

Abstract
Adipokine dysregulation and insulin resistance are two hallmark sequelae attributed to the current clinical definition of metabolic syndrome (MetS) that are also linked to atherosclerotic vascular disease. Here, we critically discuss the underlying pathophysiological mechanisms and the interplay between the two sequelae. Adipokine dysregulation is involved with decreased nitric oxide, vascular inflammation, and insulin resistance in itself to promote atherosclerosis. Insulin resistance is involved with endothelial dysfunction by direct and indirect mechanisms that also promote vascular inflammation and atherosclerosis. These mechanisms are discussed in atherosclerosis irrespective of MetS, and to evaluate the possibility of synergism in MetS. High retinol binding protein-4 (RBP-4) and low cholesterol efflux in MetS may provide evidence of possible synergism and elevated atherosclerotic risk. An adverse adipokine panel that includes fetuin-A and adiponectin can potentially assess atherosclerotic risk in even those without MetS. Genetic possibilities may exist in atherosclerotic vascular diseases secondary to insulin resistance.
Keywords: Adipokines, Atherosclerosis, Endothelial dysfunction, Inflammation, Insulin resistance, Metabolic syndrome
Introduction
In a joint statement from the American Diabetes Association (ADA) and European Association for The Study of Diabetes (EASD) in 2005, the metabolic syndrome (MetS) is argued to be imprecisely defined and with a considerable doubt that it is a risk factor for cardiovascular disease.1 However, the MetS, which has been linked to obesity, is characterized by a cluster of risk factors for atherosclerosis such as hypertension, dyslipidemia and elevated blood glucose, and has also been claimed to be an independent risk factor for cardiovascular disease and stroke.2,3 Currently, the understanding of this cluster of risk factors as a ‘syndrome’ is subject to debate, with increased risk attributable to the syndrome to be controversial, compared to the individual risk of each factor. Nevertheless, adipokine dysregulation and insulin resistance, two hallmark sequelae presumably resulting from the MetS cluster, have been linked to vascular inflammation and endothelial dysfunction, and increased levels of inflammatory markers, collectively increasing the risk of atherosclerosis and harmful vascular remodeling.4 While many non-vascular sequelae have been attributed to MetS, the purpose of this critical review is twofold. The first is to discuss the pathophysiological mechanisms of adipokine dysregulation and insulin resistance, two primary issues believed to be working in concert in MetS, in relation to atherosclerotic vascular disease (AVD) from our presumed understanding of MetS. The second is to utilize our knowledge from this pathophysiology to evaluate and refine diagnostic routes for AVD from the independent identification of these two hallmark sequelae, but also whether the possibility of true synergism for AVD risk in MetS exists. As the prevalence of obesity increases in the United States, and therefore potentially adipocyte dysfunction, insulin resistance and adipokine dysregulation will become an even greater health care problem that will require aggressive screening of patients with optimized treatment. Similarly, the co-occurrence of the risk factors within the MetS diagnostic cluster that promote these sequelae, whether independently or synergistically, can be appreciated by the prevalence of MetS in the general adult population that has increased from 25.3% (1988–1994) to 35% (2007–2012) in the United States.5
Historical Perspective
The term “metabolic syndrome” first appeared in literature in the 1950s when researchers identified risk factors for the progression and development of type 2 diabetes.6 The current concept of metabolic syndrome began to take shape in the 1970s. In 1977, Haller7 and Singer8 used the term “metabolic syndrome” in association with obesity, diabetes mellitus, hyperlipoproteinemia, hyperuricemia, steatohepatitis and atherosclerosis. One year later, Gerald Phillips9 recognized that there is an array of risk factors associated with myocardial infarction, such as glucose intolerance, hyperinsulinemia, hyperlipidemia and hypertension. It was hypothesized that sex factors are accountable for this relationship. In 1988, Reaven6 reevaluated this hypothesis and proposed that insulin resistance was the underlying cause of hyperinsulinemia, hyperlipidemia and hypertension (i.e., the metabolic syndrome). More recently, the term “metabolic syndrome” has been used interchangeably with several other terms such as insulin resistance syndrome, Reaven’s syndrome and syndrome X. These all refer to a combination of disorders that increase an individual’s risk of cardiovascular disease and diabetes, with adipokine dysregulation and insulin resistance implicated at the heart of the disorder.
Diagnostic Criteria for MetS
The diagnostic criteria for MetS was developed to improve understanding of the link between insulin resistance and vascular disease. At first, there was no consensus on a definition for metabolic syndrome. Currently, there are five definitions of the MetS as described by the World Health Organization (WHO), International Diabetes Federation (IDF), American Association of Clinical Endocrinologists (AACE), European Group for the Study of Insulin Resistance (EGIR) and National Cholesterol Education Program (NCEP). In 2002, the NCEP Adult Treatment Panel III (NCEP ATP III) proposed a revised definition of the MetS that could be easily measured in clinical practice.10 Emphasizing the risk of cardiovascular disease, ATP III criteria define the MetS as the presence of any three of five traits described in Table 1, and is the most widely used.11 While many patients with type 2 diabetes may have similar traits, the co-occurrence with MetS is believed to confer a greater risk for macrovascular over microvascular complications.12 The interplay of these criterion of MetS on AVD (Figure 1) is believed to evolve into two major events – adipokine dysregulation and insulin resistance, each of which may occur individually and not in the context of a syndrome at all.
Table 1:
Major Criteria to Define Metabolic Syndromen
| Criterion | Characteristics of the criterion |
|---|---|
| Abdominal obesity (waist circumference) |
men >102 cm (40 in.) women >88 cm (35 in.) |
| Fasting Serum triglycerides | ≥150 mg/do OR drug treatment for elevated triglycerides |
| Fasting Serum HDL cholesterol (HDL-C) | <40 mg/dl in men <50 mg/dl in women OR drug treatment for low HDL-C |
| Blood pressure | ≥130/85 mmHg OR drug treatment for elevated blood pressure |
| Fasting plasma glucose | ≥100 mg/dl OR drug treatment for elevated blood glucose |
Figure 1:
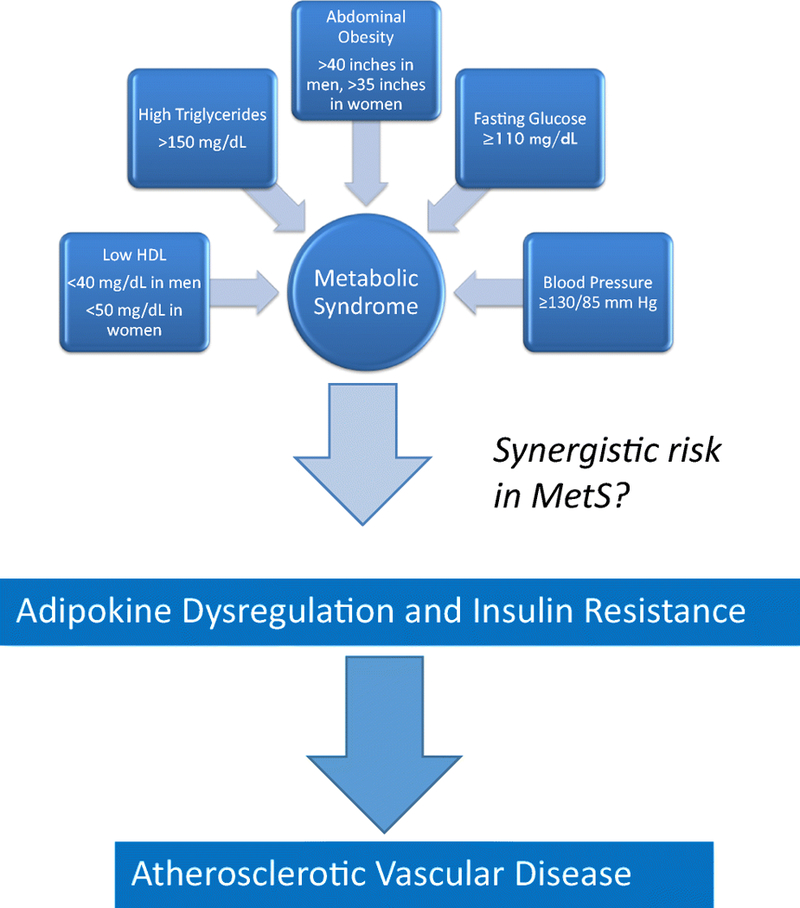
Schematic diagram showing an association between metabolic syndrome and atherosclerotic vascular disease
Pathophysiology of the Adipokine Dysregulation and Atherosclerotic Vascular Disease
The role of adipokine dysregulation in promoting atherosclerosis is briefly summarized in Figure 2. A group of adipose tissue generated cytokines, collectively termed as adipokines that have both local and systemic effects, promote atherosclerosis. Furthermore, the dysregulation of adipokines has been implicated in obesity, type 2 diabetes, and MetS, where overproduction comes secondary to increased adiposity.13 On the contrary, the reduction in certain adipokines, such as adiponectin, has been linked to insulin resistance and decreased production of nitric oxide in vascular endothelial cells, thus promoting atherosclerosis. However, the control of various adipokine production remains unknown and an interest for future research and intervention.
Figure 2:
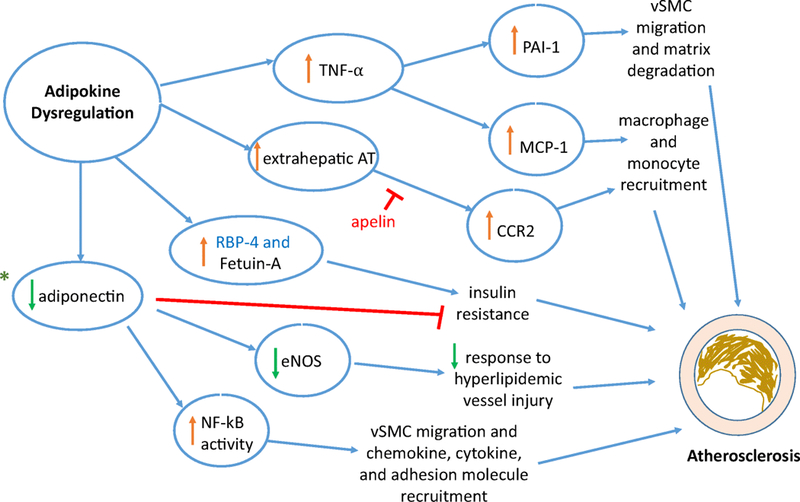
Mediators involved due to adipokine dysregulation leading to the pathophysiology in the development of atherosclerosis.
Adipokines ➔ tumor necrosis factor – alpha (TNF-⍺); extrahepatic angiotensin (AT); retinol-binding protein-4 (RBP-4); fetuin-A, adiponectin, plasminogen activator inhibitor-1 (PAI-1); Mediators Impacted ➔ monocyte chemoattractant protein (MCP-1); C-C chemokine receptor type 2 (CCR2) signaling; endothelial nitric oxide synthase (eNOS); nuclear factor-kappa light-chain-enhancer of activated B cells; *Diagnostic targets ➔ genetic predisposition: adiponectin (AdipoQ gene) polymorphisms and specific microRNAs of adipokine dysregulation promoting atherosclerosis; Therapeutic target ➔ apelin; MetS link ➔ RBP-4; vSMC - vascular smooth muscle cells
Tumor necrosis factor-alpha (TNF-α) is one adipokine that is seen in greater serum levels in patients with the diagnostic profile of MetS and provides evidence for inflammation that underlies atherosclerosis and promotes insulin resistance independently.14 Similarly, TNF-α has also been associated with stages of early atherosclerosis in humans.15 Within ApoE- deficient mice, the dysregulation of TNF-α upregulates the expression levels of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and monocyte chemoattractant protein-1 (MCP-1) implicating an atherogenic role with scavenger receptor class A (SRA) expression and oxidized low-density lipoprotein (LDL) uptake in macrophages.16 MCP-1 potentiates the atherogenic role with the recruitment of monocytes/macrophages into the arterial vessel wall and has been shown to be elevated in patients with coronary artery disease (CAD).17 In the continuation of a chronic inflammatory state, TNF-α can also induce synthesis of another adipokine, plasminogen activator inhibitor - 1 (PAI-1) leading to a pro-thrombotic state that can independently promote atherosclerosis through smooth muscle migration and changes in matrix degradation within vasculature.18 PAI-1 is an inhibitor of plasminogen activators (urokinase and tissue types) and vitronectin, and exhibits circadian release that may explain the occurrence of myocardial infarction and stroke in the early morning hours.19 Other inducers of PAI-1 exist with strong evidence among many of the MetS ATP III defining criteria (mean arterial pressure (MAP), high-density lipoprotein (HDL) cholesterol, triglycerides, and fasting plasma glucose), in addition to very low-density lipoprotein (VLDL) cholesterol and angiotensin II, offering a potential link between MetS and atherosclerosis.20–22
Hypertension is associated with lower wall shear stress, which is known to be associated with the development of atherosclerosis and vascular remodeling secondary to inflammation of the vessel wall with cell proliferation and thrombosis.23–26 As an adipokine as well, the major extrahepatic production of angiotensinogen (AT) by adipose tissue is increased in obesity and provides another link between adipocyte dysfunction or MetS, with vascular sequelae and subsequent hypertension.27 Additionally, the relationship of AT from adipose tissue and atherosclerosis seems to be mediated mostly by the angiotensin II (Ang II) intermediate.27–28 Supported by Ldlr−/−mice with a lack of bone marrow C-C chemokine receptor type 2 (CCR2), Ang II is believed to promote the differentiation of monocytes from progenitor cells and upregulate CCR2, which recruits the inflammatory milieu that promotes atherosclerosis.29–31 Furthermore, in understanding such hypertension to be obesity-related, differential activation of the renin-angiotensin system (RAS) within the microvasculature of visceral adipose tissue may underlie the systemic hypertension seen in obesity or MetS.32 In a study of obese human subjects undergoing elective bariatric surgery, visceral arterioles in hypertensives had significantly greater Ang-II mediated vasoconstriction than normotensives, and was selectively greater than subcutaneous arterioles. Antagonistic to the actions of AT, apelin (secreted by mature adipocytes with a G-protein coupled apelin receptor - AJP) is a lesser known adipokine that has been implicated in MetS (in the context of adipokine dysregulation) with the ability to counteract Ang II signaling seen in atherosclerotic mice models.33–35 However, higher levels of apelin have been discovered in clinical MetS and obesity, and may reflect endothelial damage with vasoconstriction via apelin receptors in vascular smooth muscles.36 Nevertheless, such variability in physiologic response is not fully appreciated in the context of adipocyte dysregulation, with or without MetS, and is worth inquiry given the associations of the apelinergic system with type 2 diabetes mellitus, hypertension, and heart failure.37–38
In the Third Generation Cohort of the Framingham Heart Study, higher levels of adipokines, retinol-binding protein-4 (RBP-4) and fetuin-A, marked future cardiometabolic risk and the incidence of clinical MetS.39 RBP-4 is a protein with retinol transport function that has been implicated in insulin resistance, which can promote vascular inflammation.40 However, the mechanism of this connection between RBP-4 and insulin resistance is less known. Fetuin-A is another adipokine protein that also promotes insulin resistance through inhibition of insulin receptor’s tyrosine kinase activity and is likewise also associated with vascular disease.41–42 Notably from this Framingham Heart study, RBP4 levels were elevated independently of obesity and may potentially be a unique biomarker of MetS, providing evidence of such a syndrome as well. Therefore, in the absence of obesity, this may raise the possibility of overlapping mechanisms of insulin resistance by other MetS criteria and RBP-4, if not interrelated. Furthermore, other components of the adverse adipokine profile noted in incident MetS within the study were higher levels of fetuin-A and lower levels of adiponectin, the latter of which was notably lower in metabolically healthy obese patients (those without MetS) in comparison.
Adiponectin is an adipokine protein that interacts with distinct G-protein coupled receptors (GPCRs), AdipoR1 and AdipoR2, where its binding associates with intracellular protein adaptor protein phosphotyrosine interacting with plekstrin homology domain and leucine zipper 1 (APPL1) to improve insulin sensitivity and promote anti-inflammatory response, fatty acid oxidation and increased endothelial nitric oxide synthase (eNOS) vasodilatory activity.43 Focusing on the anti-atherosclerotic potential of adiponectin, the inhibition of downstream components of nuclear factor-kappa beta (NF-kB) and other factors may explain the anti-inflammatory mechanism as noted in apoE-deficient mice.44 A second anti-atherosclerotic mechanism of adiponectin may also be explained by its ability to improve endothelial function by the upregulation of eNOS and the inhibition of inducible NOS (iNOS) activity in vasculature to limit hyperlipidemic vessel injury.45 Adiponectin has also been implicated in limiting further plaque progression with decreased adhesion capacity to vascular endothelial cells, decreased migration of smooth muscle cells, and decreased oxidation of lipids within macrophages (foam cells).46–47
Quite the contrary to adiponectin, leptin, while also involved in energy homeostasis, is speculated to be involved in the pathogenesis of atherosclerosis through enhanced inflammatory cytokine production (perhaps through stimulating a Th1 phenotype in helper cells)48, vascular smooth muscle cell migration and proliferation (mediated by mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3-K) activation)49–50, vessel wall angiogenesis, and increased oxidative stress in endothelial cells (with reduction in nitric oxide (NO) bioactivity and increased expression of MCP-1).51 However, in a recent meta-analysis, high leptin levels were not associated with risks of cardiovascular disease or stroke and may reflect the dominant role played by weight in the leptin pathway, as indicated by the findings of the Framingham Heart Study as well.39,52 Unlike the evidence surrounding adiponectin, the implications of leptin, beyond MetS, to link adipocyte dysregulation and AVD are less well known.
Insulin Resistance and Endothelial Dysfunction in Atherosclerotic Vascular Disease
The role of insulin resistance in promoting atherosclerosis is briefly summarized in Figure 3. Seen in diabetes and likewise considered in MetS, endothelial dysfunction is one of the earliest events of insulin resistance promoting AVD and is an area of therapeutic intervention that is not well-understood.53 Stemming from adipokine dysregulation, hyperinsulinemia and hyperglycemia associated with increased insulin resistance facilitates the release of vasoconstrictors and inflammatory markers in promoting endothelial dysfunction. Additionally, dyslipidemia abnormalities secondary to insulin resistance, or as part of the MetS cluster further potentiate endothelial dysfunction.54 Endothelial homeostasis is a balance between mediators that promote vasodilation (e.g., NO or prostacyclin (PGI2) and vasoconstriction (e.g, Ang II or ET-1), both of which are altered by insulin.55–56 Endothelial cell dysfunction is an initial step in atherosclerosis that involves a phenotypic change within cells that alters vascular tone and redox balance, in addition to acute and chronic inflammatory control responsible for hemostasis and thrombosis.57 Likewise, as reviewed by Gimbrone and Cardena (2017), the link between atherosclerosis with endothelial proinflammatory activation and endothelial cell dysfunction is mediated by: (i) Selective adhesivity of VCAM-1 for mononuclear leukocyte and lymphocyte recruitment, (ii) Intrinsic capacity of activated vascular endothelium to secrete chemokines, disrupting a balance between inflammatory mediators (e.g, IL-1) and anti-inflammatory mediators (e.g., specialized pro-resolving mediators [SPMs]), (iii) Upregulation of endothelial NF-kB, which may promote changes in endothelial chromatin, and (iv) Distinct hemodynamic forces with dysregulation of specific shear stress response elements (e.g, NO), which can serve as a focal risk factor. However, genetic considerations that might surround resolution deficits long after endothelial cell dysfunction are warranted.
Figure 3: Insulin Resistance and Development of Atherosclerosis.
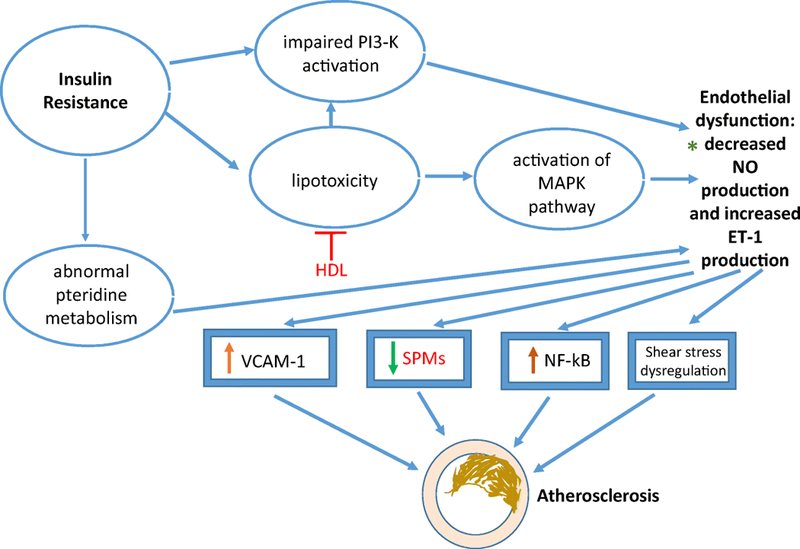
Signaling pathways ➔ phosphatidylinositol 3-kinase (PI3-K); mitogen-activated protein kinase (MAPK); Mediators ➔ nitric oxide (NO); endothelin-1 (ET-1), vascular cell adhesion molecule-1 (VCAM-1), nuclear factor-kappa light-chain-enhancer of activated B cells, specialized pro-resolving mediators (SPMs); *Diagnostic targets ➔ genetic predisposition to atherosclerosis: genetic variation in haplotype tagging single nucleotide polymorphisms (htSNPs) at the eNOS locus & specific microRNAs of insulin resistance promoting atherosclerosis; Therapeutic targets ➔ upregulation of high-density lipoprotein (HDL) to induce anti-oxidative effect and reduce lipotoxicity and SPM delivery to resolve inflammation associated with insulin resistance.
As a part of the normal function of insulin, activation of membrane-bound eNOS and the subsequent synthesis of NO by insulin binding allows for vasodilation.55,58 This action is mediated by the activation of the PI3-K/Akt pathway and phosphorylation of eNOS, promoting the conversion of L-arginine to L-citrulline and NO. However, in insulin resistance seen in cardiometabolic disorders such as MetS, the response is the opposite, where vasoconstriction ensues for reasons that are multifactorial.59 Abnormal pteridine metabolism has been linked to decreased NO production and endothelial dysfunction in insulin-resistant patients.60 The mechanism is believed to involve tetrahydrobiopterin depletion (BH4), an activating cofactor of NOS, and elevation of 7,8-dihydrobiopterin (BH2), an inactivating cofactor of NOS, leading to a decrease in NOS activity and impairment of vasodilation with increased superoxide anion generation.61 Furthermore, the hyperglycemia that ensues from insulin resistance impairs activation of PI3-K and phosphorylation of eNOS by activation of the hexosamine biosynthesis pathway, leading to modification in insulin proteins, decreased NO signaling by O-Glc-N-acylation of insulin receptor substrate-1 (IRS-1) adaptor protein and formation of advanced glycation end products (AGEs) that stimulate reactive oxygen species (ROS).62,63 Additionally, the production of AGEs inhibit the PI3-K/Akt pathway by activation of protein kinase C (PKC) (through increased synthesis of diacylglycerol), and the subsequent ROS produced activate NF-kB and inflammatory mediators via stimulation of inhibitor of nuclear factor kappa-B kinase subunit beta (IKKB kinase-β).64,65 PKC activation promotes production of both pro-thrombotic and growth factors, and induces the formation of the vasoconstrictor, endothelin-1 (ET-1).59 The importance of insulin resistance affecting these NO mechanisms in MetS is supported by findings of genetic variation at the eNOS locus by haplotype tagging single nucleotide polymorphisms (htSNPs) in patients meeting criteria for the diagnosis of MetS, perhaps indicating a genetic susceptibility for endothelial dysfunction in this context.66
Insulin resistance is also linked to endothelial dysfunction through lipotoxicity, which is potentiated by dyslipidemia (particularly, low HDL-C), a component of the MetS cluster. Lipotoxicity can stem from accumulation of harmful lipids in obesity secondary to metabolic stress from nutrient excess, and the adipokine dysregulation and insulin resistance discussed here.67 In addition to inhibiting the PI3-K/Akt pathway via inactivation of IRS-1/2, the presence of these harmful free fatty acids stimulates the MAPK pathway and stimulates ROS production, including pro-inflammatory and pro-thrombotic mediator production via NADPH oxidase stimulation.68–70 Additionally, activation of the MAPK pathway, which is also activated from hyperinsulinemia secondary to insulin resistance, increases ET-1 production in the midst of decreased NO production, creating a balance that is offset between high MAPK pathway and low PI3-K/Akt pathway.71–72 ET-1 expression by both insulin resistance and free fatty acids is also potentiated by ROS activation of NF-kB.73 To note, in addition to vasoconstriction via endothelin A (ET-A) receptors on vascular smooth muscle cells, ET-1 induces mitogenic activity that causes proliferation of smooth muscle cells and vessel proteins to promote atherosclerosis.74–75 With respect to the impact of dyslipidemia potentiating lipotoxicity, low HDL-C in hyperlipidemic patients were shown to have higher levels of VCAM-1 and ICAM-1 contributing to endothelial damage.76 Similar to hyperinsulinemia, oxidized LDL-C contributes to a reduction in NO production but is mediated by upregulation of arginase I, decreasing L-arginine availability to eNOS.77 Therefore, the importance of HDL with atherosclerosis, which prevents lipoprotein oxidation, can be appreciated for its protection against both endothelial dysfunction and subsequent atherosclerosis.
Evidence of a Syndrome and Diagnostic Considerations for AVD
As discussed above, MetS is a clinical diagnosis that consists of the clustering of cardiovascular disease factors (ATP III criteria) with debate on whether their cardiovascular risk is collectively greater than the summation of their individual contributions.78–80 While a paucity of data exists in relation to this gap in knowledge, the current clinical approach is to address each risk factor among the cluster individually.1 Current diagnostic assessment has yet to attribute a higher risk profile for any AVD with MetS and would be necessary if synergism exists. Nevertheless, as discussed with the Framingham Heart Study, an elevated RBP4 was associated with MetS independent of obesity and may be evidence of a syndrome process in relation to insulin resistance, and likewise a diagnostic possibility deserving further evaluation. Furthermore, in MetS, adiponectin may also offer some clarity despite lower levels of it with adipocyte dysfunction in metabolically healthy obesity.
Contrary to the Framingham study and in support for the diagnostic value of adiponectin in MetS, Hung et al. found that low circulating levels were associated with pro-inflammatory markers, insulin resistance and MetS independent of obesity.81 Outside of considering adipokine dysregulation and insulin resistance, in a prospective study among those with clinically defined MetS, the individual components of the ATP III criteria were synergistically associated with reduced cholesterol efflux capacity, a mechanism and potential diagnostic marker by which macrophages regulate cholesterol metabolism and homeostasis to prevent cholesterol accumulation.82 Among the first to show an escalation in risk among the diagnostic cluster of MetS, this study also showed that reduced cholesterol efflux capacity is seen in atherosclerotic plaques across various vascular beds, providing a link between cardiometabolic disease and AVD. With the evidence taken together here, the possibility that MetS is a true syndrome is sufficient to investigate the magnitude and etiology of possible risk escalation, or synergism, that would be greater than the sum of individual ATP III criteria.
Outside of the context of a syndrome, the adverse adipokine profile seen in metabolically healthy obese subjects of the Framingham included high levels of RBP4, fetuin-A and low levels of adiponectin that may have relevance to assess AVD risk among adipokine dysregulation without a syndrome process. Irrespective of MetS, several pro-inflammatory and pro-thrombotic mediators exist with adipokine dysregulation and insulin resistance (Figures 2 and 3) and require prospective and temporal evaluation for refined diagnostic and prognostic utilization, respectively, with AVD. Likewise, AVD stemming from adipokine dysregulation or insulin resistance, may be better served with adjunctive anti-inflammatory treatment that could complement current anti-thrombotic efforts with AVD. Finally, genetic possibilities may exist that underlie these two atherogenic metabolic processes and are a worthy endeavor diagnostically. For example, a genomic polymorphism has been noted in the adiponectin (AdipoQ) gene (locus on chromosome 3q27) related to dysregulation causing low levels.83 Additionally, as noted earlier, genetic variation (htSNPs) at the eNOS locus may explain susceptibility to endothelial dysfunction relative to insulin resistance and subsequent atherosclerosis.66 Likewise, the role of specific microRNAs, which are posttranscriptional regulators, have been shown to be expressed or downregulated during atherogenic processes secondary to adipokine dysregulation, and endothelial dysfunction secondary to insulin resistance, respectively.84–85
Conclusion
With the debate of whether MetS is truly a syndrome process with synergism and advanced risk, the hallmark sequelae implicated in the disease of adipokine dysregulation and insulin resistance are significant derangements that also work independently with atherosclerotic vascular disease. The pathophysiology learned from our attempts to understand MetS further, offer diagnostic potential to atherosclerotic vascular disease. Nevertheless, the possibility that the MetS diagnostic cluster accounts for a higher atherogenic risk than the individual ATP III risk factors responsible for adipokine dysregulation and insulin resistance is a warranted endeavor and should be the focus for this ‘syndrome’ in the future.
Acknowledgments
Funding: The research work of DK Agrawal is supported by research grants R01 HL120659 and R01HL144125 from the National Institutes of Health, USA. The content of this review article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Compliance with Ethical Standards
Conflict of Interest: All authors have read the journal’s policy on disclosure of potential conflicts of interest. Author C has received grants from the National Institutes of Health, State of Nebraska, and Dialysis Clinic Inc. Other authors have no other relevant affiliations or financial or non-financial involvement with any organization or entity with financial or non-financial interest or conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. Author A (MS) declares that he has no conflict of interest. Author B (SKS) declares that he has no conflict of interest. Author C (DKA) declares that he has no conflict of interest.
Ethical approval: This article does not contain any studies with human participants and/or animals performed by any of the authors.
No writing assistance was utilized in the production of this manuscript.
References:
- 1.Kahn R The Metabolic Syndrome: Time for a Critical Appraisal: Joint Statement From the American Diabetes Association and the European Association for the Study of Diabetes: Response to Citrome et al., Giugliano and Esposito, Cheta, and Psaty et al. Diabetes Care 2006;29(1):177–178. doi: 10.2337/diacare.29.1.177-a. [DOI] [PubMed] [Google Scholar]
- 2.Engin A The Definition and Prevalence of Obesity and Metabolic Syndrome. Obesity and Lipotoxicity Advances in Experimental Medicine and Biology 2017:1–17. doi: 10.1007/978-3-319-48382-5_1. [DOI] [PubMed] [Google Scholar]
- 3.Thorn LM, Forsblom C, Waden J, et al. Metabolic Syndrome as a Risk Factor for Cardiovascular Disease, Mortality, and Progression of Diabetic Nephropathy in Type 1 Diabetes. Diabetes Care 2009;32(5):950–952. doi: 10.2337/dc08-2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan QA, Sola S, Khan BV. The metabolic syndrome: inflammation and endothelial dysfunction. Hospital Physician 2006;42:26–37. [Google Scholar]
- 5.Preventing Chronic Disease Centers for Disease Control and Prevention. https://www.cdc.gov/pcd/issues/2017/16_0287.htm. Published September 20, 2017. Accessed September 2, 2018.
- 6.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988;37:1595–607. [DOI] [PubMed] [Google Scholar]
- 7.Haller H. Epidemiology and associated risk factors of hyperlipoproteinemia. Z Gesamte Inn Med 1977;32:124–8. [PubMed] [Google Scholar]
- 8.Singer P Diagnosis of primary hyperlipoproteinemias. Z Gesamte Inn Med 1977;32:129–33. [PubMed] [Google Scholar]
- 9.Phillips GB. Relationship between serum sex hormones and glucose, insulin, and lipid abnormalities in men with myocardial infarction. Proc Natl Acad Sci U S A 1977;74:1729–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002;106:3143–421. [PubMed] [Google Scholar]
- 11.Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Forceon Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World HeartFederation; International Atherosclerosis Society; and International Association for the Study of Obesity. Obesity and metabolism 2010; (1):63. doi: 10.14341/2071-8713-5281. [DOI] [PubMed] [Google Scholar]
- 12.Cull CA, Jensen CC, Retnakaran R, Holman RR. Impact of the Metabolic Syndrome on Macrovascular and Microvascular Outcomes in Type 2 Diabetes Mellitus: United Kingdom Prospective Diabetes Study 78. Circulation 2007;116(19):2119–2126. doi: 10.1161/circulationaha.107.733428. [DOI] [PubMed] [Google Scholar]
- 13.Maury E, Brichard S. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Molecular and Cellular Endocrinology 2010;314(1):1–16. doi: 10.1016/j.mce.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 14.Mohammadi M, Gozashti MH, Aghadavood M, Mehdizadeh MR, Hayatbakhsh MM. Clinical Significance of Serum IL-6 and TNF-α Levels in Patients with Metabolic Syndrome. Reports of Biochemistry & Molecular Biology 2017;6(1):74–79. [PMC free article] [PubMed] [Google Scholar]
- 15.Skoog T Plasma tumour necrosis factor-α and early carotid atherosclerosis in healthy middle-aged men. European Heart Journal 2002;23(5):376–383. doi: 10.1053/euhj.2001.2805. [DOI] [PubMed] [Google Scholar]
- 16.Ohta H, Wada H, Niwa T, et al. Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis 2005;180(1):11–17. doi: 10.1016/j.atherosclerosis.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 17.Martinovic I, Abegunewardene N, Seul M, et al. Elevated Monocyte Chemoattractant Protein-1 Serum Levels in Patients at Risk for Coronary Artery Disease. Circulation Journal 2005;69(12):1484–1489. doi: 10.1253/circj.69.1484. [DOI] [PubMed] [Google Scholar]
- 18.Alessi M-C, Poggi M, Juhan-Vague I. Plasminogen activator inhibitor-1, adipose tissue and insulin resistance. Current Opinion in Lipidology 2007;18(3):240–245. doi: 10.1097/mol.0b013e32814e6d29. [DOI] [PubMed] [Google Scholar]
- 19.Scheer FA, Shea SA. Human circadian system causes a morning peak in prothrombotic plasminogen activator inhibitor-1 (PAI-1) independent of the sleep/wake cycle. Blood 2014;123:590–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kodaman N, Aldrich MC, Sobota R, et al. Plasminogen Activator Inhibitor‐1 and Diagnosis of the Metabolic Syndrome in a West African Population. Journal of the American Heart Association 2016;5(10). doi: 10.1161/jaha.116.003867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alessi MC, Juhan-Vague I. Contribution of PAI-1 in cardiovascular pathology. Arch Mal Coeur Vaiss 2004;97:673–678. [PubMed] [Google Scholar]
- 22.Alessi MC, Juhan-Vague I. PAI-1 and the metabolic syndrome: links, causes, and consequences. Arterioscler Thromb Vasc Biol 2006;26:2200–2207 [DOI] [PubMed] [Google Scholar]
- 23.Zarins CK, Giddens DP, Bharadvaj BK, Sottiurai VS, Mabon RF, Glagov S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circulation Research 1983;53(4):502–514. doi: 10.1161/01.res.53.4.502. [DOI] [PubMed] [Google Scholar]
- 24.Gnasso A, Irace C, Carallo C, et al. In Vivo Association Between Low Wall Shear Stress and Plaque in Subjects With Asymmetrical Carotid Atherosclerosis. Stroke 1997;28(5):993–998. doi: 10.1161/01.str.28.5.993. [DOI] [PubMed] [Google Scholar]
- 25.Ku DN, Giddens DP, Zarins CK, Glagov S. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis, Thrombosis, and Vascular Biology 1985;5(3):293–302. doi: 10.1161/01.atv.5.3.293. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Cho K, Kim J, et al. Wall shear stress in hypertensive patients is associated with carotid vascular deformation assessed by speckle tracking strain imaging. Clinical Hypertension 2014;20(1):10. doi: 10.1186/2056-5909-20-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harrison DG, Guzik TJ, Goronzy J, Weyand C. Is hypertension an immunologic disease? Current Cardiology Reports 2008;10(6):464–469. doi: 10.1007/s11886-008-0073-6. [DOI] [PubMed] [Google Scholar]
- 28.Yvan-Charvet L, Quignard-Boulangé A. Role of adipose tissue renin–angiotensin system in metabolic and inflammatory diseases associated with obesity. Kidney International 2011;79(2):162–168. doi: 10.1038/ki.2010.391. [DOI] [PubMed] [Google Scholar]
- 29.Swirski FK, Nahrendorf M, Etzrodt M, et al. Identification of Splenic Reservoir Monocytes and Their Deployment to Inflammatory Sites. Science 2009;325(5940):612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukuda D, Sata M, Ishizaka N, Nagai R. Critical Role of Bone Marrow Angiotensin II Type 1 Receptor in the Pathogenesis of Atherosclerosis in Apolipoprotein E Deficient Mice. Arteriosclerosis, Thrombosis, and Vascular Biology 2007;28(1):90–96. doi: 10.1161/atvbaha.107.152363. [DOI] [PubMed] [Google Scholar]
- 31.Ishibashi M Bone Marrow-Derived Monocyte Chemoattractant Protein-1 Receptor CCR2 Is Critical in Angiotensin II-Induced Acceleration of Atherosclerosis and Aneurysm Formation in Hypercholesterolemic Mice. Arteriosclerosis, Thrombosis, and Vascular Biology 2004;24(11). doi: 10.1161/01.atv.0000143384.69170.2d. [DOI] [PubMed] [Google Scholar]
- 32.Angiotensin II- mediated vasoconstriction of the visceral adipose tissue vasculature is linked to systemic hypertension in obesity. The FASEB Journal. https://www.fasebj.org/doi/abs/10.1096/fasebj.31.1_supplement.684.6. Accessed September 4, 2018.
- 33.Chun HJ, Ali ZA, Kojima Y, et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. Journal of Clinical Investigation January 2008. doi: 10.1172/jci34871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mutlak SS, Ali VS, Hussein RM. Apelin and some Biomarkers in Females with Metabolic Syndrome. Biomedical and Pharmacology Journal 2018;11(1):247–253. doi: 10.13005/bpj/1369. [DOI] [Google Scholar]
- 35.Angelova P, Kamenov Z, Tsakova A. Apelin and Testosterone Levels in Men with Metabolic Syndrome. Open Journal of Endocrine and Metabolic Diseases 2014;04(02):35–43. doi: 10.4236/ojemd.2014.42004. [DOI] [Google Scholar]
- 36.Katugampola SD, Maguire JJ, Matthewson SR, Davenport AP. [125I]-(Pyr1)Apelin-13 is a novel radioligand for localizing the APJ orphan receptor in human and rat tissues with evidence for a vasoconstrictor role in man. British Journal of Pharmacology 2001;132(6):1255–1260. doi: 10.1038/sj.bjp.0703939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuba K, Zhang L, Imai Y, et al. Impaired Heart Contractility in Apelin Gene Deficient Mice Associated With Aging and Pressure Overload. Circulation Research 2007;101(4). doi: 10.1161/circresaha.107.158659. [DOI] [PubMed] [Google Scholar]
- 38.Yue P, Jin H, Aillaud M, et al. Apelin is necessary for the maintenance of insulin sensitivity. American Journal of Physiology-Endocrinology and Metabolism 2010;298(1). doi: 10.1152/ajpendo.00385.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zachariah JP, Quiroz R, Nelson KP, et al. Prospective Relation of Circulating Adipokines to Incident Metabolic Syndrome: The Framingham Heart Study. Journal of the American Heart Association 2017;6(7). doi: 10.1161/jaha.116.004974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Q, Graham TE, Mody N, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005;436(7049):356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 41.Rauth G, Poschke O, Fink E, Eulitz M, Tippmer S, Kellerer M, Haring HU, Nawratil P, Haasemann M, Jahnen-Dechent W, Muller-Esterl W. The nucleotide and partial amino acid sequences of rat fetuin. Identity with the natural tyrosine kinase inhibitor of the rat insulin receptor. Eur J Biochem 1992;204:523–529 [DOI] [PubMed] [Google Scholar]
- 42.Weikert C, Stefan N, Schulze MB, Pischon T, Berger K, Joost HG, Haring HU, Boeing H, Fritsche A. Plasma fetuin-A levels and the risk of myocardial infarction and ischemic stroke. Circulation 2008;118:2555–2562 [DOI] [PubMed] [Google Scholar]
- 43.Ruan H, Dong LQ. Adiponectin signaling and function in insulin target tissues. Journal of Molecular Cell Biology 2016;8(2):101–109. doi: 10.1093/jmcb/mjw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Chen Q, Pu H, et al. Adiponectin improves NF-κB-mediated inflammation and abates atherosclerosis progression in apolipoprotein E-deficient mice. Lipids in Health and Disease 2016;15(1). doi: 10.1186/s12944-016-0202-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li R, Wang W-Q, Zhang H, et al. Adiponectin improves endothelial function in hyperlipidemic rats by reducing oxidative/nitrative stress and differential regulation of eNOS/iNOS activity. American Journal of Physiology-Endocrinology and Metabolism 2007;293(6). doi: 10.1152/ajpendo.00462.2007. [DOI] [PubMed] [Google Scholar]
- 46.Ouchi N, Kihara S, Arita Y, et al. Novel Modulator for Endothelial Adhesion Molecules : Adipocyte-Derived Plasma Protein Adiponectin. Circulation 1999;100(25):2473–2476. doi: 10.1161/01.cir.100.25.2473. [DOI] [PubMed] [Google Scholar]
- 47.Arita Y Adipocyte-Derived Plasma Protein Adiponectin Acts as a Platelet-Derived Growth Factor-BB-Binding Protein and Regulates Growth Factor-Induced Common Postreceptor Signal in Vascular Smooth Muscle Cell. Circulation 2002;105(24):2893–2898. doi: 10.1161/01.cir.0000018622.84402.ff. [DOI] [PubMed] [Google Scholar]
- 48.Martín-Romero C, Santos-Alvarez J, Goberna R, Sánchez-Margalet V. Human Leptin Enhances Activation and Proliferation of Human Circulating T Lymphocytes. Cellular Immunology 2000;199(1):15–24. doi: 10.1006/cimm.1999.1594. [DOI] [PubMed] [Google Scholar]
- 49.Oda A, Taniguchi T, Takahash A, et al. Leptin stimulates rat aortic smooth muscle cell proliferation and migration. Kobe J Med Sci 2001;47:141–150. doi: 10.1016/s0021-9150(97)89646-3. [DOI] [PubMed] [Google Scholar]
- 50.Kraemer R, Nguyen H, March KL, Hempstead B. NGF Activates Similar Intracellular Signaling Pathways in Vascular Smooth Muscle Cells as PDGF-BB But Elicits Different Biological Responses. Arteriosclerosis, Thrombosis, and Vascular Biology 1999;19(4):1041–1050. doi: 10.1161/01.atv.19.4.1041. [DOI] [PubMed] [Google Scholar]
- 51.Maingrette F, Renier G. Leptin Increases Lipoprotein Lipase Secretion by Macrophages: Involvement of Oxidative Stress and Protein Kinase C. Diabetes 2003;52(8):2121–2128. doi: 10.2337/diabetes.52.8.2121. [DOI] [PubMed] [Google Scholar]
- 52.Yang H, Guo W, Li J, et al. Leptin concentration and risk of coronary heart disease and stroke: A systematic review and meta-analysis. Plos One 2017;12(3). doi: 10.1371/journal.pone.0166360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su Y, Liu X-M, Sun Y-M, Wang Y-Y, Luan Y, Wu Y. Endothelial Dysfunction in Impaired Fasting Glycemia, Impaired Glucose Tolerance, and Type 2 Diabetes Mellitus. The American Journal of Cardiology 2008;102(4):497–498. doi: 10.1016/j.amjcard.2008.03.087. [DOI] [PubMed] [Google Scholar]
- 54.Kolovou GD. Pathophysiology of dyslipidaemia in the metabolic syndrome. Postgraduate Medical Journal 2005;81(956):358–366. doi: 10.1136/pgmj.2004.025601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janus A, Szahidewicz-Krupska E, Mazur G, Doroszko A. Insulin Resistance and Endothelial Dysfunction Constitute a Common Therapeutic Target in Cardiometabolic Disorders. Mediators of Inflammation 2016;2016:3634948. doi: 10.1155/2016/3634948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davignon J Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004;109(23_suppl_1). doi: 10.1161/01.cir.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 57.Deedwania PC. Mechanisms of endothelial dysfunction in the metabolic syndrome. Current Diabetes Reports 2003;3(4):289–292. doi: 10.1007/s11892-003-0019-8. [DOI] [PubMed] [Google Scholar]
- 58.Kahn Nighat N., Acharya K, Bhattachary S. Nitric Oxide: The “Second Messenger” of Insulin. IUBMB Life (International Union of Biochemistry and Molecular Biology: Life) 2000;49(5):441–450. doi: 10.1080/152165400410308. [DOI] [PubMed] [Google Scholar]
- 59.Lee SK, Khambhati K, Bhargava A, Engels MC, Sandesara PB, Quyyumi AA. Endothelial Dysfunction and Metabolic Syndrome. Hypertens J 2017; 3(2):72–80. [Google Scholar]
- 60.Shinozaki K, Hirayama A, Nishio Y, et al. Coronary endothelial dysfunction in the insulin-resistant state is linked to abnormal pteridine metabolism and vascular oxidative stress. Journal of the American College of Cardiology 2001;38(7):1821–1828. doi: 10.1016/s0735-1097(01)01659-x. [DOI] [PubMed] [Google Scholar]
- 61.Shinozaki K, Nishio Y, Okamura T, et al. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in aortas of insulin-resistant rats. Circ Res 2000;87: 566–73. [DOI] [PubMed] [Google Scholar]
- 62.Prato SD. Role of glucotoxicity and lipotoxicity in the pathophysiology of Type 2 diabetes mellitus and emerging treatment strategies. Diabetic Medicine 2009;26(12):1185–1192. doi: 10.1111/j.1464-5491.2009.02847.x. [DOI] [PubMed] [Google Scholar]
- 63.Musicki B, Kramer MF, Becker RE, Burnett AL. Inactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. Proceedings of the National Academy of Sciences 2005;102(33):11870–11875. doi: 10.1073/pnas.0502488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hançer NJ, Qiu W, Cherella C, Li Y, Copps KD, White MF. Insulin and Metabolic Stress Stimulate Multisite Serine/Threonine Phosphorylation of Insulin Receptor Substrate 1 and Inhibit Tyrosine Phosphorylation. Journal of Biological Chemistry 2014;289(18):12467–12484. doi: 10.1074/jbc.m114.554162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Puyvelde KV, Mets T, Njemini R, Beyer I, Bautmans I. Effect of advanced glycation end product intake on inflammation and aging: a systematic review. Nutrition Reviews 2014;72(10):638–650. doi: 10.1111/nure.12141. [DOI] [PubMed] [Google Scholar]
- 66.Gonzalez-Sanchez JL, Martinez-Larrad MT, Saez ME, Zabena C, Martinez-Calatrava MJ, Serrano-Rios M. Endothelial Nitric Oxide Synthase Haplotypes Are Associated with Features of Metabolic Syndrome. Clinical Chemistry 2006;53(1):91–97. doi: 10.1373/clinchem.2006.075176. [DOI] [PubMed] [Google Scholar]
- 67.Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. Journal of Lipid Research 2016;57(12):2099–2114. doi: 10.1194/jlr.r066514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wende AR, Symons JD, Abel ED Mechanisms of lipotoxicity in the cardiovascular system. Current Hypertension Reports 2012;14(6):517–531. doi: 10.1007/s11906-012-0307-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Li H, Bao Y, Zhang X, Yu Y Free fatty acids induce endothelial dysfunction and activate protein kinase C and nuclear factor-κB pathway in rat aorta. International Journal of Cardiology 2011;152(2):218–224. doi: 10.1016/j.ijcard.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 70.Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000;49(11):1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 71.Hsueh WA, Quiñones MJ. Role of endothelial dysfunction in insulin resistance. The American Journal of Cardiology 2003;92(4):10–17. doi: 10.1016/s0002-9149(03)00611-8. [DOI] [PubMed] [Google Scholar]
- 72.Ferri C, Pittoni V, Piccoli A, et al. Insulin stimulates endothelin-1 secretion from human endothelial cells and modulates its circulating levels in vivo. The Journal of Clinical Endocrinology & Metabolism 1995;80(3):829–835. doi: 10.1210/jc.80.3.829. [DOI] [PubMed] [Google Scholar]
- 73.Mathew M, Tay E, Cusi K. Elevated plasma free fatty acids increase cardiovascular risk by inducing plasma biomarkers of endothelial activation, myeloperoxidase and PAI-1 in healthy subjects. Cardiovascular Diabetology 2010;9(1):9. doi: 10.1186/1475-2840-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sihvola RK, Pulkkinen VP, Koskinen PK, Lemström KB. Crosstalk of endothelin-1 and platelet-derived growth factor in cardiac allograft arteriosclerosis. Journal of the American College of Cardiology 2002;39(4):710–717. doi: 10.1016/s0735-1097(01)01782-x. [DOI] [PubMed] [Google Scholar]
- 75.Goto K, Miyauchi T. New expansion of endothelin research: Perspectives for clinical application of endothelin-receptor antagonists. Folia Pharmacologica Japonica 2003;121(2):91–101. doi: 10.1254/fpj.121.91. [DOI] [PubMed] [Google Scholar]
- 76.Lupattelli G, Marchesi S, Lombardini R, et al. Mechanisms of high-density lipoprotein cholesterol effects on the endothelial function in hyperlipemia. Metabolism 2003;52(9):1191–1195. doi: 10.1016/s0026-0495(03)00157-4. [DOI] [PubMed] [Google Scholar]
- 77.Wang W, Hein TW, Zhang C, Zawieja DC, Liao JC, Kuo L. Oxidized Low-Density Lipoprotein Inhibits Nitric Oxide-Mediated Coronary Arteriolar Dilation by Up-regulating Endothelial Arginase I. Microcirculation 2010;18(1):36–45. doi: 10.1111/j.1549-8719.2010.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wen CP, Chan HT, Tsai MK, Cheng TY, Chung WS, Chang YC, Hsu HL, Tsai SP, Tsao CK, Man Wai JP, Hsu CC. Attributable mortality burden of metabolic syndrome: comparison with its individual components. Eur J Cardiovasc Prev Rehabil 2011;18:561–573. [DOI] [PubMed] [Google Scholar]
- 79.Samaras K, Crawford J, Baune BT, Campbell LV, Smith E, Lux O, Brodaty H, Trollor JN, Sachdev P. The value of the metabolic syndrome concept in elderly adults: is it worth less than the sum of its parts? J Am Geriatr Soc 2012;60:1734–1741. [DOI] [PubMed] [Google Scholar]
- 80.Godsland IF, Lecamwasam K, Johnston DG. A systematic evaluation of the insulin resistance syndrome as an independent risk factor for cardiovascular disease mortality and derivation of a clinical index. Metabolism 2011;60:1442–1448. [DOI] [PubMed] [Google Scholar]
- 81.Hung J, Mcquillan BM, Thompson PL, Beilby JP. Circulating adiponectin levels associate with inflammatory markers, insulin resistance and metabolic syndrome independent of obesity. International Journal of Obesity 2008;32(5):772–779. doi: 10.1038/sj.ijo.0803793. [DOI] [PubMed] [Google Scholar]
- 82.Gall J, Frisdal E, Bittar R, et al. Association of Cholesterol Efflux Capacity With Clinical Features of Metabolic Syndrome: Relevance to Atherosclerosis. Journal of the American Heart Association 2016;5(12). doi: 10.1161/jaha.116.004808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ohashi K et al. (2004) Adiponectin I164T mutation is associated with the metabolic syndrome and coronary artery disease. J. Am. Coll. Cardiol 43, 1195–1200 [DOI] [PubMed] [Google Scholar]
- 84.Virtue A, Johnson C, Lopez-Pastraña J, et al. MicroRNA-155 Deficiency Leads to Decreased Atherosclerosis, Increased White Adipose Tissue Obesity, and Non-alcoholic Fatty Liver Disease. Journal of Biological Chemistry 2016;292(4):1267–1287. doi: 10.1074/jbc.m116.739839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mirra P, Nigro C, Prevenzano I, et al. The role of miR-190a in methylglyoxal-induced insulin resistance in endothelial cells. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2017;1863(2):440–449. doi: 10.1016/j.bbadis.2016.11.018. [DOI] [PubMed] [Google Scholar]