

Abstract
Recently, there has been a major shift in the field of psychiatry towards the exploration of complex relationships between blood-based biomarkers and the pathophysiology of psychiatric and neuropsychiatric disorders. However, issues with study reproducibility, validity and reliability have hindered progress towards the identification of clinically relevant biomarkers for psychiatry. The achievement of laboratory validity is a crucial first step for the posterior development of clinical validity. There is evidence that the variability observed in blood-based research studies may be minimised with the implementation of standardised pre-analytical methods and uniform clinical protocols (i.e., pre-venipuncture). It has been documented that errors made in the pre- analytical phase account for 46–68.2% of laboratory testing errors. Thus, standardising clinical assessment, ethical procedures and pre-analytical phase of clinical research is essential for the reproducibility, validity and reliability of blood marker assessment, and reducing the risk of invalid test results. Various other areas of research have already moved towards guidelines for the standardised collection of blood-based biomarkers. Here we aim to provide a set of guidelines that we believe would improve biomarker research: (1) pre-venipuncture information and documentation, (2) ethics of participant consent and (3) pre-analytical methods. Ultimately, we hope this will assist study planning and will improve data comparison across studies allowing for the discovery of biomarkers in psychiatry with both laboratorial and clinical validity.
Keywords: Biomarkers, guidelines, reliability, standardisation, validity
The challenge of standardisation in psychiatric biomarker research
Studies searching for the discovery of blood-based biomarkers in the fields of psychiatry, neurology and neuropsychiatry have increased exponentially (Fernandes et al. 2017; Gwinn et al. 2017; Lewczuk et al. 2018). However, reproducibility, validity and reliability (all essential for achieving laboratory validity) have been a major roadblock towards the identification of clinically relevant biomarkers for psychiatric disorders. In addition, there is an inherent confounding factor of participants with mental illnesses at times being unable to consent at either initial or follow-up blood collection time points. Several of the discrepancies, and also variability, could be considerably mini-mised if researchers would consider using a standardised pre-analytical method and uniformed clinical protocols (i.e., pre-venipuncture). Similarly, variance in collection and veracity of clinical information, which is normatively correlated with assay data, is a source of error.
The most common form of blood withdrawal employed in research is through venipuncture. Studies have shown that the majority of variations and errors in blood-based diagnostics and other areas of clinical research occur in the pre-venipuncture and pre-analytical (i.e., before the analysis of the biomarker) phases (Rai and Vitzthum 2006). It has been documented that 46–68.2% of laboratory testing errors can be accounted for by pre-analytical errors (Plebani 2012). Pre-venipuncture and analytical phases involve all the processes that are performed prior to blood processing and analysis including but not limited to: documentation of patient demographic and clinical information, sample collection and adherence to specific timeframe, processing, transportation and storage (Plebani 2012). Some common blood analytical samples include plasma, serum, white blood cells (WBCs), red blood cells (RBCs), DNA and RNA (including microRNA) (Rai and Vitzthum 2006). Standardizing the pre-analytical phase of clinical research is essential for the reproducibility validity, and reliability of blood marker measurements, and reducing the risk of invalid test results. Similarly, minimising pre-analytical errors in blood-based biomarkers that will be the basis for the future development of blood-based clinical tests is important, as parameters will support decisions on interventions used on patients. Ideally, access to a data management resource to support standardisation and data-sharing modules could enable data entry, quality control, data access, data query, biosample access and account management, which could be central hub for coordinating date-related activities integral to a biomarker programme.
Other areas of research, such as Alzheimer’s disease (O’Bryant et al. 2015), have already moved towards guidelines for the standardised collection of body fluids, including blood and cerebrospinal fluid-based biomarkers (Lewczuk et al. 2006). Due to the complex differences in blood-based biomarker collection across different countries, here we aim to suggest a set of steps that we believe would help improving biomarker research: (1) pre-venipuncture information and documentation, (2) ethics of participant consent and (3) pre-analytical methods. Ultimately, we hope that these suggestions will assist study planning and will improve data comparison across studies allowing for the discovery of future clinically relevant and reliable biomarkers for psychiatric disorders. We postulate that the proposed guidelines offer guidance on critical procedures; however, they cannot be considered universal guidelines as each country will have to adjust to their legal and procedural requirements.
1. General ethics and pre-venipuncture information guidelines
Ethical and legal requirements can vary in each institution below we offer a set of general guidelines that would have to be adjusted following institutions requirements.
1.1. General ethical guidelines for obtaining participant consent
Research personnel must understand, implement and follow ethical guidelines specified by their local governing Institutional Review Board (IRB) or Research Ethics Board. Prior to enrolment, all consent forms should be approved by the governing IRB for each site that will be recruiting participants. Researchers must facilitate this process and ensure that potential participants are provided with all the information required both orally and in written form for informed and conscious consent to being involved in a research study before their enrolment. This exchange is necessary to facilitate a thorough understanding of the participant’s involvement in the research study. The study consent forms should provide the potential study participant with adequate information about the study and its future course. If the participant is a child then the parents or guardians need to be informed and approve the participation, while in adolescent participants it is both the participant and the parents/guardians who need to be informed and approve the participation. In cases of participants with mental disorders who are unable to make their own decision, as with children, then the parents or guardians need to be informed and approve the participation.
In particular, participants should be informed about the purpose of the study, its visit structure, procedures and whether these are performed for research purposes, clinical diagnosis as for blood analytics or both. Risks of these procedures, alternatives to participation and the possibility to withdraw participation at any time point, how investigators will handle confidential information (e.g., coding semi-anonymized or fully-anonymized) and samples, and how accidental release of this information may induce financial or legal harm (e.g., how it may affect insurance eligibility or, in the case of substance abuse, alert legal authorities) should also be addressed. Often, participant samples can be useful for future studies both within the scope of the original study (e.g., to study a particular disease) or for different purposes (e.g., to assess variability of a metabolite in a population). When the objective is to create a blood biobank, participants may agree to permit use of samples in multiple studies and to the possibility for the samples to be shipped outside of the institute for analysis (either restricting their use within the scope of the original study or allowing their use without limitations) and access to their stored clinical samples with-out additional consent, or may agree to be contacted to give permission of their use in future studies. In addition, they may be given the option to allow the use of their stored samples in future studies only if they get de-identified (i.e., assigned a code that cannot be traced to their personally identifiable information). In these cases, especially when de-identified samples are used, a second IRB-approved consent form may not be required, although this may vary according to the local ethics committee. More specifically, the informed consent should bear in mind the following considerations.
1.1.1. Study design
In general, the principal investigator (PI) will be required to design clinical studies in which risk of harm or discomfort to participants is minimised. Due to the inherently vulnerable nature of the subject population of persons with mental illnesses, in particular when dealing with child and adolescent populations, but, also in aged probands, particular attention needs to be paid to the design of clinical and epidemiological studies, especially to avoid increasing the risk of symptom provocation. These include, but are not limited to, case-control studies, placebo-controlled studies, washout studies and challenge studies.
1.1.2. Risk assessment
The PI will need to ensure that the clinical study information is transparent by explaining study procedures and clarifying for participants the potential benefits and risks of the research. The PI will be required to explain the potential benefits of the research for the individual (in the case of interventions of proven clinical benefit) and for society in general (generating generalisable knowledge, which may lead to treatment of diseases). In addition, alternatives to participation (e.g., availability of standard treatments) should be provided. This information is to be made available both orally and in written form for the participant (and in some cases parents or legal guardians) to make an informed decision about participation in the study.
Another vital consideration is the capacity to consent, which is of particular importance in psychiatric research, as a mental illness may impair this competence. To this end the participant’s competence to consent must be assessed before any study procedures take place and include the foundational elements of consent of information, decisional capacity and autonomy (Roberts and Roberts 1999). We now describe a standardised procedure for obtaining informed participant consent.
1.2. Standardized procedure for obtaining informed participant consent
In general, in the process of obtaining informed consent researchers will present all salient information to potential participants in a genuine dialogue in reference to the written consent document whereby they will concurrently test the participant’s understanding of the study and its risks and enable opportunities for questions. One also needs to avoid overwhelming the person with excessive information and, in particular, scientific jargon. This will require researchers to provide a clear yet comprehensive explanation of all aspects of the clinical study before obtaining informed consent from participants. This includes ensuring that the participant unequivocally understands the intervention-related medical information and comprehends significant consequences, as well as individually appreciating the significance of the clinical study. A good practice to ensure understanding is to ask participants to repeat the information back in their own words and the reason for their choice and/or alternatives. For children, alternative options (often appreciated by parents) are short cartoon videos that explain the study and the procedures, as well as the participant’s rights.
As part of the standardized process of obtaining informed consent, prospective participants should be asked to endorse a participant information form and the participant consent form (PCF). These forms should contain succinct and plain language enabling the research to be explained at the level of understanding of the prospective participants to enhance their understanding of their role within the study. More specifically, these forms should include a brief explanation of the aims and objectives of the clinical study, what authorization/approval exists for the research, and what methods, techniques and procedures will be used to conduct the research. The dynamic and continuous nature of the research process, including the potential future use of samples in conjunction with other information collected in the study need to be addressed in the process of obtaining consent. Furthermore, the demands on the participant are to be clearly defined, including time and travel requirements, inconvenience, monetary costs, number of interviews, meetings, test sessions and overall duration of the research project. Lastly, participants will then be given time for reflection and consultation under non-coercive conditions before a decision to participate in the study is reached. A lack of dissent will not be considered equivalent to consent.
In addition, the participant should be given the opportunity to withdraw at any time, without need for explanation or having any clinical disadvantages, from the clinical study. Throughout the duration of the study, the consent dialogue should continue, as new questions are addressed and new information is shared. For longitudinal studies and long-term interventional studies, formal written consent should be re-obtained during regular visits, every year or as frequently as mandated by the IRB. If there are any changes to the agreed terms (importantly, new information about risks and benefits or new alternatives to participation), researchers will need to notify participants in advance and give them the option of continuing their participation or enabling withdrawal. In the case of withdrawal, the participant should be given the option of withdrawing permission to use previously collected data and samples or allow their continuous use (e.g., in an intent-to-treat analysis or a sensitivity analysis of enrolled participants that did not complete).
1.3. General guidelines for obtaining consent for clinical and research studies as part of collaborations within national and international consortia
All participants will be explicitly asked to agree to the use of their biological material and resultant data being shared as part of collaborations within national and international consortia, as well storage in a biobank for future studies (within or outside the scope of the initial research). To this end the PCF will need to clearly require consent for sharing across sites of (a) specimen and/or (b) data.
We suggest that the consent should be specific for the five main types of biomarkers and resulting data that aim to be shared including, but not necessarily limited to:
-
a.
(epi)genomic biomarkers based on DNA analyses or others
-
b.
transcriptomic biomarkers based on RNA and microRNA analyses
-
c.
proteomic biomarkers based on protein profiles
-
d.
metabolic and lipidomic biomarkers based on metabolite analyses (intermediates and products of metabolism)
-
e.
extracellular vesicle (EV)-based biomarkers.
-
f.
immortalized cells or reprogrammed cells (e.g., induced pluripotent stem cells) for modelling
Consent will also be required to be specific for the different types of biological specimen being shared. This includes blood (RBCs and WBCs, serum, plasma or whole blood), and other types of samples. If the study plans to generate either immortalised lymphocytes or induced pluripotent stem cells and derived organelles, consent should be based on an explanation of stem cell research and the goals of the study, and the risks associated should be clearly identified.
2. General ethical guidelines for pre-analytical methods
2.1. Collection of demographical information from participants
Researchers must ensure conduct of research methodology in which anonymity for research participants is upheld, while the identifiable or potentially identifiable information provided by participants is treated with confidentially. All data must be non-identifiable and must not be re-identifiable. All researchers are to be adequately trained in the handling of personal information in accordance with Good Clinical Practice, in addition to the respective country’s code of conduct for research and fair information practice.
Various participant-related factors influence the results from blood marker measurements. Standardized documentation can help account for and minimise these influences. Documentation should include demographical information, anthropometric data, clinical information, life style and rating scales in order to identify sources of potential noise and/or physiological variance. Demographical and anthropometric characteristics, use of drugs of abuse, including alcohol and smoking status, are examples of uncontrollable variables that may potentially contribute to noise on blood-based biomarker assessments.
The following is a minimum list of sociodemographic variables that, in our view and experience, are necessary for pre-venipuncture standardisation: demographic characteristics, including, but not limited to, age at the time of blood withdrawal (in years), gender, race, ethnicity (consider acquiring place of birth of participants and both parents if known).
Anthropometric data and indices (height, weight, BMI, body surface area, abdominal girth, etc.).
Time of withdrawal (many analytes have circadian variations).
Day of female menstrual cycle and use of contraceptive.
Fasting status or not and number of hours since the last meal (conventionally, at least 8 h since last meal).
Diet (type of food and beverage consumed prior to sample collection should be documented using an approved and standardised collection tool).
Smoking status: categorical variable yes/no, and number of cigarettes/day. If smoking status is ‘no’, differentiate between past smoker and never smoker, and length of abstinence period if past smoker.
Alcohol and other drugs of abuse in addition to smoking. These should be characterised in the same way as for smoking.
The following is a list of desirable clinical information recommended for pre-venipuncture standardisation:
Current medication at the time of blood withdrawal including dose and regimen. For the best results, medication lists should be reconciled taking into account the participant’s electronic medical record, input from the family, and/or from the participant directly. If the participant is currently not on psychiatric medication, it should be specified whether the participant is drugnaïve (i.e., never used psychiatric medication, with the exception of benzodiazepines) or drug-free (definition of drug-free might differ in different studies; the most commonly employed definition is out of psychiatric medication, with the exception of benzodiazepines, for at least 2 weeks).
Length of illness in years or months. Specify if the current episode at the time of blood withdrawal is the first episode of the disease.
Length of the current psychiatric episode in days (for instance, length in days of the current mood episode or of the psychotic episode).
Age of onset or/and age of first hospitalisation and, if available, the age of prodromal symptoms.
-
Medical comorbidities, to be determined in accordance to each study design.
-
•
Note: comorbidities are noted to have specific interference on blood-based biomarkers and should be taken into consideration.
-
•
Exercise as a categorical variable: never, once a month, once a week or more than once a week. Specify if the exercise is predominantly aerobic, anaerobic, or both.
History of suicide attempts: categorical variable yes/no.
History of family history of neuropsychiatric disorders: categorical variable yes/no; if yes, describe the disorder specifying the relative (i.e., mother, brother, son, etc.), and its age of onset.
Severity of symptoms according to appropriate scales for each disorder. It is advisable that the scales are rated according to the symptoms present at the moment of the application of the clinical protocol and not according the worst period of symptoms in the last 2 weeks.
If it is necessary to share any of the above-mentioned data with collaborators, these should be sent with codes only, and no link to the identity of participants should be shared. It is recommended that data should be sent using an encrypted-locked file.
2.2. Participant preparation, blood collection and processing
Depending on the blood constituent and analyte being measured, particular patient preparation, sample collection, processing and storage conditions are required. Also, contingent to these factors, some additional laboratory steps may be needed for accurate measurement. Due to these differing requirements, it is not possible to propose general pre-analytical methods applicable for all analytes. In contrast, it is necessary to develop standardisation methods outlining pre-analytical procedures specific to the analytes being measured (Rai and Vitzthum 2006).
Participant preparation:
As a general, and not universal, guideline, often the participant should be advised to eat a small meal before beginning a 9–10-h fasting phase the day before the blood withdrawal. It is important to note that not all measurements require participants to be fasted. During this period, it is recommended that participants do not eat, drink (with the exception of water), or participate in strenuous physical activities until the blood withdrawal. Choosing between fasting- and non-fasting is dependent on the biomarker being measured. However, if the aim is to establish a blood biobank, it is advisable that the participant is fasting.
Blood collection:
There are many different variables that can impact blood marker measurement results within the blood collection procedure. Needle gauge, single- or multi-draw needles and container material can affect test results (Rai and Vitzthum 2006). Appropriate sample handling and blood collection procedures must be followed in order to minimise error in blood marker measurements.
The following set of guidelines are offered as an example and based on our group’s experiences. Blood collection should be completed ideally at the same time of the day across participants (to minimise circadian variability) and on the same day as the collection of anthropometric, clinical, psychometric/neuropsychological and neuroimaging data. If this is logistically not possible, the blood collection should be done no more than 24 h before or after the other assessment (Haenisch et al. 2016). In studies analysing the effect of an intervention on biomarkers, ideally, the blood collection should be done prior to the beginning of the intervention. Under most collection conditions, 21- gauge needles should be used for adults (Bowen et al. 2010). Smaller needles have been associated with increased clotting and occlusion, while larger needles may cause haemolysis by disturbing laminar blood flow. Haemolysis can cause an increase in release of haemoglobin and various intracellular analytes, thus interfering with many analytical assays (Bowen et al. 2010). If the quantity of blood collection is higher than 20ml (the maximum quantity possible to collect using a syringe), it is preferable to do the blood withdrawal using a blood collection set. For biobank establishment, we believe that it would make sense to collect at least 30–45 ml of blood. All blood withdrawal tubes should be labelled with study ID number and date and time of collection, or barcodes. The ID numbers or barcodes should be stored securely and properly matched to demographical and clinical characteristics on the institution’s password-protected computers.
Note: in this general guideline, we focus on blood collection and processing; however, if the study is collecting other types of samples, such as cerebrospinal fluid, tissue specimens, urine, saliva, faeces or hair, similar identification standards should be applied. This is a general guideline; each study should revise the protocol in light of any specific requirements of the biomarker included in the study. Also, each laboratory should consider establishing quality-control processes. For recommendation on such protocols please see quality-control recommendations by the National Institute of Neurological Disorders and Stroke BioSEND biorepository, which includes, for example, haemoglobin assessment for quality control of serum and plasma samples, and the DNA/RNA Integrity Number (i.e., electrophoresis) and/or 260/280 and 260/230 ratios (i.e., spectrophotometric) to determine RNA and DNA quality (Gwinn et al. 2017).
Blood processing:
The volumes and types of specimens suggested below are presented as examples and based on our group’s previous experience of blood fraction storage in a biobank; volumes can be adjusted for each study. However, we encourage each study to collect and store all blood fractions as suggested below (see the graphical description of points A-H in Figure 1).
Figure 1.
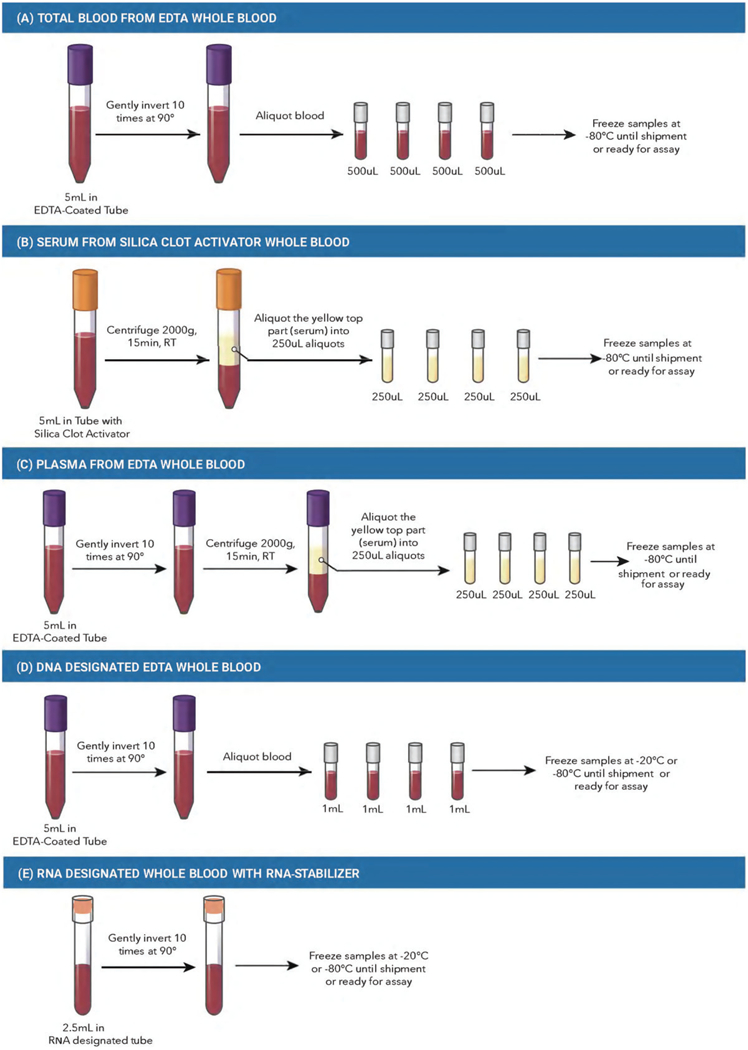
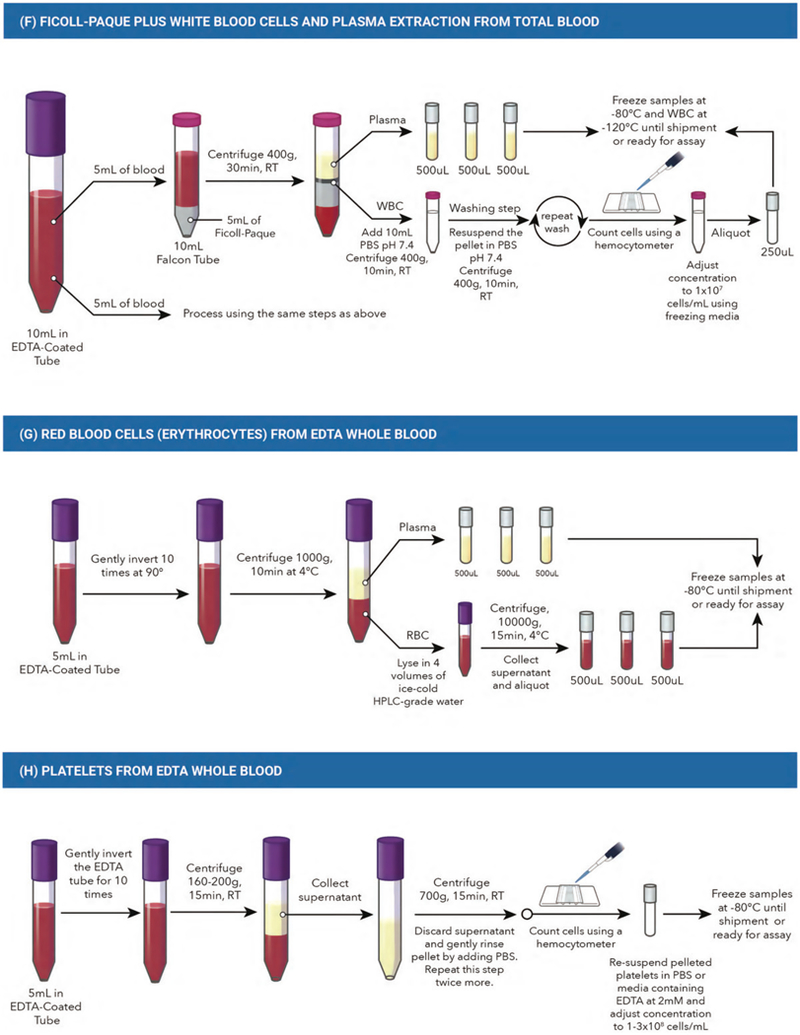
Blood Processing. (A) Total blood from EDTA whole blood. (B) Serum from silica clot activator whole blood. C) Plasma from EDTA whole blood. (D) DNA designated EDTA whole blood. (E) RNA-designated whole blood with PAXgene collection tube. (F) Ficoll-Paque + white blood cells and plasma extraction from total blood. G) Red blood cells (erythrocytes) from EDTA whole blood. (H) Platelets from EDTA whole blood. RT, room temperature; PBS, phosphate-buffered saline; EDTA, ethylenediaminetetra-acetic acid. The tube colours depicted in the figure are representative of the colours in North America, but may not be representative of appropriate tube colours across all countries and/or institutions. This figure depicts an example of blood processing methods. However, we are aware that there are other successful biobanks with different specifications for processing. This protocol has been validated by the authors of this guideline.
The biospecimen (i.e., serum, plasma, WBCs, etc.) labels should contain only the study and participant ID, specimen type and time point collected, or use of barcodes. After laboratory analyses, coded data should be stored securely on the institution’s password-protected computers and properly matched to participant demographical and clinical characteristics. We highlight the importance of using software that can create a log for each time of data change, data adjustment or data added, and the researcher who did it.
Total blood from EDTA whole blood:
Collect at least 5 ml of blood using a vacutainer tube with spray-coated, or equivalent, K2EDTA (tube size: 13 × 75). Mix the blood well by inverting the tube 10 times at 90° in aliquots of 500 μl in 1.5 ml low-binding protein Eppendorf tubes or cryovials with O-ring-sealed screw leads. Total blood should be kept frozen at −80 °C until assayed (see Figure 1(A)).
Serum from silica clot activator whole blood:
Collect at least 5 ml of blood using vacutainer tubes containing silica clot activator, polymer gel, silicone-coated interior, or equivalent (tube size: 13 × 75, 3.5 ml withdrawal). Centrifuge samples for 15 min at 2,000 × g at room temperature. Remove serum from the top layer and aliquot 250 μl in 1.5-ml low-binding protein Eppendorf tubes or cryovials with O-ring-sealed screw-leads. For biobanking purposes, in which serum and plasma will be stored for prolonged periods, the use of low-binding protein Eppendorfs or cryovials is recommended, as this will decrease the binding of molecules of interest (i.e., proteins and/or metabolites) to the surface of the tube, while the O-ring-sealed screw leads prevent evaporation. Serum should be kept frozen at −80 °C until assayed. Roughly, 1ml of blood produces 0.5 ml of serum (Olson et al. 2013), although the quantity can vary according to the participant (see Figure 1B). Note: If processing serum tubes immediately after collection, no coagulation is necessary. If you are unable to process serum within 20 min of collection, allow fresh blood to coagulate naturally for 2–3 h at room temperature before placing on ice to prevent large clots from forming prior to sample extraction. (Haenisch et al. 2016). If samples cannot be extracted immediately after coagulation, samples may be stored in a fridge at 4 °C for 12 h. At room temperature, samples must be processed no more than 4h after the blood withdrawal.
Plasma from EDTA whole blood:
Collect at least 5 ml of blood using a vacutainer tube with spray-coated, or equivalent, K2EDTA (tube size: 13×75). Mix the blood well by inverting the tube 10 times at 90°. Centrifuge samples for 15min at 2,000 × g at room temperature. Remove plasma from the top layer and aliquot 250 μl in 1.5 ml low-binding Eppendorf or cryovial tubes with O-ring-sealed screw leads. As for serum, for biobanking purposes, in which plasma usually will be stored for prolonged periods, it is recommended to use low-binding protein Eppendorf or cryovials. Plasma should be kept frozen at −80 °C until assayed (see Figure 1(A)). Roughly, 1 ml of blood produces 0.5 ml of plasma, although the quantity can vary according to the participant. As for serum, at room temperature the blood sample should be processed in the first 4h after the blood withdrawal (see Figure 1(C)).
A special note should be made for extraction of EVs from stored plasma or serum. To date, two notable consensus papers have been published with recommendations on pre-analytical factors, collection, types of tubes and additives (with optimum choices heavily dependent on the type of biomarkers (protein, RNA) that will be pursued), storage, freeze-thaw cycles and EV isolation options after thawing (1, 3). Fortunately for EV biomarker studies, EVs are remarkable stable during storage and resistance to a limited number of freeze thaws cycles (2, 4), whereas EV phosphoproteins remain stable over years in storage (5).
DNA designated EDTA whole blood:
Collect 5 ml of blood using a vacutainer tube with spray-coated, or equivalent, K2EDTA (tube size: 13×75). Mix the blood well by inverting the tube 10 times at 90°. Aliquot 1 ml of total blood in 1.5 ml low-binding protein Eppendorf tubes or cryovials with O-ring-sealed screw leads. Store total blood samples at −20 or −80 °C until assayed (see Figure 1(D)).
RNA designated whole blood with RNA-stabiliser collection tube:
Collect 2.5 ml of blood in the RNA- stabiliser Blood RNA Tube (tube size: 16 × 100 mm). Mix the blood well by inverting the tube 10 times at 90°. Store the RNA-stabiliser tube at −20 °C (for less than 6 months) or −80 °C (for over 6 months up to years) until assayed. When collecting blood for RNA, a blood collector set must be employed and not a syringe. If the sample for RNA is the only one to be collected, a small quantity of blood must be drawn to a ‘discard tube’. If other blood specimens are being collected, the sample for RNA must be the LAST one collected (see Figure 1(E)).
WBCs from EDTA whole blood:
For isolation of WBCs, collect 10 ml of blood using EDTA interiorcoated vacutainer tubes. Mix the blood well by inverting the tube 10 times at 90°. Take two-15 ml falcon tubes and, in each tube, carefully layer 5 ml of blood on 5 ml of density gradient medium (i.e., Ficoll-Paque plus, 71–7167-00 AG, GE-Healthcare). Centrifuge each tube for 40min, at 400 × g at room temperature. Extract the layer of WBCs, which appears as a ring in the middle of the tube, using a long pipette tips (i.e., Gel-Loading Pipet Tips, Flat tip, 0.2 mm) and transfer to another 15-ml conical tube containing 10 ml of phosphate-buffered saline (PBS), pH 7.4. Centrifuge tubes for 10min at 400 × g at room temperature. Wash the WBC pellet by removing the supernatant and re-suspending the pellet in 10 ml of PBS, followed by centrifuging the tube for 10min at 400 × g at room temperature. Repeat the washing process. Count cells using a haemocytometer and add freezing media (Roswell Park Memorial Institute medium media + 10% dimethyl sulfoxide [DMSO]) to store samples at 1 × 107 cells/ml. Aliquot 250 μl into 1.5-ml cryovials (i.e., cryogenic vial, 1.8 ml, internal thread, round bottom, starfoot, free standing). For long-term storage, plasma and WBCs should be kept frozen at −80 °C until assayed. Alternatively, commercially available complete cell preparation tubes containing anticoagulant, liquid density medium and an inert gel barrier) can be used for WBC isolation. In this case, processing may begin with direct centrifugation of each tube for 40 min, 400 × g at room temperature, followed by sample washing and the addition of freezing media for storage at −80 °C until assayed. See Figure 1(F).
RBCs (erythrocytes) lysates from EDTA whole blood:
Collect at least 5 ml of blood using a vacutainer tube with spray-coated, or equivalent, K2EDTA (tube size: 13 × 75). Mix the blood well by inverting the tube 10 times at 90°. Centrifuge the blood at 1,000 × g for 10min at 4 °C. Pipette off the top yellow plasma layer. Remove the white buffy layer (leukocytes) and discard. Lyse the RBCs in 4 volumes of ice- cold HPLC-grade water and then centrifuge at 10,000 × g for 15min at 4 °C. Collect the supernatant (erythrocyte lysate) in 1.5 ml low-binding Eppendorf tubes or cryovials. If not assaying the same day, store samples at −80 °C (see Figure 1(G)).
Platelets from EDTA whole blood:
Collect at least 5 ml of blood using a vacutainer tube with spraycoated K2EDTA (tube size: 13×75). Mix the blood well by inverting the tube 10 times at 90°. Spin EDTA whole blood at 160–200 × g at room temperature for 15 min. Carefully collect the supernatant (note: the supernatant contains small platelets in suspension; if all platelets are required, spin at 2,200 × g for 15 min), which will contain the plasma rich in platelets, and transfer it to a new centrifuge tube. Centrifuge plasma rich in platelets at 700 × g for 15 min. Discard the supernatant and gently resuspend the pellet by adding PBS. Repeat rinse twice more. Resuspend pelleted platelets in PBS or media containing EDTA at 2 mM (resuspension solution must be calcium free to avoid activation and degranulation). Before storage, count platelets using a haemocytometer and adjust the concentration to 1 −3 × 108 platelets/ml in PBS or media. If not assaying the same day, store samples at −80 °C in low-binding protein Eppendorf tubes or Nunc cryovials. See Figure 1(H).
Table 1 shows, as an example, the tube type used in the blood collections, and the blood fractionation and analytes that can be acquired from it.
Table 1.
Examples of blood fraction (sample type) and analytes that can be acquired from different source of blood withdrawal tubes.
| Coating | Sample Type | Analyte Example |
|---|---|---|
| EDTA | Plasma, DNA | Cytokines Genome Wide |
| WBCs RBCs | Association Studies Mitochondrial Electron Transport Chain Activity | |
| Total Blood | ATP Synthase Activity Hemoglobin A1c | |
| Citrate | Plasma RBCs | Lithium Level Coagulation |
| Clot activator gel | Serum | BDNF |
| No coating or gel | Serum | BDNF |
| Sodium fluoride (NaHF2) | Total Blood | Glucose |
| RNA stabiliser | RNA, microRNA | Transcriptomics |
2.3. Storage, sample shipment and data management
All patient specimens should be kept in a secure biobanking facility including state-of-the-art lockable fridge or freezers in a designated room, accessible only to authorised research personnel. Each storage facility should include −20 °C and −80 °C freezers and liquid nitrogen tanks that contain magnetic access cards, which ensure proper security and monitoring of the personnel accessing the samples, the temperature and power alarms and power backup contingencies. Freezers should be equipped with temperature records and alarms to call cell phones if the temperature drops or if any other maintenance is necessary, a CO2 backup system to maintain the temperature down to −70 °C in the case of power failure, a bar code printer and reader to facilitate sample identification and creation of electronic records of sample storage, and a dedicated computer to archive and create biobank information. A low-temperature alarm at −90 °C and a high-end temperature alarm at −65 °C is highly recommended. Biospecimen resource personnel should monitor and record storage conditions, number of freeze-thaw cycles and number of equipment failures, including temperature deviations, via an independent temperature monitor. Validation of freezer units, including temperature mapping of the freezer interior, is also recommended to ensure uniform temperature throughout the storage unit, as is regular defrosting. Each biospecimen should be stored in the appropriate conditions outlined above.
Challenges for adequate biobanking include, but are not limited to, storage space for samples, an efficient system for sample identification and a protected mechanism for sample storage, including temperature and access monitoring. In biobanks in which very large amounts of samples will be stored, whenever possible, the employment of a biobank using robotic selection of samples are preferred; however, this option is currently limited by the high costs associated with it.
If the study includes sample shipment for biomarkers analyses to collaborators or service facilities, it is recommended to ship samples overnight, if over-seas, from Monday to Thursday, to minimise sample loss, using 2 kg of dry ice for every 100 microcentrifuge tubes (2 ml).
Any information generated should be maintained within a locked electronic database by the research team. The database needs to be securely stored using an institution password, with adequate electronic protection including data backups on geographically discrete servers, firewalls and antivirus software in lock-restricted access offices. Specimen data should be backed up in a password-protected folder on the institution’s network whereby passwords need to be regularly changed in accordance with the institution’s security policy. On completion of the research project, unidentified samples should be disposed of according to state and federal legislation and the regulations applicable to the disposal of human materials and biohazardous waste. Since biological material can suffer degradation with long-term storage, even when at −80°C, it is important to check the literature, performing the laboratory assays to determine whether the particular analyte being assessed suffers degradation, and, if so, to what extent. As an example, RNA can show degradation at 5 years. For −80 °C freezers, aliquots frozen in RNA-stabilizing solutions is a consideration. Proteins such as cytokines and brain-derived neurotrophic factor can start to present degradation after 1 year of storage at −80°C (Shabihkhani et al. 2014). Biomolecules and notably EVs also can degrade with increasing freeze-thaw cycles, and, therefore, that should be avoided.
3. Summary and future perspectives
In order for the reliability and signal-to-noise ratio of research exploring blood-based biomarkers in psychiatry to advance, there is a strong need for the implementation of guidelines that standardise pre-analytical methods across various research facilities. The standardisation procedures outlined in this paper aim to minimise laboratory-testing errors and allow for cross-validation across research centres. In addition, standardisation aims to improve reproducibility and laboratory validity of research exploring the complex relationships between blood-based biomarkers and psychiatric disease pathophysiology. Blood-based biomarkers have the potential to provide decision-guided evidence for determining clinical diagnoses, prognoses, stage and disease activity, and of predicting treatment responses for psychiatric disorders. These guidelines provide the minimum information required to minimise pre-analytical error propagation in psychiatry research (Figure 2). More importantly, it is imperative that in addition to pre-analytic steps, that analytic and post-analytic steps are carefully designed in a study-specific manner and executed in order to obtain robust results with laboratory validity that have the potential to be used for the development of future blood tests with clinical validity.
Figure 2.
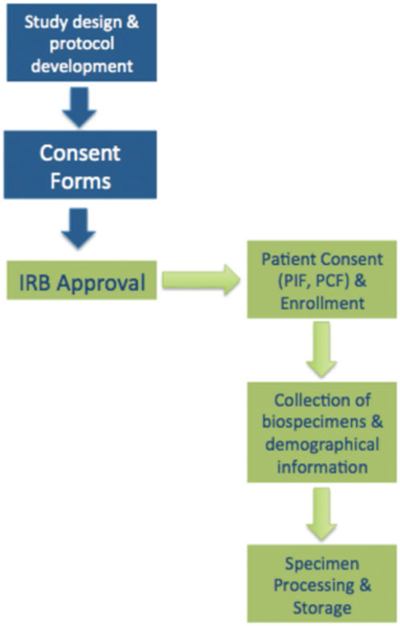
Summary flowchart for collection, processing and storing of blood samples in clinical studies: *participant information form (PIF), participant consent form (PFC).
Acknowledgements
ACA is funded by Canadian Institute of Health Research [MOP-133439], and Ministry of Research and Innovation of Canada [ERA-14-10-022].
MB is supported by a NHMRC Senior Principal Research Fellowship (APP1059660 and APP1156072).
SHK is supported by the Ontario Brain Institute to carry out the work of Can-Bind.
PL has received support from the Innovative Medicines Initiative Joint Undertaking under EMIF grant agreement n° 115372, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme [FP7/2007-2013] and EFPIA companies’ in kind contribution. He has received consultation and/or lecture honoraria from IBL International, Fujirebio Europe, AJ Roboscreen, Virion/serion GmbH, Immungenetics, and Roche.
Footnotes
Statement of interest
None to declare.
References
- Bowen RAR, Hortin GL, Csako G, Otañez OH, Remaley AT. 2010. Impact of blood collection devices on clinical chemistry assays. Clin Biochem. 43:4–25. [cited 2016 Nov 13] Available from: http://linkinghub.elsevier.com/retrieve/pii/S0009912009004391 [DOI] [PubMed] [Google Scholar]
- Fernandes BS, Williams LM, Steiner J, Leboyer M, Carvalho AF, Berk M. 2017. The new field of “precision psychiatry.” BMC Med. 15:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn K, David KK, Swanson-Fischer C, Albin R, Hillaire-Clarke CS, Sieber BA, Lungu C, Bowman FD, Alcalay RN, Babcock D, et al. 2017. Parkinson’s disease biomarkers: perspective from the NINDS Parkinson’s Disease Biomarkers Program. Biomarkers Med. 11:451–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haenisch F, Cooper JD, Reif A, Kittel-Schneider S, Steiner J, Leweke FM, Rothermundt M, van Beveren NJM, Crespo-Facorro B, Niebuhr DW, et al. 2016. Towards a blood-based diagnostic panel for bipolar disorder. Brain Behav Immun. 52:49–57. [DOI] [PubMed] [Google Scholar]
- Lewczuk P, Kornhuber J, Wiltfang J. 2006. The German Competence Net Dementias: standard operating procedures for the neurochemical dementia diagnostics. J Neural Transm. 113:1075–1080. [DOI] [PubMed] [Google Scholar]
- Lewczuk P, Riederer P, O’Bryant SE, Verbeek MM, Dubois B, Visser PJ, Jellinger KA, Engelborghs S, Ramirez A, Parnetti L, et al. 2018. Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: An update of the Consensus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry. World J Biol Psychiatry. 19:244–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Bryant SE, Gupta V, Henriksen K, Edwards M, Jeromin A, Lista S, Bazenet C, Soares H, Lovestone S, Hampel H, et al. 2015. Guidelines for the standardization of preanalytic variables for blood-based biomarker studies in Alzheimer’s disease research. Alzheimer’s Dement. 11:549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JE, Ryu E, Johnson KJ, Koenig BA, Maschke KJ, Morrisette JA, Liebow M, Takahashi PY, Fredericksen ZS, Sharma RG, et al. 2013. The Mayo Clinic Biobank: a building block for individualized medicine. Mayo Clin Proc. 88: 952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plebani M 2012. Quality indicators to detect pre-analytical errors in laboratory testing. Clin Biochem Rev. 33:85–88. [PMC free article] [PubMed] [Google Scholar]
- Rai AJ, Vitzthum F. 2006. Effects of preanalytical variables on peptide and protein measurements in human serum and plasma: Implications for clinical proteomics. Expert Rev Proteomics. 3:409–426. [DOI] [PubMed] [Google Scholar]
- Roberts LW, Roberts B. 1999. Psychiatric research ethics: an overview of evolving guidelines and current ethical dilemmas in the study of mental illness. Biol Psychiatry. 46: 1025–1038. [DOI] [PubMed] [Google Scholar]
- Shabihkhani M, Lucey GM, Wei B, Mareninov S, Lou JJ, Vinters HV, Singer EJ, Cloughesy TF, Yong WH. 2014. The procurement, storage, and quality assurance of frozen blood and tissue biospecimens in pathology, biorepository, and biobank settings. Clin Biochem. 47: 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]