

Abstract
Polarized, non-overlapping, regularly spaced, tiled organization of radial glial cells (RGCs) serves as a framework to generate and organize cortical neuronal columns, layers, and circuitry. Here, we show that mediator of cell motility 1 (Memo1) is a critical determinant of radial glial tiling during neocortical development. Memo1 deletion or knockdown leads to hyperbranching of RGC basal processes and disrupted RGC tiling, resulting in aberrant radial unit assembly and neuronal layering. Memo1 regulates microtubule (MT) stability necessary for RGC tiling. Memo1 deficiency leads to disrupted MT minus-end CAMSAP2 distribution, initiation of aberrant MT branching, altered polarized trafficking of key basal domain proteins such as GPR56, and thus aberrant RGC tiling. These findings identify Memo1 as a mediator of RGC scaffold tiling, necessary to generate and organize neurons into functional ensembles in the developing cerebral cortex.
Keywords: Memo1, Progenitors, Corticogenesis, Radial glia, Microtubule minus end, MADM, Autism
Graphical Abstract
eTOC Blurb
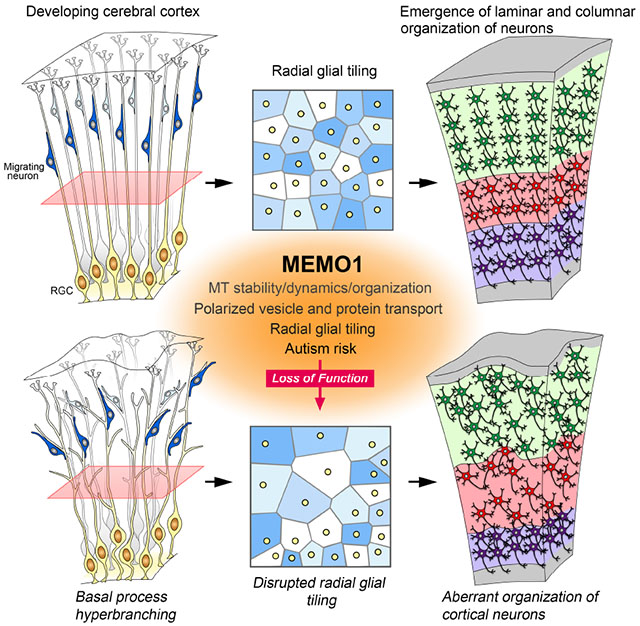
Tiled radial glial cells (RGCs) provide an instructive template for the formation of the cerebral cortex. Memo1 regulates RGC tiling underlying radial unit formation and neuronal laminar organization. Mutations in MEMO1 and resultant cortical malformations may contribute to autism risk.
Introduction
Polarized radial glial cells (RGCs) provide a template for the formation of the cerebral cortex. The polarized epithelial morphology of a RGC is characterized by a periventricular soma anchored to the ventricular surface with a short apical process and a long, thin basal process extending across the width of the cortical wall and attached to the pial membrane. Radial glial basal processes are non-randomly arranged, regularly interspaced, and do not overlap, thus forming a characteristically tiled RGC scaffold (Hansen et al., 2010; Misson et al., 1991a&b, Rakic, 1972). Tiled RGCs divide symmetrically and asymmetrically to expand the pool of progenitors and to generate neurons, respectively. During neurogenic period, RGCs asymmetrically divide to generate cortical neurons and neurogenic intermediate progenitors or outer RGCs (oRGCs). Newborn neurons use RGCs to guide their migration and placement in the developing cerebral cortex (Rakic, 1972). Migration of clonally related neurons along radial glial basal processes contributes to the formation of functional cortical columns (or radial units) and laminar organization of neurons (Jones and Rakic, 2010; Maruoka et al., 2017; Rakic, 1988, 2007, Yu et al., 2009, 2012). The tiled organization of RGCs thus serves as a blueprint to guide the formation of cortical columns and neuronal layers, the fundamental units of cortical neuronal circuitry (Evsyukova et al., 2013; Gao et al., 2014; Molnár, 2013; Noctor et al., 2001; Yu et al., 2009, 2012).
Abnormalities in RGC development, organization, proliferation, and neuron-RGC interactions lead to aberrant generation, placement and connectivity of neurons in human cerebral cortex. This is an underlying cause of cortical malformations (e.g. microcephaly and lissencephaly) and complex neurodevelopmental disorders such as epilepsy, schizophrenia, and autism spectrum disorder (ASD) (Hu et al., 2014; Kwan et al., 2012; Stoner et al., 2014; Sun and Hevner, 2014). Although the functions of radial glial progenitors in the developing cerebral cortex depend on the systematically tiled organization of RGCs, we are yet to fully understand the mechanisms regulating this arrangement of radial progenitors and how this tiled organization of RGCs enables the construction of columnar and laminar units of cortical neurons.
Microtubules (MT) are crucial for the establishment and maintenance of polarized cell morphology. MTs, built from repeating αβ-tubulin heterodimers, undergo distinct patterns of post translational modifications (tyrosination, acetylation, glutamylation, glycylation) to enable them to adopt specific subcellular functions (Garnham and Roll-Mecak, 2012; Song and Brady, 2015). While stable MTs, marked by acetylation, act as substrates essential for morphological maintenance and vesicle transport, dynamic MTs, which are tyrosinated, allow transient morphological remodeling. The balance between stable and dynamic MTs is orchestrated by MT-associated proteins and post-translational modification of tubulin (Akhmanova and Steinmetz, 2015). MT plus-ends are captured at the cell periphery by Adenomatous polyposis coli (APC), CAP-Gly domain containing linker proteins (CLIPs) and cytoplasmic linker associated proteins (CLASPs), while the minus-ends are tethered to the γ-tubulin ring complex (γ-TuRC) and centrosomes or Golgi apparatus, thus generating directional MT arrays in cells (Akhmanova and Steinmetz, 2015; Muroyama and Lechler, 2017). Appropriate regulation of MT dynamics and array formation is fundamentally important for radial progenitor development, organization, and function during corticogenesis (Cappello et al., 2012; Eom et al., 2011; Rasin et al., 2007; Yokota et al., 2009).
Mediator of cell motility 1 (Memo1), an adaptor protein at the hub of multiple signaling pathways critical for radial progenitor development (e.g., Neuregulin1 (NRG1)-ErbB2/ErbB4, GSK3, and APC), can modulate MT dynamics necessary for cellular morphological plasticity (Jereb et al., 2018; Marone et al., 2004; Zaoui et al., 2010). Memo1 signaling thus provides a unique avenue to decipher the mechanisms that determine the tiled organization of radial glial progenitors during development and their impact on the neuronal organization in the cerebral cortex. Further, de novo mutations in MEMO1 have recently been identified in autism probands (De Rubeis et al., 2014; Iossifov et al., 2014; Nguyen et al., 2017), suggesting a potential connection between Memo1 dysfunction and neurodevelopmental disorders. We therefore examined Memo1’s role in the tiling of radial progenitors and the resultant functional organization of neurons in the cerebral cortex.
Results
Memo1 Expression in the developing cerebral cortex
In situ hybridization and immunohistochemical analysis reveal prominent expression of Memo1 in the ventricular zone (VZ) and the cortical plate (CP) of the mouse embryonic cerebral cortex (E16) (Figures S1A–B). In vitro, Memo1 is distributed throughout the RGC, including the endfeet and basal process (Figure S1C). RC2+ radial progenitors (Figure S1D) and Tbr2+ intermediate progenitors express Memo1 (Figure S1E). Further, Memo1 expression pattern is highly conserved between mouse and human. mRNA expression profiling of the human fetal brain (15, 16, and 21 post conception weeks; http://brainspan.org/) indicates that MEMO1 expression is enriched in the VZ and CP of human embryonic cortex (Figure S1F). The prominent expression pattern of Memo1 in RGCs of the developing cerebral cortex suggests a potential role for Memo1 in radial progenitor organization.
Generation of Memo1 conditional allele
To define the role of Memo1 in radial progenitor organization during corticogenesis, we generated a conditional Memo1 allele in which exon 2 of Memo1 gene was flanked by loxP sites (Figures S2A–B). Memo1Lox/Lox mice were then crossed with Emx1-Cre or hGFAP-Cre lines to delete Memo1 from radial progenitors in the embryonic cortex. The Emx1-Cre or hGFAP-Cre transgenic lines enable Cre recombinase-mediated deletion of Memo1 allele in cortical progenitors from E10.5 and E13.5, respectively (Gorski et al., 2002; Yokota et al., 2009). Emx1-Cre is selective to cerebral cortex whereas hGFAP-Cre is active in other CNS regions including cerebellar Bergmann glia. A reduction of Memo1 protein level in the embryonic cerebral cortices is evident following Cre-mediated inactivation (Figures S2C–D).
Generation of RG-Brainbow mice
To analyze the morphology and organization of embryonic RGCs, it is necessary to visualize individual RGCs, sparsely and distinguishably, from neighboring radial progenitors. Towards this goal, we generated a radial progenitor-specific Brainbow mice (RG-Brainbow) to differentially label RGCs in a Cre-dependent manner in the embryonic cortex (Figure S2E–H, S3).
Effect of Memo1 deletion on RGC organization in the developing cerebral cortex
To examine if Memo1 function is required for the tiled organization and function of RGCs in the developing neocortex, we generated Memo1Lox/Lox; RG-Brainbow; Emx1-Cre (Memo1 cKOEmx) and Memo1Lox/Lox; RG-Brainbow; hGFAP-Cre (Memo1 cKOhGFAP) mice and analyzed their brains at E16 and P0. In embryonic cortices of control mice (controlEmx [Memo1Lox/+; RG-Brainbow; Emx1-Cre] or controlhGFAP [Memo1Lox/+; RG-Brainbow; hGFAP-Cre]), typical RGCs with single basal processes terminally branching at the pial surface were evident (Figure 1A–1L). In contrast, radial processes of Memo1 cKOEmx and Memo1 cKOhGFAP RGCs branch extensively (Figure 1A–1L). oRGCs also showed excessively branched basal processes in Memo1 cKOEmx cortex (Figure S4A–F). Immunolabeling with RGC-specific RC2 antibodies revealed similar increases in the aberrantly branched and misoriented radial fibers in Memo1 cKOEmx and Memo1 cKOhGFAP cortices (Figures 1M–1P). Radial glial endfeet branching is increased in Memo1 cKO and their pial attachment is perturbed (Figures 1I). Furthermore, acute inactivation of Memo1 in the RGCs, using Memo1-specific shRNA (Marone et al., 2004), disrupted the polarity and branching of RGC basal process (Figure 2). Live imaging indicates that, in contrast to control RGCs that have a single and stable basal process, Memo1-deficient RGCs display multiple, continuously extending/retracting branches with aberrant directionality (Figures 2A–2C and Movie S1). These data indicate that Memo1 is a critical determinant of the growth and organization of primary basal processes underlying radial progenitor tiling.
Figure 1: Disrupted organization of Memo1-deficient RGCs.

(A-D) Radial progenitor morphology in Memo1Lox/+; RG-Brainbow; Emx1-Cre (control) (A and C) and Memo1Lox/Lox; RG-Brainbow; Emx1-Cre (Memo1 cKOEmx) (B and D) mice at E16 (A and B) and P0 (C and D). High-magnification images of basal processes (yellow boxes) are shown in right panels. Control RGCs show single polarized basal processes (arrow, A, C). Mutant RGCs display extensive branching in their basal processes (arrowheads, B, D). (E-H) Representative images of RG basal processes in control (E and G) and Memo1 cKOEmx (F and H) cortices at E16 (E and F) and P0 (G and H). (I) Quantification of the basal process branching (top), endfeet branches per RGC (bottom), and % RGCs with pial attachment (bottom). Inset (I’) shows representative images RGC endfeet in control and Memo1 cKOEmx cortices at E16. Yellow dotted lines indicate the pial surface. Memo1 cKO RGC endfeet branches not attached to the pial surface are indicated by cyan arrowheads. (I”) Pial membrane is intact in both control and Memo1 cKOEmx cortices. For the basal process branching, branch number / 100 μm was quantified and used as branching index. Pial attachment was calculated by dividing the number of RGCs without pial attachment by the total number of RGCs counted. Data shown are mean ± SEM from 33 (control) and 37 (Memo1 cKOEmx) cells [E16] and 52 (control) and 51 (Memo1 cKOEmx) cells [P0] from 5 or more mice per group. (J-L) Radial progenitor morphology in P0 Memo1Lox/+; RG-Brainbow; hGFAP-Cre (J) and Memo1Lox/Lox; RG-Brainbow; hGFAP-Cre (Memo1 cKOhGFAP, K and L) cortices. High-magnification images of basal processes from J and K are shown in J’ and K’. While control RGCs show single polarized basal processes (arrow, J’), mutant RGCs display extensive basal process branching (arrowheads, K’, L). (M-P) RC2 immunostaining of E16 (M and N) and P0 (O and P) dorsal cortices. High-magnification images of basal process array (boxes) are shown in bottom panels. Memo1 mutants show disrupted arrayed organization and branching of basal processes (arrowheads) while control has regularly interspaced basal process array (arrows). (Q-T) The VZ of E16 (Q and R) and P0 (S and T) control (Q and S) and Memo1 cKOEmx (R and T) cortex showing aberrant spatial arrangement of radial progenitors in Memo1 cKO. (U) Co-labeling of E16 Memo1Lox/+; Emx1-Cre and Memo1Lox/Lox; Emx1-Cre dorsal cortices with anti-Pax6 and Tbr2 antibodies. Pax6+ and Tbr2+ progenitors delaminate and form ectopias at the IZ in the Memo1-deficient cortex (arrowheads). (V and W) Co-immunolabeling of E16 dorsal cortices with anti-Pax6 and anti-BrdU (V) or anti-Tbr2 and anti-BrdU (W) antibodies after pulse labeling of proliferating progenitor populations with BrdU (50 mg/kg, 1 hr. before sacrifice). (X) Quantification of progenitor proliferation and delamination. Data shown are mean ± SEM (n=3 mice/genotype). Memo1 cKOEmx cortex shows increased number of ectopic progenitors that remain proliferative. Student’s t-test; *P < 0.05 and ***P < 0.001. Scale bar: A, B (18 μm), C, D, I”, M-P (30 μm), E-H, J-L (8 μm), I’ (15 μm), Q-T (10 μm), U (35 μm), and V, W (20 μm). See also Figures S1–4 and S6–8.
Figure 2: Effect of Memo1 knockdown on RGCs.

(A and B) Real-time imaging of RGCs following acute inactivation of Memo1. Control (A) or Memo1 shRNA (B) were co-electroporated with BLBP-GFP at E14 and BLBP-GFP+ RGCs were live-imaged at E15. Basal processes of Memo1-deficient RGCs lose their polarity and randomly sprouted multiple branches along the primary basal process. Time elapsed is indicated in hours and minutes. (C) ShRNA-mediated knockdown of Memo1. Knockdown efficacy of shRNAs (fold change compared to control) was examined by quantitative real-time PCR. Data shown are mean ± SEM (n=3 for each condition). (D) Quantification of the basal process branching events / hour. Data shown are mean ± SEM (n=13 cells from 3 mice for each condition). (E and F) Representative examples of the morphology of control (E) and Memo1-deficient (F) RGCs. shMemo1-treated RGCs do not form polarized basal processes and show excessive branching. (G) Quantification of the basal process branching. Data shown are mean ± SEM (n=14 cells from 3 mice for each condition). Student’s t-test; **P < 0.01 and ***P < 0.001. Scale bar: A, B, D, and E (10 μm). See also Supplemental Movie 1.
The excessive basal process branching altered the tiling of RGC scaffold in Memo1-deficient neocortex. RC2-immunolabeling of E16 horizontal sections revealed regularly interspaced array of RGC basal processes in control cortex and the lack of this tiled organization in Memo1 cKOEmx cortex (Figures 3A–B). The Voronoi domain analysis of RG basal processes in horizontal sections showed that the area of Voronoi cells surrounding each basal process, indicative of tiling and regularity, is altered and non-uniform in Memo1 cKOEmx neocortex (Figures 3B–C). Together, these observations further suggest that the regular, tiled organization of RG basal processes is perturbed in the absence of Memo1 function.
Figure 3: Altered RGC tiling, neuron-radial glial interactions, and radial migration in Memo1 cKO neocortex.

(A) Horizontal sections of cortex were used to analyze RGC scaffold tiling. (B) RC2 immunolabeling of horizontal sections from control and Memo1 cKOEmx cortices (E16), showing altered tiling of RGC basal processes in Memo1 cKO (top). Voronoi domains of RC2+ fibers (bottom) from control and Memo1 cKO cortices were plotted to illustrate the difference in the RGC basal process tiling. Each Voronoi cell is color coded according to its area. (C) Quantification of Voronoi domains. Data shown are mean ± SEM (n=4 mice/genotype). (D-G) MADM-based clonal analysis of Memo1 cKO radial columns. Isolated RGCs in MADM-control or MADM-Memo1Lox/Lox cortices were electroporated with Cre-expression vectors at E14 and single radial units containing mother RGC and its progenies were visualized at E16 (D, right). Representative images of MADM-control (E) and MADM-Memo1 cKO (F) radial units. MADM-Memo1 cKO cells are pseudocolored. Distance between neuronal progenies and the mother RGC’s basal process is increased in the MADM-Memo1 cKO clones. Quantification of neuronal distribution along RGC basal processes (G, left) and the average neuron-RG distance (G, right). Data shown are mean ± SEM from 30 (MADM-control) and 37 (MADM-Memo1 cKO) radial units from 4 mice/genotype. (H-K) Visualization of neuronal migration along RGC basal processes. Migrating neurons in E16 cortex were visualized by pulse labeling with FLASH Tag. FLASH Tag+ neurons (cyan) attached to Brainbow+ RGCs are shown (H). Neurons not in contact with the RGC process are pseudocolored (gray). Representative images of migrating neurons (arrowheads) in control (I) and Memo1 cKOEmx (J). Multiple neurons associated with the primary RG basal processes in control (I, arrowheads). However, in Memo1 cKO RGCs, many neurons are attached to the aberrantly branched processes (J, arrows) as well as to the main basal process (J, arrowheads), resulting in altered trajectories of neuronal migration. (K) Quantification of changes in the percentage of RGCs with neurons on basal process branches. Data shown are mean ± SEM from 109 (control) and 116 (Memo1 cKO) cells from 4 mice/genotype. (L-R) Live imaging of neuronal migration in control and Memo1 cKOEmx embryonic cortex. Control (L) and Memo1 cKOEmx (M) neurons at each time point are pseudo colored. Time is indicated in minutes. (N-O) Trajectories of 16 representative control (N) and Memo1 cKO (O) neurons are indicated. Neurons in Memo1 cKO cortex migrate in non radial directions. (P-R) Quantification of changes in radial migration of Memo1 cKO neurons. Quantification of average speed (P) and pause duration (Q). (R) Measurement of changes in the angle of migration trajectory (a) towards the pial surface (radial: 80°-100° non-radial: <80°or >100°). Data shown are mean ± SEM (30 cells from 3 mice/group). Student t-test; *P < 0.05, **P < 0.01 and ***P < 0.001. Scale bar: B (5.8 μm), D (140 μm), E, F, H (23.4 μm), and I, J (11.7 μm), L-M (40 μm), and N-O (20 μm). See also Figures S2–5 and Supplemental Movie 2.
Memo1 deletion also affected the spatial organization of RGC soma in the VZ. Somata of RGCs are regularly interspaced in the VZ of E16 and P0 control cortex (Figure 1Q and 1S). However, in the Memo1 cKOEmx cortex, mosaic organization of RGCs is lost and they formed abnormal clusters within the VZ (Figure 1R and 1T). Further, compared to controls, a substantial number of Pax6+ progenitors and Tbr2+ intermediate progenitors were ectopically located in the IZ of the Memo1 cKOEmx cortex (Figure 1U). Bromodeoxyuridine (BrdU) pulse labeling indicates that these delaminated Memo1-deficient Pax6+ and Tbr2+ progenitors remain proliferative and Memo1 deletion did not affect their overall rate of proliferation (% proliferation of delaminated Pax6+ progenitors: Control 45.6±6.8, Memo1 cKO 48.4±2.3; % proliferation of delaminated Tbr2+ progenitors: Control 35.7±5.2, Memo1 cKO 41.6±1.2; Figures 1V–1X). However, Brainbow expression and GFAP immunostaining revealed an increase of cortical astrocytes in Memo1-deficient cerebral cortex (Figure S4G–N), suggesting disrupted progression of radial progenitor development and aberrant astrogliogenesis in Memo1 cKOEmx or hGFAP neocortex. Taken together, these findings suggest a fundamental role for Memo1 in the appropriate organization of RGCs in the developing neocortex.
Disrupted neuron-radial glial interactions in Memo1 cKO
We next examined the effect of defective RGC tiling on neuron-RGC interactions in Memo1 cKOEmx cortex. We used Mosaic Analysis with Double Markers (MADM)-based lineage tracing system (Beattie et al., 2017; Hippenmeyer et al., 2010; Zong et al., 2005) to evaluate the radial unit relationship between a clonally related RGC and its neuronal progenies. We expressed Cre recombinase in isolated RGCs of E14 MADM-11GT/TG; Memo1Lox/+ (MADM-control) or MADM-11GT/TG; Memo1Lox/Lox (MADM-Memo1 cKO) dorsal cortex. Since we applied MADM as a lineage tracer without linking Memo1-floxed allele with the TG or GT MADM cassettes (Beattie et al., 2017), all of the MADM-labeled (green, red, or yellow) clones in MADM-control or MADM-Memo1 cKO cortices are Memo1Lox/+ or Memo1Lox/Lox, respectively. This isolated radial unit labeling results from deletion of Memo1 in a single RGC. At E16, we analyzed MADM-positive, Memo1-deleted clones for spatial distribution of neuronal progenies along the basal process of the mother RGCs (Figure 3D). Compared to control, neurons in MADM-Memo1 cKO clones distributed significantly away from the main basal process of the mother RGC (Figures 3E–3G), suggesting altered radial trajectory of migrating newborn neurons in Memo1 cKOEmx cortex. To further clarify the altered neuron-RGC interactions, we visualized newborn neurons migrating along RGC basal processes with FLASH tag (Telley et al., 2016) in control and Memo1 cKOEmx embryonic cortex (E16). In control cortices, FLASH tag-labeled arrays of neurons were attached to RGC basal processes, indicative of the characteristic RGC scaffold-guided radial neuronal migration (Figures 3H–I). In contrast, in Memo1 cKOEmx cortices, migrating neurons associated not only with the RGC’s primary basal process but also with its aberrantly branched, lateral processes (Figures 3J–K), indicating aberrant neuronal trajectories in the Memo1 cKOEmx cortex. Correspondingly, live imaging of migrating neurons in the intermediate zone of embryonic cortices show that majority of control neurons migrate in a radial trajectory, whereas neurons in Memo1 cKO cortex often branch, pause during migration and move in non-radial trajectories (Figure 3L–R, Movie S2).
Laminar organization of neurons in Memo1 cKO
Consistent with disrupted RGC scaffold tiling, neuron-RGC interactions, and migration, we found that laminar organization of neurons is altered in Memo1 cKO cortex. Cux1+ or Pou3f2+ neurons, which are normally present in layers II-IV or layers II-III and V, respectively, ectopically localize to layer I in Memo1 cKOEmx cortex (Figures 4A, 4B, and 4D). Ctip2+ Layer V neurons extend into upper layers (Figures 4A and 4D), and Tbr1+ layer VI is thinner (Figures 4C–D). In contrast to the abnormal layering of radially migrating projection neurons, laminar distribution of tangentially migrating inhibitory interneurons was not significantly affected in Memo1 cKOEmx neocortex (Figure S5).
Figure 4: Aberrant neuronal layer organization in Memo1-deficient neocortex.

(A-D). P30 cerebral cortices from control and Memo1 cKOEmx mice were immunolabeled with Cux1 and Ctip2 (A), Pou3f2 (B) and Tbr1 (C) antibodies to illustrate abnormal neuronal layering in Memo1 cKO neocortex (somatosensory area). Quantification of neuronal distribution (D). Data shown are mean ± SEM (n=3 mice/genotype). (E-J) 3D nearest neighbor distance (NND) analysis of neuronal columnar organization. 3D reconstruction of the Cux1+ (E), Ctip2+ (G), and Tbr1+ (I) layers obtained by volumetric confocal scanning of somatosensory cortex area. Regularity of neuronal distribution in each plane (x-y, x-z, and y-z) was evaluated by NND analysis. Radial arrangement of neurons (arrowheads; E, G, I) are altered in Memo1 cKO. Quantification of the changes in the average distance between neurons (F, H, J). Data shown are mean ± SEM (n=4 mice/genotype). Student’s t-test; *P < 0.05, **P < 0.01, and ***P < 0.001. Scale bar: A-C (50 μm). See also Figures S4–8.
Overall density of different layer specific cortical neurons was not affected (Cux1: Control 1072.8±46.9, Memo1 cKO 1067.4±26.7; Ctip2: Control 604.5±29.6, Memo1 cKO 588.4±33.3; Pou3f2: Control 493.8±23.2, Memo1 cKO 501.2±28.9; Tbr1: Control 661.2±43.9, Memo1 cKO 681.5±59.9; cells/mm2, n=3 brains/group), indicating the generation and survival of neurons were not altered in Memo1 cKOEmx.
To analyze if the perturbed tiled organization of RGCs affects the columnar distribution of neurons in the Memo1-deficient cerebral cortex, we carried out 3D nearest neighbor distance (NND) analysis on radially aligned neuronal columns extracted from volumetric confocal scans of different neuronal layers. The average inter-neuronal distance (x, y, z) of Cux1+, Ctip2+, and Tbr1+ neurons in the Memo1 cKOEmx cortex is altered (Figures 4E–J), indicating that Memo1-inactivation dysregulates the columnar organization of cortical neurons.
Focal inactivation of Memo1 in RGCs by electroporation of Cre into Memo1Lox/Lox embryonic cortices resulted in foci of disrupted neuronal layering (Figure S6). The degree of layer malformation was Cre dose-dependent and correlated with the severity of the perturbed RGC array (Figure S6), further supporting our hypothesis that function of Memo1 in RGCs is necessary for neuronal laminar organization.
To examine if Memo1 cell autonomously affects radial migration and placement of neurons, we selectively deleted Memo1 in new born cortical neurons using the Nex-Cre line (Graus-porta et al., 2001). NeurOn-specific deletion of Memo1 (Memo1Lox/Lox; Nex-Cre [Memo1 cKONex]) did not affect neuronal migration and placement (Figure S7).
Together, these observations suggest that disrupted RGC tiling leads to aberrant radial trajectories of neuronal migration in Memo1 cKO, thus contributing to the abnormal laminar and columnar organization of neurons in the Memo1-deficient neocortex.
Effect of Memo1 deletion on progenitors and organization of hippocampus, cerebellum, and adult SVZ
Radial progenitor-like cells are also necessary for the development of other brain structures such as hippocampus and cerebellum. We thus investigated the effect of Memo1 inactivation on these structures. Memo1 cKOEmx and Memo1 cKOhGFAP hippocampus is markedly small and hypoplastic (Figures S8A–H and S8Q–X). In the hippocampal neurogenic niche (DG), radial glia-like progenitors (also known as type I cells) are positioned along the subgranular zone (SGZ) and extend a single basal process through the granular cell layer (GCL) to terminally branch at the molecular layer (ML) (Figures S8A, S8C, S8E, and S8G). In contrast, cell soma of Memo1-deficient hippocampal progenitors are ectopically placed outside of the SGZ and sprout multiple, branched primary processes (Figures S8B, S8D, S8F, and S8H).
In cerebellum, Memo1-deficient Bergmann glial cells are often delaminated and their processes are abnormally branched and retracted from the pial surface (Figures S8I–S8N’). Further, ectopic NeuN+ neuronal clusters are present near the pial surface (Figures S8N and S8N’), presumably due to the disrupted Bergman glial-guided migration of granule neurons. Calbindin+ dendrites of Purkinje cells were excluded from such neuronal ectopias (data not shown), suggesting that cerebellar organization and circuitry is perturbed in the Memo1 cKOhGFAP cerebellum.
Lastly, in the adult SVZ neurogenic progenitor niche, radial glia-like progenitors (i.e. B cells) localize beneath the ventricular surface and possess long, thin radial processes (Figure S9O). In contrast, Memo1-deficient SVZ progenitors have misdirected, branched radial fibers and are often detached from the ventricular surface (Figure S8P).
Collectively, these data demonstrate a fundamental role for Memo1 in the organization and function of radial glial-like progenitors in non-cortical brain regions and adult neurogenic niches.
Disrupted microtubule organization in Memo1-deficient RGCs
What are the cellular mechanisms underlying Memo1’s role as a regulator of RGC tiling? Balanced deployment of stable and dynamic MT subpopulations, which are post-translationally modified by acetylation and tyrosination, respectively, is critical for building the polarized MT network necessary to maintain the tiled RG scaffold. Immunoblotting analyses of Memo1 cKO cortex reveal a reversal of the tubulin modification pattern in Memo1 cKO cortex. Memo1 deletion reduced MT acetylation and increased tyrosination (Figures 5A–B). MT glutamylation, a characteristic of stable MT population, is also decreased in Memo1 cKO cortex (Figures 5A–B). In isolated cortical RGCs, the level of tubulin tyrosination in the elongated basal process exhibits a sharp decline towards the tip. In contrast, high tyrosination of MTs is sustained at the tip in Memo1-deficient RGCs (Figures 5C–D). These results suggest that Memo1-inactivation augments unstable, dynamic, tyrosinated MT population particularly in the radial processes of RGCs. Real time observation of MT fibers within radial glial processes show that MT filaments in radial process tips in Memo1-deficient RGCs are highly dynamic and branch rapidly when compared to MT filaments in control RGCs (Figure 5E). Further, Memo1 cKO RGCs have significantly higher number of loop-like MT structures protruding from the main MT bundle in the basal processes (Figures 5F–G), indicative of defective anchoring and fasciculation of MT fibers in the Memo1-deficient RGCs.
Figure 5: Altered MT stability and dynamics in Memo1-deficient RGCs.

(A) Dorsal cortical lysates from Memo1Lox/+; Emx1-Cre and Memo1Lox/Lox; Emx1-Cre mice (P0) were analyzed for tubulin post-translational modifications. An inversion of the MT acetylation/tyrosination ratio is evident in the Memo1-deficient cortex. (B) Quantification of tubulin modification. The intensities of acetylated, tyrosinated, and glutamylated tubulin bands were measured and normalized to those of β-actin. Data shown are mean ± SEM (n=5 mice/genotype). (C) Tubulin acetylation and tyrosination in RGCs in vitro. Outlined areas show sustained tubulin tyrosination at the tip of the Memo1 cKO RGC basal process. RGC identity of these cells was validated by BLBP immunolabeling (insets). (D) Quantification of MT stability along radial glial processes. Tyrosination/acetylation ratio was measured along radial glial processes. Data shown are mean ± SEM from 21 (control) and 20 (Memo1 cKO) cells from 5 mice/genotype. (E) Real time observation of MT dynamics in RGC processes. MTs labeled with SiR-tubulin in Memo1 cKO RGCs show highly dynamic behavior when compared to controls. Time elapsed is indicated in minutes and MTs from each time point are pseudo colored. (F) MT bundles within RGC processes visualized with SiR-tubulin, showing aberrant MT fasciculation and MT looping defects (arrowheads) in Memo1-deficient RGCs. (G) Quantification of loop-like MT structures. Data shown are mean ± SEM from 18 (control) and 19 (Memo1 cKO) cells from 4 mice/genotype. (H) Real time imaging of MT plus- and minus-ends labeled with EB1-DsRed and GFP-CAMSAP2, respectively, in radial processes. The movement of plus- and minus-ends are shown in kymographs (right). (I) Quantification of the velocity and directionality of EB1- and CAMSAP2-positive ends. Data shown are mean ± SEM (n=25 cells from 5 mice/genotype). (J) Distribution of EB1- and CAMSAP2-positive MT ends in radial processes, showing increased minus-end labeled MTs in Memo1 cKO RGC processes. (K) Quantification of plus and minus-end labeled MTs. Data shown are mean ± SEM (n=15 cells from 4 mice/genotype). (L) Time lapse images of EB1-DsRed (yellow arrowheads) and GFP-CAMSAP2 (cyan arrowheads) along SiR-tubulin-labeled MTs in the basal processes, showing initiation of EB1-tagged MT branches from CAMSAP2+ minus-end in Memo1 cKO RGCs. (M) Quantification of MT branching events (MT branching/100 μm/min). Data shown are mean ± SEM (n=20 cells from 5 mice/genotype). (N) Co-immunoprecipitation of Memo1 and CAMSAP2 in P0 cortical lysates. (O) Proximity biotinylation assays with HEK293T cells. Strong biotinylation of CAMSAP2 by BirA-Memo1 indicate their intracellular association. (P-R) CAMSAP2 overexpression leads to extensive RGC basal process branching in RGCs. (P-Q) RGCs in E14 cortex were co-electroporated with BLBP-DsRed and GFP (P) or CAMSAP2-GFP (Q), and analyzed at E16. While control RGCs display characteristic polarized basal processes, CAMSAP2-expressing RGCs show excessive basal process branching (white arrowheads). (R) Quantification of the basal process branching. Branch number /100 μm was quantified and used as branching index. Data shown are mean ± SEM (n=26 cells from 4 mice for each condition). Student’s t-test; *P < 0.05, **P < 0.01, and ***P < 0.001. Scale bar: C (5 μm), E, H, L (0.8 μm), F (1.5 μm), J (1 μm), and P, R (10 μm). See also Figure S9 and Supplemental Movie 3.
To further examine the effect of Memo1-inactivation on MT array organization in RGC basal processes, we labeled MT plus- and minus-ends in control and Memo1-deficient RGCs with EB1-DsRed and GFP-CAMSAP2, respectively (Jiang et al., 2014; Tirnauer and Bierer, 2000). In the radial process of Memo1 cKOEmx RGCs, EB1 comets moved at slower speed (control, 0.051±0.0039 μm/sec; Memo1 cKOEmx, 0.039±0.0031 μm/sec) and showed a higher frequency of retrograde movement (control, 15.2±3.54%; Memo1 cKOEmx, 31 ±4.51 %) when compared to control (Figures 5H–I and Movie S3). Average length of MT plus end extension is reduced in mutant RGCs (control, 1.80±0.11 μm; Memo1 cKOEmx, 1.25±0.09 μm). Consistent with MT instability in Memo1-deficient RGCs, MT fibers in Memo1 cKO RGCs show increased MT catastrophe frequency (control, 0.113±0.013 min−1; Memo1 cKOEmx, 0.168±0.013 min−1; n=20 cells from 4 mice). Whilst the velocity and directionality of CAMSAP2 comets were not significantly altered, Memo1 cKO RGCs display increased number of CAMSAP2+ puncta in the radial processes (Figures 5J–K), suggesting that normal organization of MT arrays is disrupted in the absence of Memo1. Since CAMSAP2 can organize non-centrosomal MT nucleation sites (Akhmanova and Steinmetz, 2015; Martin and Akhmanova, 2018), increased CAMSAP2 foci in basal processes may cause atypical MT nucleation. Consistent with this possibility, CAMSAP2-nucleated EB1 comets that branch from the main MT fiber are frequently detected in Memo1 cKO, but not control, basal processes (Figures 5L–M), indicating that deregulated non-centrosomal MT nucleation may lead to basal process hyperbranching. Correspondingly, in MT nucleation assays, centrosome-mediated MT re-nucleation was unaffected (Figure S9). Further, an interaction between Memo1 and CAMSAP2 was detected by co-immunoprecipitation in cortical lysates and in proximity labeling assays with BirA-Memo1 (Figures 5N–O), indicating that Memo1 may associate with CAMSAP2 to regulate its subcellular localization and MT organizing function in RGCs. The loss of which in Memo1 cKO lead to aberrant CAMSAP2 distribution. Ectopic expression of CAMSAP2 in developing RGCs caused extensive branching in their basal processes (Figure 5P–R), consistent with the hypothesis that deregulated CAMSAP2 expression underlies inappropriate extension and branching of RGC basal processes. Together, these observations suggest that Memo1 functions to maintain appropriate MT organization in RGCs necessary for their tiling.
Polarized vesicular trafficking in Memo-deficient RGCs
Polarized, directional protein transport in the basal processes is essential to generate specialized cellular compartments in RGCs and thus their polarized and tiled morphology. Since intracellular vesicle trafficking system is highly dependent on MT networks, we examined the effect of Memo1 loss on polarized trafficking along RGC basal processes. First, to visualize molecular cargo transporting vesicles, transferrin receptor (TfR)-GFP was expressed in RGCs and TfR-GFP+ vesicle movement along SiR-Tubulin-labeled MTs in basal processes was monitored. Compared to control, TfR-GFP+ vesicles in Memo1 cKOEmx RGCs showed slower movement and an increased percentage of immobile, pausing vesicles (Figures 6A–6D and Movie S4). To examine if the altered polarized vesicle movement affects the transport of cell surface proteins necessary for the polarized morphology of RGCs, we expressed superecliptic pHluorin-tagged GPR56 (pH-GPR56) in control and Memo1 cKO RGCs. GPR56 is required for the RGC morphogenesis, enriched in the basal endfeet, and GPR56 mutations cause bilateral frontoparietal polymicrogyria in humans (Piao et al., 2004).
Figure 6: Disrupted MT-dependent vesicle trafficking in Memo1 cKO RGCs.

(A-D) Tracking of TfR-GFP+ vesicle movement in radial glial processes. The movement of TfR-GFP+ vesicles on MTs in basal process tips (yellow box, A) were traced and shown in kymographs (B and C). Examples of anterograde, retrograde, and pausing vesicle movements are indicated (blue, purple, and green arrows, respectively, B). Quantification of the velocity and directionality of TfR-GFP+ vesicles (D). Data shown are mean ± SEM (n=15 cells from 5 mice/genotype). (E-I) Live imaging of pH-GPR56 at radial glial process tips. Time lapse images of pH-GPR56 exocytic events from control (E), Memo1 cKOEmx (F), and Memo1-rescued (Memo1+ Memo1 cKOEmx) RGCs (G). Time elapsed is indicated in seconds. pH-GPR56+ vesicles from each time point are pseudo colored and merged in rightmost panels. Quantification of the polarized release of GPR56 (events/100 μm/min) (H) and branching index (branching/100 μm/min) (I). Data shown are mean ± SEM (n=22 cells from 4 mice for each condition). Student’s t-test; *P < 0.05 and ***P < 0.001. Scale bar: A (2 μm) and E, F, G (1 μm). See also Figure S9 and Supplemental Movie 4.
pHluorin is detectable only when exposed to the extracellular space and thus pHluorin tagging enables the detection of polarized transport and cell surface release of GPR56 at RGC tips. Live imaging of pH-GPR56 at the basal process tips reveals that Memo1 cKOEmx RGCs show decreased frequency of transport and insertion of GPR56 in basal ends of RGCs (Figures 6E, 6F, 6H). Re-expression of Memo1 in Memo1 cKOEmx RGCs rescued the GPR56 release rate as well as the hyperbranching phenotype (Figures 6F–I). These results suggest that disrupted MT organization in Memo1 cKO RGC processes affects polarized vesicle and protein trafficking and thus may contribute to the disrupted RGC tiling in Memo1 cKO cortex.
Characterization of MEMO1 mutation in autism patients
Whole exome sequencing of the Simons Simplex Collection (SSC) of ASD patients identified a de novo nonsense mutation of MEMO1 in an autism proband (Fischbach and Lord, 2010; Iossifov et al., 2014). This proband is characterized with intellectual disabilities, developmental delay, autistic features, and low IQ. The mutation in this proband is located at the splice donor site in the intron 2 of MEMO1 gene (Figure 7A, c.143+1G>A). To examine how this intronic mutation affects MEMO1 expression, we performed RT-PCR analyses using RNA isolated from the patient-derived and unaffected sibling-derived lymphocytes. RT-PCR revealed the abnormal inclusion of intron 2 sequences in MEMO1 mRNA in the patient-derived cells (Figure 7B). Sequencing of the amplicon from the patient sample demonstrated an inclusion of the 297 bp fragment of intron 2 between exon 2 and 3 in the patient MEMO1 mRNA (Figure 7C), indicating that c.143+1G>A splice donor site mutation activates a cryptic donor site within the intron 2 and alters MEMO1 mRNA sequence in the affected individuals (Figure 7A and 7C). This splicing error introduces a premature stop codon in the MEMO1 transcript (p.His49*, Figure 7C). Importantly, full length human MEMO1 can ameliorate the radial process branching defect in Memo1 cKO RGCs in vivo, whereas the truncated MEMO1 His49* mutant does not rescue this defect (Figures 7D–H), confirming that the ASD-associated MEMO1 mutation results in functional loss of MEMO1.
Figure 7: ASD-associated intronic mutation disrupts MEMO1 mRNA processing.

(A) MEMO1 pre-mRNA structure (top). Exonic and intronic sequences are shown in uppercase and lowercase letters, respectively. The c.143+1G>A substitution and cryptic splice donor site are indicated. Green arrowheads indicate primers designed for RT-PCR analyses. Sequences encompassing the 3’ end of exon 2 (blue box), intron 2 (yellow box), and 5’ end of exon 3 (purple box) (bottom) are covered. (B) RT-PCR analyses using unaffected sibling (S)- and affected patient (P)-derived lymphocytes. An amplicon in the PCR with intron-specific primers indicates that the c.143+1G>A mutation causes aberrant inclusion of intron 2 sequences. (C) Consequences of MEMO1 splicing error. Resultant MEMO1 mRNA (top) and protein (bottom) sequences are shown. The splicing error leads to the inclusion of 297 bp of the 5’ end of intron 2, which generates a premature stop codon and creates a short MEMO1 protein variant (p.His49*, 48 amino acids). (D-H) Mutant MEMO1 does not rescue RG tiling defects. E14.5 cortices from control or Memo1 cKO mice were focally electroporated with BLBP-DsRed and GFP, hMEMO1-GFP or hMEMO1 His49*-GFP and analyzed 48 hours later (D-G). High-magnification images of basal processes are shown in right panels. Full length human MEMO1, but not His49* mutant, can rescue the basal process hyperbranching phenotype in Memo1 cKO. Arrows and arrowheads in D-G indicate normal and branched basal processes, respectively. Quantification of basal process branching (H). Data shown are mean ± SEM (n=3 mice for each condition). (I) Cultured human NPCs were transfected with BLBP-GFP and co-immunolabeled with anti-BLBP antibodies. BLBP+ cells in vitro show polarized RG morphology with single basal processes. (J) ShRNA-mediated knockdown of MEMO1. HEK293T cells were transfected with control scrambled shRNA vector or two pools of shRNAs against human MEMO1. Knockdown efficacy of shRNAs (fold change compared to control) was examined by quantitative real-time PCR. Data shown are mean ± SEM (n=3 for each condition). (K) Human NPCs were co-transfected with control shRNA or MEMO1 shRNA and BLBP-GFP. High-magnification images of RG basal processes (box) are shown in bottom panels. MEMO1 knockdown causes extensive basal process branching (arrowheads). (L) Quantification of the basal process branching. Data shown are mean ± SEM from 94 (shControl), 70 (shMEMO1 #1), and 66 (shMEMO1 #2) cells. One-way ANOVA with Tukey’s multiple comparison test; *P < 0.05, **P < 0.01, and ***P < 0.001. Scale bar: D-G (50 μm), I (25 μm) and K (20 μm). See also Figure S10.
To further examine the conserved function of MEMO1 on human RGC development, we knocked down MEMO1 in human neural progenitor cell (hNPC) derived RGCs (Stein et al., 2014), using validated human MEMO1-specific shRNAs. BLBP+ human RGCs show characteristic polarized morphology with a thin, long basal process (Figures 7I). In contrast, MEMO1-deficient RGC basal processes are highly branched (Figures 7J–L). These results indicate a conserved role for MEMO1 as a mediator of RGC organization and function in the developing brain.
Discussion
Tiled RGC scaffold serves as an instructive framework to form radial columns and laminar organization of neurons in the neocortex. However, little is known about the molecular logic of radial glial tiling and how it drives the appropriate formation of the cerebral cortex. Here, we demonstrate that Memo1 functions as a determinant of radial progenitor tiling. Mutations in MEMO1 and resultant cortical malformation may contribute to ASD (Figure S10).
Radial progenitor tiling and neuronal organization
Radial glial progenitors directly or indirectly generate neurons of all six layers of the cerebral cortex in the course of their sequential divisions (Beattie et al., 2017; Gao et al., 2014; Kwan et al., 2012). Once generated, most neurons migrate radially, using basal processes of radial progenitors as migratory guides, resulting in the construction of radial columns and laminar organization of neurons (Jones and Rakic, 2010; Rakic, 1972, 1988). Radial columns of neurons show synchronized activity and are a defining feature of cortical areas (Maruoka et al., 2017; Yu et al., 2009). Furthermore, neuronal laminar organization enables the generation of cortico-cortical and subcortical circuitry. Neuronal columnar and laminar organization thus contributes to the formation of functional circuits in the cerebral cortex. The tiled array of radial progenitors provides a blueprint to build this circuitry. Disruptions in the tiling of RGCs can interrupt the radial flow of neurons, resulting in misplacement of neurons and a spectrum of neurodevelopmental diseases, including lissencephaly, pachygyria, polymicrogyria, and ASD (Piao et al., 2004; Rakic, 1988; Stoner et al., 2014; Zhang et al., 2016). Memo1 deletion in radial progenitors results in disrupted tiling characterized by hyperbranching and irregular distribution of basal processes and by reduced endfeet-pial membrane contacts, leading to perturbed columnar and laminar organization of cortical neurons.
Microtubule dynamics and RGC tiling
Appropriate MT organization is required for radial glial scaffold formation and hence brain development. A number of MT regulators were found to be critical for the polarity of RGCs, including MT-associated proteins [APC, EFHC1, DCL] and signal transducers [RhoA, CDC42, GSK3β] (Cappello et al., 2012; De Nijs et al., 2009; Muroyama and Lechler, 2017; Nakagawa et al., 2017; Yokota et al., 2009, 2010). These MT organizers regulate tubulin post-translational modifications, stability, bundle formation, and plus/minus-end dynamics. Mutations or deletions affecting their functions disrupt the cortical progenitor niche, thus highlighting the significance of a fine-tuned MT network for organizing the radial progenitor niche and function in the developing brain. Further, weighted gene co-expression network analysis (WGCNA) of the prenatal human brain transcriptome assigns MEMO1, with several MT-associated proteins (e.g., CLASP2, CLIP1, CAMSAP½, and APC), to a gene cluster, in which MT modification/transport-related genes are enriched (Miller et al., 2014). This suggests the highly conserved role for Memo1 in the MT-network regulation during mammalian neocortical development.
Memo1’s regulation of MT in part involves MT minus-end targeting protein CAMSAP2. While significant inroads have been made in our understanding of MT-centrosome dynamics in progenitor proliferation in particular, the relevance of non-centrosomal MT and MT minus-end dynamics in progenitor development and functions remain unknown. Our work points to an important role for MT minus-end linked CAMSAP2 regulation and acentrosomal MT dynamics in the tiling of radial progenitors. Recent studies on CAMSAP/Patronin/Nezha family of proteins revealed that minus-end-specific deposition of CAMSAPs protects MT minus-end from depolymerization and thus is essential for stabilization of the non-centrosomal MT population in cells (Hendershott and Vale, 2014; Jiang et al., 2014, 2017; Martin and Akhmanova, 2018; Tanaka et al., 2012). CAMSAP-mediated stabilization of MT minus-ends may then facilitate non-centrosomal MT nucleation and outgrowth (Jiang et al., 2014; Tanaka et al., 2012).
Compared to many non-polarized mammalian cell lines (e.g. CHO, COS-7, and HeLa cells) with the centrosome-centered MT network system, non-centrosomal MTs are abundant in polarized cells such as epithelial cells and neurons (Akhmanova and Steinmetz, 2015). In epithelial cells, CAMSAP protects minus-ends of non-centrosomal MT arrays organized along the apico-basal axis, thus contributing to the establishment of appropriate cellular compartmentalization and polarity (Toya et al., 2016). During neuronal differentiation, de novo non-centrosomal MTs, nucleated locally from Golgi outposts or CAMSAP-stabilized minus-end foci, drive dendrite morphogenesis and complexity (Ori-McKenney et al., 2012; Yau et al., 2014). Our findings show that an appropriate MT minus-end organization and non-centrosomal MTs are essential for polarized RGC morphology and tiling. Memo1 deletion increases CAMSAP2-positive MT minus-ends in RGC basal processes, which may serve as ectopic MT nucleation sites, leading to basal process hyperbranching and perturbed RGC tiling. Consistent with this, ectopic CAMSAP2 misexpression leads to RGC hyperbranching.
Further, our observations indicate the dynamism in cytoskeletal regulation necessary for progenitor function in cerebral cortex. Disrupting MT cytoskeleton does not always lead to progenitor proliferation defects and not all MT cytoskeletal defects are the same. The effect of Memo1 on minus-end MT proteins and the acentrosomal MTs indicate that the MT cytoskeletal mechanisms underlying radial progenitor polarity and organization are perhaps distinct from the ones that underlie progenitor division.
The functional importance of this MT regulatory system is highlighted by the genetic links between CAMSAPs and neurodevelopmental disorders. A copy number variation (CNV) that causes CAMSAP2 duplication was identified in an ASD patient with intellectual disability (Leblond et al., 2012). Single nucleotide polymorphisms (SNPs) in CAMSAP2 gene were reported as susceptibility loci for epilepsy (Guo et al., 2012). Further, disruption of MT minus-end dynamics through ASPM-katanin complex can cause microcephaly in humans (Jiang et al., 2017).
Diversity of cellular mechanisms relevant to RGC tiling
Disrupted MT stability in Memo1-deficient RGCs can affect RGC functions indirectly as well. MT-dependent cargo trafficking is one of the intracellular events through which MTs may impact tiled radial progenitor scaffold. RGCs extend two processes (apical and basal) with distinct molecular properties, especially in cell surface adhesion molecules. While basal process tip requires extracellular matrix receptors (e.g. GPR56, dystroglycan, integrinβ1) to attach to pia and maintain pial basement membrane integrity (Graus-porta et al., 2001; Li et al., 2008; Moore et al., 2002), cadherins are enriched at apical endfeet to create adherens junctions between neighboring progenitors (Rasin et al., 2007). Dysfunctions of basal and apical anchorage cause basal process retraction and progenitor delamination, respectively, resulting in the disrupted brain cytoarchitecture (Kim et al., 2010; Higginbotham et al., 2013; Weimer et al., 2009). Similar to neuronal axon-dendrite protein sorting system (Hirokawa et al., 2010), the apicobasal molecular sorting appears to be fundamental for radial progenitor organization and is driven by MT-dependent directional vesicle transport. MT dysregulation leading to disrupted polarized trafficking of apico-basal proteins such as GPR56 may have contributed to the progenitor delamination and untiling following Memo1 deletion.
Whilst Memo1 regulates RGC tiling primarily through intracellular MT network organization, cell-cell interactions also play a role in patterned spatial organization of cells or cellular processes in the central nervous system (CNS). Distinct classes of neurons in the retina are known to display highly ordered spatial distribution (Huckfeldt et al., 2009; Kay et al., 2012). The fundamental role of Cajal-Retzius cells in neuronal layer formation and circuitry relies on their distinct spatial distribution on the cortical surface (de Frutos et al., 2016). The neuronal dendritic arborization is streamlined by a self-avoidance mechanism to achieve the non-overlapping growth of dendrites (Jan and Jan, 2010). A shared mechanism in these different types of cellular tiling in the CNS is contact repulsion mediated by cell surface molecules such as MEGF10/11, Eph/ephrin, and Slit/Robo (Gibson et al., 2014; Huckfeldt et al., 2009; Kay et al., 2012; Riccomagno et al., 2014). Consistent with this, developing RGCs show highly dynamic inter-radial glial interactions along their basal processes (Yokota et al., 2010), suggesting that neighboring RGCs may also sense and organize each other to optimize the regular spacing of basal processes within the radial glial scaffold. Identifying cell surface mediators of inter-radial glial interactions will be necessary to further validate this hypothesis.
RGC tiling, Memo1, and neurodevelopmental disorders
We found that an ASD-associated MEMO1 mutation (c.143+1G>A) causes defective mRNA splicing, resulting in a truncated MEMO1. A second MEMO1 splice donor variant was also found recently in a case ([c.762+1G>C]; De Rubeis et al., 2014; Satterstrom et al., 2018). Disrupted laminar architecture of neurons in Memo1 cKO cortex resembles neocortical patch-like neuronal disorganization found in children with ASD (Stoner et al., 2014). The human MEMO1 gene (ENSG00000162959) is poorly tolerant of loss-of-function exonic variation (Lek et al., 2016). It is notable for its association with autism, with autism cases accumulating more MEMO1 rare deleterious variants than controls (q-value=0.096; Nguyen et al., 2017). Further, phenotypic data of CNVs reported in the DECIPHER database (https://decipher.sanger.ac.uk) indicate that duplication or deletion of the genomic region harboring MEMO1 gene may cause wide range of neurodevelopmental disorders, including ASD, intellectual disability, and seizures as well as macrocephaly and microcephaly. Previous studies have highlighted projection neuron or interneuron dysfunction as well as progenitor proliferation defects as potential neurodevelopmental triggers of ASD phenotype (de la Torre-Ubieta et al., 2016). Our analyses of Memo1 shed light on the potential pathogenic connection between defective RGC tiling and neurodevelopmental disorders. Future identification of additional human mutations that affect RGC scaffold tiling will help further assess the organizational impact of radial progenitor tiling on cortical circuit formation and function.
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the lead contact, E. S. Anton (anton@med.unc.edu).
Experimental Model and Subject Details
Mice
This study is based on data from mice at various developmental stages ( embryonic days 12, 14, 16, 18, postnatal days 0, 7, and 30) and includes both males and females. Mice were cared for according to the University of North Carolina’s animal care and use committee guidelines. Light/dark cycle in the vivarium is 7/7 hours. Animals were housed in groups of 3 adults per cage. Memo1 was conditionally inactivated in cortical progenitors by crossing Memo1Lox/Lox mice with Memo1Lox/+; Cre+ mice. Two different Cre lines, hGFAP-Cre (Zhuo et al., 2001) and Emx1-Cre (Guo et al., 2000), were used to inactivate Memo1 in radial progenitors. Memo1Lox/Lox; hGFAP-Cre and Memo1Lox/Lox; Emx-Cre mice were also crossed with RG-Brainbow; hGFAP-Cre or RG-Brainbow; Emx1-Cre mice to generate the respective Memo1Lox/Lox; RG-Brainbow; Cre+ animals. To generate Memo1Lox/Lox; GFP-Centrin2; Emx1-Cre mice, Memo1Lox/Lox; Emx1-Cre were bred with GFP-Centrin2 mice (Higginbotham et al., 2004). To generate MADM-11GT/TG; Memo1Lox/Lox mice, MADM-11TG/TG; Memo1Lox/+ mice were crossed with MADM-11GT/GT; Memo1Lox/Lox mice (Hippenmeyer et al., 2010). Nex-Cre line (Goebbels et al., 2006) was used for postmitotic neuron-specific inactivation of Memo1.
Construction of Memo1 conditional allele and generation of Memo1 floxed mice
Memo1Lox/Lox allele was generated using BAC recombineering technology as described in Bouvier and Cheng, 2009. A mouse C57 BAC clone RP23-53D2 containing Memo1 gene was obtained from RP23 BAC library (UNC BAC engineering Core; clone ID in NCBI CloneDB: 586810). Memo1 genomic DNA was then subcloned into pBS-DT vector. An orphan loxP site was inserted into intron 1 in pBS-DS-Memo1 followed by the insertion of a FRT-PGK-Neo-FRT-loxP cassette into the AccI site of intron 2. Purified Memo1 cKO constructs were linearized with NotI digestion, electroporated into a 129 ES cell line (E14/Tg2a.4) and selected with G418 at the UNC Animal Model Core Facility. Targeted ES cells were screened by genomic PCR and then confirmed by genomic Southern. FRT floxed Neo cassette was removed by transient expression of flpe in the targeted ES cells. After blastocyte injection, chimeric mice were checked for germline transmission. The Memo1Lox/Lox lines are maintained on C57Bl6 background.
Generation of GLAST1-Brainbow1.1M transgenic mice
GLAST1-Brainbow1.1M transgene was generated using BAC recombineering as described in Bouvier and Cheng, 2009. pCMV-Brainbow-1.1M was a gift from Dr. J. Lichtman (Harvard University). Brainbow1.1M with FRT-Neo was inserted at GLAST exon 2 (ATG exon) within C57 mouse BAC (RP23-63O21). Purified BAC was used for pronuclear microinjection of fertilized eggs from the CBA X C57Bl6 mouse strain at the UNC Animal Model Core Facility. Fertilized ova were subsequently implanted into pseudo-pregnant females and offspring were analyzed for the insertion of the BAC construct. Founder lines were identified by Southern blot analysis of tail DNA. An [α-32P] dCTP labeled DNA probe was generated from a 1300 or 1113 bp BgIII-BgIII fragment of the Brainbow1.1M region of the BAC construct and used to probe the DNA blot. Independent breeding lines were established from founders and subsequent offspring were analyzed by PCR using the following two primer sets, 5’ GGA AAT GTG GGT GCT TGG TCT C 3’ and 5’ TCG TAA GGT TTG CCT GTG CC 3’ (p1); 5’ TGG ATA CTT TCT CGG CAG GAG C 3’ and 5’ AAA CGA CTC ACC CAC AAT GAC AG 3’ (p2) complimentary to 5’ and 3’ arms of the GLAST1-Brainbow1.1M transgene, respectively. The GLAST1-Brainbow1.1M (RG-Brainbow) mice are maintained on C57Bl6 background.
Human neural progenitor cell culture
The fetal tissue (GW15-18) harvesting and neurosphere collection were done as described previously (Konopka et al., 2012; Stein et al., 2014) and cells were derived from frozen pHNPC stocks. Cells from both males and females were used. Cells were kept in proliferation media [Neurobasal A (Invitrogen) supplemented with 10% BIT (Stem Cell Technologies), Primocin (Invivogen), GlutaMAX (Gibco), and heparin (1 μg/mL; Sigma)] with freshly added EGF, FGF2, PDGF (each at 20 ng/mL; Invitrogen), and LIF (2 ng/mL; Invitrogen) and passaged when confluent on polyrnothine/fibronectin coated plates. Prior to transfection, NPCs are P5-P10 and were plated onto polyornithine/laminin coated German glass coverslips at ~80,000 cells/cm2. Cells were maintained at 37°C/ 5% CO2.
Cell lines
HEK293T cells (authenticated by American Type Culture Collection [ATCC]) were maintained in DMEM/10% FBS at 37°C and 5 % CO 2.
Method Details
DNA constructs
pCAG-BirA was generated by cloning BirA (amplified by PCR from pAAV-BirA [gift from Dr. S. Soderling, Duke University]) into AgeI/NotI sites of pCAG-GFP to replace GFP. pCAG-MEMO1-BirA was constructed by cloning mouse MEMO1 fragment into pCAG-BirA. For pCAG-SEP-GPR56 (pH-GPR56), human GPR56 amplified from GPR56-Tango (Kroeze et al., 2015) was cloned into SpeI/NotI sites of pCI-SEP-GluR1 (Addgene #24000) to replace GluR1. SEP-GPR56 was then amplified and inserted into XbaI/NotI sites of pCAG-GFP to replace GFP. For pCAG-hMEMO1-GFP, cDNA fragment of human MEMO1 was inserted into KpnI/AgeI sites of pCAG-GFP to make hMEMO1-GFP. For pCAG-hMEMO1 (His49*)-GFP, truncated human MEMO1 cDNA corresponding to its 1-48 amino acids was inserted into KpnI/AgeI sites of pCAG-GFP.
Memo1 shRNA
shRNA against mouse Memo1 was generated using Memo1 specific target sequence TGCATTTGCCTTATACAGCT. The target sequence oligos were subcloned into pCGLH vector (Yokota et al., 2007). shRNA against human Memo1 was obtained from the UNC Gene Therapy Center. shRNA target sequences used were as follows: CCTTCCTTAATTTCAACTCAT (1), CCTCTGTATGACCTTCGTATT (2), TGGAGCTCTGAGTGAGTCAAA (3), and CCGTCTATTACCCGGAGAATT (4). Two pools (1.5 μg/μl) of shRNAs [shMemo1 #1: (1-2), and shMemo1 #2: (3-4)], containing equivalent amount of different shRNAs, were used. Non-silencing scrambled shRNA was used as control. shRNAs were validated as described in Nakagawa et al., 2017.
In utero electroporation
Lateral ventricles of E14 embryos were electroporated with pBLBP-EGFP, pBLBP-DsRed, EB1-DsRed, GFP-CAMSAP2 (generous gift from Dr. A. Akhmanova, Utrecht University), TfR-GFP, Memo1 or pH-GPR56 as described earlier (Nakagawa et al., 2017; Yokota et al., 2009). Cortices were harvested at E15 or E16 for live imaging or in vitro RGC culture. For live imaging, cortices were coronally sectioned (250 μm) in a vibratome (Leica VT 1000S), mounted on cell culture inserts (0.4 μm, Millicell, Merck Millipore) in a glass bottom tissue culture dish (glass thickness 0.17 mm, FluoroDish, World Precision Instruments, Inc) and maintained in DMEM/10% fetal bovine serum (FBS). Slices were then imaged using a Zeiss LSM 780 microscope equipped with live cell incubation system.
Live Imaging of Neuronal Migration
E14 embryos in pregnant dams were electroporated following ventricular injection of pCAG-EGFP DNA (0.5 μg/μl). At E16, cortices were removed from the embryos, embedded in 3% low-melting-point agarose in complete Hank’s Balanced Salt Solution, coronally sectioned (250 μm), mounted on Millicell-CM membrane filters (Millipore), placed in glass-bottom FluoroDish chambers (World Precision Instruments, Inc.) and maintained in DMEM/10% FBS at 37°C and 5 % CO 2. GFP-expressing neurons were repeatedly imaged using a Zeiss LSM780 inverted confocal microscope attached to a live cell incubation chamber. Zeiss LSM Image Browser or ImageJ Software was used for quantification of migration patterns.
FLASH tag labeling of progenitors and neurons
To fluorescently pulse-label ventricular progenitors and their daughter neurons (i. e. FLASH tagging), 0.5 μl of CytoTell Blue (AAT Bioquest) was injected into lateral ventricles of E14 embryos (Telley et al., 2016). FLASH Tag-incorporated cells were imaged after 24 or 48 hours.
Live cell imaging
For the live cell imaging of EB1-DsRed, GFP-CAMSAP2, TfR-GFP and pHluorin-GPR56 in RGCs, cortical progenitors from E15 Memo1Lox/Lox; Emx1-Cre animals were dissociated and plated on poly-L-lysine/laminin coated glass bottom dishes (Yokota et al., 2007). Cells were maintained at 37°C/ 5% CO2. Two hours before live imaging, cells were incubated with 50 nM SiR-Tubulin (Lukinavicius et al., 2014) to label the microtubules. Cells were then imaged repeatedly at 3 second intervals for up to 5 minutes. Vesicle dynamics (i.e., the percentage of vesicles displaying anterograde or retrograde movement, and the speed of vesicle movement) and pHluorin-GPR56 release events were analyzed using ImageJ.
Immunohistochemistry
Immunohistochemical labeling of embryonic/postnatal brain sections or isolated cortical cells was performed as described earlier (Higginbotham et al., 2013; Yokota et al., 2009). The following primary antibodies were used: acetylated tubulin (mouse, T6793, Sigma-Aldrich), α-tubulin (mouse, T6199, Sigma-Aldrich), BLBP (rabbit, ab32423, Abcam), BrdU (mouse, 347580, Becton Dickinson), Ctip2 (rat, ab18465, Abcam), Cux1 (rabbit, sc-13024, Santa Cruz Biotechnology), GABA (rabbit, A2052, Sigma-Aldrich), GFAP (rabbit, Z0334, DAKO), GFP (chicken, ab13970, Abcam), laminin (rabbit, L9393, Sigma), MEMO1 (rabbit, ab124901, Abcam), NeuN (mouse, MAB377, Chemicon), phospho-Vimentin (mouse, ab22651, Abcam), Pou3f2 (mouse, sc-393324, Santa Cruz Biotechnology), PTPRZ1 (rabbit, HPA015103, Sigma-Aldrich), PV (mouse, MAB1572, Millipore), RC2 (mouse IgM, Developmental Studies Hybridoma Bank, University of Iowa), RFP (rabbit, 600-401-379, Rockland), SST (rat, MAB354, Millipore), Tbr1 (rabbit, ab31940, Abcam), Tbr2 (rat, 14-4875, eBioscience), Tuj1 (mouse, 1409, STEMCELL technologies) and tyrosinated tubulin (rat, ab6160, Abcam). Appropriate Cy2, Cy3 or Alexa dye-conjugated secondary antibodies (Jackson ImmunoResearch, Molecular Probes) were used to detect primary antibody binding. DAPI (Sigma-Aldrich, D9542) was used as a nuclear counterstain.
Proximity biotinylation assay
HEK293T cells (authenticated by ATCC) were transfected with BirA or BirA-Memo1. Cells were maintained in DMEM/10% FBS at 37°C and 5 % CO2. 36 hr after transfection, cells were incubated with 5 μM biotin for 12 hr and harvested in lysis buffer [10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100] supplemented with protease and phosphatase inhibitor cocktail (Thermo Scientific). Lysates were centrifuged at 14,000 rpm for 15 min. Biotinylated proteins were precipitated from the cleared supernatants by incubation with NeutrAvidine agarose resin (Thermo Scientific) for 3 hr and analyzed by immunoblotting.
Analysis of MT dynamics
To measure the MT plus- and minus-end dynamics, we co-electroporated EB1-DsRed and GFP-CAMSAP2 into E14 control or Memo1 cKOEmx cortices. RGCs in the VZ region were dissociated and cultured 24 hours later. Cells were maintained at 37°C/ 5% CO2. EB1+ plus- and CAMSAP2+ minus-ends of MT in basal processes were imaged every 3 seconds for 5 minutes. The average velocity and directionality of EB1+ and CAMSAP2+ comets within basal processes were measured using ImageJ software. The average length and duration of MT plus-end extension were also quantified from EB1+ comet movements. EB1+ and CAMSAP2+ puncta in basal processes were quantified in fixed RGCs, whose MT fibers were labeled with anti-α-tubulin antibodies. To measure MT catastrophe frequency, MT fibers in RGCs were visualized with SiR-tubulin (Lukinavicius et al., 2014) and live-imaged every 4 seconds for 5 minutes. The catastrophe frequency was calculated as the number of transitions from growth or pause to shortening per minute (Watanabe et al., 2015).
Immunoblotting
Protein extracts from P0 dorsal cortex were prepared and immunoblotted as described previously (Higginbotham et al., 2013). The following primary antibodies were used: acetylated tubulin (mouse, T6793, Sigma-Aldrich), β-actin (mouse, 8226, Abcam), CAMSAP2 (rabbit, 17880-1-AP, Proteintech), glutamylated tubulin (rabbit, AB3201, Millipore), HA (mouse, A01244, Genscript), Memo1 (mouse [Marone et al., 2004]; rabbit, ab124901 [Abcam]; rabbit, HPA057952 [Sigma-Aldrich]), and tyrosinated tubulin (rat, ab6160, Abcam).
Quantification and Statistical Analysis
General statistical Analysis
Excel and R language were used for data analysis. Statistical significance was determined by two-tailed Student’s t-test for comparisons between two groups and by ANOVA with Tukey-Kramer’s test for comparisons among multiple groups. All experiments were independently repeated for at least 3 or more times. No a priori power analyses were performed. All data are expressed as means ± standard error of the mean (SEM). Statistical details, including p values, are indicated in text or figure legends.
Quantification of RG basal processes tiling
For the quantification of tiling of the RG basal processes, we performed Voronoi domain analysis using RC2-immunolabeled horizontal sections (50 μm thickness) from E16 dorsal cortex. Horizontal sections were prepared to obtain cross sectional images of basal process arrays. Sections at the CP and IZ levels were determined by cellular density and neuronal distribution as revealed by DAPI and Cux1 immunolabeling, respectively (CP: cell dense and Cux1+ neurons enriched, IZ: cell sparse with fewer Cux1+ neurons). Images were obtained from the central region of the hemisphere, where the RC2+ basal processes are organized vertical to the plane of section (Misson et al., 1991). The Voronoi domain of each basal process was drawn and quantified by ImageJ software.
Quantification of MADM-labeled clones
For the quantification of MADM-labeled clones at E16, isolated RGC clones located in the dorsal region of the cortex were confocally imaged (serial Z axis scans) and analyzed. A line perpendicular to the RG basal process was drawn from RG process to the center of the soma of neuronal progeny. This length was measured as the distance between RG basal process and neuronal progeny.
Quantification of Flash Tag labeled cells
In Flash Tag labeling experiments, percentage of RGCs with neurons on basal process branches was counted. Attachment of each Flash Tag+ neuron to the Brainbow+ RG basal process was evaluated using 3D reconstructed confocal images of RG basal process and Flash Tag+ neurons.
Quantification of the final placement of neurons
To measure the final placement of neurons, somatosensory region of the cerebral cortex (P30) was divided into ten equal bins from the ventricular surface (VS) to the pial surface (PS) and the number of respective layer marker+ neurons in each bin was counted and presented as the percentage of total number. Cell counts were performed in sections corresponding to the somatosensory cortex. For 3D nearest neighbor distance (NND) analysis of neuronal organization, 3D reconstructed volumes of Cux1+, Ctip2+, and Tbr1+ layers were obtained by volumetric confocal scanning. Regularity of neuronal distribution in each plane (x-y, x-z, and y-z) was assessed by NND analysis with ImageJ and averaged.
Supplementary Material
Supplemental movie 1 (Related to Figure 2): Time-lapse images of control and Memo1-deficient BLBP+ radial progenitors in embryonic cerebral cortex. Time is indicated in hours and minutes.
Supplemental movie 2 (Related to Figures 3 and 4): Live imaging of migrating neurons in the intermediate zone of control and Memo1 cKOEmx cortex. Cyan arrowheads point to radially migrating neurons (control). Memo1 cKO neurons (purple arrowheads) branch and migrate non radially. Time is indicated in hours and minutes.
Supplemental movie 3 (Related to Figure 6): Time-lapse images of EB1-DsRed and GFP-CAMSAP2 in the basal process of control and Memo1 cKOEmx RGCs. Arrowheads point to anterogradely and retrogradely moving EB1 (red) or CAMPSAP2 (green) comets. Microtubules (blue) are labeled in left panels. Time is indicated in seconds.
Supplemental movie 4 (Related to Figure 7): Time-lapse images of TfR-GFP vesicles in the basal process of control and Memo1 cKOEmx RGCs. Arrowheads point to anterogradely (magenta) and retrogradely (white) moving vesicles. Time is indicated in seconds.
Figure S1 (Related to Figure 1): Memo1 expression in the developing cerebral cortex.
Figure S2 (Related to Figures 1 and 3): Generation of floxed Memo1 and RG-Brainbow mice.
Figure S3 (Related to Figures 1 and 3): Brainbow expression in radial progenitors and astrocytes during brain development.
Figure S4 (Related to Figures 1, 3, and 4): Aberrant outer RGCs and increased number of cortical astrocytes in Memo1 cKO neocortex.
Figure S5 (Related to Figures 3 and 4): Interneuron placement is not altered in Memo1 cKOEmx cortex.
Figure S6 (Related to Figures 1 and 4): Cre electroporation mediated inactivation of Memo1 leads to RGC abnormalities and disrupted neuronal layering.
Figure S7 (Related to Figures 1 and 4): Neuron-specific inactivation of Memo1 does not affect neuronal placement and laminar organization.
Figure S8 (Related to Figures 1, 2, 3 and 4): Memo1 deletion leads to hippocampal and cerebellar malformations and abnormal SVZ organization.
Figure S9 (Related to Figures 5 and 6): Centrosomal MT nucleation in Memo1-deficient RGCs.
Figure S10 (Related to Figures 1–7): Mechanisms and significance of radial glial tiling.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken polyclonal anti-GFP | Abcam | Cat# ab13970; RRID: AB_300798 |
| Rabbit polyclonal anti-RFP | Rockland | Cat# 600-401-379; RRID: AB_2209751 |
| Mouse anti-acetylated tubulin | Sigma-Aldrich | Cat# T6793, RRID: AB_477585 |
| Mouse anti-α tubulin | Sigma-Aldrich | Cat# T6199, RRID: AB_477583 |
| Mouse anti-BrdU | Becton-Dickinson | Cat# 347580, RRID: AB_10015219 |
| Rabbit polyclonal anti-MEMO1 | Abcam | Cat# ab124901: RRID: AB_10974792 |
| Mouse monoclonal anti-Pou3f2 | Santa Cruz | Cat# sc-393324: RRID: AB_2737347 |
| Rabbit polyclonal anti-PTPRZ1 | Sigma-Aldrich | Cat# HPA015103, RRID: AB_1855946 |
| Mouse monoclonal anti-phospho-Vimentin | Abcam | Cat# ab22651: RRID: AB_447222 |
| Mouse RC2 | Developmental Studies Hybridoma Bank, U. Iowa | RRID: AB_531887 |
| Mouse anti-classIII β-tubulin | STEMCELL Technologies | Cat# 1409: RRID: AB_215509 |
| Rat anti-Tbr2 | eBioscience | Cat# 14-4875: RRID: AB_11042577 |
| Rabbit polyclonal anti-Tbr1 | Abcam | Cat# ab31940: RRID: AB_2200219 |
| Rabbit polyclonal antil-laminin | Sigma | Cat# L9393: RRID: AB_477163 |
| Rabbit polyclonal anti-BLBP | Abcam | Cat# ab32423: RRID: AB_880078 |
| Rabbit polyclonal anti-Cux1 | Santa Cruz | Cat# sc-13024: RRID: AB_2261231 |
| Mouse monoclonal anti-PV | Millipore | Cat# MAB1572: RRID: AB_2174013 |
| Rat monoclonal anti-SST | Millipore | Cat# MAB354: RRID: AB_2255365 |
| Rabbit polyclonal anti-GABA | Sigma-Aldrich | Cat# A2052: RRID: AB_ 477652 |
| Rat anti-Ctip2 | Abcam | Cat# ab18465: RRID: AB_2064130 |
| Rat anti-tyrosinated tubulin | Abcam | Cat# ab6160: RRID: AB_305328 |
| Rabbit polyclonal anti-GFAP | DAKO | Cat# Z0334: RRID: AB_10013382 |
| Mouse monoclonal anti-NeuN | Millipore | Cat# MAB377: RRID: AB_2298772 |
| AlexaFluor goat anti-mouse 488 | Invitrogen | Cat# A32723 |
| AlexaFluor goat anti-mouse Cy3 | Invitrogen | Cat# A10521 |
| AlexaFluor goat anti-mouse 647 | Invitrogen | Cat# A21235 |
| AlexaFluor goat anti-rabbit 488 | Invitrogen | Cat# A11034 |
| AlexaFluor goat anti-rabbit Cy3 | Invitrogen | Cat# A10520 |
| AlexaFluor goat anti-rabbit 647 | Invitrogen | Cat# A32733 |
| AlexaFluor goat anti-rat 568 | Invitrogen | Cat# A11077 |
| AlexaFluor goat anti-rat 647 | Invitrogen | Cat# A21247 |
| AlexaFluor goat anti-chicken 488 | Invitrogen | Cat# A11039 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DAPI | Sigma-Aldrich | Cat# D9542 |
| CytoTell Blue (Flash Tag) | AAT Bioquest | Cat# 22251 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-3216 |
| Human neural progenitor cells | Stein et al., 2014 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Memo1Lox/Lox | This paper | N/A |
| Mouse: RG-Brainbow | This paper | N/A |
| Mouse: FVB-Tg(GFAP-cre)25Mes/J | The Jackson Laboratory | JAX: 004600 |
| Mouse: B6.129S2-Emx1tm1(cre)Krj/J | The Jackson Laboratory | JAX: 005628 |
| Mouse: Neurod6tm1(cre)Kan | Goebbels et al., 2006 | MGI:2668659 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX: 007909 |
| Mouse: Igs2tm1(ACTB-EGFP,-tdTomato)Luo/J | The Jackson Laboratory | JAX: 013749 |
| Mouse: Igs2tm2(ACTB-tdTomato,-EGFP)Luo/J | The Jackson Laboratory | JAX: 013751 |
| Recombinant DNA | ||
| Plasmid: pBLBP-GFP | Schmid et al., 2006 | N/A |
| Plasmid: pBLBP-DsRed2 | Schmid et al., 2006 | N/A |
| Plasmid: pCAG-hMEMO1 (WT)-GFP | This paper | N/A |
| Plasmid: pCAG-hMEMO1 (His49*)-GFP | This paper | N/A |
| Plasmid: pcDNA3-Myc-MEMO1 | Marone et al.,2004 | N/A |
| Plasmid: pcDNA-Cre | Masuda et al., 1998 | N/A |
| Plasmid: pCAG-MEMO1-BirA | This paper | N/A |
| Plasmid: pCAG-BirA | This paper | N/A |
| Plasmid: pCAG-SEP-GPR56 | This paper | N/A |
| Plasmid: pCAG-EB1-DsRed | Barth et al., 2002 | N/A |
| Plasmid: pEGFP-C1-CAMSAP2 | Jiang et al., 2014 | N/A |
| Plasmid: pBa-TfR-GFP | Burack et al., 2000 | Addgene #45060 |
| Software and Algorithms | ||
| Excel | https://products.office.com/en-us/excel | |
| R (version 3.3.3) | R Core Team | https://www.r-project.org/ |
| ImageJ | https://imagej.nih.gov/ij/ | |
| Zeiss LSM Image Browser | https://www.zeiss.com/microscopy/us/website/downloads/lsm-image-browser.html | |
Highlights.
Radial glial tiling regulates neuronal organization during cortical formation
Memo1 is a determinant of radial glial cell (RGC) tiling
Memo1 modulates microtubule stability and minus end protein, CAMSAP2, function
Disrupted radial glial tiling may contribute to cortical malformations in ASD
Acknowledgements
This research was supported by NIH grant MH060929 to E.S.A. and by the confocal imaging core of an NINDS institutional center core grant (P30NS045892) and UNC IDDRC grant (U54HD079124). NN was supported by the Osamu Hayaishi Memorial Scholarship. We thank Pasko Rakic, Yukako Yokota, Anthony LaMantia and Troy Ghashghaei for insightful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Akhmanova A, and Steinmetz MO (2015). Control of microtubule organization and dynamics: two ends in the limelight. Nat. Rev. Mol. Cell Biol 16, 711–726. [DOI] [PubMed] [Google Scholar]
- Barth AIM, Siemers KA, and Nelson WJ (2002). Dissecting interactions between EB1, microtubules and APC in cortical clusters at the plasma membrane. J. Cell Sci 115, 1583–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie R et al. (2017). Mosaic analysis with double markers reveals distinct sequential functions of Lgl1 in neural stem cells. Neuron 94, 517–533. [DOI] [PubMed] [Google Scholar]
- Bouvier J, and Cheng JG (2009). Recombineering-Based Procedure for Creating Cre/loxP Conditional Knockouts in the Mouse. Curr. Protoc. Mol. Biol January;Chapter 23:Unit 23.13. doi: 10.1002/0471142727.mb2313s85. [DOI] [PubMed] [Google Scholar]
- Burack MA, Silverman MA, and Banker G (2000). The role of selective transport in neuronal protein sorting. Neuron 26, 465–472. [DOI] [PubMed] [Google Scholar]
- Cappello S et al. (2012). A Radial Glia-Specific Role of RhoA in Double Cortex Formation. Neuron 73, 911–924. [DOI] [PubMed] [Google Scholar]
- de Frutos CA et al. (2016). Reallocation of Olfactory Cajal-Retzius Cells Shapes Neocortex Architecture. Neuron 92, 435–448. [DOI] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Won H, Stein JL, and Geschwind DH (2016). Advancing the understanding of autism disease mechanisms through genetics. Nat. Med 22, 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom T-Y et al. (2011). Direct visualization of microtubules using a genetic tool to analyse radial progenitor-astrocyte continuum in brain. Nat. Commun 2, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evsyukova I, Plestant C, and Anton ES (2013). Integrative mechanisms of oriented neuronal migration in the developing brain. Annu. Rev. Cell Dev. Biol 29, 299–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach GD, and Lord C (2010). The Simons Simplex Collection: A Resource for Identification of Autism Genetic Risk Factors. Neuron 68, 192–195. [DOI] [PubMed] [Google Scholar]
- Gao P et al. (2014). Deterministic progenitor behavior and unitary production of neurons in the neocortex. Cell 159, 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnham CP, and Roll-Mecak A (2012). The chemical complexity of cellular microtubules: tubulin post-translational modification enzymes and their roles in tuning microtubule functions. Cytoskeleton (Hoboken). 69, 442–463.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DA et al. (2014). Dendrite Self-Avoidance Requires Cell-Autonomous Slit/Robo Signaling in Cerebellar Purkinje Cells. Neuron 81, 1040–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, and Nave K-A (2006). Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 44, 611–621. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JLR, and Jones KR (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci 22, 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-porta D et al. (2001). β1-Class Integrins Regulate the Development of Laminae and Folia in the Cerebral and Cerebellar Cortex. Neuron 31, 367–379. [DOI] [PubMed] [Google Scholar]
- Guo H et al. (2000). Specificity and Efficiency of Cre-Mediated Recombination in Emx1-cre Knock-in Mice. Biochem. Biophys. Res. Commun 273, 661–665. [DOI] [PubMed] [Google Scholar]
- Guo Y et al. (2012). Two-stage genome-wide association study identifies variants in CAMSAP1L1 as susceptibility loci for epilepsy in Chinese. Hum. Mol. Genet 21, 1184–1189. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PRL, and Kriegstein AR (2010). Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561. [DOI] [PubMed] [Google Scholar]
- Hendershott MC, and Vale RD (2014). Regulation of microtubule minus-end dynamics by CAMSAPs and Patronin. Proc. Natl. Acad. Sci. U. S. A 111, 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham H, Bielas S, Tanaka T, and Gleeson JG (2004). Transgenic mouse line with green-fluorescent protein-labeled Centrin 2 allows visualization of the centrosome in living cells. Transgenic Res 13, 155–164. [DOI] [PubMed] [Google Scholar]
- Higginbotham H et al. (2013). Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat. Neurosci 16, 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S et al. (2010). Genetic Mosaic Dissection of Lis1 and Ndel1 in Neuronal Migration. Neuron 68, 695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, and Tanaka Y (2010). Molecular Motors in Neurons: Transport Mechanisms and Roles in Brain Function, Development, and Disease. Neuron 68, 610–638. [DOI] [PubMed] [Google Scholar]
- Hu WF, Chahrour MH, and Walsh CA (2014). The Diverse Genetic Landscape of Neurodevelopmental Disorders. Annu. Rev. Genomics Hum. Genet 15, 195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckfeldt RM et al. (2009). Transient neurites of retinal horizontal cells exhibit columnar tiling via homotypic interactions. Nat. Neurosci 12, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan Y, and Jan LY (2010). Branching out: mechanisms of dendritic arborization. Nat. Rev. Neurosci 11, 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jereb S et al. (2018). Differential 3’ Processing of Specific Transcripts Expands Regulatory and Protein Diversity Across Neuronal Cell Types. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Hua S, Mohan R, et al. (2014). Microtubule minus-end stabilization by polymerization-driven CAMSAP deposition. Dev. Cell 28, 295–309. [DOI] [PubMed] [Google Scholar]
- Jiang K et al. (2017). Microtubule minus-end regulation at spindle poles by an ASPM–katanin complex. Nat. Cell Biol 19, 480–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG, and Rakic P (2010). Radial columns in cortical architecture: It is the composition that counts. Cereb. Cortex 20, 2261–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay JN, Chu MW, and Sanes JR (2012). MEGF10 and MEGF11 mediate homotypic interactions required for mosaic spacing of retinal neurons. Nature 483, 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S et al. (2010). The apical complex couples cell fate and cell survival to cerebral cortical development. Neuron 66, 69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka G et al. (2012). Modeling the functional genomics of autism using human neurons. Mol. Psychiatry 17, 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze WK et al. (2015). PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol 22, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Sestan N, and Anton ES (2012). Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 139, 1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Won H, Stein JL, and Geschwind DH (2016). Advancing the understanding of autism disease mechanisms through genetics. Nat. Med 22, 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CS et al. (2012). Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of Autism spectrum disorders. PLoS Genet 8, e1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S et al. (2008). GPR56 Regulates Pial Basement Membrane Integrity and Cortical Lamination. J. Neurosci 28, 5817–5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavicius G et al. (2014). Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 11, 731–733. [DOI] [PubMed] [Google Scholar]
- Marone R, Hess D, Dankort D, Muller WJ, Hynes NE, and Badache A (2004). Memo mediates ErbB2-driven cell motility. Nat. Cell Biol 6, 515–522. [DOI] [PubMed] [Google Scholar]
- Martin M, and Akhmanova A (2018). Coming into Focus: Mechanisms of Microtubule Minus-End Organization. Trends Cell Biol 28, 574–588. [DOI] [PubMed] [Google Scholar]
- Maruoka H, Nakagawa N, Tsuruno S, Sakai S, Yoneda T, and Hosoya T (2017). Lattice system of functionally distinct cell types in the neocortex. Science (80-. ). 358, 610–615. [DOI] [PubMed] [Google Scholar]
- Miller JA et al. (2014). Transcriptional landscape of the prenatal human brain. Nature 508, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misson J-P, Takahashi T, and Caviness VS (1991a). Ontogeny of radial and other astroglial cells in murine cerebral cortex. Glia 4, 138–148. [DOI] [PubMed] [Google Scholar]
- Misson J-P, Austin CP, Takahashi T, Cepko CL, and Caviness VS (1991b). The Alignment of Migrating Neural Cells in Relation to the Murine Neopallial Radial Glial Fiber System. Cereb. Cortex 1, 221–229. [DOI] [PubMed] [Google Scholar]
- Molnar Z (2013) Cortical Columns In: RUBENSTEIN JLR and RAKIC P (ed.) Comprehensive Developmental Neuroscience: Neural Circuit Development and Function in the Brain, Volume 3, 109–129 Amsterdam: Elsevier [Google Scholar]
- Moore SA et al. (2002). Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 418, 422–425. [DOI] [PubMed] [Google Scholar]
- Muroyama A, and Lechler T (2017). Microtubule organization, dynamics and functions in differentiated cells. Development 144, 3012–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa N et al. (2017). APC sets the Wnt tone necessary for cerebral cortical progenitor development. Genes Dev 31, 1679–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HT et al. (2017). Integrated Bayesian analysis of rare exonic variants to identify risk genes for schizophrenia and neurodevelopmental disorders. Genome Med 9, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nijs L et al. (2009). EFHC1 interacts with microtubules to regulate cell division and cortical development. Nat. Neurosci 12, 1266–1274. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, and Kriegstein AR (2001). Neurons derived from radial glial cells establish radial units in neocortex. Nature 409, 714–720. [DOI] [PubMed] [Google Scholar]
- Ori-McKenney KM, Jan LY, and Jan Y-N (2012). Golgi outposts shape dendrite morphology by functioning as sites of acentrosomal microtubule nucleation in neurons. Neuron 76, 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao X et al. (2004). G Protein–Coupled Receptor–Dependent Development of Human Frontal Cortex. Science (80-. ). 303, 2033–2036. [DOI] [PubMed] [Google Scholar]
- Rakic P (1972). Mode of cell migration to the superficial layers of fetal monkey neocortex. J. Comp. Neurol 145, 61–83. [DOI] [PubMed] [Google Scholar]
- Rakic P (1988). Specification of cerebral cortical areas. Science (80-. ). 241, 170–176. [DOI] [PubMed] [Google Scholar]
- Rakic P (2007). The radial edifice of cortical architecture: From neuronal silhouettes to genetic engineering. Brain Res. Rev 55, 204–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasin M-R et al. (2007). Numb and Numbl are required for maintenance of cadherin-based adhesion and polarity of neural progenitors. Nat. Neurosci 10, 819–827. [DOI] [PubMed] [Google Scholar]
- Riccomagno MM et al. (2014). Cas Adaptor Proteins Organize the Retinal Ganglion Cell Layer Downstream of Integrin Signaling. Neuron 81, 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom et al. (2018). Novel genes for autism implicate both excitatory and inhibitory cell lineages in risk. bioRxiv. doi: 10.1101/484113. [DOI] [Google Scholar]
- Schmid RS, Yokota Y, and Anton ES (2006). Generation and characterization of brain lipid-binding protein promoter-based transgenic mouse models for the study of radial glia. Glia 53, 345–351. [DOI] [PubMed] [Google Scholar]
- Song Y, and Brady ST (2015). Post-translational modifications of tubulin: Pathways to functional diversity of microtubules. Trends Cell Biol 25, 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein JL et al. (2014). A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron 83, 69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner R et al. (2014). Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med 370, 1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, and Hevner RF (2014). Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nat. Rev. Neurosci 15, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Meng W, Nagae S, and Takeichi M (2012). Nezha/CAMSAP3 and CAMSAP2 cooperate in epithelial-specific organization of noncentrosomal microtubules. Proc. Natl. Acad. Sci. U. S. A 109, 20029–20034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telley L et al. (2016). Sequential transcriptional waves direct the differentiation of newborn neurons in the mouse neocortex. Science (80-. ). 351, 1443–1446. [DOI] [PubMed] [Google Scholar]
- Tirnauer JS, and Bierer BE (2000). EB1 Proteins Regulate Microtubule Dynamics, Cell Polarity, and Chromosome Stability. J. Cell Biol 149, 761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toya M et al. (2016). CAMSAP3 orients the apical-to-basal polarity of microtubule arrays in epithelial cells. Proc. Natl. Acad. Sci. U. S. A 113, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T et al. (2015). TTBK2 with EB1/3 regulates microtubule dynamics in migrating cells through KIF2A phosphorylation. J. Cell Biol 210, 737–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer JM, Yokota Y, Stanco A, Stumpo DJ, Blackshear PJ, and Anton ES (2009). MARCKS modulates radial progenitor placement, proliferation and organization in the developing cerebral cortex. Development 136, 2965–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau KW et al. (2014). Microtubule minus-end binding protein CAMSAP2 controls axon specification and dendrite development. Neuron 82, 1058–1073. [DOI] [PubMed] [Google Scholar]
- Yokota Y, Ring C, Cheung R, Pevny L, and Anton ES (2007). Nap1-regulated neuronal cytoskeletal dynamics is essential for the final differentiation of neurons in cerebral cortex. Neuron 54, 429–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota Y et al. (2009). The adenomatous polyposis coli protein is an essential regulator of radial glial polarity and construction of the cerebral cortex. Neuron 61, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota Y et al. (2010). Cdc42 and Gsk3 modulate the dynamics of radial glial growth, inter-radial glial interactions and polarity in the developing cerebral cortex. Development 137, 4101–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y-C, Bultje RS, Wang X, and Shi S-H (2009). Specific synapses develop preferentially among sister excitatory neurons in the neocortex. Nature 458, 501–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y-C et al. (2012). Preferential electrical coupling regulates neocortical lineage-dependent microcircuit assembly. Nature 486, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaoui K, Benseddik K, Daou P, Salaün D, and Badache A (2010). ErbB2 receptor controls microtubule capture by recruiting ACF7 to the plasma membrane of migrating cells. Proc. Natl. Acad. Sci. U. S. A 107, 18517–18522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X et al. (2016). Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 166, 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Espinosa JS, Su HH, Muzumdar MD, and Luo L (2005). Mosaic Analysis with Double Markers in Mice. Cell 121, 479–492. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, and Messing A (2001). hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31, 85–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental movie 1 (Related to Figure 2): Time-lapse images of control and Memo1-deficient BLBP+ radial progenitors in embryonic cerebral cortex. Time is indicated in hours and minutes.
Supplemental movie 2 (Related to Figures 3 and 4): Live imaging of migrating neurons in the intermediate zone of control and Memo1 cKOEmx cortex. Cyan arrowheads point to radially migrating neurons (control). Memo1 cKO neurons (purple arrowheads) branch and migrate non radially. Time is indicated in hours and minutes.
Supplemental movie 3 (Related to Figure 6): Time-lapse images of EB1-DsRed and GFP-CAMSAP2 in the basal process of control and Memo1 cKOEmx RGCs. Arrowheads point to anterogradely and retrogradely moving EB1 (red) or CAMPSAP2 (green) comets. Microtubules (blue) are labeled in left panels. Time is indicated in seconds.
Supplemental movie 4 (Related to Figure 7): Time-lapse images of TfR-GFP vesicles in the basal process of control and Memo1 cKOEmx RGCs. Arrowheads point to anterogradely (magenta) and retrogradely (white) moving vesicles. Time is indicated in seconds.
Figure S1 (Related to Figure 1): Memo1 expression in the developing cerebral cortex.
Figure S2 (Related to Figures 1 and 3): Generation of floxed Memo1 and RG-Brainbow mice.
Figure S3 (Related to Figures 1 and 3): Brainbow expression in radial progenitors and astrocytes during brain development.
Figure S4 (Related to Figures 1, 3, and 4): Aberrant outer RGCs and increased number of cortical astrocytes in Memo1 cKO neocortex.
Figure S5 (Related to Figures 3 and 4): Interneuron placement is not altered in Memo1 cKOEmx cortex.
Figure S6 (Related to Figures 1 and 4): Cre electroporation mediated inactivation of Memo1 leads to RGC abnormalities and disrupted neuronal layering.
Figure S7 (Related to Figures 1 and 4): Neuron-specific inactivation of Memo1 does not affect neuronal placement and laminar organization.
Figure S8 (Related to Figures 1, 2, 3 and 4): Memo1 deletion leads to hippocampal and cerebellar malformations and abnormal SVZ organization.
Figure S9 (Related to Figures 5 and 6): Centrosomal MT nucleation in Memo1-deficient RGCs.
Figure S10 (Related to Figures 1–7): Mechanisms and significance of radial glial tiling.