

Abstract
Sjögren syndrome (SS), a chronic autoimmune disorder causing dry mouth, adversely affects the overall oral health in patients. Activation of innate immune responses and excessive production of type I interferons (IFNs) play a critical role in the pathogenesis of this disorder. Recognition of nucleic acids by cytosolic nucleic acid sensors is a major trigger for the induction of type I IFNs. Upon activation, cytosolic DNA sensors can interact with the stimulator of interferon genes (STING) protein, and activation of STING causes increased expression of type I IFNs. The role of STING activation in SS is not known. In this study, to investigate whether the cytosolic DNA sensing pathway influences SS development, female C57BL/6 mice were injected with a STING agonist, dimethylxanthenone-4-acetic acid (DMXAA). Salivary glands (SGs) were studied for gene expression and inflammatory cell infiltration. SG function was evaluated by measuring pilocarpine-induced salivation. Sera were analyzed for cytokines and autoantibodies. Primary SG cells were used to study the expression and activation of STING. Our data show that systemic DMXAA treatment rapidly induced the expression of Ifnb1, Il6, and Tnfa in the SGs, and these cytokines were also elevated in circulation. In contrast, increased Ifng gene expression was dominantly detected in the SGs. The type I innate lymphoid cells present within the SGs were the major source of IFN-γ, and their numbers increased significantly within 3 d of treatment. STING expression in SGs was mainly observed in ductal and interstitial cells. In primary SG cells, DMXAA activated STING and induced IFN-β production. The DMXAA-treated mice developed autoantibodies, sialoadenitis, and glandular hypofunction. Our study demonstrates that activation of the STING pathway holds the potential to initiate SS. Thus, apart from viral infections, conditions that cause cellular perturbations and accumulation of host DNA within the cytosol should also be considered as possible triggers for SS.
Keywords: innate immunity, TMEM173, vadimezan, interferon-α, interferon-β, sialadenitis
Introduction
The etiopathogenesis of Sjögren syndrome (SS), a chronic autoimmune disorder mainly affecting the exocrine glands, especially the salivary and lacrimal glands, is not clear. The disease shows a strong female predominance and manifests clinically during the postmenopausal years (Brito-Zerón et al. 2016). It is clear that genetic susceptibility, environmental factors such as viral infections, dysregulated immune responses, and sex hormones strongly influence the development of SS.
A characteristic feature of SS is an elevated type I interferon (IFN)–responsive gene signature, which is indicative of heightened type I IFN production and innate immune activation (Kiripolsky et al. 2017). Engagement of endosomal Toll-like receptors (TLR3, TLR7, and TLR9), cytosolic RNA sensors (RIG-1, MDA5), and a multitude of cytosolic DNA sensors hold the potential to induce the expression of type I IFN (Roers et al. 2016). Previous work from our laboratory has demonstrated that poly (IC), a dsRNA mimic of viral nucleic acids, induces acute salivary gland (SG) dysfunction and accelerates the development of SS in mouse model systems (Deshmukh et al. 2009; Nandula et al. 2011). Poly (IC) induces the production of type I IFN and proinflammatory cytokines through the activation of TLR3 and the cytosolic RNA sensors (Cheng and Xu 2010). These findings support the hypothesis that viral infections of SGs might be responsible for initiating SS.
DNA viruses belonging to the Herpesviridae family, particularly Epstein-Barr virus and human herpes virus 6, have long been suspected as etiological factors for SS (Brito-Zerón et al. 2016). The nucleic acids from these viruses are recognized by DNA sensors present in the nucleus as well as in the cytoplasm. Many of these sensors converge downstream and activate the stimulator of interferon gene (STING) protein (Barrat et al. 2016). STING is an endoplasmic reticulum (ER)–associated protein encoded by the TMEM173 gene (Ishikawa and Barber 2008). The major pathway for STING activation involves the recognition of cytosolic DNA by the enzyme cyclic GMP-AMP synthase (cGAS), which leads to the synthesis of the STING agonist 2′3′-cGAMP (Ablasser et al. 2013). Upon binding its agonist, activated STING translocates from the ER to a perinuclear ER intermediate Golgi complex, where it recruits a serine/threonine-protein kinase, Tank Binding Kinase 1, (TBK1) (Tanaka and Chen 2012). Activated TBK1 phosphorylates interferon regulatory factor 3 (IRF3), which dissociates from this complex, enters the nucleus, and induces the transcription of type I IFNs. In addition, STING has also been shown to activate the nuclear factor (NF)–κB pathway and induce the transcription of proinflammatory cytokines (Abe and Barber 2014).
The nucleic acids activating the cGAS-STING-IRF3 pathway could be of microbial origin, or they could be from the host mitochondria and nucleus. In the absence of appropriate functioning of cytoplasmic DNA-degrading enzymes, DNA can accumulate in the cytosol and lead to excessive type I IFN production with pathological consequences (Yan 2017). In humans, hyperactivation of STING has been associated with immune disorders such as Aicardi-Goutières syndrome, chilblain lupus, and STING-associated vasculopathy with onset in infancy (SAVI). Although the role of STING activation in SS has not been formally investigated, indirect evidence suggests its possible involvement in this disorder. The interferon-inducible protein 16 (IFI16) is a DNA sensor protein that binds STING and is also critical for the activation of STING (Almine et al. 2017). SS patients show considerable upregulation in the expression of IFI16 (Alunno, Caneparo, Carubbi, Bistoni, Caterbi, Bartoloni, et al. 2015; Alunno, Caneparo, Carubbi, Bistoni, Caterbi, Gariglio, et al. 2015). The aberrant expression of IFI16 in SS patients has been suspected to be the cause for the production of anti-IFI16 autoantibodies, and their presence is associated with markers of severe disease (Baer et al. 2016). Thus, to investigate whether STING pathway activation is involved in the pathogenesis of SS, in this study, we activated STING with its agonist dimethylxanthenone-4-acetic acid (DMXAA) and investigated the development of SG disease in mice.
Materials and Methods
Mice
All mouse experiments were approved by the Institutional Animal Care and Use Committee, involved humane practices, and followed ARRIVE guidelines. Female C57BL/6 (B6) mice were obtained from Jackson Laboratories and maintained under specific pathogen-free conditions. Female mice (10 to 12 wk old) were injected subcutaneously with (20 mg/kg body weight) DMXAA (Bio-Techne) dissolved in 5% sodium bicarbonate solution. For long-term experiments, the mice received 2 injections (days 0 and 21). The dose of DMXAA was based on previously published literature (Bellnier et al. 2003; Wang et al. 2009; Peng et al. 2011). Control mice were injected similarly with the vehicle alone.
Saliva Measurement
Pilocarpine-induced saliva was measured as described previously (Bagavant et al. 2018). Briefly, mice were injected with pilocarpine (0.375 mg/kg body weight), and saliva was collected by placing a highly absorbent piece of sponge (Salimetrics) in the animal’s mouth for 15 min. The weight of saliva produced (mg) was calculated as the difference between the dry and wet sponge weights.
Gene Expression Analysis
Expression levels of different genes in submandibular SG and primary SG cells were analyzed by real-time polymerase chain reaction (PCR) employing TaqMan assays (Applied Biosystems) as described previously (Nandula et al. 2011).
Immunohistochemistry
Submandibular SGs were collected in 10% buffered formalin. Paraffin-embedded tissue sections were stained with hematoxylin and eosin (H&E) and evaluated for inflammatory cell infiltrates as described previously (Nandula et al. 2011). For analysis of STING expression, 3-µm sections were deparaffinized and rehydrated, and antigen retrieval was carried out at acidic pH. The slides were incubated overnight with rabbit anti-STING (Cell Signaling Technology). Control sections were incubated with an equal concentration of purified rabbit IgG. SignalStain boost IHC reagent (Cell Signaling Technology) was used to detect bound rabbit antibodies, and the reaction was developed with diaminobenzidine (DAB).
Cytokine Analysis
Enzyme-linked immunosorbent assay (ELISA) kits were used to estimate serum levels of IFN-β (BioLegend) and IFN-α (Life Technologies). The levels of serum interleukin (IL)–6, IFN-γ, and tumor necrosis factor (TNF)–α were measured by using a Luminex bead-based multiplex assay (R&D Systems).
Cells
Primary SG cells were harvested from submandibular SGs of 10- to 12-wk-old female B6 mice and STING−/− mice (Ishikawa and Barber 2008) and cultured in conditioned media as described by Baker et al. (2008). At all time points, ROCK inhibitor Y-27632 at a concentration of 10 µM was used in the culture medium (Liu et al. 2017). The epithelial nature of the cells was confirmed by staining for pan-cytokeratin. Cells at passages 2 to 4 were used for all experiments.
Innate Lymphoid Cell Analysis
Single-cell suspensions from submandibular SGs were prepared and stained with the combination of antibodies described by Gasteiger et al. (2015; Appendix Table 1). Zombie aqua fixable viability kit (BioLegend) was used for live/dead cell discrimination. A cocktail of PE-labeled antibodies to CD3, CD5, CD19, TCRγδ, TCRβ, FCεRI, and CD8α was used to exclude lineage (Lin)–positive cells in the CD45 gate. For detecting IFN-γ production, SG single-cell suspensions were treated with ionomycin (1 µg/mL) and either 0.3 nM or 1 nM phorbol myristate acetate (PMA) for 6 h in the presence of Brefeldin A. The cells were then stained for surface markers as above, followed by intracellular staining for IFN-γ. Data were acquired on an LSR II flow cytometer equipped with Diva software (BD Biosciences) and analyzed with FlowJo (FlowJo LLC).
Statistical Analyses
To determine statistical significance, 2-tailed unpaired Student’s t test was used. P < 0.05 at 95% confidence interval was considered significant. For samples not passing normality test, the Mann-Whitney test was used. One-way analysis of variance (ANOVA) was used to compare more than 2 groups of experimental conditions. Correlation analysis was carried out using Pearson test. All analyses were carried out using GraphPad Prism (GraphPad Software).
Results
Proinflammatory Effects of DMXAA Treatment on SGs
DMXAA is an agonist for murine STING (Prantner et al. 2012; Conlon et al. 2013), and it induces strong in vitro and in vivo proinflammatory responses (Shirey et al. 2011). Thus, to investigate the effects of systemic STING activation on SGs, expression levels of different proinflammatory cytokine genes in submandibular glands were measured (Fig. 1). Compared to the vehicle-treated mice, DMXAA-injected mice showed significant increases in the expression of Il6 (115X), Tnfa (20X), Ifng (23X), Ifnb1 (6X), and Il12p40 (25X). The differences in expression levels of Il12p35 (0.66X) and Il18 (2.1X) were statistically not significant. A considerable upregulation was observed in the expression of type I IFN responsive gene Mx1 (250X).
Figure 1.

Systemic dimethylxanthenone-4-acetic acid (DMXAA) treatment induces proinflammatory cytokine gene expression within the salivary gland (SG). Real-time polymerase chain reaction (PCR) analysis of gene expression in submandibular SG from DMXAA-treated group shows significant upregulation in the expression of Il6, Tnfa, Ifng, Ifnb1, Il12p40, and Mx1. The numbers in the right corner with “x” show mean fold increase in gene expression level in DMXAA-treated mice over vehicle-treated mice. Statistical significance was determined by 2-tailed Mann-Whitney test, and P < 0.05 was considered significant.
Since DMXAA treatment is known to cause a spike in systemic cytokine levels (Wang et al. 2009), levels of circulating type I IFNs and proinflammatory cytokines were also measured (Appendix Fig. 1). A significant increase in levels of circulating IFN-α, IFN-β, IL-6, and TNF-α was seen in the DMXAA group. Although circulating IFN-γ levels were not significantly different between the 2 groups, 1 of 6 mice treated with DMXAA showed some elevation. Collectively, these data demonstrate that in addition to the expected systemic responses, DMXAA treatment also induced inflammatory mediators within the SGs.
STING Expression in Murine Submandibular Glands
The elevated Ifnb1 gene expression in submandibular glands was suggestive of STING activation within the tissue. However, information on the expression of STING and its function in SG has been lacking. Therefore, immunohistochemical staining for STING expression was performed in formalin-fixed, paraffin-embedded submandibular SG sections from normal female B6 mice (Fig. 2). Cells in the interstitial regions, in between the SG acini, showed the most prominent staining for STING. Weaker staining was also seen in the cytoplasm of the ductal epithelium. Acinar cells failed to show significant STING expression. No staining was observed in sections obtained from STING−/− mice (right panel) and section from B6 mice incubated with isotype control (left panel).
Figure 2.

Expression of stimulator of interferon genes (STING) in murine submandibular glands. Submandibular salivary gland (SG) obtained from 8-wk-old female B6 (wild-type [WT]) and STING−/− (knockout [KO]) mice were used for the analysis of STING expression. Formalin-fixed, paraffin-embedded sections were stained with rabbit-anti-STING (center and right panels) or with rabbit IgG as isotype control. STING expression is predominantly seen in interstitial cells and SG ductal cells.
The expression of STING in cells of SG origin was further confirmed in cultured murine primary SG epithelial cells by Western blots (Appendix Fig. 2). Although in this study we have not investigated STING expression in human SG, STING protein was readily detected in the immortalized A253.1 cell line from human submaxillary gland.
STING Activation in Primary SG Cells
Having confirmed that STING is expressed in cultured primary SG cells, we investigated whether it can be activated in these cells. To investigate STING activation, murine primary SG cells were treated in vitro with DMXAA. By 2 h posttreatment, RNA expression for Ifnb1 was significantly upregulated in DMXAA-treated cells (Fig. 3A), and they secreted IFN-β (Fig. 3B). Primary SG cells cultured from STING−/− mice did not upregulate the expression of Ifnb1 (Fig. 3C). In the canonical pathway, activation of STING leads to the phosphorylation of TBK1 and IRF3. Both of these proteins were phosphorylated in DMXAA-treated primary SG cells (Appendix Fig. 3). Collectively, these data demonstrate for the first time that the STING-TBK1-IRF3 axis can be activated in SG cells, and it leads to the production of IFN-β.
Figure 3.
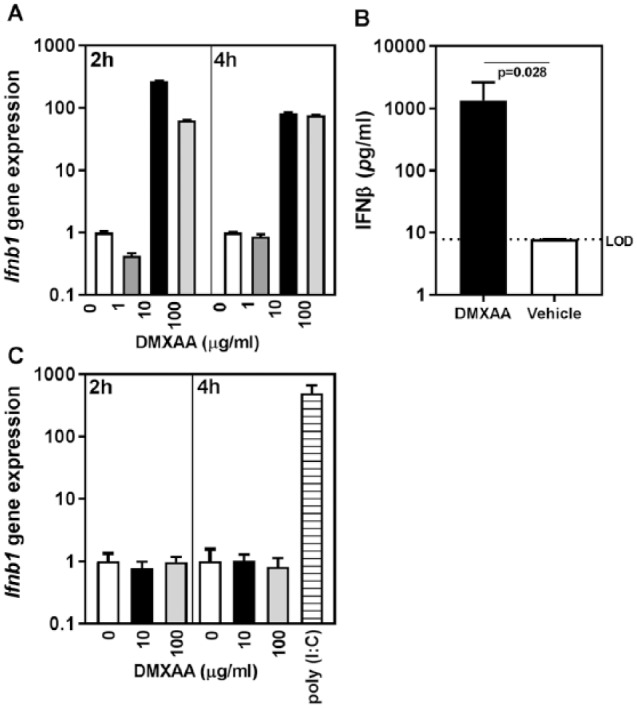
Stimulator of interferon genes (STING) pathway activation in primary salivary gland (SG) cells. (A) dimethylxanthenone-4-acetic acid (DMXAA) induces Ifnb1 gene expression in primary SG cells. TaqMan real-time polymerase chain reaction assays were used for the analysis of Ifnb1 gene expression in primary SG cells treated with different concentrations of DMXAA for 2 and 4 h. Gapdh was used as the housekeeping gene and the relative gene expression calculated as 2−(ΔCt). Data from 2 independent experiments are shown as mean ± SD fold change in gene expression over no drug control (0), which was considered a baseline. (B) Supernatants from either DMXAA (10 µg/mL, 4 h) or vehicle-treated primary SG cells were used to estimate interferon (IFN)–β by enzyme-linked immunosorbent assay. Data are shown as mean ± SD of IFN-β pg/mL levels determined in 2 independent experiments (4 biological replicates). Mann-Whitney test was used to determine the statistical significance. LOD, limit of detection. (C) DMXAA does not induce Ifnb1 upregulation in primary SG cells from STING−/− mice. Data are shown as mean ± SEM from 3 independent experiments. Poly (IC) at a concentration of 1 µg/mL was used as a positive control.
DMXAA Treatment Affects SG Innate Lymphoid Cell 1 Population
Within 4 h of DMXAA treatment, a considerable increase in the expression of Ifng was seen in submandibular glands (Fig. 1). This upregulated expression of Ifng was also evident 3 d following DMXAA treatment (Appendix Fig. 4). By this time, the expression of type I IFN responsive gene Mx1 had returned to the baseline. The rapid induction of Ifng in SGs was suggestive of the involvement of innate cells, specifically innate lymphoid cell 1 (ILC1), in this process. Therefore, submandibular SGs were studied by flow cytometry for ILC1s, identified as NK1.1+ cells within the CD45+Lin−, live lymphocyte gate (Fig. 4). By day 3 after injection, DMXAA-treated mice showed a significant increase in the frequency of NK1.1+ cells. Based on the expression of CD49a and CD49b, the NK1.1+ cells were further classified into tissue ILC1 (CD49a+, CD49b−), SG-specific ILC1 (CD49a+, CD49b+), and natural killer (NK) cells (CD49a−, CD49b+) (Cortez and Colonna 2016). As shown in Figure 4, the predominant increase in NK1.1+ cells following DMXAA treatment was accounted for by the increase in the SG-specific ILC1s. The other 2 subsets, tissue ILC1 and NK cells, were not significantly different between the DMXAA-treated mice and vehicle-treated controls.
Figure 4.

Dimethylxanthenone-4-acetic acid (DMXAA) treatment increases innate lymphoid cell 1 (ILC1) in salivary glands (SGs). Submandibular SGs obtained on day 3 after DMXAA and vehicle treatment were analyzed for changes in ILC1s. (A) Representative flow plots showing NK1.1 cells in the live CD45+ lin− gate. NK1.1 cells were further classified as tissue ILC1 (CD49a+CD49b−), SG ILC1 (CD49a+CD49b+), and conventional natural killer (NK) cells (CD49a−CD49b+). (B) Frequencies of NK1.1+ subsets in DMXAA- and vehicle-treated mice. Data are pooled from 2 independent experiments and are presented as frequency of the live cell gate. Mann-Whitney test was used to determine statistical significance. A similar trend was observed in an additional experiment performed in a different cohort of mice.
To determine whether ILC1s were the source of the local IFN-γ expression, SG cell suspensions were stimulated with PMA and ionomycin in the presence of Brefeldin A. Intracellular staining for IFN-γ showed that all the NK1.1+ cell subsets readily produced IFN-γ compared to vehicle-treated controls at low PMA concentrations (Appendix Fig. 5). At higher doses of PMA, both DMXAA- and vehicle-treated groups showed comparable IFN-γ production in the NK cell (CD49a− CD49b+) subset. However, even at the higher PMA concentration, frequencies of IFN-γ producing cells in both tissue ILC1 and SG-specific ILC1 subsets were higher in the DMXAA-treated mice.
Treatment of Mice with DMXAA Induces SG Disease
To investigate the consequences of STING activation on SG disease, mice were treated twice (days 0 and 21) with DMXAA or the vehicle. Mice were euthanized at 2 time points (1 and 2 mo post treatment), and submandibular SGs were analyzed for the presence of inflammatory cell infiltrates. Representative pictures of H&E-stained submandibular glands from unaffected vehicle controls and DMXAA mice with multiple lymphocytic foci are shown in Figure 5A. In the DMXAA-treated group, 10 of 21 (47.6%) mice showed evidence for sialoadenitis, whereas only 1 of 17 (5.8%) vehicle-treated mice had inflammatory foci within their SGs (Fisher exact test P = 0.009) (Fig. 5B). The differences in the incidence and severity of sialoadenitis in SGs analyzed at 1 mo or at 2 mo posttreatment were statistically not significant.
Figure 5.

Dimethylxanthenone-4-acetic acid (DMXAA)–treated mice develop salivary gland (SG) disease. (A) Representative images of hematoxylin and eosin–stained sections of formalin-fixed, paraffin-embedded submandibular SGs from either vehicle-treated (left panel) or DMXAA-treated (right panel) mice. Arrows point out the inflammatory cell infiltrates. (B) Severity of sialoadenitis in submandibular SGs obtained from mice euthanized between 1 and 2 mo posttreatment was scored by an observer blinded to experimental details. Two-tailed, Mann-Whitney test was used to determine the statistical significance. (C) DMXAA-treated mice show evidence of glandular dysfunction. Pilocarpine-induced saliva was measured in vehicle- and DMXAA-injected groups of mice 4 wk after the initial treatment. The mean amount of saliva in DMXAA-treated mice was significantly lower than the vehicle-treated and untreated groups of mice. One-way analysis of variance with Holm-Sidak’s post-test was used to determine statistical significance and P < 0.05 was considered significant. (D) The severity of sialoadenitis showed an inverse correlation with the amount of saliva produced. Open circles represent DMXAA-treated mice, and open squares represent vehicle-treated mice. Spearman correlation test was used for analysis.
To determine whether the presence of inflammatory cell infiltrates within the submandibular glands affected glandular function, pilocarpine-induced saliva production was measured. As shown in Figure 5C, compared with the untreated and the vehicle-treated mice, DMXAA-treated mice produced a significantly reduced amount of saliva (P = 0.001). Furthermore, the loss of function inversely correlated with the degree of inflammation within the SGs (Fig. 5D). Surprisingly, although a higher trend in severity of lacrimal gland inflammation was seen in DMXAA-treated mice, it did not reach statistical significance (Appendix Fig. 6).
Discussion
In this study, to address the possible involvement of cytosolic DNA sensor pathways in the pathogenesis of SS, we activated STING with its well-established agonist DMXAA. Our data demonstrate that mice treated with DMXAA had elevated expression of type I IFNs and proinflammatory cytokines, both systemically and within the SGs, and the mice developed some of the critical features of the human disease (i.e., SG inflammation and hypofunction).
DMXAA is a flavone acetic acid derivative that was developed as an antitumor agent (Zhou et al. 2002). It binds to murine STING in a way similar to that of the endogenous agonist (Gao et al. 2013) and activates the TBK1-IRF3 axis to induce potent IFN-β production. In addition, engagement of STING by DMXAA also activates the canonical and the noncanonical NF-κB pathways and thereby induces the expression of proinflammatory cytokines. In our study, within 4 h, systemic treatment of mice with DMXAA caused a significant increase in the levels of circulating IFN-α, IFN-β, IL-6, and TNF-α, as well as increased the expression of proinflammatory cytokine genes within the SGs.
Within 3 d, the number of ILC1s in SGs of DMXAA-treated mice increased significantly. This might be through the rapid division of these cells, since the expansion of ILC1s in nonlymphoid organs is mainly dependent on the tissue resident pool (Gasteiger et al. 2015). The ILC1s were the main source of rapidly upregulated Ifng expression within the SGs. A recent study has demonstrated that type I IFNs, produced following infection with murine cytomegalovirus, activates tissue resident ILC1s to rapidly produce IFN-γ (Weizman et al. 2017). While this activation of ILC1s helps in the early control of viral infections, our study suggests the possibility of their involvement in inducing SG disease. The production of both type I and type II IFNs within the SGs following STING activation caused significantly upregulated levels of IFN-induced chemokines Ccl2, Cxcl9, and Cxcl10 (Appendix Fig. 7). These chemokines can attract CCR2+ and CXCR3+ cells and could initiate the formation of lymphocytic foci within the SGs. Indeed, 4 wk after DMXAA treatment, the submandibular SGs showed the presence of lymphocytic foci, and the mice had developed glandular hypofunction. Furthermore, the severity of lymphocytic infiltration inversely correlated with saliva production.
SG epithelial cells are considered to play an important role in the etiopathogenesis of SS (Manoussakis and Kapsogeorgou 2010). DNA viruses infecting these cells can activate innate immune responses and initiate inflammatory cell infiltration. However, for this thesis to work, the STING pathway has to be functional within the salivary gland cells. Thus, the expression and function of STING in salivary gland cells were investigated in depth. Our study shows for the first time that STING is expressed in ductal cells. This observation was confirmed by using primary SG cells. The treatment of these cells with DMXAA induced significantly upregulated expression of Ifnb1 and Tnfα, and the cells secreted IFN-β. Furthermore, transfection of primary SG cells with cGAMP, the canonical endogenous ligand for STING, readily induced IFN-β production (Appendix Fig. 8). Collectively, these data establish for the first time that the STING-TBK1-IRF3 axis is active in SG cells and induces type I IFN production.
There are several spontaneous and induced mouse models for SS, and each of them recapitulates certain features of the disorder (Park et al. 2015). The experimental model described in this study satisfies the current classification criteria established for primary SS (Shiboski et al. 2017). The mice show the presence of lymphocytic infiltrates within the SGs, and they develop glandular hypofunction. These 2 features together meet the cutoff score required for positive classification of SS. However, there are some limitations of this model in mimicking the complete disease in patients. Anti-Ro and anti-La antibodies were not detectable in the sera of DMXAA-treated mice. Instead, the mice showed elevated levels of antinuclear antibodies (Appendix Fig. 9), a feature more common in lupus patients. Thus, we investigated the mice for development of kidney disease or other organ involvement. None of the mice showed evidence for glomerulonephritis (Appendix Fig. 10), although several mice showed the presence of inflammatory cells in the lungs. Considering that some SS patients show lung involvement as an extraglandular manifestation (Roca et al. 2017), it is possible that our model system represents a group of SS patients with concomitant involvement of SGs and lungs.
The elevated type I IFN gene signature in SS patients has led to the hypothesis that viral infections might be responsible for initiating SS (Kivity et al. 2014; Lucchesi et al. 2014). The demonstration of hepatitis delta virus in the SGs of SS patients (Weller et al. 2016) provides strong support to the thesis that viral infection of SGs may influence the pathogenesis of SS. Alternatively, endogenous DNA accumulating in the cytosol holds the potential to initiate SS. SGs from SS patients show elevated expression of long interspersed nuclear element 1 (LINE-1) (Mavragani et al. 2016, 2017). Thus, LINE-1, as well as leaky nuclear and mitochondrial DNA, can be potentially involved in the etiopathogenesis of SS. It is plausible that SS patients may be genetically prone to such defects, which might manifest during aging, explaining the late onset of the disease.
Author Contributions
J. Papinska, contributed to data acquisition, analysis, and interpretation, drafted the manuscript; H. Bagavant, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; G.B. Gmyrek, M. Sroka, contributed to data acquisition and analysis, critically revised the manuscript; S. Tummala, contributed to conception and design, critically revised the manuscript; K.A. Fitzgerald, contributed to conception, design, and data acquisition, critically revised the manuscript; U.S. Deshmukh, contributed to conception, design, data analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, DS_10.1177_0022034518760855 for Activation of Stimulator of Interferon Genes (STING) and Sjögren Syndrome by J. Papinska, H. Bagavant, G.B. Gmyrek, M. Sroka, S. Tummala, K.A. Fitzgerald and U.S. Deshmukh in Journal of Dental Research
Acknowledgments
The authors thank Louise Williamson for her administrative assistance in the preparation of this manuscript and the OMRF Imaging Core Facility for providing excellent technical support for the acquisition of images.
Footnotes
This work was supported by the National Institute of Dental and Craniofacial Research (DE025030) and the Oklahoma Center for Advancement of Science & Technology (OCAST HR15-151).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
A supplemental appendix to this article is available online.
References
- Abe T, Barber GN. 2014. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol. 88(10):5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner KP, Ludwig J, Hornung V. 2013. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 498(7454):380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almine JF, O’Hare CAJ, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, et al. 2017. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun. 8:14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alunno A, Caneparo V, Carubbi F, Bistoni O, Caterbi S, Bartoloni E, Giacomelli R, Gariglio M, Landolfo S, Gerli R. 2015. Interferon gamma-inducible protein 16 in primary Sjögren’s syndrome: a novel player in disease pathogenesis? Arthritis Res Ther. 17:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alunno A, Caneparo V, Carubbi F, Bistoni O, Caterbi S, Gariglio M, Bartoloni E, Landolfo S, Gerli R. 2015. Interferon gamma-inducible protein 16 (IFI16) and anti-IFI16 antibodies in primary Sjögren’s syndrome: findings in serum and minor salivary glands. Reumatismo. 67(3):85–90. [DOI] [PubMed] [Google Scholar]
- Baer AN, Petri M, Sohn J, Rosen A, Casciola-Rosen L. 2016. Association of antibodies to interferon-inducible protein-16 with markers of more severe disease in primary Sjögren’s syndrome. Arthritis Care Res. 68(2):254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagavant H, Trzeciak M, Papinska J, Biswas I, Dunkleberger ML, Sosnowska A, Deshmukh US. 2018. A method for the measurement of salivary gland function in mice. J Vis Exp. 131:e57203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker OJ, Camden JM, Redman RS, Jones JE, Seye CI, Erb L, Weisman GA. 2008. Proinflammatory cytokines tumor necrosis factor-alpha and interferon-gamma alter tight junction structure and function in the rat parotid gland Par-C10 cell line. Am J Physiol Cell Physiol. 295(5):C1191–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat FJ, Elkon KB, Fitzgerald KA. 2016. Importance of nucleic acid recognition in inflammation and autoimmunity. Annu Rev Med. 67:323–336. [DOI] [PubMed] [Google Scholar]
- Bellnier DA, Gollnick SO, Camacho SH, Greco WR, Cheney RT. 2003. Treatment with the tumor necrosis factor-alpha-inducing drug 5,6-dimethylxanthenone-4-acetic acid enhances the antitumor activity of the photodynamic therapy of RIF-1 mouse tumors. Cancer Res. 63(22):7584–7590. [PubMed] [Google Scholar]
- Brito-Zerón P, Baldini C, Bootsma H, Bowman SJ, Jonsson R, Mariette X, Sivils K, Theander E, Tzioufas A, Ramos-Casals M. 2016. Sjögren syndrome, Nat Rev Dis Primer. 2:16047. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Xu F. 2010. Anticancer function of polyinosinic-polycytidylic acid. Cancer Biol Ther. 10(12):1219–1223. [DOI] [PubMed] [Google Scholar]
- Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, Rathinam VA, Monks B, Jin T, Xiao TS. 2013. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol. 190(10):5216–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez VS, Colonna M. 2016. Diversity and function of group 1 innate lymphoid cells. Immunol Lett. 179:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh US, Nandula SR, Thimmalapura PR, Scindia YM, Bagavant H. 2009. Activation of innate immune responses through Toll-like receptor 3 causes a rapid loss of salivary gland function. J Oral Pathol Med. 38(1):42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, et al. 2013. Structure-function analysis of STING activation by c(G(2′,5′)pA(3′,5′)p) and targeting by antiviral DMXAA. Cell. 154(4):748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. 2015. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 350(6263):981–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signaling. Nature. 455(7213):674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivity S, Arango MT, Ehrenfeld M, Tehori O, Shoenfeld Y, Anaya JM, Agmon-Levin N. 2014. Infection and autoimmunity in Sjögren’s syndrome: a clinical study and comprehensive review. J Autoimmun. 51:17–22. [DOI] [PubMed] [Google Scholar]
- Kiripolsky J, McCabe LG, Kramer JM. 2017. Innate immunity in Sjögren’s syndrome. Clin Immunol. 182:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, Dakic A, Simic V, Zheng YL, Sripadhan P, Chen C, et al. 2017. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc. 12(2):439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi D, Pitzalis C, Bombardieri M. 2014. EBV and other viruses as triggers of tertiary lymphoid structures in primary Sjögren’s syndrome. Expert Rev Clin Immunol. 10(4):445–455. [DOI] [PubMed] [Google Scholar]
- Manoussakis MN, Kapsogeorgou EK. 2010. The role of intrinsic epithelial activation in the pathogenesis of Sjögren’s syndrome. J Autoimmun. 35(3):219–224. [DOI] [PubMed] [Google Scholar]
- Mavragani CP, Sagalovskiy I, Guo Q, Nezos A, Kapsogeorgou EK, Lu P, Liang Zhou J, Kirou KA, Seshan SV, Moutsopoulos HM, et al. 2016. Expression of long interspersed nuclear element 1 retroelements and induction of type I interferon in patients with systemic autoimmune disease. Arthritis Rheumatol. 68(11):2686–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavragani CP, Nezos A, Sagalovskiy I, Seshan S, Kirou KA, Crow MK. 2017. Defective regulation of L1 endogenous retroelements in primary Sjögren’s syndrome and systemic lupus erythematosus: role of methylating enzymes. J Autoimmun. pii:S0896-8411(17)30610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandula SR, Scindia YM, Dey P, Bagavant H, Deshmukh US. 2011. Activation of innate immunity accelerates sialoadenitis in a mouse model for Sjögren’s syndrome-like disease. Oral Dis. 17(8):801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YS, Gauna AE, Cha S. 2015. Mouse models of primary Sjögren’s syndrome. Curr Pharm Des. 21(18):2350–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Monie A, Pang X, Hung CF, Wu TC. 2011. Vascular disrupting agent DMXAA enhances the antitumor effects generated by therapeutic HPV DNA vaccines. J Biomed Sci. 18:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prantner D, Perkins DJ, Lai W, Williams MS, Sharma S, Fitzgerald KA, Vogel SN. 2012. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)–dependent innate immune pathways and is regulated by mitochondrial membrane potential. J Biol Chem. 287(47):39776–39788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca F, Dominique S, Schmidt J, Smail A, Duhaut P, Lévesque H, Marie I. 2017. Interstitial lung disease in primary Sjögren’s syndrome. Autoimmun Rev. 16(1):48–54. [DOI] [PubMed] [Google Scholar]
- Roers A, Hiller B, Hornung V. 2016. Recognition of endogenous nucleic acids by the innate immune system. Immunity. 44(4):739–754. [DOI] [PubMed] [Google Scholar]
- Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, Rasmussen A, Scofield H, Vitali C, Bowman SJ, et al. ; International Sjögren’s Syndrome Criteria Working Group. 2017. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis. 76(1):9–16. [DOI] [PubMed] [Google Scholar]
- Shirey KA, Nhu QM, Yim KC, Roberts ZJ, Teijaro JR, Farber DL, Blanco JC, Vogel SN. 2011. The anti-tumor agent, 5,6-dimethylxanthenone-4-acetic acid (DMXAA), induces IFN-beta-mediated antiviral activity in vitro and in vivo. J Leukoc Biol. 89(3):351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Chen ZJ. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 5(214):ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LCS, Thomsen L, Sutherland R, Reddy CB, Tijono SM, Chen CJJ, Angel CE, Dunbar PR, Ching LM. 2009. Neutrophil influx and chemokine production during the early phases of the antitumor response to the vascular disrupting agent DMXAA (ASA404). Neoplasia. 11(8):793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, Degli-Esposti MA, Leslie CS, Sun JC, Sullivan O’ TE. 2017. ILC1 confer early host protection at initial sites of viral infection. Cell. 171(4):795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller ML, Gardener MR, Bogus ZC, Smith MA, Astorri E, Michael DG, Michael DA, Zheng C, Burbelo PD, Lai Z, et al. 2016. Hepatitis delta virus detected in salivary glands of Sjögren’s syndrome patients and recapitulates a Sjögren’s syndrome-like phenotype in vivo. Pathog Immun. 1(1):12–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N. 2017. Immune diseases associated with TREX1 and STING dysfunction. J Interferon Cytokine Res. 37(5):198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Kestell P, Baguley BC, Paxton JW. 2002. 5,6-dimethylxanthenone-4-acetic acid (DMXAA): a new biological response modifier for cancer therapy. Invest New Drugs. 20(3):281–295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, DS_10.1177_0022034518760855 for Activation of Stimulator of Interferon Genes (STING) and Sjögren Syndrome by J. Papinska, H. Bagavant, G.B. Gmyrek, M. Sroka, S. Tummala, K.A. Fitzgerald and U.S. Deshmukh in Journal of Dental Research