

Abstract
Objective:
Previous studies demonstrated that deficiency of angiotensin converting enzyme 2 (ACE2) augmented angiotensin II-induced atherosclerosis and abdominal aortic aneurysm (AAA) formation in hypercholesterolemic mice. Effects of ACE2 deficiency could arise from increased concentrations of its substrate, AngII, or decreased concentrations of its product, angiotensin-(1-7) (Ang-(1-7)). Infusion of Ang-(1-7), a Mas receptor (MasR) ligand, to hypercholesterolemic male mice reduced AngII-induced atherosclerosis, suggesting a protective role of the Ang-(1-7)/MasR axis. However, it is unclear if endogenous Ang-(1-7) acts at MasR to influence AngII-induced vascular diseases. The purpose of this study was to define the role of MasR deficiency on AngII-induced atherosclerosis and AAA formation/severity in hypercholesterolemic male mice.
Methods:
MasR+/+ and −/− male mice on a low density lipoprotein receptor-deficient (Ldlr−/−) or apolipoprotein E-deficient (Apoe−/−) background were infused with AngII at either 600 or 1,000 ng/kg/min by osmotic minipump for 28 days. Atherosclerosis was quantified at study endpoint as percent lesion surface area of the aortic arch in Ldlr−/− mice. Abdominal aortic internal diameters were quantified by ultrasound and maximal external AAA diameters were quantified at study endpoint. Blood pressure was quantified by either radiotelemetry and a tail-cuff based technique. Serum cholesterol concentrations and vascular tissue characterization were examined at study endpoint.
Results:
MasR deficiency did not influence body weight, systolic blood pressure at baseline and during AngII infusion, or serum cholesterol concentrations in either Apoe−/− or Ldlr−/− mice. MasR deficiency increased AngII-induced atherosclerosis in aortic arches of Ldlr−/− mice (P<.05), associated with increased oxidative stress and apoptosis in aortic root sections (P<.05). MasR deficiency also augmented internal and external AAA diameters and increased aortic ruptures of both Ldlr−/− and Apoe−/− mice (P<.05). These effects were associated with increased elastin breaks and T-lymphocyte and macrophage accumulation into abdominal aortas of AngII-infused MasR-deficient mice (P<.05).
Conclusions:
These results demonstrate that MasR deficiency augmented AngII-induced atherosclerosis and AAA rupture through mechanisms involving increased oxidative stress, inflammation, and apoptosis, suggesting that MasR activation may provide therapeutic efficacy against vascular diseases.
Keywords: Mas receptor, angiotensin-(1-7), aneurysm, atherosclerosis, rupture
TOC Summary:
Mas receptor deficiency significantly increases angiotensin II-induced atherosclerosis and aortic aneurysm ruptures in hyperlipidemic male mice. The authors suggest that this receptor plays an important role in angiotensin II-induced vascular diseases.
Introduction
Vascular diseases, including atherosclerosis and abdominal aortic aneurysms (AAAs), are prevalent disorders that contribute to significant morbidity and mortality. Unfortunately, there are currently no effective medical therapies for AAAs, despite a high risk of rupture and death. While statins are widely used to reduce development of atherosclerosis, there are limited therapies that regress established atherosclerotic lesions. Infusion of angiotensin II (AngII), the primary peptide of the renin angiotensin system (RAS), into hypercholesterolemic male mice (low density lipoprotein receptor (Ldlr−/−)-deficient or apolipoprotein E (Apoe−/−)-deficient)) augments atherosclerosis and results in the formation of AAAs.1, 2 Blockade of the production of AngII through angiotensin converting enzyme inhibitors or angiotensin type 1 receptor (AT1R) antagonists have proven effective to reduce development of atherosclerosis and AAAs in experimental models.3, 4 However, RAS blockade through these conventional pathways has not shown clinical efficacy against development or progression of atherosclerosis or AAAs.5–9
The monocarboxypeptidase enzyme angiotensin converting enzyme 2 (ACE2) catabolizes AngII to form angiotensin-(1-7) (Ang-(1-7)), a peptide that binds to the G-protein coupled receptor, MasR.10–12 The ACE2/Ang-(1-7)/MasR axis has been suggested to counterbalance the actions of AngII on angiotensin type 1 receptors (AT1Rs). We demonstrated previously that whole body ACE2 deficiency augmented AAA formation in AngII-infused Ldlr−/− mice.13 Conversely, pharmacologic activation of ACE2 reduced AngII-induced AAAs.13 Furthermore, overexpression of ACE2 by adenovirus in Apoe−/− mice lowered the incidence of AngII-induced AAAs.14 These findings were supported by results localizing ACE2 to human abdominal aortic tissue.13 Similar protective effects of ACE2 have been implicated in experimental atherosclerosis, as ACE2 deficiency augmented development of diet-induced atherosclerosis in Ldlr−/− male mice.15 Further, co-infusion of AngII with Ang-(1-7) reduced atherosclerosis in Ldlr−/− mice.15 Taken together, these results support an emerging role for ACE2 and its product Ang-(1-7) as pathways providing protection from atherosclerosis and AAA formation. However, since ACE2 manipulation influences not only production of Ang-(1-7) but also catabolism of AngII, it is unclear if vascular protective effects result from reduced AngII/AT1R or through Ang-(1-7) activation of MasR.
The MasR was described originally as an AngII receptor since the peptide induced calcium transients in a MasR-transfected neural cell line.16 Fourteen years later, Ang-(1-7) was identified to bind MasR and elicit relaxation of endothelium-containing aortic rings.10 Ang-(1-7) mediated MasR activation resulted in improvement of vascular function by increasing nitric oxide bioavailability and reducing reactive oxygen species generation.17, 18 Conversely, MasR-deficient mice exhibit endothelial dysfunction and increased vascular ROS production.19 In this study, we defined effects of MasR deficiency on AngII-induced atherosclerosis and AAA formation. We hypothesized that MasR deficiency increases AngII-induced atherosclerosis and augments formation and severity of AngII-induced AAAs.
Methods
Mice.
The MasR was deleted by insertion of a lacZ gene into exon 1 of the MasR gene (Knockout Mouse Project, UC Davis). Clones were made through neomycin selection and embryos were injected into pseudo-pregnant females (C57B1/6N, Taconic). Mouse genotyping was performed on DNA extracted from tail or ear clips by PCR using the presence and/or absence of the MasR and lacZ genes (MasR forward, 5’-GTCCTCTACTTGCTGTACTACGAG-3’; reverse, 5’-GTTGGCGCTGCTGTTGATC-3’; LacZ forward, 5’-GGTAAACTGGCTCGGATTAGGG-3’; reverse, 5’-TTGACTGTAGCGGCTGATGTTG-3’). All studies were performed in wildtype or MasR-deficient litter mate mice bred to either an Ldlr- or Apoe-deficient background (The Jackson Laboratory, Bar Harbor, ME). Males (Ldlr−/−, 5-6 months of age; Apoe−/−, 2 months of age) were fed either standard mouse laboratory diet (Apoe−/−, Teklad, TD.2918) or a Western diet (Ldlr−/−, Teklad, TD.88137) beginning 1 week prior to implantation of osmotic minipumps and throughout the remainder of each study. Body weights were quantified weekly. Osmotic micropumps (Alzet, Model 1004) were implanted subcutaneously to infuse AngII at either 600 (Ldlr−/−) or 1,000 (Apoe−/−) ng/kg/min to MasR+/+ and −/− mice on each background strain for 28 days. At study endpoint, mice were anesthetized (ketamine/xylazine, 100:10 mg/kg, ip) and blood was obtained by cardiac puncture to quantify serum cholesterol and plasma Ang-(1-7) concentrations. Aortas were harvested for quantification of atherosclerosis and AAAs. All experiments involving mice conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by Institutional Animal Care and Use Committees at the University of Kentucky or the University of Dusseldorf (AZ. 8.87-50.10.34.08.216 and AZ 84–02.04.2012.A250).
Quantification of blood pressure by a tail-cuff based technique.
Systolic blood pressures (SBP) were quantified in MasR+/+ (n=11) and MasR−/− mice (n=9) on an Ldlr−/− background for 4 consecutive days using a Visitech blood pressure monitoring system before (day 0) and during (week 3) AngII infusions. Blood pressure data were averaged over the days of recording for each genotype.
Quantification of blood pressure by radiotelemetry.
Radiotelemetry was used to quantify blood pressure in conscious, unrestrained Apoe−/− and Apoe−/− Mas−/− mice. Prior to implantation of osmotic minipumps, mice were anesthetized and pressure-sensing catheters (PA-C10, Data Sciences International, s’Hertogenbosch, the Netherlands) were implanted via the left common carotid artery as described previously.20 After a recovery period of 7 days, blood pressure was monitored continuously to collect baseline (5 days) pressures and following infusion of AngII. Blood pressure data were averaged over the days of recording for each mouse within each genotype.
Quantification of serum and plasma components.
Serum was collected using serum separator tubes (BD Microtainers, 20,000g for 4 min). Cholesterol concentrations in sera were quantified as described previously15 using a Wako Cholesterol E kit (Wako Diagnostics). Complete blood (20 μl) counts were quantified using MiniCollect tubes (Greiner bio-one) followed by analysis using a Hemavet 950FS (Erba Diagnostics). Plasma (50 μl) Ang-(1-7) concentrations were quantified as described previously13 using a commercial ELISA (Pennisula Labs, San Carlos, CA) according to the manufacturer’s instructions.
Quantification of AAAs by in vivo ultrasound and ex vivo maximal AAA diameter.
In vivo internal abdominal aortic diameters were quantified by ultrasound using the B-mode of a VisualSonics ultrasound imaging system (Vevo 2100) with a 55-MHz probe. Ultrasound was performed on anesthetized mice at day 0 and 27 of AngII infusion. Images were analyzed by observers who were blinded to the study design. At study endpoint, aortas from anesthetized mice were anterograde perfused (PBS, 5 ml) through the left ventricle and placed in 10% (wt/vol) formalin. After fixation (24-48 hours), aortas were cleaned of adherent tissue and mounted on black wax and imaged using a dissecting microscope (Nikon, Model SMZ800) with camera attachment (Nikon, Model DS-Fi1) and image analysis software (Nikon Elements, V.3.1) for measurement of maximal external abdominal aortic diameters.
Quantification of immune cell infiltration and elastin fragmentation
To quantify abdominal aortic immune cell infiltration and elastin fragmentation, abdominal aortas were fixed with 4% (wt/vol) paraformaldehyde, paraffin-embedded, and sectioned at 5 μm. Cross sections of maximum diameter and adjacent sections (three preceding and three succeeding sections at 30 μm intervals in each direction) were used for Movat staining, as well as for F4/80 and CD3 immunohistochemistry. For F4/80 staining, slides were incubated with Proteinase K (S3020, Dako, Glostrup, Denmark) for 3 minutes. For CD3 staining, slides were incubated with antigen retrieval buffer (pH 9) (S2367, Dako, Carpinteria, CA, USA, pH 9; for 20 minutes at 98°C) and then at room temperature for 30 minutes. Afterwards, all slides were incubated with 3% (vol/vol) H2O2 (10 minutes) and then with horse serum (Vector, MP-7401 Burlingame, CA, USA; 20 minutes). Slides were incubated with rat anti-F4/80 antibody (1:100, MCA497RT, Bio-Rad, Oxford, UK) or rabbit anti-CD3 antibody (IS503, Dako, Glostrup, Denmark) overnight (4°C). After washing (Dako, S0809, Glostrup, Denemark), slides were incubated either with ImmPRESS anti-rabbit IgG HRP (Vector, MP-7401, Burlingame, CA, USA) or with ImmPRESS anti-rat IgG HRP (Vector, MP-7444, Burlingame, CA, USA) for 30 minutes (room temperature). Immunostaining was visualized using 3,3’-diaminobenzidine (DM827, Agilent, Ratingen, Germany). After washing (10 minutes), slides were dried and mounted (Roti©-Mount HP68.1, Karlsuhe, Germany) with coverslips.
To identify elastin fragmentation, Movat staining was performed on paraffin-embedded aortic sections. Slides were fixed in Bouin’s solution (No.10132 Sigma-Aldrich; 50°C, 10 minutes) and stained with 5% (vol/vol) sodium thiosulfate (No.217263 Sigma-Aldrich; 5 minutes), 1% (vol/vol) alcian blue (No.5268 Sigma-Aldrich; 15 minutes), and alkaline alcohol (No.6899 Sigma-Aldrich; 10 minutes). Then, slides were incubated (20 minutes) with Movat Weigert’s solution which was prepared from a 2% (wt/vol) alcohol hematoxylin, ferric chloride and iodine stock solution in ratios of 3:2:1. Afterwards, slides were incubated with crocein scarlet-acid/fuchsin solution (Crocein Scarlett, No.0531 Chempur; Acid Fuchsin, No.8129 Sigma-Aldrich; 1 minute), 5% (vol/vol) phosphotungstic acid (No.4006 Sigma-Aldrich; 5 minutes) and 1% (vol/vol) acetic acid (5 minutes). Slides were washed in between each of these steps, and then dehydrated in 95% ethanol, followed by 100% ethanol (1 minute). Slides were immersed in alcohol saffron (8 minutes), 100% ethanol (1 minute, twice), and then xylol (5 minutes, twice). Finally, tissue sections were mounted in mounting medium (Roti-Mount HP68.1) and covered. Chemicals were purchased from Sigma, Chempur, Microm and Carl-Roth. Images were acquired at 100× and 400× magnification and quantification was performed using analysis software (ImageJ 1.37v software, NIH). CD3 or F4/80 positive cells were calculated as a percentage of positive staining per cross section area and elastic fragmentation was quantified as elastin breaks per cross section. Quantification of the results were performed by two observers blinded to the experimental design.
En face quantification of atherosclerotic lesion surface area in aortic arch.
Aortic arches from Apoe−/− and Ldlr−/− mice infused with AngII were pinned on black wax and a 3-mm line was drawn from the left subclavian artery. Aortic lesions were then outlined using image analysis software (Nikon Elements, V3.1) and lesion areas were determined for the entire arch. Lesion surface area (sum of all lesions) was expressed as a percentage of the aortic arch.
Characterization of aortic root sections.
Aortic roots were sectioned as described previously21 and sections were stained with Oil Red O for neutral lipids within atherosclerotic lesions. Apoptosis of smooth muscle cells was quantified in aortic root tissue sections using a terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) kit (Roche, Inc.). Two to three tissue sections were averaged from each mouse (n = 3 mice/group) from the midpoint of the aortic semilunar valves. TUNEL positive cells were counted and expressed as a percentage of cell number (DAPI-stained). Oxidative stress was quantified in aortic root sections (2-3 sections averaged from n = 3 mice/group) using dihydroethidium (DHE) staining. Images were obtained at exposures of 150 milliseconds with a gain of 1.4×, and then analyzed by intensity-frequency histogram (intensities of 250-2500) expressed as area under the curve (AUC). Quantification of results were performed by two observers blinded to the experimental design.
Statistics
Data are reported as mean ± standard error (SEM). Data were tested for normal distribution and equal variance. For Table 1 and Figures 1 and 2, data were analyzed by unpaired Student’s t-tests. For Figure 3, Student’s t-tests were performed, except for AAA incidence (Figures 3C,D), a Fischer’s exact test was performed. Also, for survival curves (Figures 3G,H), a Gehan-Breslow-Wilcoxon test was performed. For Figure 4, Student’s t-tests were performed. P ≤ 0.05 was considered statistically significant. Statistical analyses were performed using Graphpad Prism v.5.0 (Graphpad Software).
Table I.
Body weights, baseline and endpoint systolic and diastolic blood pressure, serum cholesterol concentrations, plasma Ang-(1-7) concentrations, and complete blood counts in MasR+/+ and MasR−/− male mice. Systolic blood pressures of Apoe−/− mice were quantified by radiotelemetry. Systolic blood pressures of Ldlr−/− mice were quantified by tail-cuff. ND indicates not determined.
| MasR+/+ Ldlr−/− | MasR−/− Ldlr−/− | MasR+/+ Apoe−/− | MasR−/− Apoe−/− | |
|---|---|---|---|---|
| Body weight (g) | 34 ± 0.8 | 31 ± 1.3 | 28 ± 0.7 | 31 ± 0.6 |
| Baseline systolic blood pressure (mmHg) | 112 ± 4 | 110 ± 4 | 129 ± 8 | 124 ± 7 |
| Endpoint systolic blood pressure (mmHg) | 141 ± 6 | 143 ± 2 | 158 ± 13 | 162 ± 8 |
| Baseline diastolic blood pressure (mmHg) | 90 ± 2 | 84 ± 6 | 99 ± 3 | 95 ± 4 |
| Endpoint diastolic blood pressure (mmHg) | 112 ± 7 | 110 ± 1 | 133 ± 8 | 133 ± 5 |
| Serum cholesterol (mg/dl) | 1624 ± 130 | 2204 ± 418 | 326 ± 21 | 349 ± 25 |
| Plasma Ang-(1-7) (ng/mL) | 0.1 ± 0.01 | 0.1 ± 0.02 | ND | ND |
| WBCs (K/μL) | 4.0 ± 0.5 | 3.7 ± 0.2 | ND | ND |
| Platelets (K/μL) | 606 ± 35 | 604 ± 40 | ND | ND |
| Hematocrit (%) | 38 ± 0.7 | 35 ± 1.5 | ND | ND |
Figure 1.
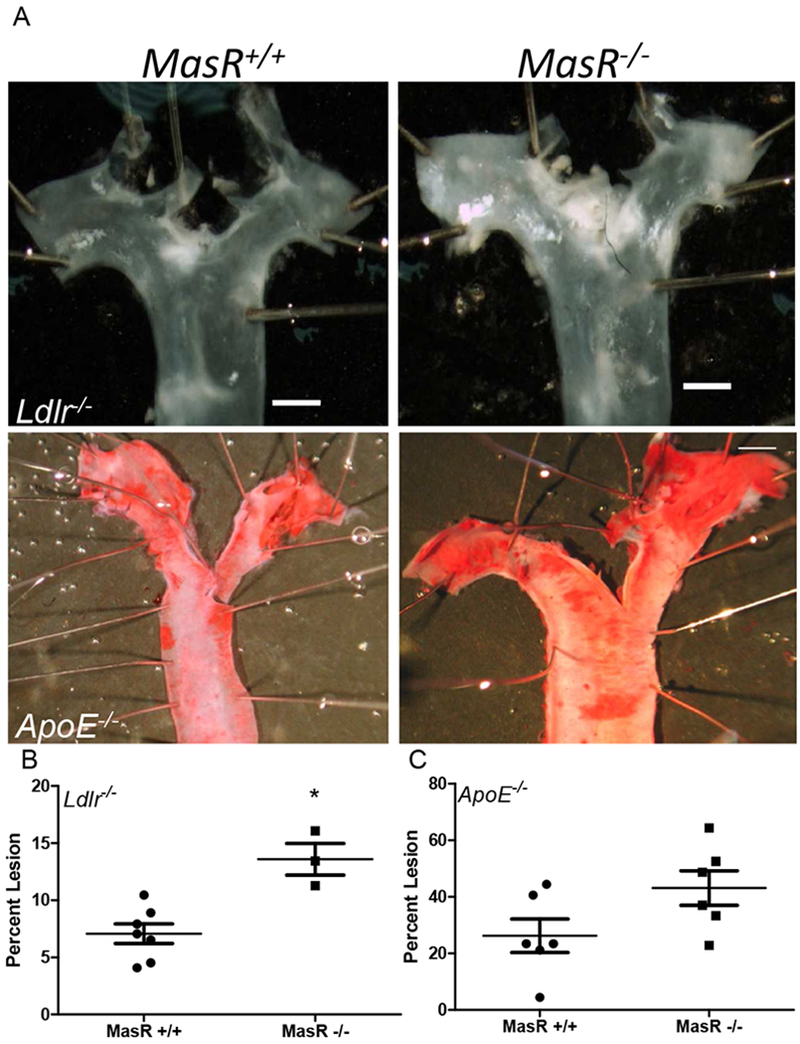
MasR deficiency increased AngII-induced atherosclerosis in Ldlr−/− and Apoe−/−male mice. En face analysis of aortic arches of MasR+/+ and MasR−/−/Ldlr−/− and Apoe−/− mice. Figure 1A, Representative images of the aortic arch from mice of each genotype (scale bar represents 5 mm). Figure 1B and 1C, Percent atherosclerosis lesion surface area in Ldlr−/− and Apoe−/− mice. Data are mean ± SEM from n=3-7 mice/group; *, P<0.05.
Figure 2.
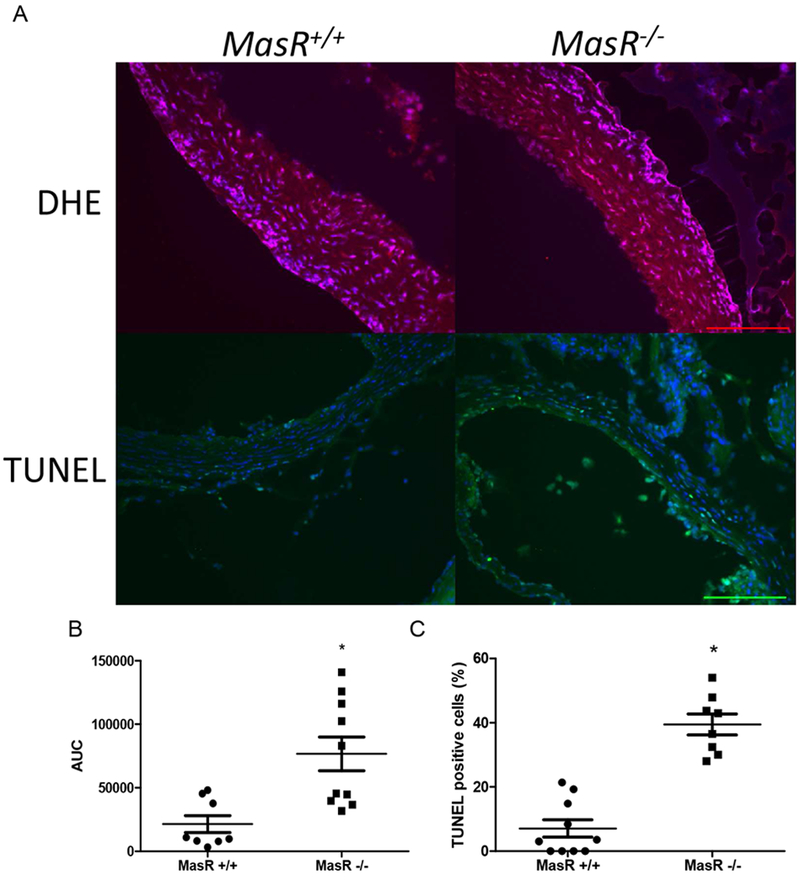
Elevations in oxidative stress and apoptosis in aortic sinus tissue sections from MasR-deficient Ldlr−/− mice infused with AngII. Figure 2: Representative DHE staining (red, A) with quantification (B) from 2-3 aortic sinus tissue sections from n = 3 mice/genotype. Representative TUNEL staining (green, A) with quantification (C) of percent TUNEL positive cells in aortic sinus sections from mice of each genotype. Scale bar represents 50 microns. Data are mean ± SEM from n=3 sections/mouse, n=3 mice/genotype; *, P<0.05.
Figure 3.
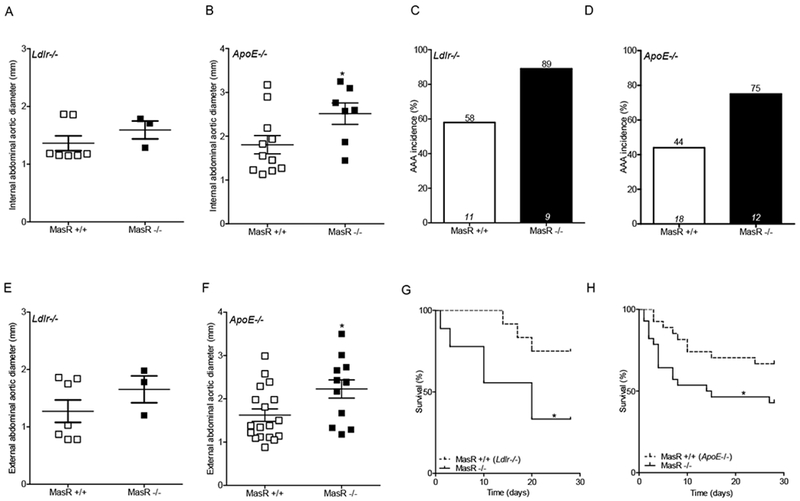
MasR deficiency augmented formation and severity of AngII-induced AAAs. A,B: In vivo ultrasound abdominal aortic lumen diameters on day 27 of AngII infusion in Ldlr−/− (A; n=3-7 mice/group) and Apoe−/− (B, n=7-11 mice/group) mice. C,D: AAA incidence in AngII-infused Ldlr−/− (C) and Apoe−/− (D) mice. Numbers of mice/genotype are illustrated within columns for each group, while percent incidence is illustrated above each column. E,F: Maximal external AAA diameters of AngII-infused Ldlr−/− (E) and Apoe−/− (F) mice. G,H: Percent survival over days of AngII infusion in Ldlr−/− (G) and Apoe−/− (H) mice. Data are mean ± SEM. *, P<0.05.
Figure 4.
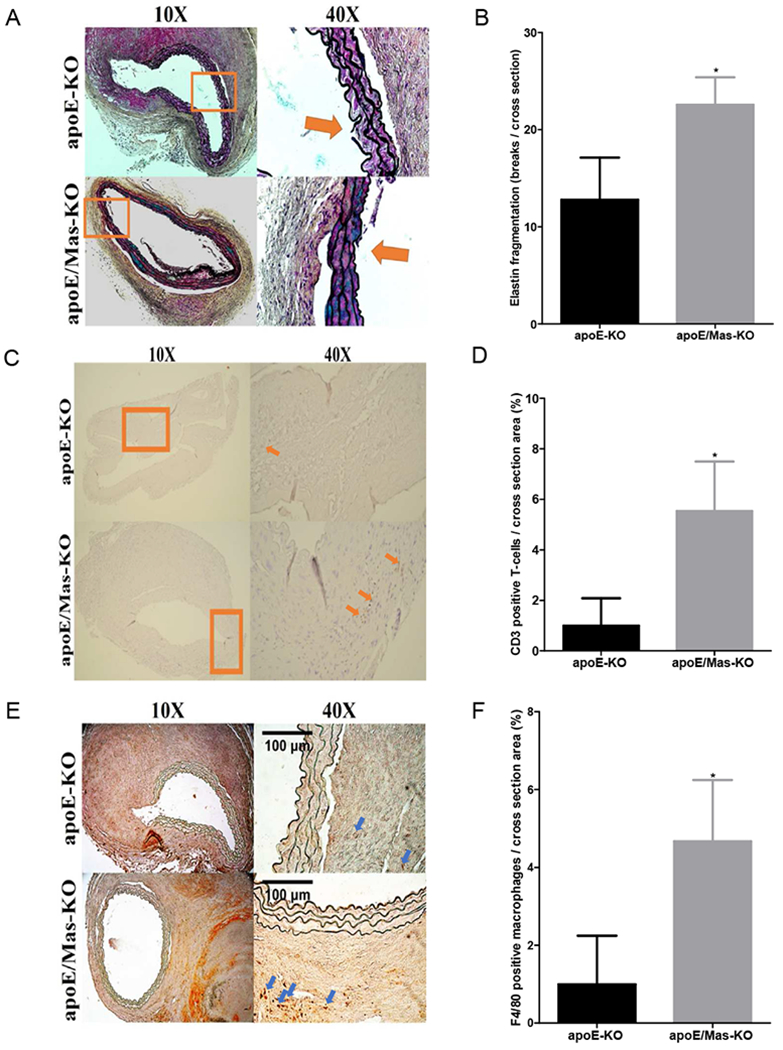
MasR deficiency increased elastin fragmentation and immune cell accumulation in AAA tissue sections. A, Representative Movat staining in AAA tissue sections from MasR+/+ or MasR−/− (KO) Apoe−/− mice infused with AngII. Nuclei and elastin stain black, smooth muscle cells stain red, blue staining indicates ground substance, and yellow staining indicates collagen. Boxes at lower magnification (100×) are magnified at 400× to illustrate fragmentation of elastin. B, Quantification of elastin fragmentation in AAA tissue sections from n=5-7 mice/genotype. C, Representative T-lymphocyte (CD3) immunostaining in AAA tissue sections from mice of each genotype. D, Quantification of CD3 positive T-lymphocyte staining per section. E, Representative macrophage (F4/80) immunostaining in AAA tissue sections from mice of each genotype. F, Quantification of F4/80 positive macrophage staining per section. Scale bar represents 100 microns. Data are mean ± SEM. *, P<0.05.
Results
MasR deficiency had no effect on body weight, systolic blood pressure, or serum cholesterol concentrations in hypercholesterolemic male mice
MasR deficiency had no effect on body weight, baseline blood pressure, blood pressure during infusion of AngII, blood cell counts, or plasma concentrations of Ang-(1-7)(Table I). Similarly, MasR deficiency did not influence serum cholesterol concentrations in Apoe−/− or Ldlr−/− AngII-infused mice; however, serum cholesterol concentrations were markedly higher in Ldlr−/− compared to Apoe−/− mice, regardless of genotype (Table I).
MasR deficiency augmented AngII-induced atherosclerosis in Ldlr−/− and Apoe−/− male mice
To investigate the effect of MasR on atherosclerosis, we quantified atherosclerotic lesion surface area on intimal surfaces of the aortic arches of AngII infused MasR−//− Ldlr−/−, MasR+/+/Ldlr−/−, MasR−/−/Apoe−/− and Mas+/+/Apoe−/−. Atherosclerotic lesion surface area was increased significantly in AngII-infused MasR−/− compared to MasR+/+/Ldlr−/− mice (Figure 1A,B; P<0.05). There was a trend for increased atherosclerotic lesion surface area of MasR−/−/Apoe−/− mice compared to control; however, the effect was not significant (Figure 1A,C; P=0.07). To delineate potential mechanisms for effects of MasR deficiency, we quantified DHE staining in aortic sinus tissue sections which was increased significantly in MasR−/− compared to MasR+/+//Ldlr−/− mice (Figure 2A,B; P<0.05). Similarly, TUNEL apoptotic staining was increased markedly in aortic sinus tissue sections from MasR−/− compared to MasR+/+/Ldlr−/− mice (Figure 2A,C; P<0.05). In contrast to aortic arch atherosclerosis, there was no difference in lesion area (quantified by Oil Red O staining) of aortic sinus sections from MasR+/+ compared to MasR−/−/Ldlr−/− mice (MasR+/+, 26,459 ± 7717 μm2; MasR−/−, 24,281 ± 9659 μm2, P=.61).
MasR deficiency promoted the formation and severity of AngII-induced AAAs in Ldlr−/− and Apoe−/− male mice
Abdominal aortic lumen diameters (day 27 of AngII infusion) were increased significantly in Apoe−/−, but not in Ldlr−/− MasR-deficient mice infused with AngII (Figure 3A,B; Supplemental Figure 1; P<0.05), potentially due to the small number of AngII-infused Ldlr−/− mice (n=3) that survived the study protocol, or due to the lower dose of AngII infusion in Ldlr−/− mice. AAA incidence increased (by 31%) in both Ldlr−/− and Apoe−/− MasR-deficient mice compared to wildtype mice (Figure 3C,D). AAA severity was augmented by MasR deficiency, as evidenced by significant increases in external abdominal aortic diameters of Apoe−/− but not Ldlr−/−/MasR-deficient mice (Figure 3E,F; P<0.05). Percent survival was significantly lower in both Apoe−/− and Ldlr−/− MasR-deficient mice infused with AngII (Figure 3G,H; P<0.05). The incidence of aneurysm rupture was higher in MasR-deficient mice on an Ldlr−/− (MasR+/+, 36%; MasR−/−, 67%) compared to Apoe−/− (MasR+/+, 39%; MasR−/−, 42%) background.
Since aneurysm rupture was increased in MasR-deficient mice, we quantified elastin fragmentation in AAA tissue sections from AngII-infused Apoe−/−/MasR+/+ and −/− mice. Elastin fragmentation within the aortic media increased significantly in AAA sections from MasR−/− compared to MasR+/+ mice (Figure 4A,B; P<0.05). Moreover, immunostaining for T-lymphocytes (CD3) and macrophages (F4/80) increased significantly in AAA tissue sections from AngII-infused MasR−/− compared to MasR+/+ mice (Figure 4C-F; P<0.05).
Discussion and Conclusion
Results from this study demonstrate that deficiency of MasR augments AngII-induced atherosclerosis and AAA formation and severity in hypercholesterolemic male mice (Figure 5). Effects of MasR deficiency to augment AngII-induced vascular diseases were observed without the need for exogenous infusion of its ligand, Ang-(1-7). These results suggest that endogenous Ang-(1-7), acting at MasR, protects against AngII-induced vascular diseases. Similar effects of MasR deficiency were observed in two different hypercholesterolemic murine strains, albeit with differences in the severity of effect. For example, compared to Apoe−/− MasR-deficient mice, Ldlr−/− mice with MasR deficiency had 5-fold higher serum cholesterol concentrations and more pronounced aneurysm rupture. Increased oxidative stress, inflammation, apoptosis, and elastin fragmentation may contribute to effects of MasR deficiency to augment AngII-induced vascular diseases. These results suggest that activation of the MasR axis may provide an alternative mode of intervening within the RAS for treatment of vascular diseases.
Figure 5.
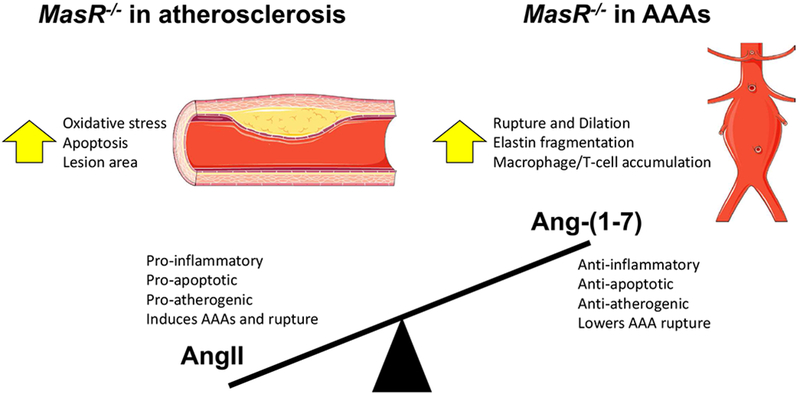
MasR deficiency increases atherosclerosis and AAAs in hyperlipidemic male mice. Summary of findings in both atherosclerosis and AAAs with MasR-deficient mice. With MasR deficiency, the balance of angiotensin peptides favors a pro-inflammatory, pro-apoptotic, pro-atherogenic environment with increased AAA formation and rupture.
In this study, blood pressure did not appear to be a variable that contributed to effects of MasR deficiency to promote AngII-induced atherosclerosis and AAAs, as there was no effect of MasR deficiency on baseline or AngII-induced hypertension. A lack of effect of MasR deficiency on baseline blood pressures in the present study is consistent with previous reports.22 However, others have reported that MasR deficiency augmented baseline blood pressure when mice are bred on an FVB/N background.23 Similar conflicting results have been reported for effects of ACE2 deficiency on baseline blood pressure in experimental mice.15, 24, 25 Taken together, it is unclear whether Ang-(1-7) regulates baseline blood pressure through a MasR-dependent mechanism. Moreover, results from this study do not support a role for MasR in the regulation of hypertension induced by infusion of exogenous AngII to male hypercholesterolemic mice. Notably, since augmented AngII-induced atherosclerosis and AAAs were observed in MasR-deficient mice that had similar blood pressures as wildtype controls, these results suggest that blood pressure is not a primary mechanism for effects of MasR deficiency to promote AngII-induced vascular diseases.
Previous studies demonstrated that deletion of components of the traditional RAS, such as AT1R, reduced diet-induced atherosclerosis in experimental mice.1–3, 26, 27 Conversely, whole body deletion of ACE2 promoted diet-induced atherosclerosis in Ldlr−/− male mice15; however, it is unclear whether effects of ACE2 deficiency to promote atherosclerosis arose from increased AngII concentrations, or from reductions in Ang-(1-7). We and others have demonstrated that infusion of exogenous Ang-(1-7) reduced diet- and AngII-induced atherosclerosis in experimental mice.15, 28–32 In this study, whole body deficiency of MasR augmented AngII-induced atherosclerosis in Ldlr−/− and Apoe−/− mice, suggesting that previous studies using exogenous Ang-(1-7) infusion to reduce atherosclerosis resulted from activation of MasR. Moreover, our results extend previous findings since augmented atherosclerosis was observed in MasR-deficient mice that were not infused with exogenous Ang-(1-7), suggesting protective effects from endogenous Ang-(1-7) activation of MasR. Moreover, similar to our findings, recent studies demonstrated that MasR deficiency augmented atherosclerosis when Apoe−/− mice were fed a high fat diet.32, 33 Similar to observations from the present study, oxidative stress and impaired endothelial function were observed in aortas from MasR-deficient mice.32 Taken together, these results suggest a protective role for endogenous Ang-(1-7) through MasR activation in development of AngII-induced atherosclerosis.
In addition to AngII-induced atherosclerosis, AAAs were more severe in MasR-deficient mice, with aneurysm ruptures occurring in 67% of MasR-deficient Ldlr−/− mice. Previous studies from our laboratory demonstrated that deficiency of ACE2, the enzyme responsible for formation of Ang-(1-7), also augmented AngII-induced AAAs, while pharmacologic activation of ACE2 reduced AAA formation and severity.13 It was unclear whether effects of ACE2 manipulation resulted from alterations in AngII or Ang-(1-7). Results from the present study extend these findings by demonstrating that MasR deficiency augmented the severity of AngII-induced AAAs, suggesting that ACE2-mediated production of Ang-(1-7) results in MasR activation to protect against AAA formation and severity. The underlying mechanisms how MasR activation mediates these effects are unclear. AAA formation is a multi-factorial disease that can be influenced by inflammation 34–36. In the present study, markers for oxidative stress, inflammation, apoptosis and elastin fragmentation were increased in aortas of MasR deficient mice suggesting a direct link between MasR activation, inflammation and vascular injury in the development of AAA. In this regard, it has been shown that the MasR expressed on macrophages and the vasculature influence polarization and migration of macrophages as well as macrophage-mediated T-cell activation and vascular inflammation.18, 33, 37. In line with this observation, other studies have also shown that the Ang-(1-7) receptor agonist, AVE 0991, also lowers pro-inflammatory macrophages within atherosclerotic lesions and perivascular adipose tissue of Apoe−/− mice.38, 39
In conclusion, MasR deficiency promotes AngII-induced atherosclerosis and AAA formation and severity. These studies suggest that activation of the MasR pathway may have efficacy against vascular pathologies associated with elevations in AngII.
Supplementary Material
Clinical relevance paragraph.
Results from this study suggest a novel mode of intervening in the RAS to treat vascular diseases, namely by activating MasR as the protective arm of the system. Our results demonstrate that when Mas receptors are absent, AngII-induced atherosclerosis and AAAs are exacerbated. These results suggest that the endogenous ligand of MasR, Ang-(1-7), acts at MasR to counterbalance vascular disease-promoting effects of AngII. Since the development of atherosclerosis and severity of AAAs were augmented by MasR deficiency, then these results suggest that activation of the MasR pathway may reduce the progressive growth of these dangerous vascular diseases.
Type of Research:
Experimental study using an angiotensin-II induced atherosclerosis model in mice
Key Findings:
Mas receptor deficiency increased angiotensin-II-induced atherosclerosis as well as increased aortic rupture.
Take Home Message:
Mas receptor activation may be a new therapeutic target to prevent or treat vascular disease.
Acknowledgements
We would like to acknowledge KOMP for generation of the vectors, ES cells, and mice for this project (U01HG004085, U01HG004080). We would also like to acknowledge NIH (HL107326, LA Cassis). Lars Christian Rump, Johannes Stegbauer and Guang Yang are supported by the Deutsche Forschungsgemeinschaft (Grant IRTG1902).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105(11): 1605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cassis LA, Rateri DL, Lu H, Daugherty A. Bone marrow transplantation reveals that recipient AT1a receptors are required to initiate angiotensin II-induced atherosclerosis and aneurysms. Arteriosclerosis, thrombosis, and vascular biology. 2007;27(2):380–6. [DOI] [PubMed] [Google Scholar]
- 3.Lu H, Balakrishnan A, Howatt DA, Wu C, Charnigo R, Liau G, et al. Comparative effects of different modes of renin angiotensin system inhibition on hypercholesterolaemia-induced atherosclerosis. Br J Pharmacol. 2012;165(6):2000–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayek T, Attias J, Smith J, Breslow JL, Keidar S. Antiatherosclerotic and antioxidative effects of captopril in apolipoprotein E-deficient mice. J Cardiovasc Pharmacol. 1998;31(4):540–4. [DOI] [PubMed] [Google Scholar]
- 5.Investigators O, Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, et al. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358(15):1547–59. [DOI] [PubMed] [Google Scholar]
- 6.Hackam DG, Thiruchelvam D, Redelmeier DA. Angiotensin-converting enzyme inhibitors and aortic rupture: a population-based case-control study. Lancet (London, England). 2006;368(9536):659–65. [DOI] [PubMed] [Google Scholar]
- 7.Sweeting MJ, Thompson SG, Brown LC, Greenhalgh RM, Powell JT. Use of angiotensin converting enzyme inhibitors is associated with increased growth rate of abdominal aortic aneurysms. J Vasc Surg. 2010;52(1):1–4. [DOI] [PubMed] [Google Scholar]
- 8.Kristensen KE, Torp-Pedersen C, Gislason GH, Egfjord M, Rasmussen HB, Hansen PR. Angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers in patients with abdominal aortic aneurysms: nation-wide cohort study. Arteriosclerosis, thrombosis, and vascular biology. 2015;35(3):733–40. [DOI] [PubMed] [Google Scholar]
- 9.Lederle FA, Noorbaloochi S, Nugent S, Taylor BC, Grill JP, Kohler TR, et al. Multicentre study of abdominal aortic aneurysm measurement and enlargement. Br J Surg. 2015;102(12):1480–7. [DOI] [PubMed] [Google Scholar]
- 10.Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100(14):8258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275(43):33238–43. [DOI] [PubMed] [Google Scholar]
- 12.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87(5):E1–9. [DOI] [PubMed] [Google Scholar]
- 13.Thatcher SE, Zhang X, Howatt DA, Yiannikouris F, Gurley SB, Ennis T, et al. Angiotensin-converting enzyme 2 decreases formation and severity of angiotensin II-induced abdominal aortic aneurysms. Arteriosclerosis, thrombosis, and vascular biology. 2014;34(12):2617–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao Q, Dong X, Chen X, Yan F, Wang X, Shi H, et al. ACE2 Inhibits Angiotensin II-Induced Abdominal Aortic Aneurysm in Mice. Hum Gene Ther. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thatcher SE, Zhang X, Howatt DA, Lu H, Gurley SB, Daugherty A, et al. Angiotensin-converting enzyme 2 deficiency in whole body or bone marrow-derived cells increases atherosclerosis in low-density lipoprotein receptor−/− mice. Arteriosclerosis, thrombosis, and vascular biology. 2011;31(4):758–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jackson TR, Blair LA, Marshall J, Goedert M, Hanley MR. The mas oncogene encodes an angiotensin receptor. Nature. 1988;335(6189):437–40. [DOI] [PubMed] [Google Scholar]
- 17.Stegbauer J, Potthoff SA, Quack I, Mergia E, Clasen T, Friedrich S, et al. Chronic treatment with angiotensin-(1-7) improves renal endothelial dysfunction in apolipoproteinE-deficient mice. Br J Pharmacol. 2011;163(5):974–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Potthoff SA, Fahling M, Clasen T, Mende S, Ishak B, Suvorava T, et al. Angiotensin-(1-7) modulates renal vascular resistance through inhibition of p38 mitogen-activated protein kinase in apolipoprotein E-deficient mice. Hypertension. 2014;63(2):265–72. [DOI] [PubMed] [Google Scholar]
- 19.Xu P, Costa-Goncalves AC, Todiras M, Rabelo LA, Sampaio WO, Moura MM, et al. Endothelial dysfunction and elevated blood pressure in MAS gene-deleted mice. Hypertension. 2008;51(2):574–80. [DOI] [PubMed] [Google Scholar]
- 20.Stegbauer J, Chen D, Herrera M, Sparks MA, Yang T, Konigshausen E, et al. Resistance to hypertension mediated by intercalated cells of the collecting duct. JCI Insight. 2017;2(7):e92720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daugherty A, Whitman SC. Quantification of atherosclerosis in mice. Methods Mol Biol. 2003;209:293–309. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Shoemaker R, Powell D, Su W, Thatcher S, Cassis L. Differential effects of Mas receptor deficiency on cardiac function and blood pressure in obese male and female mice. Am J Physiol Heart Circ Physiol. 2017;312(3):H459–H68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rabello Casali K, Ravizzoni Dartora D, Moura M, Bertagnolli M, Bader M, Haibara A, et al. Increased vascular sympathetic modulation in mice with Mas receptor deficiency. J Renin Angiotensin Aldosterone Syst. 2016;17(2):1470320316643643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurley SB, Allred A, Le TH, Griffiths R, Mao L, Philip N, et al. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J Clin Invest. 2006; 116(8):2218–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–8. [DOI] [PubMed] [Google Scholar]
- 26.Lu H, Rateri DL, Feldman DL, Charnigo RJ Jr, Fukamizu A, Ishida J, et al. Renin inhibition reduces hypercholesterolemia-induced atherosclerosis in mice. J Clin Invest. 2008;118(3):984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daugherty A, Rateri DL, Lu H, Inagami T, Cassis LA. Hypercholesterolemia stimulates angiotensin peptide synthesis and contributes to atherosclerosis through the AT1A receptor. Circulation. 2004;110(25):3849–57. [DOI] [PubMed] [Google Scholar]
- 28.Thomas MC, Pickering RJ, Tsorotes D, Koitka A, Sheehy K, Bernardi S, et al. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ Res. 2010;107(7):888–97. [DOI] [PubMed] [Google Scholar]
- 29.Lovren F, Pan Y, Quan A, Teoh H, Wang G, Shukla PC, et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am J Physiol Heart Circ Physiol. 2008;295(4):H1377–84. [DOI] [PubMed] [Google Scholar]
- 30.Zhang C, Zhao YX, Zhang YH, Zhu L, Deng BP, Zhou ZL, et al. Angiotensin-converting enzyme 2 attenuates atherosclerotic lesions by targeting vascular cells. Proc Natl Acad Sci U S A. 2010;107(36):15886–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tesanovic S, Vinh A, Gaspari TA, Casley D, Widdop RE. Vasoprotective and atheroprotective effects of angiotensin (1-7) in apolipoprotein E-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2010;30(8):1606–13. [DOI] [PubMed] [Google Scholar]
- 32.Yang G, Istas G, Hoges S, Yakoub M, Hendgen-Cotta U, Rassaf T, et al. Angiotensin-(1-7)-induced Mas receptor activation attenuates atherosclerosis through a nitric oxide-dependent mechanism in apolipoproteinE-KO mice. Pflugers Arch. 2018;470(4):661–7. [DOI] [PubMed] [Google Scholar]
- 33.Hammer A, Yang G, Friedrich J, Kovacs A, Lee DH, Grave K, et al. Role of the receptor Mas in macrophage-mediated inflammation in vivo. Proc Natl Acad Sci U S A. 2016;113(49):14109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pyo R, Lee JK, Shipley JM, Curci JA, Mao D, Ziporin SJ, et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105(11):1641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Vickle-Chavez SJ, Tung WS, Absi TS, Ennis TL, Mao D, Cobb JP, et al. Temporal changes in mouse aortic wall gene expression during the development of elastase-induced abdominal aortic aneurysms. J Vasc Surg. 2006;43(5):1010–20. [DOI] [PubMed] [Google Scholar]
- 36.Rush C, Nyara M, Moxon JV, Trollope A, Cullen B, Golledge J. Whole genome expression analysis within the angiotensin II-apolipoprotein E deficient mouse model of abdominal aortic aneurysm. BMC Genomics. 2009;10:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Souza LL, Costa-Neto CM. Angiotensin-(1-7) decreases LPS-induced inflammatory response in macrophages. J Cell Physiol. 2012;227(5):2117–22. [DOI] [PubMed] [Google Scholar]
- 38.Jawien J, Toton-Zuranska J, Gajda M, Niepsuj A, Gebska A, Kus K, et al. Angiotensin-(1-7) receptor Mas agonist ameliorates progress of atherosclerosis in apoE-knockout mice. J Physiol Pharmacol. 2012;63(1):77–85. [PubMed] [Google Scholar]
- 39.Skiba DS, Nosalski R, Mikolajczyk TP, Siedlinski M, Rios FJ, Montezano AC, et al. Anti-atherosclerotic effect of the angiotensin 1-7 mimetic AVE0991 is mediated by inhibition of perivascular and plaque inflammation in early atherosclerosis. Br J Pharmacol. 2017;174(22):4055–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.