, 4: complement 3, 4, ANA: anti-nuclear antibody, RF: rheumatoid factor, SS-A: Sjögren’s-syndrome-related antigen A, SS-B: Sjögren’s-syndrome-related antigen B, PR3-ANCA: proteinase-3 anti-neutrophil cytoplasmic antibody, MPO-ANCA: myeloperoxidase anti-neutrophil cytoplasmic, sIL-2R: soluble interleukin-2 receptor, IL-6: interleukin-6, NR: Normal Range.
After admission, the patient’s platelet count decreased gradually, ascites and pleural effusion increased, and renal insufficiency showed progression (blood urea nitrogen 46.7 mg/dL and creatinine 2.16 mg/dL). We conducted a bone marrow biopsy, which revealed severely hypocellular marrow. A whole body 18-F-fluorodeoxyglucose-positron emission tomography/computed tomography scan showed 18-F-fluorodeoxyglucose uptake by the left cervical and submandibular lymph nodes (Fig. 1).
Fig. 1.
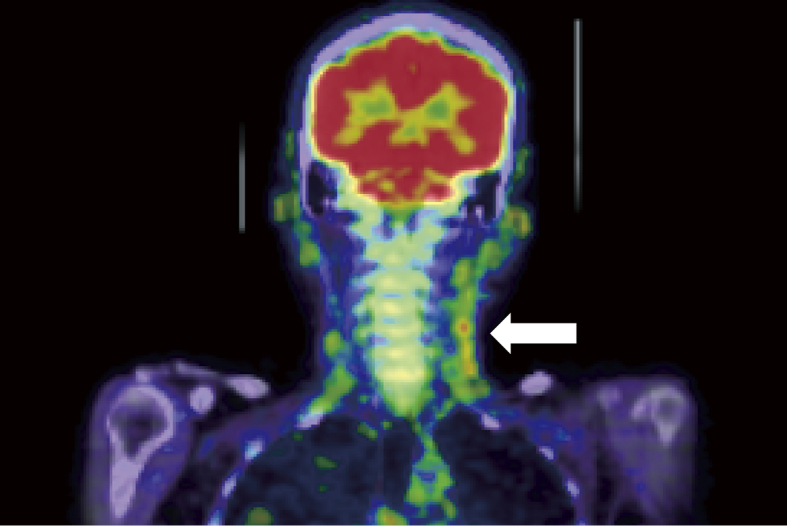
Positron emission tomography
Whole body 18-F-fluorodeoxyglucose-positron emission tomography findings at 11 days after admission. 18-F-fluorodeoxyglucose uptake was observed in the left cervical and submandibular lymph nodes (arrow).
We performed a cervical lymph node biopsy after prophylactic platelet transfusion, and began pulse therapy with methylprednisolone (1000 mg daily for 3 days) from day 16 because his general condition had worsened. The size of the lymph node was normal (8×5 mm). Although the number of germinal centers was preserved, all germinal centers were remarkably atrophic and contained high endothelial venules (Fig. 2A). High endothelial venules observed in interfollicular zones were surrounded by plasma cell infiltration (Fig. 2B).
Fig. 2.
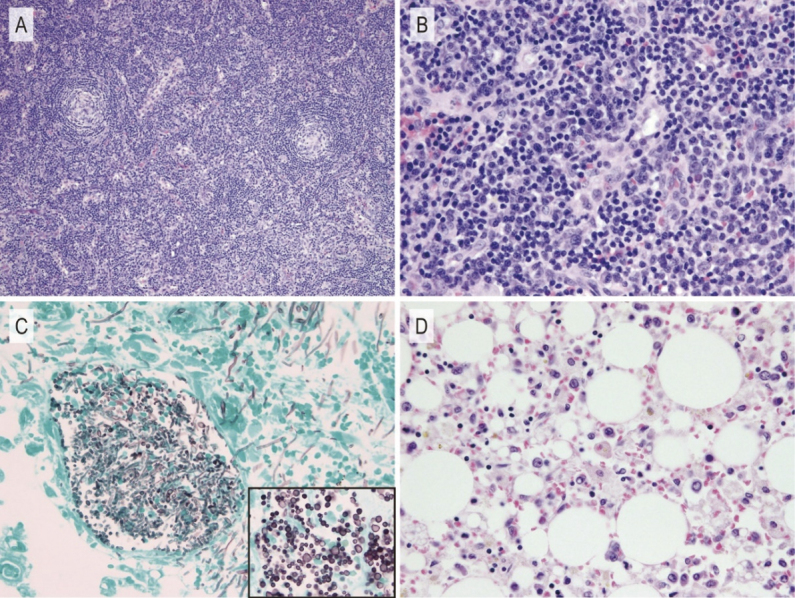
Pathological findings of the present case
Cervical lymph node biopsy sample on admission (A: ×100; B: ×400). Germinal centers are atrophic (A). Plasma cell infiltration is observed in the interfollicular zone around the high endothelial venules (B). Lung tissue from autopsy sample (C: ×400). Candida proliferated in the blood vessels and grew as pseudohyphae and yeast (inset). Bone marrow tissue from autopsy sample (D: ×400). The marked infiltration of macrophages and hemophagocytosis suggests hemophagocytic lymphohistiocytosis.
These findings were consistent with TAFRO syndrome (2). The serum level of interleukin-6 was 72.7 pg/mL (<4.0 pg/mL). Human herpes virus-8 was not found in the serum. Overall, based on the diagnostic criteria of TAFRO syndrome (4), the patients fulfilled the three major categories (anasarca, thrombocytopenia, and systemic inflammation) and two minor categories (Castleman’s disease-like features on lymph node biopsy and progressive renal insufficiency). We diagnosed the patient as TAFRO syndrome. After pulse therapy, the thrombocytopenia and massive ascites had not improved. The patient required multiple blood and platelet transfusions, but the beneficial effect was temporary. Tocilizumab (8 mg/kg body weight) was initiated on day 21 in addition to 1 mg/kg/day of prednisolone; however, the patient’s anasarca and renal function worsened. The patient was then transferred to the intensive care unit, put on a ventilator, and given continuous hemofiltration because of his deteriorating hemodynamic status. Five consecutive blood cultures taken during this period were sterile, indicating that he did not develop infection, although his TAFRO syndrome activity was still high. We restarted pulse therapy from day 25, added intravenous immunoglobulin from day 27, and initiated tocilizumab (8 mg/kg body weight) on day 29. However, the patient’s condition, including thrombocytopenia and massive ascites, deteriorated gradually. After the second administration of tocilizumab, he developed gastrointestinal bleeding and methicillin-resistant Staphylococcus aureus sepsis, which was treated with meropenem and teicoplanin. He developed bacterial peritonitis, and his ascitic fluid cultures showed Stenotrophomonas maltophilia on day 37; therefore, we changed antibiotics (daptomycin and tazobactam/piperacillin with ciprofloxacin added at a later timepoint). He developed gastrointestinal perforation on day 46 (Fig. 3).
Fig. 3.
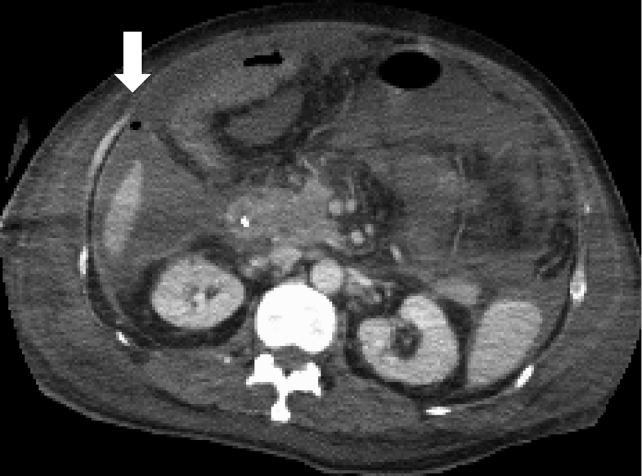
Computed tomography images
Computed tomography findings at 46 days after admission. The abdominal computed tomography scan shows free air in the ascites (arrow), which indicates gastrointestinal perforation.
His general condition made the perforation inoperable. On day 51, candidemia (blood cultures showed Candida glabrata and Candida lusitaniae) was observed and his β-D-glucan level was elevated to 193.3 pg/mL; therefore, we administered micafungin and trimethoprim-sulfamethoxazole (to treat Stenotrophomonas maltophilia on the basis of antimicrobial susceptibility testing), but he eventually died on day 57 (Fig. 4).
Fig. 4.

Clinical course after admission
The clinical course of the patient receiving intensive immunosuppressive drugs. After the introduction of methylprednisolone and tocilizumab, C-reactive protein began to decrease but did not reach <1.0 mg/dL. Thrombocytopenia deteriorated gradually despite multiple platelet transfusions. The patient developed methicillin-resistant Staphylococcus aureus sepsis, gastrointestinal bleeding, and peritonitis caused by Stenotrophomonas maltophilia treated by antibiotics. He eventually developed gastrointestinal perforation, leading to death.
Abbreviations: CRP: C-reactive protein, CPFX: Ciprofloxacin, DAP: Daptomycin, GI: gastrointestinal, IVIG: intravenous immunoglobulin, LVFX: Levofloxacin, MCFG: Micafungin, MEPM: Meropenem, mPSL: methylprednisolone, MRSA: methicillin-resistant Staphylococcus aureus, PIPC/TAZ: Piperacillin/Tazobactam, PLT: platelet, PSL: prednisolone, ST: Sulfamethoxazole Trimethoprim, TCZ: tocilizumab, TEIC: Teicoplanin, WBC: white blood cell.
An autopsy revealed large amounts of pleural effusion and ascites (1.5 L, 1.5 L, 5 L for left pleural effusion, right pleural effusion, and ascites, respectively). Disseminated candidiasis was histologically observed in multiple organs including bilateral lungs, pleura, the gastrointestinal tract, peritoneum, liver, both kidneys, heart, diaphragm, and thyroid gland. Candida had invaded the blood vessel walls and proliferated in the blood vessels (Fig. 2C) growing as yeast and pseudohyphae (Fig 2C, inset); however, we could not identify specific Candida species. In the bone marrow, the infiltration of macrophages was prominent and hemophagocytosis was observed. These features suggested hemophagocytic lymphohistiocytosis (Fig. 2D). The small and large intestines were ischemic and partially necrotic, but macroscopic perforation was not detected. Lymph node samples taken during autopsy indicated they were of normal size and findings suggestive of TAFRO syndrome were not observed.
DISCUSSION
During the course of treatment, the patient’s condition (including thrombocytopenia and massive ascites) deteriorated, and we were unable to decrease the immunosuppressive treatment. Based on the autopsy finding, the cause of the patient’s death was proven to be disseminated candidiasis. In the postmortem lymph node samples, the histological features suggestive of TAFRO syndrome had disappeared, suggesting that the treatment of TAFRO syndrome itself was successful. The thrombocytopenia was caused by secondary hemophagocytic lymphohistiocytosis syndrome, which might be induced by candidiasis and sepsis because of over-immunosuppression. Although we should have avoided over-immunosuppression, it is unknown what kind of clinical index is useful to judge the efficacy of treatment for TAFRO syndrome. We searched PubMed and the ICHUSHI web (a Japanese document database hosted by the Japan Medical Abstract Society) between May 2010 and September 2017 using “TAFRO syndrome” as a keyword. The exclusion criteria included: 1) not consistent with the 2015 diagnostic criteria for TAFRO syndrome as determined by the All Japan TAFRO Syndrome Research Group in the Research Program for Intractable Disease of the Ministry of Health, Labor and Welfare (MHLW) Japan4; 2) histological diagnosis was not provided (to exclude malignancies including lymphoma)— we defined histological diagnosis as atrophic germinal centers with penetrating hyalinized vessels and plasma cell proliferation after consultation with our pathologist (MN)14; 3) could not determine the start date of therapy and values provided were difficult to assess; and 4) not written in English or Japanese. We retrieved a total of 46 articles, 22 of which included 23 cases that met the inclusion criteria (Table 2). We investigated which clinical indicators showed clinical improvement after treatment. We checked platelet count, CRP, and anasarca (pleural effusion and ascites). We assessed the day on which CRP and platelet count began to improve. In addition, we recorded the days on which platelet count exceeded 100,000/µL, CRP was below 1.0 mg/dL, and anasarca resolved, because these points were important predictors for the improvement of TAFRO syndrome. Anasarca resolved and platelet counts recovered were noted in all survivors; however, there were no improvements in platelet count or anasarca in fatal cases, including our case.9 Of note, the improvement in platelet count and anasarca did not occur until several weeks after the initial therapy in most patients who showed improvement. The improvement in platelet count and resolution of anasarca are not acute phase indicators of TAFRO syndrome; therefore, they should not influence the strategy for managing TAFRO syndrome after initial therapy. CRP levels improved immediately after initial therapy. The possibility of complications should be considered if CRP initially declines but does not continue to be in the normal range, even during the administration of tocilizumab, as shown here and as previously reported.26-28,30 Our case showed that CRP decreased without reaching < 1.0 mg/dL, and that thrombocytopenia and anasarca did not improve until death. At autopsy, we could not identify any characteristics of TAFRO syndrome. Thrombocytopenia was caused by hemophagocytic syndrome. The clinical course of our case suggests that the incomplete decline of CRP was caused by complications rather than incomplete treatment for TAFRO syndrome. We could not determine the patient’s clinical index, which is useful when adjusting treatment with immunosuppressive drugs. The important finding from this study is that clinicians should pay attention to the balance between insufficient treatment and over-immunosuppression to avoid treatment-related complications, including opportunistic infection and adverse effects related to individual immunosuppressive therapy, such as gastrointestinal perforation associated with tocilizumab.33
Table 2.
Characteristics of reported cases with TAFRO syndrome*

*We defined day 0 as initial therapy.
†The items were defined as follows: PLT improved: platelet count started to increase, PLT >100,000/µL: platelet count exceeds 100,000/µL, CRP improved: CRP started to decrease, CRP <1.0 mg/dL: CRP below1.0 mg/dL, Anasarca disappeared: pleural effusion and ascites had disappeared.
‡The patient was diagnosed with TAFRO syndrome after antibiotic therapy for cholangitis and infective endocarditis.
§0 day indicates CRP improved soon after initial therapy.
PLT: platelet, CRP:C-reactive protein, M: male, F: female, NA: not available, mPSL: methylprednisolone, DEX: dexamethasone, PSL: prednisolone, IVIG: intravenous immunoglobulin, TCZ: tocilizumab, GC: Glucocorticoid, Cy A: cyclosporine A, RTX: rituximab, PE: plasma exchange, ETP: etoposide, CHOP: cyclophosphamide, adriamycin, vincristine and prednisolone, CHOEP: cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisolone, IVCY: intermittent pulse intravenous cyclophosphamide therapy, R-CVP: rituximab, cyclophosphamide, vincristine and prednisolone, CEPP: cyclophosphamide, etoposide, procarbazine, and prednisolone, CMV: Cytomegalovirus, PCP: Pneumocystis pneumonia, AHRU: acute hemorrhagic rectal ulcer, CNS: coagulase negative staphylococci, GI: Gastrointestinal, T: treatment, Ref: reference.
In conclusion, we report the case of a Japanese man with TAFRO syndrome treated with intensive immunosuppression therapy who subsequently died of disseminated candidiasis and secondary hemophagocytic lymphohistiocytosis syndrome. A retrospective review of TAFRO syndrome suggests that platelet count and anasarca take several weeks to improve after initial therapy.
ACKNOWLEDGMENTS
We thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
CONFLICT OF INTERESTS
The authors declared no conflict of interests for this article.
Abbreviations
- CRP
C-reactive protein
REFERENCES
- 1.Kawabata H, Takai K, Kojima M, et al. Castleman-Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly : a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop. 2013;53(1):57–61. [DOI] [PubMed]
- 2.Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220–226. doi: 10.1002/ajh.24242. [DOI] [PubMed]
- 3.Takai K, Nikkuni K, Shibuya H, Hashidate H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. Rinsho ketsueki. 2010;51(5):320–325. [Article in Japanese] [PubMed]
- 4.Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. 2016;103(6):686–692. doi: 10.1007/s12185-016-1979-1. [DOI] [PubMed]
- 5.Behnia F, Elojeimy S, Matesan M, Fajgenbaum DC. Potential value of FDG PET-CT in diagnosis and follow-up of TAFRO syndrome. Ann Hematol. 2017;96(3):497–500. doi: 10.1007/s00277-016-2875-8. [DOI] [PubMed]
- 6.Simons M, Apor E, Butera JN, Treaba DO. TAFRO syndrome associated with EBV and successful triple therapy treatment: case report and review of the literature. Case Rep Hematol. 2016;2016:4703608. [DOI] [PMC free article] [PubMed]
- 7.Kawabata H, Kadowaki N, Nishikori M, et al. Clinical features and treatment of multicentric castleman’s disease: a retrospective study of 21 Japanese patients at a single institute. J Clin Exp Hematop. 2013;53(1):69–77. [DOI] [PubMed]
- 8.Takai K, Nikkuni K, Momoi A, Nagai K, Igarashi N, Saeki T. Thrombocytopenia with reticulin fibrosis accompanied by fever, anasarca and hepatosplenomegaly: a clinical report of five cases. J Clin Exp Hematop. 2013;53(1):63–68. [DOI] [PubMed]
- 9.Masaki Y, Nakajima A, Iwao H, et al. Japanese variant of multicentric castleman’s disease associated with serositis and thrombocytopenia: a report of two cases: is TAFRO syndrome (Castleman-Kojima disease) a distinct clinicopathological entity? J Clin Exp Hematop. 2013;53(1):79–85. [DOI] [PubMed]
- 10.Tadokoro A, Kanaji N, Hara T, et al. An uncharted constellation: TAFRO syndrome. Am J Med. Sep. 2016;129(9):938–941. doi: 10.1016/j.amjmed.2016.05.010. [DOI] [PubMed]
- 11.Allegra A, Rotondo F, Russo S, et al. Castleman-Kojima disease (TAFRO syndrome) in a Caucasian patient: a rare case report and review of the literature. Blood Cells Mol Dis. 2015;55(3):206–207. doi: 10.1016/j.bcmd.2015.06.013. [DOI] [PubMed]
- 12.Ozawa Y, Yamamoto H, Yasuo M, et al. Two patients with TAFRO syndrome exhibiting strikingly similar anterior mediastinal lesions with predominantly fat attenuation on chest computed tomography. Respir Investig. 2017;55(2):176–180. doi: 10.1016/j.resinv.2016.10.003. [DOI] [PubMed]
- 13.Awano N, Inomata M, Sonoda Y, et al. A case of multicentric castleman’s disease of mixed-type, which showed constellation of symptoms, i.e., thrombocytopenia, anasarca, anemia, fever, myelofibrosis, and lymphadenopathy. J Clin Exp Hematop. 2013;53(1):101–105. [DOI] [PubMed]
- 14.Carbone A, Pantanowitz L. TAFRO syndrome: an atypical variant of KSHV-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):171–172. doi: 10.1002/ajh.24274. [DOI] [PubMed]
- 15.Kawashima M, Usui T, Okada H, et al. TAFRO syndrome: 2 cases and review of the literature. Mod Rheumatol. 2017;27(6):1093–1097. doi: 10.3109/14397595.2015.1059982. [DOI] [PubMed]
- 16.Inoue M, Ankou M, Hua J, Iwaki Y, Hagihara M, Ota Y. Complete resolution of TAFRO syndrome (thrombocytopenia, anasarca, fever, reticulin fibrosis and organomegaly) after immunosuppressive therapies using corticosteroids and cyclosporin A: a case report. J Clin Exp Hematop. 2013;53(1):95–99. [DOI] [PubMed]
- 17.Yamaga Y, Tokuyama K, Kato T, et al. Successful treatment with cyclosporin A in tocilizumab-resistant TAFRO syndrome. Intern Med. 2016;55(2):185–190. doi: 10.2169/internalmedicine.55.4710. [DOI] [PubMed]
- 18.Suzuki K, Nakamura K, Kasuya T, et al. A case of TAFRO syndrome successfully treated with intravenous cyclophosphamide therapy. Nihon Naika Gakkai Zasshi. 2016;105: 2417–2423. [Article in Japanese] [PubMed]
- 19.Iwaki N, Sato Y, Takata K, et al. Atypical hyaline vascular-type castleman’s disease with thrombocytopenia, anasarca, fever, and systemic lymphadenopathy. J Clin Exp Hematop. 2013;53(1):87–93. [DOI] [PubMed]
- 20.Fujiwara S, Mochinaga H, Nakata H, et al. Successful treatment of TAFRO syndrome, a variant type of multicentric Castleman disease with thrombotic microangiopathy, with anti-IL-6 receptor antibody and steroids. Int J Hematol. 2016;103(6):718–723. doi: 10.1007/s12185-016-1978-2. [DOI] [PubMed]
- 21.Ozawa T, Kosugi S, Kito M, et al. Efficacy of rituximab for TAFRO syndrome, a variant type of multicentric Castleman’s disease. Rinsho Ketsueki. 2014;55(3):350–355. [Article in Japanese] [PubMed]
- 22.Nagai Y, Ando S, Honda N, et al. TAFRO syndrome showing cholangitis on liver biopsy. Rinsho Ketsueki. 2016;57(12):2490–2495. doi: 10.11406/rinketsu.57.2490. [DOI] [PubMed]
- 23.Tedesco S, Postacchini L, Manfredi L, et al. Successful treatment of a Caucasian case of multifocal Castleman’s disease with TAFRO syndrome with a pathophysiology targeted therapy: a case report. Exp Hematol Oncol. 2015;4(1):3. doi: 10.1186/2162-3619-4-3. [DOI] [PMC free article] [PubMed]
- 24.Nara M, Komatsuda A, Itoh F, et al. Two Cases of thrombocytopenia, anasarca, fever, reticulin fibrosis/renal failure, and organomegaly (TAFRO) syndrome with high serum procalcitonin levels, including the first case complicated with adrenal hemorrhaging. Intern Med. 2017;56(10):1247–1252. doi: 10.2169/internalmedicine.56.7991. [DOI] [PMC free article] [PubMed]
- 25.Sakashita K, Murata K, Inagaki Y, Oota S, Takamori M. An anterior mediastinal lesion in TAFRO syndrome showing complete remission after glucocorticoid and tocilizumab therapy. Respirol Case Rep. 2016;4(5):e00173. doi: 10.1002/rcr2.173. [DOI] [PMC free article] [PubMed]
- 26.Kubokawa I, Yachie A, Hayakawa A, et al. The first report of adolescent TAFRO syndrome, a unique clinicopathologic variant of multicentric Castleman’s disease. BMC Pediatr. 2014;14:139. doi: 10.1186/1471-2431-14-139. [DOI] [PMC free article] [PubMed]
- 27.Kawabata H, Kotani S, Matsumura Y, et al. Successful treatment of a patient with multicentric Castleman’s disease who presented with thrombocytopenia, ascites, renal failure and myelofibrosis using tocilizumab, an anti-interleukin-6 receptor antibody. Intern Med. 2013;52(13):1503–1507. [DOI] [PubMed]
- 28.Hiramatsu S, Ohmura K, Tsuji H, et al. Successful treatment by rituximab in a patient with TAFRO syndrome with cardiomyopathy. Nihon Rinsho Meneki Gakkai Kaishi. 2016;39(1):64–71. doi: 10.2177/jsci.39.64. [DOI] [PubMed]
- 29.Yasuda S, Tanaka K, Ichikawa A, et al. Aggressive TAFRO syndrome with reversible cardiomyopathy successfully treated with combination chemotherapy. Int J Hematol. 2016;104(4):512–518. doi: 10.1007/s12185-016-2025-z. [DOI] [PubMed]
- 30.Konishi Y, Takahashi S, Nishi K, et al. Successful treatment of TAFRO syndrome, a variant of multicentric Castleman’s disease, with cyclosporine A: possible pathogenetic contribution of interleukin-2. Tohoku J Exp Med. 2015;236(4):289–295. doi: 10.1620/tjem.236.289. [DOI] [PubMed]
- 31.Takasawa N, Sekiguchi Y, Takahashi T, Muryoi A, Satoh J, Sasaki T. A case of TAFRO syndrome, a variant of multicentric Castleman’s disease, successfully treated with corticosteroid and cyclosporine A. Mod Rheumatol. 2016:1–5. (Epub ahead of print) [DOI] [PubMed]
- 32.Sakai K, Maeda T, Kuriyama A, Shimada N, Notohara K, Ueda Y. TAFRO syndrome successfully treated with tocilizumab: a case report and systematic review. Mod Rheumatol. 2018;28(3):564–569. doi: 10.3109/14397595.2015.1120389. [DOI] [PubMed]
- 33.Xie F, Yun H, Bernatsky S, Curtis JR. Brief report: risk of gastrointestinal perforation among rheumatoid arthritis patients receiving tofacitinib, tocilizumab, or other biologic treatments. Arthritis Rheumatol. 2016;68(11):2612–2617. [DOI] [PMC free article] [PubMed]