

Summary
The enzyme carbamoyl phosphate synthetase 1 (CPS1; EC 6.3.4.16) forms carbamoyl phosphate from bicarbonate, ammonia, and ATP and is activated allosterically by N-acetylglutamate. The neonatal presentation of bi-allelic mutations of CPS1 results in hyperammonemia with reduced citrulline and is reported as the most challenging nitrogen metabolism disorder to treat. As therapeutic interventions are limited, patients often develop neurological injury or die from hyperammonemia. Survivors remain vulnerable to nitrogen overload, being at risk for repetitive neurological injury. With transgenic technology, our lab developed a constitutive Cps1 mutant mouse and report its characterization herein. Within 24 hours of birth, all Cps1−/− mice developed hyperammonemia and expired. No CPS1 protein by Western blot or immunostaining was detected in livers nor was Cps1 mRNA present. CPS1 enzymatic activity was markedly decreased in knockout livers and reduced in Cps1+/−mice. Plasma analysis found markedly reduced citrulline and arginine and markedly increased glutamine and alanine, both intermolecular carriers of nitrogen, along with elevated ammonia, taurine and lysine. Derangements in multiple other amino acids were also detected. While hepatic amino acids also demonstrated markedly reduced citrulline, arginine, while decreased, was not statistically significant; alanine and lysine were markedly increased while glutamine was trending towards significance. In conclusion we have determined that this constitutive neonatal mouse model of CPS1 deficiency replicates the neonatal human phenotype and demonstrates the key biochemical features of the disorder. These mice will be integral for addressing the challenges of developing new therapeutic approaches for this, at present, poorly treated disorder.
Keywords: Carbamoyl Phosphate Synthetase (CPS1) Deficiency, Murine Model, Hyperammonemia, Citrulline, Glutamine
Introduction
Carbamoyl phosphate synthetase 1 (CPS1; EC 6.3.4.16) is the initial and rate-limiting enzyme of the urea cycle, located primarily in hepatocytes as 20% of total mitochondrial matrix protein (Clarke 1976). It exists as a monomeric enzyme of 1462 amino acids. Its function is to catalyze carbamoyl phosphate formation in a multistep reaction that requires ammonia, bicarbonate and two molecules of ATP (Diez-Fernandez and Häberle 2017), and it is allosterically activated by N-acetylglutamate (Rubio and Grisolia 1981).
CPS1 deficiency is a rare disorder with an incidence that is estimated to be from 1 in 539,000 (Keskinen et al 2008) to 1 in 1.3 million births (Summar et al 2013). Some have described the disorder as the most difficult of the nitrogen metabolism enzyme deficiencies to treat (Ah Mew et al 2003–2015). If not recognized and treated early, CPS1 deficiency typically leads to encephalopathy and death (Maestri et al 1991). Typical human plasma analysis has shown reduced citrulline and arginine (Funghini et al 2012), and the recommended treatment is for patients to adhere to a low protein diet that also includes the supplementation of arginine and administration of nitrogen scavenging drugs (Batshaw et al 1982; Batshaw et al 2001). Present day therapies (other than liver transplantation) are only marginally effective; patients continue to be at elevated risk for subsequent hyperammonemic crises and progressive, irreversible brain damage. With the substantial advances made in gene therapy in the last 20 years, a mouse model of this disorder is essential to develop gene-based approaches to treat this condition and decrease the nitrogen-mediated central nervous system vulnerability. However, no mouse model currently exists that phenocopies neonatal-onset CPS1 deficiency seen in humans.
We generated and extensively characterized a novel constitutive Cps1 knockout mouse model as the next step toward the advancement of novel therapies for this intractable disorder. To characterize the extensive derangement of metabolism by CPS1 enzyme loss, we closely analyzed hepatic CPS1 protein and RNA, as well as both hepatic and circulating plasma amino acids. As such, we demonstrate its biochemical similarity to the human condition, important for its future use in the further study of the CPS1 deficiency and the examination of new clinically relevant therapies.
Materials and Methods
Development of the Transgenic Constitutive CPS1 Knockout Mouse
We took an approach for both homologous recombination and subsequent post germline allelic modification to develop the constitutive Cps1 mutant mice (Figure 1). The targeting vector PG00187_Z_8_A08 was obtained from EuMMCR (European Conditional Mouse Mutagenesis) (Chan et al 2007; Skarnes et al 2011) and was utilized to generate a conditional Cps1 knockout mouse as previously described (Khoja et al 2018).
Figure 1. Strategy to generate the Cps1KO (tm1b) murine model.
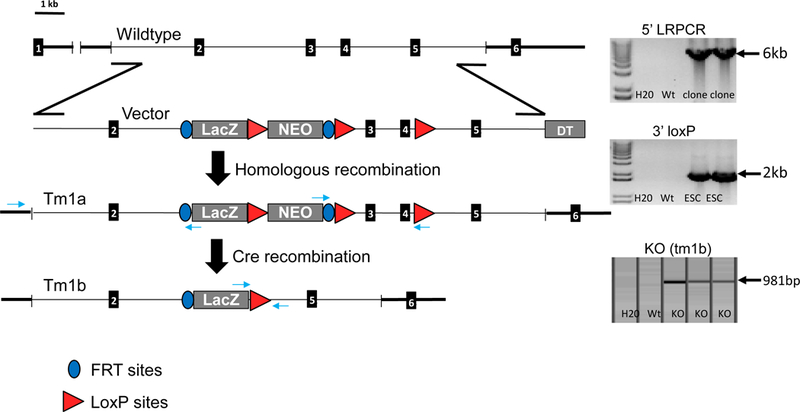
The genomic outline for this mouse is shown (left). In the top right image, 5’ LRPCR shows confirms the targeting in the N1 (tm1a) mice. With this approach, we were able to successfully introduce the gene specific forward primer 5’ in the homology arm. We also included a reverse primer in lacZ at en2/Frt junction. 3’ loxP image (middle right): In the middle image (right), loxP confirmation of the targeted N1 mice is demonstrated. This demonstrates germline transmission and that this is conditionally ready (tm1a). In the bottom right image, the demonstration of the Cre transgene positive mice confirms that successful recombination at the loxP sites has taken place. With this step a constitutive knockout is generated. With the LacZ cassette having a forward primer and a reverse in the downstream distal LoxP homology arm, determination of the Cps1 and Cre positive animals successfully recombined at the loxP sites and generated the mutant (tm1b). (Abbreviations: LRPCR refers to long range polymerase chain reaction and DTA refers to the Diphtheria Toxin A gene. The black rectangles represent exon regions of Cps1).
Heterozygous Cps1 tm1a mice were crossed to a ubiquitous Cre expressing strain C57BL/6N-Hprttm1(CMV-cre)Wtsi/Mmucd (stock number 034412 at MMRRC) in order to excise the exogenous promoter-driven neomycin cassette as well as floxed exons 3 through 4 for a lacZ expressing constitutive KO tm1b allele. DNA (50 ng) was subjected to PCR for the Cre transgene amplification product (F-primer CTTTCCTCATGCCCCAAAATCTTAC paired with R-primer GCTATCAGGACATAGCGTTGGCTAC to yield a 700 bp Cre transgene amplification product). Cre-excised Cps1 tm1b allele was subjected to PCR-amplification using F-primer GCTACCATTACCAGTTGGTCTGGTGTC paired with R-primer CCTTCCTATGCCCTTTGTGTTTCAGG to yield a Cre excised amplicon of 981 bp. Progeny of the backcrossing of Cps1 tm1b positive animals with C57BL/6N wildtype animals were screened as above. Both Flpo and Cre crosses were performed as previously described (Khoja et al 2018).
Mouse Procedures
Housing under specific pathogen-free conditions was maintained for the mice in these studies; food and water were provided ad libitum. The mice were handled according to the guidelines of the US National Institutes of Health. All experimental procedures were performed following the guidelines of our institution. Mice did suckle shortly after birth as milk spots were visible in newborn litters. Mice were euthanized if they showed symptoms of hyperammonemia which in neonates primarily consisted of inactivity compared to normal littermate controls. In general, all assays were performed with 5 mice per group. In the CPS1 enzyme assay 3 Cps1+/− and 4 Cps1+/+ mice were studied along with 5 Cps1−/− mice.
Biochemical Analysis
Plasma Ammonia
After euthanizing the mice with isoflurane, a thoracotomy was performed and the heart was transected. Blood was collected utilizing soda lime microhaematocrit tubes coated with sodium heparin (80 IU/ml) (Globe Scientific Inc., Paramus, New Jersey). Plasma was immediately collected by low-speed centrifugation at 4ºC and promptly frozen at −80ºC. Samples were stored until ready for ammonia determination and then tested in the same setting; prolonged storage was avoided, and testing was generally performed with all samples simultaneously to avoid any batch effect. Ammonia determination was performed utilizing 5 μl of plasma per sample using an Ammonia Assay Kit following the instructions of the manufacturer (ab83360, Abcam, Cambridge, MA) and optical density was performed at 570 nm as previously described (Khoja et al 2018).
Plasma Amino Acids
HPLC was performed to determine amino acid concentrations with pre-column derivatization with o-phthalaldehyde as previously described (Jones and Gilligan 1983).
Liver Amino Acids
Amino acids profile in the liver was determined as in (Nissim et al 2012). Briefly, the frozen liver tissue was ground into a fine powder and then extracted with 4% perchloric acid, neutralized with KOH, and used for measurement of amino acids. The concentration of amino acids was determined with an Agilent 1260 Infinity LC system, utilizing pre-column derivatization with o-phthalaldehyde (Nissim et al 2012; Jones and Gilligan 1983).
Immunohistochemistry
Whole livers were carefully dissected from euthanized mice, fixed in 10% neutral buffered formalin for 48 hours and stored in 70% ethanol. Routine paraffin embedding with standard procedures followed. Immunohistochemical detection of CPS1 was performed as previously described (Khoja et al 2018).
Western Blot
Liver samples were homogenized using RIPA buffer (Sigma, St. Louis, MO) along with protease inhibitor cocktail kit (ThermoFisher Scientific, Waltham, MA). Performance of Western blots was as previously reported (Khoja et al 2018).
CPS1 Enzyme Assay
CPS1 activity was assayed at 37°C by a coupled reaction where the formed carbamoyl phosphate was immediately converted to citrulline by ornithine transcarbamylase (OTC), as described (Diez-Fernandez et al 2013).
Quantitative Real-Time RT-PCR for CPS1
Total RNA from liver was extracted, cDNA synthesized, and gene expression levels quantified by real-time PCR as previously described (Khoja et al 2018). Target gene expression was normalized to that of endogenous β-Actin with fold enrichment measured using the 2-ΔΔCt method. Primers: Cps1: Forward CACCAATTTCCAGGTGACCA, Reverse TACTGCTTTAGGCGGCCTTT; β-Actin: Forward CTAAGGCCAACCGTGAAAAG, Reverse ACCAGAGGCATACAGGGACA. Conditions for qPCR: 1 min at 98°C, then cycle 40x with 15 sec at 98°C and 30 sec at 60°C.
Statistical Analysis
The data presentation is as the arithmetic mean ± standard deviation. GraphPad Prism Software 6.0 (La Jolla, CA) was utilized for statistical analyses as Student’s t-test. A value of p<0.05 was considered statistically significant.
Results
Knockout of CPS1 in murine liver leads to hyperammonemia and early postnatal death
Neonatal Cps1−/− mice (Cps1tm1b) were developed and used for these studies (Figure 1). Neonatal biallelic Cps1−/− mice were produced by mating adult Cps1+/− mice and screening pups by PCR. In progeny, we compared the observed vs the expected frequencies of the Cps1 allele using the chi-square goodness of fit test. The test statistic was computed as the sum of the (observed - expected)2/expected across the 3 groups (Cps1−/−, Cps1+/−, and Cps1+/+). Assuming there is a good fit between observed vs expected frequencies, the above test statistic is known to follow the chi-square distribution with degrees of freedom computed as number of groups minus 1. For 3 groups, the degrees of freedom = 3 – 1 = 2. Our data demonstrate an excellent fit between the observed vs the expected frequencies (chi-square = 0.21, 2 degrees of freedom, p = 0.8914) revealing a distribution of genotypes that is Mendelian (Cps1+/+ 24%, Cps1+/− 49%, Cps1−/− 27%) without a deficiency of mutant alleles in the population (Figure 2A). These findings demonstrate that there is not a loss of mutant mice because of intrauterine demise.
Figure 2. Characteristics of Neonatal CPS1-Deficient Mice.
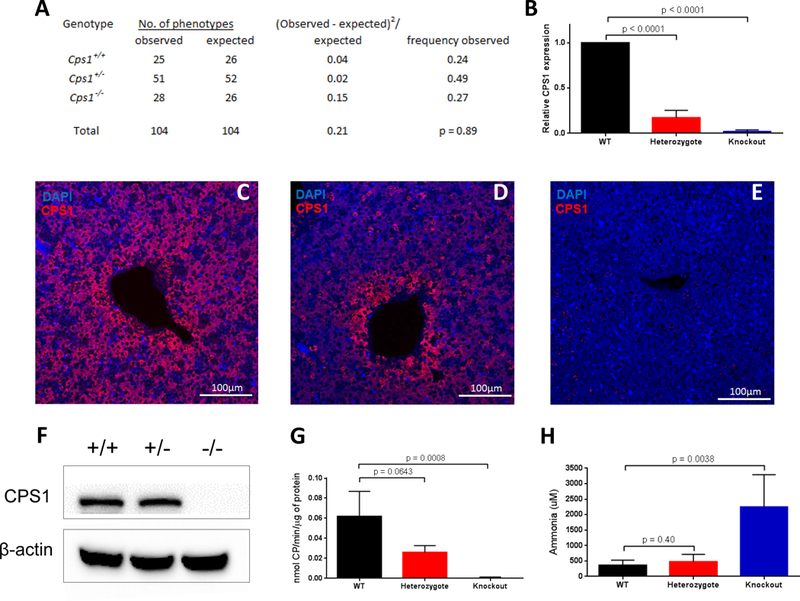
A) The genotypes are distributed among 104 progeny of CPS1 matings of heterozygote mice; B) Western blot showing hepatic CPS1 protein in wild type (+/+), heterozygote (+/−) and knockouts (−/−); representative immunostaining for CPS1 shows hepatic CPS1 expression in liver in Cps1+/+ (C) and Cps1+/− (D) mice with absent expression in knockout mouse liver (E); (F) CPS1 activity in hepatic extracts from Cps1+/+, Cps1+/− and knockout mice correspond with that of CPS1-specific mRNA (G); plasma ammonia levels of neonatal Cps1+/+, Cps1+/− and knockout mice (n=5 per group; except in the CPS1 enzyme assay [n=3 Cps1+/−, n=4 Cps1+/+ and n=5 Cps1−/−]). (Data are presented as mean ± standard deviation.) (CP = Carbamoyl Phosphate).
Quantitative real-time PCR demonstrated a lack of Cps1 hepatic mRNA in Cps1-deficient mice compared to wild types (Figure 2B) (Cps1+/+ 1.000; Cps1+/− 0.171 ± 0.083; Cps1−/− 0.021 ± 0.017). Cps1-deficient mice also completely lacked hepatic CPS1 protein as demonstrated by immunohistochemistry (Figure 2C-E) and Western blot (Figure 2F), while enzymatic activity was markedly diminished (Figure 2G). Compared to Cps1+/+ animals (360.6 ± 147.1 uM), ammonia levels in plasma of Cps1−/− mice in metabolic crisis were elevated 6-fold (2255.8 ± 927.5 uM; p = 0.0038) (Figure 2H); all Cps1−/− mice died by 24 hours after birth (n=28). There was no statistical difference in plasma ammonia between Cps1+/+ (360.6 ± 147.1 μM) and Cps1+/− (475.3 ± 214.2 μM; p=0.4) mice. Histologically, the livers of Cps1−/− mice appeared normal under H&E staining (data not shown).
Plasma amino acid analysis demonstrates hyperglutaminemia and other derangements
The plasma levels of multiple amino acids were significantly disturbed in the Cps1−/− versus Cps1+/+ mice (n=5 per group, all studies) (Figure 3). Knockout mice, assayed within 12 hours after birth, had significantly elevated plasma glutamine (1855.4 ± 862.6 μmol/L) compared to that found in Cps1+/+ mice (621.1 ± 153.2; p=0.0218). Citrulline was significantly depleted in Cps1−/− mice (1.54 ± 3.45 μmol/L) compared to the Cps1+/+ (53.4 ± 21.8 μmol/L; p=0.0003); a similarly marked reduction was found in plasma arginine (Cps1−/−: 19.5 ± 18.1 μmol/L; Cps1+/+: 79.8 ± 15.5 μmol/L; p=0.0004). Plasma ornithine was increased with a trend towards significance in Cps1−/− mice (185.0 ± 83.6 nmol/ml) compared to Cps1+/+ mice (94.4 ± 38.8 μmol/L; p=0.07).
Figure 3. Plasma Amino Acids.
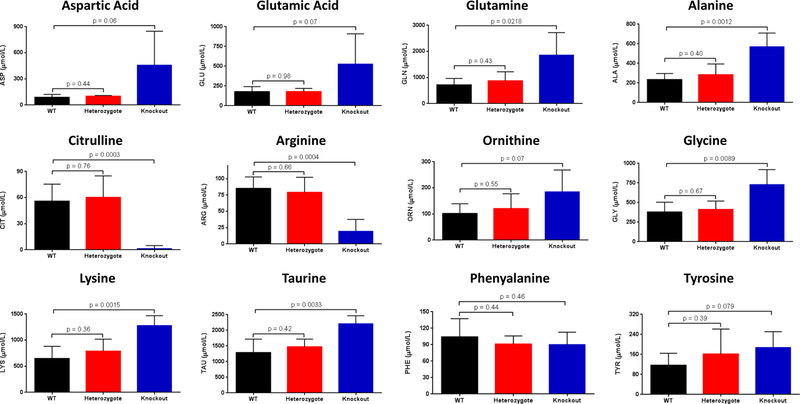
Amino acids were analyzed from plasma from constitutive CPS1 knockout mice and littermate controls within 12 hours of birth (n=5 per group; data presented as mean ± standard deviation in μmol/L). (ASP = aspartic acid, GLU = glutamic acid, GLN = glutamine, ALA = alanine, CIT = citrulline, ARG = arginine, ORN = ornithine; LYS = lysine, TAU = taurine, GLY = glycine, PHE = phenylalanine, TYR = tyrosine)
Other plasma amino acids were markedly altered concomitantly with glutamine, citrulline, arginine, and ornithine. Alanine (568.9 ± 140.7 μmol/L vs. 217.4 ± 61.5 μmol/L; p = 0.0012), glycine (729.5 ± 190.9 μmol/L vs. 338.9 ± 107.4 μmol/L; p = 0.0089), lysine (1280.0 ± 189.3 μmol/L vs. 621.2 ± 263.1 μmol/L; p = 0.0015) and taurine (2207.3 ± 256.0 μmol/L vs. 1198.8 ± 441.6 μmol/L; p = 0.0033) were all significantly elevated while aspartic acid (458.5 ± 388.9 μmol/L vs. 73.8 ± 20.2 μmol/L; p = 0.06), glutamic acid (527.1 ± 381.5 μmol/L vs. 149.2 ± 17.8 μmol/L; p = 0.08) and tyrosine (187.8 ± 63.2 μmol/L vs. 106.9 ± 49.3 μmol/L; p = 0.08) were trending toward elevated values (Cps1−/− vs Cps1+/+, respectively, all groups). Asparagine, tryptophan, methionine, phenylalanine, the branched chain and hydroxyl amino acids were not different (p > 0.5) between Cps1+/+ and Cps1−/− animals (data not shown). Comparison of Cps1+/+ plasma with that of Cps1+/− was not statistically different for all of these amino acids.
Hepatic amino acid analysis demonstrates more limited derangement
The levels of hepatic amino acids were altered but to a more limited extent compared to the plasma (n=5 per group, all studies) (Figure 4). Cps1−/− mice, similarly assayed within 12 hours of birth, had elevated hepatic glutamine (27.4 ± 5.6 nmol/mg protein) compared with levels found in Cps1+/+ mice (21.6 ± 5.5 nmol/mg protein), and there was a trend towards elevated glutamine (p = 0.13). Citrulline was nearly depleted from hepatocytes in Cps1−/− mice (0.01 ± 0.03 nmol/mg protein) compared to Cps1+/+ (2.1 ± 2.2 nmol/mg protein; p = 0.06). However, ornithine (Cps1−/−: 6.47 ± 4.46 nmol/mg protein; Cps1+/+: 5.17 ± 5.04 nmol/mg protein; p = 0.67), arginine (Cps1−/−: 0.60 ± 0.75 nmol/mg protein; Cps1+/+: 1.30 ± 1.49 nmol/mg protein; p = 0.37), and taurine (Cps1−/−: 50.89 ± 18.72 nmol/mg protein; Cps1+/+: 61.59 ± 11.67 nmol/mg protein; p = 0.30) levels were not different in the liver samples.
Figure 4. Hepatic Amino Acids.
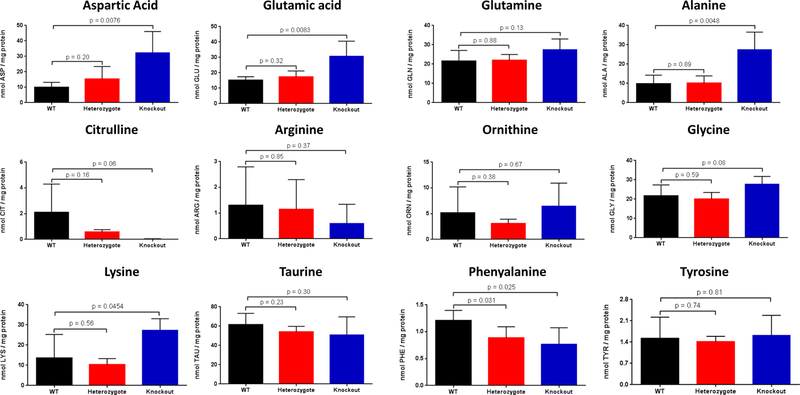
Amino acids were analyzed from liver lysates from constitutive CPS1 knockout mice and littermate controls within 12 hours of birth (n=5 per group; data presented as mean ± standard deviation in nmol/mg protein). (ASP = aspartic acid, GLU = glutamic acid, GLN = glutamine, ALA = alanine, CIT = citrulline, ARG = arginine, ORN = ornithine; LYS = lysine, TAU = taurine, GLY = glycine, PHE = phenylalanine, TYR = tyrosine)
A more limited number of other amino acids were altered compared to plasma. Alanine (27.4 ± 9.2 nmol/mg protein vs. 9.8 ± 4.5 nmol/mg protein; p = 0.0048), glycine (27.8 ± 4.0 nmol/mg protein vs. 21.7 ± 5.7 nmol/mg protein; p = 0.08), lysine (27.4 ± 5.7 nmol/mg protein vs. 13.6 ± 11.7 nmol/mg protein; p = 0.045), aspartic acid (32.3 ± 13.8 nmol/mg protein vs. 9.9 ± 3.2 nmol/mg protein; p = 0.0076), and glutamic acid (30.8 ± 9.8 nmol/mg protein vs. 15.2 ± 2.3 nmol/mg protein; p = 0.008) were all elevated while phenylalanine (0.77 ± 0.31 nmol/mg protein vs. 1.21 ± 0.19 nmol/mg protein; p = 0.026) was reduced (Cps1−/− vs Cps1+/+, respectively, all groups). Asparagine, tryptophan, methionine, isoleucine, valine and the hydroxyl amino acids showed no statistically significant differences (p > 0.25) when compared to Cps1+/+ mice (data not shown).
Discussion
Clinically two CPS1 deficiency phenotypes have been described: 1) severe neonatal onset presenting shortly after birth caused by complete loss of protein function; and 2) less severe late onset of disease seen in both children and adults caused by low/moderate protein and activity levels (Klaus et al 2009; Ono et al 2009; Funghini et al 2012). With the neonatal onset form of the disorder, patients are healthy at birth and often do well for the subsequent few days. They then develop lethargy with reduced oral intake, progressing to vomiting, hypothermia, seizures, and coma. Due to disease severity, prognosis is poor; many patients die from brain edema despite aggressive attempts to lower blood ammonia (Kurokawa et al 2007). Surviving patients suffer irreversible brain injury and continue to remain vulnerable to nitrogen overload. Present day medical and dietary therapies are limited in their efficacy and have not advanced significantly in the past 30 years.
Using standard gene replacement technology, we have developed a novel constitutive Cps1 loss of function murine model by excising exons 3–4 and introducing a stop codon in exon 5 of the knockout Cps1 allele. Genotypic analysis from matings between Cps1+/− mice by PCR demonstrates the distribution of genotypes was Mendelian, strongly suggesting that Cps1 deficiency does not result in intrauterine demise. This predominantly hepatic mitochondrial enzyme is essential for mammalian nitrogenous waste removal via the urea cycle. As expected, these Cps1−/− mice have rapid development of marked hyperammonemia in the early postnatal period resulting in 100% lethality within 24 hours of birth. Quantitative RT-PCR, Western blot, immunohistochemistry, and functional enzyme activity demonstrated a lack of CPS1 mRNA, protein, and activity respectively. In heterozygotes, while biochemically normal, we did detect a greater than expected reduction in Cps1 mRNA. The reason for such a finding is unclear but it has been reported that allelic variation can effect gene expression in human CPS1 deficiency (Klaus et a 2009). Obtaining a better understanding of this finding will be the focus of future studies with this murine model.
A constitutive Cps1-deficient mouse was reported nearly 20 years ago by another group; they targeted exon 17 and inserted a PGK-neo selection cassette into an ATP binding domain (Schofield et al 1999). However, this murine model was not submitted to a repository and is no longer available (Deignan et al 2008). In that model, double mutants of Cps1 succumbed by 36 hours suffering from marked hyperammonemia. As limited characterization was performed of this prior model, we have demonstrated in this new Cps1-deficient mouse that the biochemical features consistent with human neonatal onset CPS1 deficiency are present.
CPS1 catalyzes the first step of the urea cycle (2ATP + NH3 + HCO3- → 2ADP + HPO4 2- + NH2CO2PO32-) (Rubio 1993), converting ammonia to carbamoyl phosphate, which is then utilized by OTC to produce citrulline in the second reaction of the urea cycle. Generally, the differences in both plasma and liver amino acid profiles between Cps1−/− mice and Cps1+/+ littermates are explained by the expected disruption of nitrogen metabolism inflicted by loss of CPS1 enzyme activity. In comparison to Cps1+/+ (and less so to Cps1+/− mice), Cps1−/− mice demonstrate significant divergence in the plasma concentrations of many amino acids. These include marked reductions in plasma citrulline and arginine and increases in glutamine, alanine [both interorgan carriers of nitrogen (Nurjhan et al 1995)], aspartic acid, and glutamic acid. Similar changes are detected (albeit to a lesser extent of derangement) in the hepatic amino acids of Cps1−/− mice, with the exception of arginine which is not significantly decreased in hepatocytes as it is in the plasma.
The urea cycle perturbations caused by the lack of carbamoyl phosphate result in the expected amino acid alterations. Aspartic acid is likely increased as limited citrulline is restricting the enzymatic step of argininosuccinate synthetase to produce argininosuccinate. Plasma arginine is also likely decreased due to reduced citrulline limiting urea cycle flux. Elevated plasma glycine levels are typical of CPS1 deficiency and are a direct target of the nitrogen scavenging drug sodium benzoate. Covalent binding of benzoate reduces plasma glycine, and thus nitrogen, by forming hippurate, followed by renal excretion.
While the liver contains enzymes to metabolize all of the amino acids (with some limitation with the branched chain group) and possesses the urea cycle, interorgan amino acid transport is also a major metabolic process. This allows for distribution of amino acids to different organs and tissues to support their physiologic functions and to maintain amino acid homeostasis (Brosnan 2003). Many dietary amino acids (glutamine, threonine, leucine, lysine and phenylalanine) are also extensively metabolized in the intestine while the kidney also plays a significant role including interorgan metabolism of arginine (Wakabayashi et al 1994), glycine, glutamine, serine and citrulline (Brosnan 1987). Importantly, the presence of developmental differences in intestinal flux of amino acids between neonatal and adult animals can affect plasma levels in enzyme deficiencies. Together these may in part explain the differences in plasma and hepatic amino acids found in the Cps1−/− mice.
Initial limited investigations by our laboratory suggest that gene therapy for this neonatal onset disorder will be challenging for multiple reasons. First, the consumption of milk by neonatal Cps1−/− mice likely leads to an early rise in plasma ammonia shortly after suckling begins. Treatment after birth, even in the neonatal period, may already be too late to effectively control ammonia. For example, we have intravenously administered a helper-dependent adenoviral vector expressing Cps1 by a liver-specific promoter (Khoja et al 2018) when pups were found after birth; often milk could be visualized in the neonatal stomach. While limited and preliminary, a life extension of not greater than 1 day was detected in a small number of mice (data not shown). Second, the size of the Cps1 cDNA is challenging for potential clinical vector development. The cDNA for CPS1 is 4.5 kb, limiting which viral vectors may be developed for gene therapy. For example, adeno-associated viral (AAV) vectors, which are currently in clinical trials for certain single gene disorders including OTC deficiency, have a wild type genome size of 4.7 kb (Hastie and Samulski 2015). In development of an AAV that will express CPS1, the vector genome must also include a promoter, polyadenylation signal and inverted terminal repeats at a minimum. These together will exceed the wild type genome size and may affect packaging efficiency and yield. To date, our lab has been unable to produce a single chain AAV vector that expresses CPS1 (data not shown).
Promising delivery approaches and new vehicles other than viral vectors and neonatal administration should be considered. Development of mRNA delivered in lipid nanoparticles, with rapid onset of translation, may be a potential therapy for the neonatal onset disease (An et al 2017). Furthermore, prenatal screening does allow for the diagnosis of CPS1 deficiency before birth (Finckh et al 1998). In studies conducted by our lab (Lipshutz et al 1999; Lipshutz et al 1999; Lipshutz et al 1999; Lipshutz et al 2000; Lipshutz et al 2001; Lipshutz et al 2003) and those of others (Dejneka et al 2004; Sugano et al 2012), in utero gene administration, as an alternative to early postnatal administration, can be performed in mice and other animal models. This may present a promising avenue of therapy that could be successful in this murine model if an appropriate method of CPS1 protein expression can be developed.
With the recent identification of CPS1 variants correlating with increased plasma glycine and protective effects against atherosclerosis (Hartiala et al 2016), in addition to the role of CPS1 in vascular signaling (Summar et al 2004; Kaluarachchi et al 2018), this new model may also serve as a useful tool for the in-depth study of the relationship of short- and long-term cardiovascular effects and CPS1 enzymatic activity. Additional preliminary findings implicate CPS1 in Alzheimer’s disease and other neurological disorders (Griffin et al 2018). These new studies, combined with the more established studies of CPS1 in cancer initiation and progression (Liu et al 2011; Kim et al 2017), make the murine model described here more broadly applicable beyond the field of urea cycle disorders.
In conclusion, we have developed and characterized a novel constitutive mouse model of Cps1 deficiency that recapitulates the biochemical and physiological symptoms found in human patients. This model will be crucial to deepen the understanding of biochemical changes that occur, to develop methods to blunt toxic ammonia (and glutamine) buildup, and to investigate translatable gene- and cell-based therapies.
Synopsis:
A constitutive carbamoyl phosphate synthetase 1-deficient transgenic mouse was developed that replicates the human neonatal onset form of this disorder and will be an effective specimen for the study of hepatic gene-based and cell-based therapies for this deficiency.
Acknowledgements
Development of this mouse model would not have been possible without the technical support, products, and services of the Mouse Biology Program (MBP) at the University of California Davis. The C57BL/6N-Hprttm1(CMV-cre)Wtsi/Mmucd, RRID:MMRRC_034412-UCD (MMRRC Cre line) mouse strain was obtained from the NIH funded strain repository MMRC (Mutant Mouse Regional Resource Center). The strain was originally donated by Allan Bradley PhD from the Wellcome Sanger Institute. For these studies, we enlisted the support of the Metabolomics Core facility at The Children’s Hospital of Philadelphia (https://metabolomic.research.chop.edu/) where they performed amino acid concentration determinations of both plasma and of liver homogenates.
Funding
Jim and Ada Horwich provided funding to generate this constitutive carbamoyl phosphate synthetase deficient knockout mouse. Murine model characterization was funded by grant R21NS091654 (G.S.L.) from the United States NIH/National Institute of Neurological Disorders and Stroke (NINDS).
Funding NIH grant to GSL R21NS091654
Documentation of ICCULA Approval: Approval obtained by UCLA’s Animal Research Committee
Abbreviations:
- CPS1
Carbamoyl phosphate synthetase 1
Footnotes
Conflicts of Interest: Suhail Khoja, Matthew Nitzahn, Brian Truong, Jenna Lambert, Brandon Willis, Gabriella Allegri, Véronique Rüfenacht, Johannes Häberle declare that they have no conflict of interest. Gerald S. Lipshutz serves as a consultant to Audentes Therapeutics in a role unrelated to the studies conducted herein.
Conflict of Interest
The authors claim no conflict of interest.
References
- Ah Mew N, Lanpher B, Gropman AL, Chapman KA, Simpson KL, Summar ML (2003–2015) Urea Cycle Disorders Overview. In Editor ed. eds. Book Urea Cycle Disorders Overview Seattle, WA: University of Washington. [Google Scholar]
- An D, Schneller JL, Frassetto A, et al. (2017) Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia. Cell Rep 21: 3548–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batshaw ML, Brusilow S, Waber L, et al. (1982) Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med 306: 1387–1392. [DOI] [PubMed] [Google Scholar]
- Batshaw ML, MacArthur RB, Tuchman M (2001) Alternative pathway therapy for urea cycle disorders: twenty years later. J Pediatr 138: S46–54; discussion S54–45. [DOI] [PubMed] [Google Scholar]
- Brosnan JT (1987) The 1986 Borden award lecture. The role of the kidney in amino acid metabolism and nutrition. Can J Physiol Pharmacol 65: 2355–2362. [DOI] [PubMed] [Google Scholar]
- Brosnan JT (2003) Interorgan amino acid transport and its regulation. J Nutr 133: 2068S–2072S. [DOI] [PubMed] [Google Scholar]
- Chan W, Costantino N, Li R, et al. (2007) A recombineering based approach for high-throughput conditional knockout targeting vector construction. Nucleic Acids Res 35: e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke S (1976) A major polypeptide component of rat liver mitochondria: carbamyl phosphate synthetase. J Biol Chem 251: 950–961. [PubMed] [Google Scholar]
- Deignan JL, Cederbaum SD, Grody WW (2008) Contrasting features of urea cycle disorders in human patients and knockout mouse models. Mol Genet Metab 93: 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejneka NS, Surace EM, Aleman TS, et al. (2004) In utero gene therapy rescues vision in a murine model of congenital blindness. Mol Ther 9: 182–188. [DOI] [PubMed] [Google Scholar]
- Diez-Fernandez C, Häberle J (2017) Targeting CPS1 in the treatment of Carbamoyl phosphate synthetase 1 (CPS1) deficiency, a urea cycle disorder. Expert Opin Ther Targets 21: 391–399. [DOI] [PubMed] [Google Scholar]
- Diez-Fernandez C, Martinez AI, Pekkala S, et al. (2013) Molecular characterization of carbamoyl-phosphate synthetase (CPS1) deficiency using human recombinant CPS1 as a key tool. Hum Mutat 34: 1149–1159. [DOI] [PubMed] [Google Scholar]
- Finckh U, Kohlschutter A, Schafer H, Sperhake K, Colombo JP, Gal A (1998) Prenatal diagnosis of carbamoyl phosphate synthetase I deficiency by identification of a missense mutation in CPS1. Hum Mutat 12: 206–211. [DOI] [PubMed] [Google Scholar]
- Funghini S, Thusberg J, Spada M, et al. (2012) Carbamoyl phosphate synthetase 1 deficiency in Italy: clinical and genetic findings in a heterogeneous cohort. Gene 493: 228–234. [DOI] [PubMed] [Google Scholar]
- Griffin JWD, Liu Y, Bradshaw PC, Wang K (2018) In Silico Preliminary Association of Ammonia Metabolism Genes GLS, CPS1, and GLUL with Risk of Alzheimer’s Disease, Major Depressive Disorder, and Type 2 Diabetes. J Mol Neurosci 64: 385–396. [DOI] [PubMed] [Google Scholar]
- Hartiala JA, Tang WH, Wang Z, et al. (2016) Genome-wide association study and targeted metabolomics identifies sex-specific association of CPS1 with coronary artery disease. Nat Commun 7: 10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie E, Samulski RJ (2015) Recombinant adeno-associated virus vectors in the treatment of rare diseases. Expert Opin Orphan Drugs 3: 675–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BN, Gilligan JP (1983) o-Phthaldialdehyde precolumn derivatization and reversed-phase high-performance liquid chromatography of polypeptide hydrolysates and physiological fluids. J Chromatogr 266: 471–482. [DOI] [PubMed] [Google Scholar]
- Kaluarachchi DC, Smith CJ, Klein JM, Murray JC, Dagle JM, Ryckman KK (2018) Polymorphisms in urea cycle enzyme genes are associated with persistent pulmonary hypertension of the newborn. Pediatr Res 83: 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskinen P, Siitonen A, Salo M (2008) Hereditary urea cycle diseases in Finland. Acta Paediatr 97: 1412–1419. [DOI] [PubMed] [Google Scholar]
- Khoja S, Nitzahn M, Hermann K, et al. (2018) Conditional disruption of hepatic carbamoyl phosphate synthetase 1 in mice results in hyperammonemia without orotic aciduria and can be corrected by liver-directed gene therapy. Mol Genet Metab 124: 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hu Z, Cai L, et al. (2017) CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546: 168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus V, Vermeulen T, Minassian B, et al. (2009) Highly variable clinical phenotype of carbamylphosphate synthetase 1 deficiency in one family: an effect of allelic variation in gene expression? Clin Genet 76: 263–269. [DOI] [PubMed] [Google Scholar]
- Kurokawa K, Yorifuji T, Kawai M, et al. (2007) Molecular and clinical analyses of Japanese patients with carbamoylphosphate synthetase 1 (CPS1) deficiency. J Hum Genet 52: 349–354. [DOI] [PubMed] [Google Scholar]
- Lipshutz GS, Flebbe-Rehwaldt L, Gaensler KM (1999) Adenovirus-mediated gene transfer in the midgestation fetal mouse. J Surg Res 84: 150–156. [DOI] [PubMed] [Google Scholar]
- Lipshutz GS, Flebbe-Rehwaldt L, Gaensler KM (1999) Adenovirus-mediated gene transfer to the peritoneum and hepatic parenchyma of fetal mice in utero. Surgery 126: 171–177. [PubMed] [Google Scholar]
- Lipshutz GS, Flebbe-Rehwaldt L, Gaensler KM (2000) Reexpression following readministration of an adenoviral vector in adult mice after initial in utero adenoviral administration. Mol Ther 2: 374–380. [DOI] [PubMed] [Google Scholar]
- Lipshutz GS, Gruber CA, Cao Y, Hardy J, Contag CH, Gaensler KM (2001) In utero delivery of adeno-associated viral vectors: intraperitoneal gene transfer produces long-term expression. Mol Ther 3: 284–292. [DOI] [PubMed] [Google Scholar]
- Lipshutz GS, Sarkar R, Flebbe-Rehwaldt L, Kazazian H, Gaensler KM (1999) Short-term correction of factor VIII deficiency in a murine model of hemophilia A after delivery of adenovirus murine factor VIII in utero. Proc Natl Acad Sci U S A 96: 13324–13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipshutz GS, Titre D, Brindle M, Bisconte AR, Contag CH, Gaensler KM (2003) Comparison of gene expression after intraperitoneal delivery of AAV2 or AAV5 in utero. Mol Ther 8: 90–98. [DOI] [PubMed] [Google Scholar]
- Liu H, Dong H, Robertson K, Liu C (2011) DNA methylation suppresses expression of the urea cycle enzyme carbamoyl phosphate synthetase 1 (CPS1) in human hepatocellular carcinoma. Am J Pathol 178: 652–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestri NE, Hauser ER, Bartholomew D, Brusilow SW (1991) Prospective treatment of urea cycle disorders. J Pediatr 119: 923–928. [DOI] [PubMed] [Google Scholar]
- Nissim I, Horyn O, Nissim I, et al. (2012) Effects of a glucokinase activator on hepatic intermediary metabolism: study with 13C-isotopomer-based metabolomics. Biochem J 444: 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurjhan N, Bucci A, Perriello G, et al. (1995) Glutamine: a major gluconeogenic precursor and vehicle for interorgan carbon transport in man. J Clin Invest 95: 272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono H, Suto T, Kinoshita Y, Sakano T, Furue T, Ohta T (2009) A case of carbamoyl phosphate synthetase 1 deficiency presenting symptoms at one month of age. Brain Dev 31: 779–781. [DOI] [PubMed] [Google Scholar]
- Rubio V (1993) Structure-function studies in carbamoyl phosphate synthetases. Biochem Soc Trans 21: 198–202. [DOI] [PubMed] [Google Scholar]
- Rubio V, Grisolia S (1981) Human carbamoylphosphate synthetase I. Enzyme 26: 233–239. [DOI] [PubMed] [Google Scholar]
- Schofield JP, Cox TM, Caskey CT, Wakamiya M (1999) Mice deficient in the urea-cycle enzyme, carbamoyl phosphate synthetase I, die during the early neonatal period from hyperammonemia. Hepatology 29: 181–185. [DOI] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, et al. (2011) A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474: 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugano H, Matsumoto T, Miyake K, et al. (2012) Successful gene therapy in utero for lethal murine hypophosphatasia. Hum Gene Ther 23: 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summar ML, Gainer JV, Pretorius M, et al. (2004) Relationship between carbamoyl-phosphate synthetase genotype and systemic vascular function. Hypertension 43: 186–191. [DOI] [PubMed] [Google Scholar]
- Summar ML, Koelker S, Freedenberg D, et al. (2013) The incidence of urea cycle disorders. Mol Genet Metab 110: 179–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi Y, Yamada E, Yoshida T, Takahashi H (1994) Arginine becomes an essential amino acid after massive resection of rat small intestine. J Biol Chem 269: 32667–32671. [PubMed] [Google Scholar]