

Abstract
Reversing or slowing the aging process brings great promise to treat or prevent age‐related disease, and targeting the hallmarks of aging is a strategy to achieve this. Epigenetics affects several if not all of the hallmarks of aging and has therefore emerged as a central target for intervention. One component of epigenetic regulation involves histone deacetylases (HDAC), which include the “classical” histone deacetylases (of class I, II, and IV) and sirtuin deacetylases (of class III). While targeting sirtuins for healthy aging has been extensively reviewed elsewhere, this review focuses on pharmacologically inhibiting the classical HDACs to promote health and longevity. We describe the theories of how classical HDAC inhibitors may operate to increase lifespan, supported by studies in model organisms. Furthermore, we explore potential mechanisms of how HDAC inhibitors may have such a strong grasp on health and longevity, summarizing their links to other hallmarks of aging. Finally, we show the wide range of age‐related preclinical disease models, ranging from neurodegeneration to heart disease, diabetes to sarcopenia, which show improvement upon HDAC inhibition.
Keywords: epigenetics, geroprotector, hallmarks of aging, HDAC inhibitors, preclinical models
Subject Categories: Ageing, Chemical Biology
Glossary
- Adaptive thermogenesis
The regulated production of heat in response to environmental changes in temperature and diet.
- AMPK
5′ AMP‐activated protein kinase is a signaling protein that helps control cellular energy homeostasis.
- Ataxia
A neurological sign consisting of lack of voluntary coordination of muscle movements that can include gait abnormality, speech changes, and abnormalities in eye movements.
- Cockayne syndrome (CS)
A rare disease that is marked especially by growth and developmental failure, photosensitivity, and premature aging.
- Embryonic stem cells or ESCs
Stem cells derived from the undifferentiated inner mass cells of a human embryo.
- Epigenetics
Heritable changes to phenotype that do not alter DNA sequence including alterations in DNA methylation patterns, post‐translational modification of histones, and chromatin remodeling.
- Genotoxic
Damaging to the genetic information within a cell, causing mutations that may lead to cancer.
- Geroprotective
Influencing a pathway or aspect of the aging process, thereby prolonging age or intervening in age‐related disease.
- Hormesis
A process in which exposure to a low dose of a chemical agent or environmental factor that is damaging at higher doses induces an adaptive beneficial effect on the cell or organism.
- Hutchinson–Gilford progeria syndrome (HGPS)
Extremely rare autosomal dominant genetic disorder in which symptoms resembling aspects of aging are manifested at a very early age.
- IGF1
Insulin‐like growth factor is a hormone that plays a critical role in growth during development and has anabolic effects in adults.
- Inflammaging
Low‐grade chronic systemic inflammation established during physiological aging.
- Interleukins
Any of a class of glycoproteins produced by leukocytes for regulating immune responses.
- mTOR
Mechanistic target of rapamycin is a signaling protein that helps control cellular division and survival.
- NF‐κB
Protein complex that controls transcription of DNA, cytokine production, and cell survival.
- Sarcopenia
Degenerative loss of skeletal muscle mass and strength with aging.
- Senescence
The condition or process of deterioration with age. Cellular senescence describes the loss of a cell's ability to grow or divide.
- Spatial memory
Cognitive function that allows us for the recall of three‐dimensional objects or places.
- Steatosis
Accumulation of fat in liver cells, associated with disturbance of the metabolism.
- Synovium
A membrane that lines a joint or surrounds a tendon and releases fluid allowing for joint movement.
- Xeroderma pigmentosum
A rare hereditary defect of the enzyme system that repairs DNA after damage from ultraviolet rays, resulting in extreme sensitivity to sunlight and a tendency to develop skin cancer.
Introduction
It has become increasingly clear that epigenetics, including DNA methylation, histone modifications, and chromatin state, play a crucial role in the aging process (López‐Otín et al, 2013). For example, by assessing changes in DNA methylation patterns, a person's age can be predicted within 5 years of accuracy (Field et al, 2018). Histone modifications, including methylation and acetylation states, have been intimately linked to lifespan regulation (Maleszewska et al, 2016). Together, these modifications dictate chromatin state, affecting both gene transcription and genome stability. Epigenetic changes occurring with age provide a tantalizing therapeutic target. In contrast to DNA mutations, epigenetic alterations represent reversible changes, offering the potential for a true “rejuvenating” therapeutic intervention. Of the various epigenetic alterations occurring with age, the influence of histone acetylation, a process balanced by the activity of histone acetyltransferases (HATs) and histone deacetylases (HDACs), on lifespan regulation has been the most characterized, mainly due to the advent of HDAC inhibitors from the cancer biology field (Li & Seto, 2016).
Genes encoding HDACs are divided into four classes based on their homology to their yeast counterparts (Willis‐Martinez et al, 2010). Class I HDACs, which are most similar to the yeast RPD3 gene, include HDACs 1, 2, 3, and 8. Class II HDACs, which are most similar to the yeast HDA1 gene, include HDACs 4, 5, 6, 7, and 9. Class IV includes only one member, HDAC11, which is similar to both RPD3 and HDA1. Classes I, II, and IV are considered to form the “classical family” of HDACs, being dependent on the Zn2+ ion for their activity. HDACs of class III, rather, are dependent on NAD+ and comprise the sirtuin family of proteins. The role of sirtuin‐based pharmacological intervention in aging has been covered previously and will therefore not be part of this review (Houtkooper et al, 2012; Bonkowski & Sinclair, 2016). Likewise, reviewing the relation of all epigenetic mechanisms to healthy aging is beyond the scope of this work. The focus of this review will be limited to the classical HDACs specifically, to recapitulate the many findings related to the beneficial effects on health and aging that result from their inhibition. We aim to provide a broad overview to introduce the reader in the diverse fields involved and to facilitate deeper investigation.
Mechanisms of lifespan extension resulting from HDAC inhibition
Yeasts share many hallmarks of aging with humans, and specifically, a yeast mutant lacking the histone deacetylase gene RPD3 has a prolonged lifespan (Kim et al, 1999; Janssens & Veenhoff, 2016). Interestingly, a yeast strain lacking the HDA1 gene does not show longevity benefits, perhaps implicating class I HDACs above others in the aging process (Kim et al, 1999). In worms, three class I HDACs exist, hda‐1, ‐2, and ‐3, of which RNAi knockdown of hda‐2 and ‐3 increases lifespan (Edwards et al, 2014). This is in line with the observation that valproic acid and β‐hydroxybutyrate (BHB), both class I selective HDAC inhibitors, also increase worm lifespan (Evason et al, 2008; Edwards et al, 2014). Indeed, lifespan extension by BHB in worms depends on the HDAC genes (Edwards et al, 2014). While genetic lifespan studies with HDACs have been mainly performed in yeast and worms, research in flies has arguably contributed the most to our understanding of HDAC inhibitors as longevity drugs (Table 1 and Pasyukova & Vaiserman, 2017). Fly lifespan increased upon treatment with the class I and II HDAC inhibitors phenylbutyrate (Kang et al, 2002), and sodium butyrate (Zhao, 2005), both of which are types of short‐chain fatty acids. Trichostatin A, a hydroxamic acid targeting classes I, II, and IV HDACs, also increased fly lifespan (Tao et al, 2004), as does vorinostat (also known as SAHA), another hydroxamic acid (McDonald et al, 2013).
Table 1.
Properties of selected HDAC inhibitors
| HDACi | HDAC1 IC50 (μM) | HDAC class specificity | Structural class | Lifespan extension |
|---|---|---|---|---|
| Valproic acid | 171 | I | Short‐chain fatty acid | Worms (Evason et al, 2008) |
| Phenylbutyrate | 162 | I, II | Short‐chain fatty acid | Flies (Kang et al, 2002) |
| Butyrate | 175 | I, II | Short‐chain fatty acid | Flies (Zhao, 2005) |
| β‐hydroxybutyrate (BHB) | 5,300 | I | Ketone body | Worms (Edwards et al, 2014) |
| Trichostatin A | 0.017 | I, II, IV | Hydroxamic acid | Flies (Tao et al, 2004) |
| Vorinostat (SAHA) | 0.014 | I, II, IV | Hydroxamic acid | Flies (McDonald et al, 2013) |
| Scriptaid | 0.0064 | I | Hydroxamic acid | NA |
| Apicidin | 0.00030 | I | Cyclic peptide | NA |
| MS‐275 (Entinostat) | 0.5 | I | Benzamide | NA |
| Merck60 | 0.007 | I | Benzamide | NA |
HDAC inhibitors are as described in the text. All HDAC inhibitors listed here target at least class I HDACs, and therefore, the IC50 of HDAC1 is given as reference (though certain HDACs may have slightly higher or lower specificities to other HDACs within class I; Hu, 2003; Huber et al, 2011; Shimazu et al, 2013; Frys et al, 2015). General HDAC class inhibition is listed for added consideration (Shimazu et al, 2013; Stubbs et al, 2015; Pasyukova & Vaiserman, 2017). While all compounds inhibit HDACs, several, including the short‐chain fatty acids and ketone body, have other roles in the cell (Shimazu et al, 2013; Stubbs et al, 2015; Pasyukova & Vaiserman, 2017). The majority listed has been shown to increase lifespan in at least one model organism.
The exact means by which HDAC inhibitors extend lifespan has not been fully resolved; however, a number of possible mechanisms can be envisioned (Fig 1). One possible scenario is that HDAC inhibitors reverse the natural age‐related changes occurring in the histone acetylation landscape (Fig 1A). This is the most simple explanation for their benefits, supported by the observation that many acetylation marks on histones generally decrease with age and in certain age‐related diseases (Peleg et al, 2016). A second possible mechanism of HDAC inhibitors is that they may affect histones and nucleosomes to directly activate transcription of pro‐longevity genes (Fig 1B). This is supported by observations that an endogenous HDAC inhibitor, β‐hydroxybutyrate (BHB), can increase acetylation in the promoter of the pro‐longevity transcription factor FOXO3a resulting in its increased expression, and indeed, BHB's lifespan extending effects depend on HDAC genes (Shimazu et al, 2013; Edwards et al, 2014). A third possible mechanism through which HDAC inhibitors may increase lifespan is through hormesis (Fig 1C). In this scenario, while high doses of HDAC inhibitors may be toxic, low doses would elicit activation of protective genes to regain homeostasis, ultimately improving function (Vaiserman, 2011). This is supported by observations that flies treated with HDAC inhibitors show upregulation of heat shock protein chaperones, a class of genes that are usually upregulated under stress (Zhao, 2005). A fourth possibility is that HDAC inhibitors may regulate lifespan by modifying the acetylation state of non‐histone proteins, activating signaling cascades that promote longevity independent of histone modifications (Lu et al, 2011; Singh et al, 2010; Fig 1D).
Figure 1. Potential models whereby HDAC inhibition (HDACi) extends lifespan.
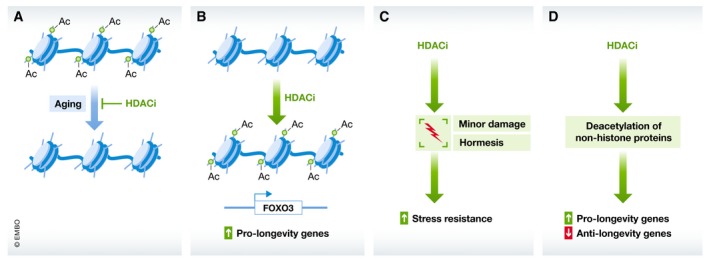
(A) HDACi may directly reverse age‐related deacetylation of chromatin, reverting the epigenome back to a more youthful state. (B) HDACi may result in acetylation of histones near pro‐longevity genes, increasing their transcription. (C) HDACi may act through a hormesis effect, causing low dose damage that activates stress resistance, resulting in a net benefit for the organism. (D) HDACi may target non‐histone proteins, activating pro‐longevity proteins, and/or de‐activating anti‐longevity proteins.
The most likely scenario is that HDAC inhibitors act through combinations of these mechanisms, dependent on the dose, cell type, and drug involved in the experiment. While the mechanism may not yet be fully resolved, their benefit to the aging process at molecular and preclinical levels is clear.
HDAC inhibitors and the hallmarks of aging
The hallmarks of aging are molecular changes associated with aging that are likely to cause the aging process and, when inhibited, should slow the onset of aging (López‐Otín et al, 2013). Nine hallmarks have been established to date (Fig 2), and these are sub‐divided into primary hallmarks (those responsible for age‐related cellular damage), antagonistic hallmarks (those that act as responses to damage), and integrative hallmarks (those that are considered culprits of age‐related phenotypes). While some hallmarks have been greatly ameliorated by treatment with HDAC inhibitors, others are only now emerging with potential links. Below, we list evidence of how HDAC inhibitors can directly and indirectly influence the nine hallmarks of aging (Fig 2).
Figure 2. Influence of HDAC inhibition on the hallmarks of aging.
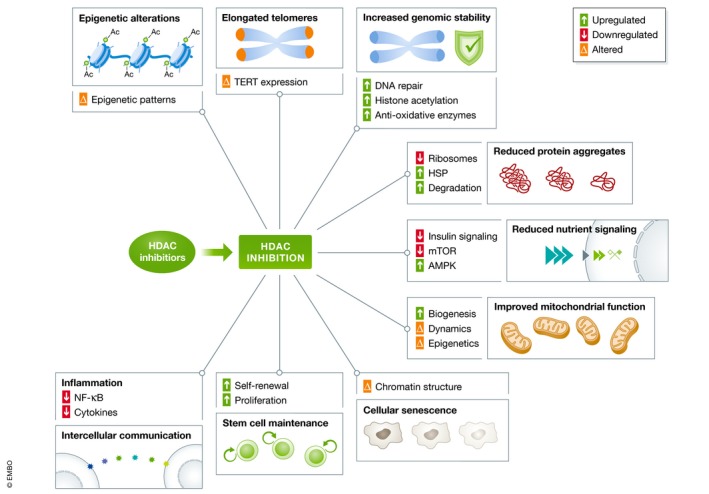
HDAC inhibitors are listed as described in the text, along with evidence for the benefits they impart at the molecular level on the hallmarks of aging; epigenetic alterations, telomere attrition, genomic instability, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication, and up‐ or downregulated processes are generally in line with beneficial changes for the health of the organism, while altered changes are indicative of potential synergies and interactions.
Primary hallmarks
Epigenetic alterations
The clearest link between the hallmarks of aging and HDAC inhibitors exists with the hallmark of “epigenetic alterations” since histone deacetylases are one of the several types of enzymes able to induce alterations in epigenetic patterns (Hirst & Marra, 2013). Many acetylation markers on histones decrease with age, including bulk histone 4 acetylation levels and histone 3 acetylation at lysine residues 18, 27, and 56, which is thought to facilitate the aging process (Feser & Tyler, 2011). Therefore, the effects of an HDAC inhibitor, which prevents HDACs from removing acetyl groups further, have the potential to directly reverse or prevent these age‐related changes. The role of epigenetic alterations in aging can be interconnected with other hallmarks, broadening the reach HDAC inhibitors have to positively benefit aging at the molecular level.
Telomere attrition
Telomeres are protective repetitive sequences located at the ends of chromosomes. These sequences can only be completely replicated by telomerase, and more specifically, the catalytic subunit telomerase reverse transcriptase (TERT), which is transcriptionally repressed in the majority of adult somatic cells (Shay & Wright, 2011). Telomere shortening has been observed during normal aging in both humans and rodents, and using animal models, a causal link has been established between telomere loss and organismal aging (López‐Otín et al, 2013).
Upon discovery that histone deacetylation is essential to the transcriptional regulation of the TERT gene (Cong & Bacchetti, 2000; Hou et al, 2002), early research attempted to elucidate the impact on telomeres. However, these resulting studies conflict in their findings. Even within the same cell types, C33A cancer cells, one study finds increased TERT expression (Takakura et al, 2001) with TSA treatment, while another finds no effect (Hou et al, 2002). In a liver cancer cell line, TSA was even shown to reduce telomerase activity (Nakamura et al, 2001). More recently, inhibition of histone deacetylation during vascular remodeling using the HDAC inhibitor scriptaid led to activation of TERT transcription but decreased TERT protein abundance (Qing et al, 2016). Telomere lengthening was shown in mouse embryonic stem cells upon treatment with sodium butyrate, without a change in Tert gene expression (Dan et al, 2015). In line with this, trichostatin A can lengthen telomeres in cloned pigs during somatic cell nuclear transfer (Kong et al, 2014). Taken together, these studies suggest that HDAC inhibitors may have distinct effects on the TERT gene depending on experimental conditions and the treated cell type. Importantly though, HDAC inhibition is connected to telomere lengthening, the main outcome necessary to reverse the hallmark of aging of telomere attrition.
Genomic instability
The accumulation of genetic damage such as mutations contributes to the aging process (López‐Otín et al, 2013). To maintain genomic integrity and stability, DNA repair mechanisms are present in the cell to restore these lesions. Since HDAC inhibitors originate from the cancer biology field, and because causing damage to DNA and impairing DNA lesion repair may be a desirable trait to reduce proliferation of cancerous cells, HDAC inhibitors have been studied for their ability to accelerate DNA damage and reduce DNA repair (Robert & Rassool, 2012).
The dual role of HDAC inhibitors in cancer versus normal cells is clear. While irreparable DNA damage occurs upon treatment with vorinostat in transformed cells, normal cells do not seem to suffer from this effect (Lee et al, 2010). This may be due to the fact that HDACs are generally overexpressed in cancerous cells compared to normal cells (Roos & Krumm, 2016), which may allow for a titrated dose to impart selectively beneficial effects. When studied in a non‐cancerous setting, sodium butyrate treatment stimulated DNA repair after UV‐irradiation in human fibroblasts (Smerdon et al, 1982). This was also the case in xeroderma pigmentosum fibroblasts, a cell type especially prone to DNA damage (Smerdon et al, 1982). Sodium butyrate also reduced hydrogen peroxide‐induced DNA damage in rat bone marrow cells, further revealing the potential for this HDAC inhibitor's anti‐genotoxic effects (El‐Shorbagy, 2017). Taken together, HDAC inhibitors may provide multiple routes to ensuring genomic stability, from boosting repair capacity in normal cells to promoting detoxification of potential DNA damaging agents.
Loss of proteostasis
The cellular homeostasis of proteins involves (i) their biogenesis by ribosomes, (ii) their folding by chaperones, and (iii) their degradation by proteasomes and autophagy. Inhibition of HDACs may benefit aging at all three steps of proteostasis. For instance, the first step to facilitate biogenesis of ribosomes is transcription of ribosomal DNA. HDAC1 modulates ribosomal DNA transcription, as HDAC1‐overexpressing cells revealed an increase in total ribosomal RNA (Meraner et al, 2008). Moreover, double treatment with the HDAC inhibitor trichostatin A and an mTOR inhibitor synergistically reduced polyribosome formation (Wilson‐Edell et al, 2014). Long‐lived model organisms are often marked by reduced polyribosome formation (Stout et al, 2013; Molenaars et al, 2018), suggesting HDAC inhibitors may act similarly through this pathway.
Once proteins are synthesized by ribosomes, multiple quality control mechanisms ensure their stability and functionality, including protein chaperones such as the heat shock proteins (HSPs). The heat shock response plays a beneficial role in lifespan regulation (Hsu et al, 2003). In relation to this, treatment with HDAC inhibitors in Drosophila resulted in altered chromatin morphology at HSP gene loci and elevated HSP expression, accompanying lifespan extension (Tao et al, 2004; Zhao, 2005). This suggests HDAC inhibitors may contribute to lifespan extension also by protein quality assurance pathways.
Finally, the last step in proteostasis involves the decomposition of proteins, performed either by proteasomal degradation or autophagy, both of which play key roles in aging and can be regulated by HDACs (Scognamiglio et al, 2008; Trüe & Matthias, 2012; Milota et al, 2013; Cellerino & Ori, 2017; Kong et al, 2017). For instance, HDAC inhibitor treatments in several cancer cell lines activate the ubiquitin–proteasome pathway, leading to increased protein degradation (Scognamiglio et al, 2008; Hakami et al, 2016; Kong et al, 2017). HDAC inhibitors also induce autophagy (Hrzenjak et al, 2008; Liu et al, 2010), as does genetic knockdown of HDAC1 (Oh et al, 2008). Taken together, these findings suggest that HDAC inhibition provides benefits at all steps required for proteostasis, and directly act to ameliorate this hallmark of aging.
Antagonistic hallmarks
Deregulated nutrient signaling
Nutrient signaling pathways, such as insulin/IGF1, mTOR, sirtuins, and AMPK, transmit cellular signals about nutrient availability to regulate the processes of growth and autophagy. Strong evidence suggests that increased growth signaling through mTOR or IGF1 accelerates aging, while their inhibition or downregulation, for example, via sirtuin or AMPK activation, extends lifespan (Houtkooper et al, 2010). Similarly, one of the most widely demonstrated lifespan lengthening interventions, calorie restriction, imparts its benefits through nutrient sensing pathways.
Calorie restriction, a treatment that increases the abundance of the endogenous HDAC inhibitor and ketone body β‐hydroxybutyrate (BHB), or administration of BHB alone, similarly increased global histone acetylation in mouse tissues and protected cells from oxidative stress and damage (Shimazu et al, 2013). Butyrate also increased phosphorylation of AMPK in both the liver and muscle of mice (Gao et al, 2009), suggesting its activation of metabolic longevity networks. Furthermore, the HDAC inhibitors apicidin and trichostatin A reduced mTOR activation (Morales et al, 2016), while an independent study showed trichostatin A and vorinostat downregulate insulin signaling (Kawada et al, 2017). This is supported by the observation that vorinostat treatment could reduce phosphorylation of the insulin receptor β (Dudakovic et al, 2013). Taken together, these studies show HDAC inhibitors can module nutrient signaling pathways in a manner beneficial to the aging process.
Mitochondrial dysfunction
With age, a number of mitochondrial regulatory factors diminish, leading to mitochondria with a decreased capacity for energy generation, as well as increased accumulation of damage and reduced mitochondrial turnover. These factors can include mutations in mtDNA, oxidation of mitochondrial proteins, destabilization of respiratory chain complexes, changes in composition of the mitochondrial membrane, alterations in dynamics, and insufficient quality control by mitophagy (López‐Otín et al, 2013). Intervening in mitochondrial biology can increase lifespan (Andreux et al, 2013; Houtkooper et al, 2013; Sun et al, 2016).
There is strong evidence that HDAC inhibitors can prevent or reverse some of this deterioration. Butyrate has been demonstrated in several studies to elevate mitochondrial biogenesis, leading to increases in oxygen consumption (Gao et al, 2009; Galmozzi et al, 2013; Walsh et al, 2015). Additionally, a variety of HDAC inhibitors lead to mitochondrial elongation by creating an imbalance in mitochondrial fission and fusion proteins, demonstrating the influence of HDACs on mitochondrial biology (Lee et al, 2012). Despite the lack of histones in mitochondrial DNA (Rebelo et al, 2011), HDAC inhibitors can also alter mtDNA epigenetics. Under long exposure to valproic acid or MS‐275, mtDNA methylation was significantly decreased, potentially due to a nuclear deacetylase inhibition of TET (ten‐eleven translocation) enzymes (Chen et al, 2012). These findings provide an interesting connection between nuclear and mitochondrial epigenetics and their potential influence on one another, and link HDAC inhibition to desirable effects for mitochondria during aging.
Cellular senescence
Inducing senescence—a quiescent, non‐dividing cell state—in cancerous cells is a desired outcome for cancer therapy, and a number of studies have shown HDAC inhibitors to cause various cancer cells to senesce (Lorenz et al, 2011; Vargas et al, 2014; Venkatesh et al, 2015). In the context of aging however, senescent cell reduction, rather than promotion, is desired, since senescent cells contribute to age‐related diseases (De Keizer, 2017). Importantly, the ability of HDAC inhibitors to cause cell senescence may be cancer specific; sodium butyrate was shown to potentiate senescence in human and rat glioma cell lines but not in normal astrocytes (Vargas et al, 2014). Furthermore, as the senescent cell state is reinforced by the chromatin state at the epigenetic level (Narita et al, 2003; Funayama & Ishikawa, 2007), there is high potential for HDAC inhibitor involvement in the phenotype. However, the potential benefit HDAC inhibitors may provide to ameliorate the hallmark of cellular senescence is a relatively unexplored field. Overexpression of HDAC1 in melanocytes induced epigenetic pathways leading to growth arrest and senescence (Bandyopadhyay et al, 2007), suggesting that a general strategy of HDAC inhibition may limit the tendency of cell senescence to occur. Indeed, endogenously high levels of HDAC1 are present in certain senescent cells, implying a hyperactivity for which inhibition may also be beneficial (Soliman et al, 2008). While HDAC1 has been highly implicated in senescence and disease, more studies are required to link other HDACs and potential benefits of HDAC inhibitors to cellular senescence (reviewed in Willis‐Martinez et al, 2010).
Integrative hallmarks
Stem cell exhaustion
The loss of the regenerative capacity of tissues is another well‐known characteristic of the aging process (López‐Otín et al, 2013). Upon aging, the loss of quiescence and self‐renewal capacity occurs in several types of stem cells (Chakkalakal et al, 2012). A potential mechanism for stem cell fate is the epigenetic modification of chromatin, such as histone acetylation state, and the chromatin status of pluripotency genes is believed to be important for stem cell identity and self‐renewal (Bibikova et al, 2008). This places drugs that modify chromatin state, such as HDAC inhibitors, as potential regulators of stem cells in aging.
Treatment of embryonic stem cells (ESCs) with trichostatin A or sodium butyrate increased their resistance to oxidant stresses, thereby promoting cell viability and decreasing apoptosis in this primal stem cell population (Chen et al, 2011). This may explain an earlier report demonstrating that treatment of ESCs with butyrate activates a self‐renewal program and that HDAC inhibitors generally support human ESC self‐renewal (Ware et al, 2009). Adding low concentrations of the HDAC inhibitors largazole or trichostatin A promoted mesenchymal stem cell proliferation, suppressing differentiation and thereby maintaining the self‐renewal capacity of the stem cells (Wang et al, 2013). Trichostatin A also upregulated several cyclins, promoting proliferative capacity in adult stem cells (Dhoke et al, 2016). Taken together, these studies suggest that HDAC inhibitors have an ability to combat the aging hallmark of stem cell exhaustion.
Altered intercellular communication
One of the main changes in intercellular communication occurring during aging is an increase in the pro‐inflammatory phenotype known as “inflammaging” (López‐Otín et al, 2013). This state is characterized by an over‐activation of the NF‐κB pathway and increases in pro‐inflammatory cytokines including interferon gamma (IFN‐gamma) and interleukins such as IL‐6 and IL‐12. This results in increased inflammation throughout the body, pro‐inflammatory tissue damage, and a reduced efficacy of the immune system (Franceschi et al, 2000, 2018). Inflammaging has been linked to many age‐related diseases including atherosclerosis, heart disease, type II diabetes, arthritis, neurodegeneration, and cancer, suggesting anti‐inflammatory agents could benefit aging and age‐related disease (Xia et al, 2016).
HDAC inhibitors have anti‐inflammatory properties, as reviewed in (Adcock, 2007). Activity of HDAC1 correlates to inflammation markers in patients with rheumatoid arthritis (Horiuchi et al, 2009). In line with this, the HDAC inhibitor MS‐275, which inhibits HDAC1 and 3, had anti‐arthritic activity and resulted in anti‐inflammatory effects in mice and rats (Lin et al, 2007). Phenylbutyrate and trichostatin A also reduced expression of tumor necrosis factor‐alpha (TNF‐alfa) in an animal model of rheumatoid arthritis (Chung et al, 2003). In line with this, a single treatment with vorinostat in mice was able to reduce circulating TNF‐alfa, IL‐1‐beta, IL‐6, and IFN‐gamma levels induced by the inflammation inducer lipopolysaccharide (Leoni et al, 2002). Furthermore, butyrate was also able to reduce NF‐κB activity in macrophages of Crohn's disease patients (Lührs et al, 2002). Taken together, these studies show the ability of HDAC inhibitors to suppress inflammatory signals, which are upregulated during aging and constitute a majority of age‐related altered intercellular communication.
Benefits of HDAC inhibitors in preclinical age‐related disease models
HDAC inhibitors have shown benefits in diverse preclinical models for age‐related diseases, likely owing to their broad effects at the molecular level as described above. Here, we review evidence that HDAC inhibitors can provide either prophylactic treatment for disease reduction, or treatment after onset, for a variety of age‐related diseases. With cancer considerations aside, most evidence to date for the application of HDAC inhibitors exists for neurodegeneration, cardio‐metabolic diseases, liver dysfunction, sarcopenia, inflammation and arthritis, and diseases of premature aging in preclinical mouse studies (Fig 3). While evidence for HDAC inhibitors as treatments is only now emerging for some of these age‐related diseases (such as liver dysfunction or sarcopenia), others have large amounts of evidence and clinical trials are underway (such as for neurodegeneration).
Figure 3. Benefits of HDAC inhibitors in preclinical models.
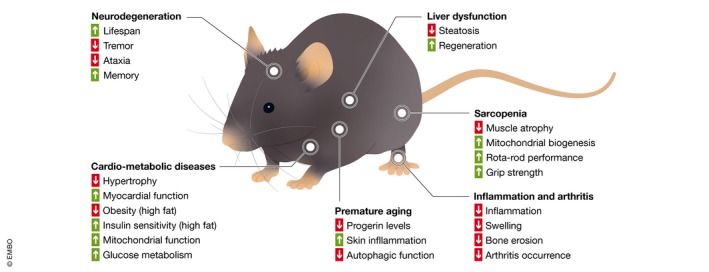
HDAC inhibition benefits a large variety of preclinical models, including those related to neurodegeneration, cardio‐metabolic deficiencies, liver dysfunctions, sarcopenia, inflammation‐related disease, and diseases of premature aging. Specifics of improvements in each condition are described in the text.
Neurodegeneration
Perhaps the most convincing case for the non‐cancer use of HDAC inhibitors can be made for treating neurodegenerative diseases, including Alzheimer's and Parkinson's. The effect on HDAC inhibitors on brain function is well known (Fischer et al, 2010) and is exemplified by the observation that treatment with the HDAC class I selective inhibitor Merck60 can alter behavior in anxiety mood tests in mice (Lewis et al, 2014). In relation to the human aging brain, histone acetylation levels are known to decline (Tang et al, 2011), suggesting HDAC inhibition may provide benefit to normal aging too. Histone acetylation levels also play a key role in neurodegenerative diseases (Gangisetty, 2018). Treatment with sodium butyrate extended mean lifespan of Atro‐118Q mice (possessing neuronal expression of a mutant human Atrophin‐1 protein containing an expanded stretch of 118 glutamines) and improved their neurodegenerative phenotypes (tremor, ataxia, and other motor defects; Ying et al, 2006). Furthermore, treatment of Alzheimer's disease transgenic mice (dual transgenic expressing mutant forms of both App and Psen1 genes) with phenylbutyrate resulted in a reduction of amyloid plaques in the cortex and hippocampus (Wiley et al, 2011). Phenylbutyrate also restored brain histone acetylation levels and reduced Tau pathology in another Alzheimer's disease transgenic mouse model (expressing mutant form of App gene; Ricobaraza et al, 2009). In line with this, treatment with the pan‐HDAC inhibitor vorinostat lead to restoration of spatial memory of another Alzheimer's disease mouse model (dual transgenic expressing mutant forms of both App and Psen1; Benito et al, 2015). Together, these findings provided justification for a human clinical trial using phenylbutyrate to treat Parkinson's disease (NCT02046434).
Cardio‐metabolic diseases
Cardio‐metabolic diseases, affecting the heart and metabolic state, such as heart disease, diabetes, and obesity, have age‐related dependencies for their occurrences and outcomes. HDAC inhibition has been shown to provide benefit to the cardio‐metabolic system in mice. Treatment with trichostatin A can reduce pressure overload‐induced cardiac hypertrophy, and results in histone acetylation of genes related to cardiac contraction, collagen deposition, inflammation, and the extracellular matrix (Ooi et al, 2015). Additionally, treatment with sodium butyrate in streptozotocin‐induced diabetic mice improved myocardial function as measured by echocardiography and reduced cardiac hypertrophy as made evident by a reduced heart/tibia ratio (Chen et al, 2015). Meanwhile, treatment with sodium butyrate of mice fed a high‐fat diet prevented development of obesity and insulin resistance, most likely due to improved mitochondrial function as made evident by improved adaptive thermogenesis and fatty acid oxidation (Gao et al, 2009). Mice on a high‐fat diet treated with sodium butyrate showed improved insulin sensitivity (Henagan et al, 2015), as did mice on a high‐fat diet treated with vorinostat (Sharma & Taliyan, 2016). Furthermore, treating naturally aged mice with sodium butyrate improved glucose metabolism as discerned from a glucose tolerance test, suggesting an age‐related benefit as well, beyond diet‐induced dysfunctions (Walsh et al, 2015). These diverse studies in mice suggest that the cardio‐metabolic system, which diminishes in function with age, benefits from HDAC inhibition.
Liver dysfunction
HDAC1 plays a crucial role in liver aging and disease with strong links emerging in regard to its role in liver dysfunction (Willis‐Martinez et al, 2010). HDAC1 is thought to inhibit liver regeneration in aged mice and liver‐specific overexpression of HDAC1 resulted in steatosis, a marker of liver aging (Wang et al, 2008). Acetylation of histone H3K9, an HDAC1 target, decreased in livers of old mice, again suggesting hyperactivity of HDAC1 with age (Kawakami et al, 2009). Together, these preliminary findings suggest that aging in the liver results in an increased HDAC activity and that HDAC inhibitors act to reverse the epigenome back toward a more youthful state, preventing steatosis and improving regenerative potential.
Sarcopenia
HDACs play key roles in regulating metabolism in skeletal muscle (Walsh & Van Remmen, 2016). For example, reduction in the level of HDAC1 or inhibition of its activity prevents muscle atrophy after nutrient deprivation (Beharry et al, 2014). Treating naturally aged mice with sodium butyrate reduced age‐related muscle atrophy and increased mitochondrial biogenesis (Walsh et al, 2015). Furthermore, in SOD1‐G93A mice, a model for oxidative damage in aging, treatment with trichostatin A ameliorated muscle atrophy and improved mouse performance in rotarod and grip strength assays (Yoo & Ko, 2011). These preliminary studies suggest that HDAC inhibition may prevent or reverse the muscle atrophy that accompanies aging.
Inflammation and arthritis
As described above in the hallmark of “altered cellular communication”, HDAC inhibitors can act as anti‐inflammatory agents, opening up their application to inflammation‐based diseases (Adcock, 2007). HDAC1 is highly expressed in the synovium of arthritis patients, which correlates to inflammation markers (Horiuchi et al, 2009). This suggests HDAC inhibition may provide benefit, and indeed, in both mouse and rat collagen‐induced arthritis models, the HDAC inhibitors vorinostat and MS‐275 had prophylactic activity against swelling and reduced bone erosion (Lin et al, 2007). MS‐275 was also able to prevent the onset of arthritis, and when treatment occurred after onset, MS‐275 prevented disease progression and joint destruction (Lin et al, 2007). Together, these findings point to HDAC inhibition as not only a means to prevent inflammation‐based age‐related disease, but also as a treatment for them.
Premature aging
Hutchinson–Gilford progeria syndrome (HGPS) is a rare human genetic disease that leads to severe premature aging, caused by mutations in the LMNA gene and characterized by an accumulation of a mutated lamin A precursor (progerin), nuclear dysmorphism, and chromatin disorganization (Columbaro et al, 2005; Arancio et al, 2014). In HGPS cells, dramatic epigenetic alterations have been reported (reviewed by Arancio et al, 2014). In both model cell lines and cells from patients with HGPS, valproic acid and TSA lowered progerin levels, which allowed for rescue of heterochromatin organization and reorganization of transcripts (Columbaro et al, 2005; Stephens et al, 2017).
Another hereditary form of premature aging, Cockayne syndrome (CS), is caused by mutations in five different genes that encode proteins involved in nucleotide excision DNA repair, causing hypersensitivity to UV radiation and loss of subcutaneous fat (Majora et al, 2018). Across CSB‐deficient human fibroblasts, Caenorhabditis elegans, and mice, treatment with SAHA enhanced alpha‐tubulin acetylation and improved autophagic function, and even rescued the skin phenotype observed in mice, suggesting it may provide a therapeutic option for CS (Majora et al, 2018). Together, these findings not only suggest that HDAC inhibitors can provide treatment for diseases of premature aging, but also provide further evidence of HDAC inhibitor efficacy as a geroprotective compound.
Conclusion
Epigenetics is a major regulator of cell fate and function and is clearly implicated in disease biology (Moosavi & Ardekani, 2016). In this review, we specifically focused on one aspect of epigenetic regulation, HDACs, and more specifically, on the pharmacological benefit of their inhibition. In this regard, HDAC inhibitors can directly target and reverse the age‐related changes of a main hallmark of aging—that of epigenetic alterations. Furthermore, as reviewed here, their influence reaches more broadly to all other hallmarks of aging, likely contributing to their ability to increase lifespan in model organisms. Interventions that reverse or slow the aging process bring great promise to promote healthy aging. HDAC inhibitors have demonstrated themselves to fill this role, providing potential treatments for age‐related diseases ranging from neurodegeneration to heart disease, diabetes to sarcopenia. While this review has specifically focused on pharmacological inhibition of HDACs, genetic knockout of HDACs has also revealed important roles for HDACs in disease biology. For example, germline knockout of either HDAC1 or HDAC2 alone results in lethality, and conditional knockout of both has in general been found to be detrimental to a variety of tissues (e.g., neuronal, cardiovascular, liver, etc; Kelly & Cowley, 2013). Therefore, despite the promising outlook of HDAC inhibitors for healthy aging, much work remains to be done to better understand their safety and how to minimize adverse side effects. Owing to their origins in the cancer biology field, many cell‐type and dose‐dependent negative effects of HDAC inhibitors on cell viability have been documented. Careful optimization of dose and drug pharmacokinetics should be made prior to pursuing any strategy in which HDAC inhibitors would be used as a prophylactic drug for healthy aging. More specifically, less‐toxic versions of current drugs may be required. Understanding of the mechanism by which HDAC inhibitors extend lifespan is noticeably limited, and many mechanistic options remain (Fig 1). Deeper study of the specific modes of action of these compounds is necessary prior to their implementation as geroprotective compounds. Finally, while all compounds discussed within this review have been demonstrated to be HDAC inhibitors, it should be noted that the majority that have been shown to produce lifespan extending effects in model organisms may also have effects other than HDAC activity in the cell (such as the short‐chain fatty acids or ketone bodies, Table 1). Therefore, further evaluation of lifespan extending effects of more potent HDAC inhibitors is also warranted. In conclusion, along with words of caution, this review provides strong molecular and preclinical evidence supporting the further development of HDAC inhibitors for the race to develop healthy aging drugs.
Conflict of interest
The authors declare that they have no conflict of interest.
Pending issues.
-
(i)
Identification of mechanism or mechanisms by which HDAC inhibitors extend lifespan.
-
(ii)
Understanding safety of HDAC inhibitors in vivo and how to minimize potential adverse effects.
-
(iii)
Dose optimization and pharmacokinetic studies of HDAC as geroprotective compounds.
-
(iv)
Development of specifically targeted or less toxic HDAC inhibitor compounds.
Acknowledgements
Work in the Houtkooper group is financially supported by an ERC Starting grant (no. 638290, https://erc.europa.eu/) and a VIDI grant from ZonMw (no. 91715305, https://www.zonmw.nl). GEJ is supported by a Federation of European Biochemical Society (FEBS, https://www.febs.org) long‐term fellowship.
EMBO Mol Med (2019) 11: e9854
See the Glossary for abbreviations used in this article.
Contributor Information
Riekelt H Houtkooper, Email: r.h.houtkooper@amsterdamumc.nl.
Georges E Janssens, Email: g.e.janssens@amsterdamumc.nl.
References
- Adcock IM (2007) HDAC inhibitors as anti‐inflammatory agents. Br J Pharmacol 150: 829–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreux PA, Houtkooper RH, Auwerx J (2013) Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov 12: 465–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancio W, Pizzolanti G, Genovese SI, Pitrone M, Giordano C (2014) Epigenetic involvement in Hutchinson‐Gilford progeria syndrome: a mini‐review. Gerontology 60: 197–203 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay D, Curry JL, Lin Q, Richards HW, Chen D, Hornsby PJ, Timchenko NA, Medrano EE (2007) Dynamic assembly of chromatin complexes during cellular senescence: implications for the growth arrest of human melanocytic nevi. Aging Cell 6: 577–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beharry AW, Sandesara PB, Roberts BM, Ferreira LF, Senf SM, Judge AR (2014) HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J Cell Sci 127: 1441–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E, Urbanke H, Ramachandran B, Barth J, Halder R, Awasthi A, Jain G, Capece V, Burkhardt S, Navarro‐Sala M et al (2015) HDAC inhibitor‐dependent transcriptome and memory reinstatement in cognitive decline models. J Clin Invest 125: 3572–3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Laurent LC, Ren B, Loring JF, Fan JB (2008) Unraveling epigenetic regulation in embryonic stem cells. Cell Stem Cell 2: 123–134 [DOI] [PubMed] [Google Scholar]
- Bonkowski MS, Sinclair DA (2016) Slowing ageing by design: the rise of NAD+ and sirtuin‐activating compounds. Nat Rev Mol Cell Biol 17: 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellerino A, Ori A (2017) What have we learned on aging from omics studies? Semin Cell Dev Biol 70: 177–189 [DOI] [PubMed] [Google Scholar]
- Chakkalakal JV, Jones KM, Basson MA, Brack AS (2012) The aged niche disrupts muscle stem cell quiescence. Nature 490: 355–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HP, Denicola M, Qin X, Zhao Y, Zhang L, Long XL, Zhuang S, Liu PY, Zhao TC (2011) HDAC inhibition promotes cardiogenesis and the survival of embryonic stem cells through proteasome‐dependent pathway. J Cell Biochem 112: 3246–3255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Dzitoyeva S, Manev H (2012) Effect of valproic acid on mitochondrial epigenetics. Eur J Pharmacol 690: 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Du J, Zhao YT, Zhang L, Lv G, Zhuang S, Qin G, Zhao TC (2015) Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc Diabetol 14: 99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YL, Lee MY, Wang AJ, Yao LF (2003) A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol Ther 8: 707–717 [DOI] [PubMed] [Google Scholar]
- Columbaro M, Capanni C, Mattioli E, Novelli G, Parnaik VK, Squarzoni S, Maraldi NM, Lattanzi G (2005) Rescue of heterochromatin organization in Hutchinson‐Gilford progeria by drug treatment. Cell Mol Life Sci 62: 2669–2678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong YS, Bacchetti S (2000) Histone deacetylation is involved in the transcriptional repression of hTERT in normal human cells. J Biol Chem 275: 35665–35668 [DOI] [PubMed] [Google Scholar]
- Dan J, Yang J, Liu Y, Xiao A, Liu L (2015) Roles for histone acetylation in regulation of telomere elongation and two‐cell state in mouse ES cells. J Cell Physiol 230: 2337–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keizer PLJ (2017) Series: current trends in aging and age‐related diseases the fountain of youth by targeting senescent cells?. Trends Mol Med 23: 6–17 [DOI] [PubMed] [Google Scholar]
- Dhoke NR, Kalabathula E, Kaushik K, Geesala R, Sravani B, Das A (2016) Histone deacetylases differentially regulate the proliferative phenotype of mouse bone marrow stromal and hematopoietic stem/progenitor cells. Stem Cell Res 17: 170–180 [DOI] [PubMed] [Google Scholar]
- Dudakovic A, Evans JM, Li Y, Middha S, McGee‐Lawrence ME, Van Wijnen AJ, Westendorf JJ (2013) Histone deacetylase inhibition promotes osteoblast maturation by altering the histone H4 epigenome and reduces akt phosphorylation. J Biol Chem 288: 28783–28791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards C, Canfield J, Copes N, Rehan M, Lipps D, Bradshaw PC (2014) D‐beta‐hydroxybutyrate extends lifespan in C. elegans . Aging 6: 621–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Shorbagy HM (2017) Potential anti‐genotoxic effect of sodium butyrate to modulate induction of DNA damage by tamoxifen citrate in rat bone marrow cells. Cytotechnology 69: 89–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evason K, Collins JJ, Huang C, Hughes S, Kornfeld K (2008) Valproic acid extends Caenorhabditis elegans lifespan. Aging Cell 7: 305–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feser J, Tyler J (2011) Chromatin structure as a mediator of aging. FEBS Lett 585: 2041–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field AE, Robertson NA, Wang T, Havas A, Ideker T, Adams PD (2018) DNA methylation clocks in aging: categories, causes, and consequences. Mol Cell 71: 882–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Mungenast A, Tsai LH (2010) Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci 31: 605–617 [DOI] [PubMed] [Google Scholar]
- Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G (2000) Inflamm‐aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 908: 244–254 [DOI] [PubMed] [Google Scholar]
- Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A (2018) Inflammaging: a new immune–metabolic viewpoint for age‐related diseases. Nat Rev Endocrinol 14: 576–590 [DOI] [PubMed] [Google Scholar]
- Frys S, Simons Z, Hu Q, Barth MJ, Gu JJ, Mavis C, Skitzki J, Song L, Czuczman MS, Hernandez‐Ilizaliturri FJ (2015) Entinostat, a novel histone deacetylase inhibitor is active in B‐cell lymphoma and enhances the anti‐tumour activity of rituximab and chemotherapy agents. Br J Haematol 169: 506–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama R, Ishikawa F (2007) Cellular senescence and chromatin structure. Chromosoma 116: 431–440 [DOI] [PubMed] [Google Scholar]
- Galmozzi A, Mitro N, Ferrari A, Gers E, Gilardi F, Godio C, Cermenati G, Gualerzi A, Donetti E, Rotili D et al (2013) Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 62: 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangisetty O (2018) Impact of epigenetics in aging and age related neurodegenerative diseases. Front Biosci 23: 1445–1464 [DOI] [PubMed] [Google Scholar]
- Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT, Ye J (2009) Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58: 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakami NY, Dusting GJ, Peshavariya HM (2016) Trichostatin A, a histone deacetylase inhibitor suppresses NADPH Oxidase 4‐Derived redox signalling and angiogenesis. J Cell Mol Med 20: 1932–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henagan TM, Stefanska B, Fang Z, Navard AM, Ye J, Lenard NR, Devarshi PP (2015) Sodium butyrate epigenetically modulates high‐fat diet‐induced skeletal muscle mitochondrial adaptation, obesity and insulin resistance through nucleosome positioning. Br J Pharmacol 172: 2782–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst M, Marra MA (2013) Next generation sequencing based approaches to epigenomics In Epigenetics and pathology: exploring connections between genetic mechanisms and disease expression, Ayyanathan K. (ed), pp 317–337. Palm Bay, FL: Apple Academic Press; [Google Scholar]
- Horiuchi M, Morinobu A, Chin T, Sakai Y, Kurosaka M, Kumagai S (2009) Expression and function of histone deacetylases in rheumatoid arthritis synovial fibroblasts. J Rheumatol 36: 1580–1589 [DOI] [PubMed] [Google Scholar]
- Hou M, Wang XB, Popov N, Zhang A, Zhao X, Zhou R, Zetterberg A, Björkholm M, Henriksson M, Gruber A et al (2002) The histone deacetylase inhibitor trichostatin A derepresses the telomerase reverse transcriptase (hTERT) gene in human cells. Exp Cell Res 274: 25–34 [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Williams RW, Auwerx J (2010) Metabolic networks of longevity. Cell 142: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13: 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497: 451–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrzenjak A, Kremser ML, Strohmeier B, Moinfar F, Zatloukal K, Denk H (2008) SAHA induces caspase‐independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J Pathol 216: 495–504 [DOI] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C (2003) Regulation of aging and age‐related disease by DAF‐16 and heat‐shock factor. Science 300: 1142–1145 [DOI] [PubMed] [Google Scholar]
- Hu E (2003) Identification of novel isoform‐selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther 307: 720–728 [DOI] [PubMed] [Google Scholar]
- Huber K, Doyon G, Plaks J, Fyne E, Mellors JW, Sluis‐Cremer N (2011) Inhibitors of histone deacetylases: correlation between isoform specificity and reactivation of HIV type 1 (HIV‐1) from latently infected cells. J Biol Chem 286: 22211–22218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens G, Veenhoff L (2016) Evidence for the hallmarks of human aging in replicatively aging yeast. Microb Cell 3: 263–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H‐L, Benzer S, Min K‐T (2002) Life extension in Drosophila by feeding a drug. Proc Natl Acad Sci USA 99: 838–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada Y, Asahara SI, Sugiura Y, Sato A, Furubayashi A, Kawamura M, Bartolome A, Terashi‐Suzuki E, Takai T, Kanno A et al (2017) Histone deacetylase regulates insulin signaling via two pathways in pancreatic β cells. PLoS One 12: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Nakamura A, Ishigami A, Goto S, Takahashi R (2009) Age‐related difference of site‐specific histone modifications in rat liver. Biogerontology 10: 415–421 [DOI] [PubMed] [Google Scholar]
- Kelly RDW, Cowley SM (2013) The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co‐stars with multiple leading parts. Biochem Soc Trans 41: 741–749 [DOI] [PubMed] [Google Scholar]
- Kim S, Benguria A, Lai CY, Jazwinski SM (1999) Modulation of life‐span by histone deacetylase genes in Saccharomyces cerevisiae . Mol Biol Cell 10: 3125–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Q, Ji G, Xie B, Li J, Mao J, Wang J, Liu S, Liu L, Liu Z (2014) Telomere elongation facilitated by trichostatin A in cloned embryos and pigs by somatic cell nuclear transfer. Stem Cell Rev 10: 399–407 [DOI] [PubMed] [Google Scholar]
- Kong LR, Tan TZ, Ong WR, Bi C, Huynh H, Lee SC, Chng WJ, Eichhorn PJA, Goh BC (2017) Belinostat exerts antitumor cytotoxicity through the ubiquitin‐proteasome pathway in lung squamous cell carcinoma. Mol Oncol 11: 965–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J‐H, Choy ML, Ngo L, Foster SS, Marks PA (2010) Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci USA 107: 14639–14644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Yoon YG, Yoo SH, Jeong NY, Jeong SH, Lee SY, Jung DI, Jeong SY, Yoo YH (2012) Histone deacetylase inhibitors induce mitochondrial elongation. J Cell Physiol 227: 2856–2869 [DOI] [PubMed] [Google Scholar]
- Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, Dona G, Fossati G, Sozzani S, Azam T et al (2002) The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci USA 99: 2995–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MC, Fass DM, Wagner FF, Hennig KM, Gale J, Hdac AS, Schroeder FA, Lewis MC, Fass DM, Wagner FF et al (2014) A selective HDAC 1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood‐related tests. PLoS One 8: e71323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Seto E (2016) HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med 6: a026831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HS, Hu CY, Chan HY, Liew YY, Huang HP, Lepescheux L, Bastianelli E, Baron R, Rawadi G, Clément‐Lacroix P (2007) Anti‐rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen‐induced arthritis in rodents. Br J Pharmacol 150: 862–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YL, Yang PM, Shun CT, Wu MS, Weng JR, Chen CC (2010) Autophagy potentiates the anti‐cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 6: 1057–1065 [DOI] [PubMed] [Google Scholar]
- López‐Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153: 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz V, Hessenkemper W, Rödiger J, Kyrylenko S, Kraft F, Baniahmad A (2011) Sodium butyrate induces cellular senescence in neuroblastoma and prostate cancer cells. Horm Mol Biol Clin Investig 7: 265–272 [DOI] [PubMed] [Google Scholar]
- Lu JY, Lin YY, Zhu H, Chuang LM, Boeke JD (2011) Protein acetylation and aging. Aging 3: 911–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lührs H, Gerke T, Müller JG, Melcher R, Schauber J, Boxberger F, Scheppach W, Menzel T (2002) Butyrate inhibits NF‐κB activation in lamina propria macrophages of patients with ulcerative colitis. Scand J Gastroenterol 37: 458–466 [DOI] [PubMed] [Google Scholar]
- Majora M, Sondenheimer K, Knechten M, Uthe I, Esser C, Schiavi A, Ventura N, Krutmann J (2018) HDAC inhibition improves autophagic and lysosomal function to prevent loss of subcutaneous fat in a mouse model of Cockayne syndrome. Sci Transl Med 10: eaam7510 [DOI] [PubMed] [Google Scholar]
- Maleszewska M, Mawer JSP, Tessarz P (2016) Histone modifications in ageing and lifespan regulation. Curr Mol Biol Rep 2: 26–35 [Google Scholar]
- McDonald P, Maizi BM, Arking R (2013) Chemical regulation of mid‐ and late‐life longevities in Drosophila . Exp Gerontol 48: 240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraner J, Lechner M, Schwarze F, Gander R, Jesacher F, Loidl P (2008) Cell cycle dependent role of HDAC1 for proliferation control through modulating ribosomal DNA transcription. Cell Biol Int 32: 1073–1080 [DOI] [PubMed] [Google Scholar]
- Milota F, Prokhorenko VI, Mancal T, Von Berlepsch H, Bixner O, Kauffmann HF, Hauer J (2013) Vibronic and vibrational coherences in two‐dimensional electronic spectra of supramolecular J‐aggregates. J Phys Chem A 117: 6007–6014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaars M, Janssens GE, Santermans T, Lezzerini M, Jelier R, MacInnes AW, Houtkooper RH (2018) Mitochondrial ubiquinone–mediated longevity is marked by reduced cytoplasmic mRNA translation. Life Sci Alliance 1: e201800082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosavi A, Ardekani AM (2016) Role of epigenetics in biology and human diseases. Iran Biomed J 20: 246–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales CR, Li DL, Pedrozo Z, May HI, Jiang N, Kyrychenko V, Cho GW, Kim SY, Wang ZV, Rotter D et al (2016) Inhibition of class I histone deacetylases blunts cardiac hypertrophy through TSC2‐dependent mTOR repression. Sci Signal 9: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Saito H, Ebinuma H, Wakabayashi K, Saito Y, Takagi T, Nakamoto N, Ishii H (2001) Reduction of telomerase activity in human liver cancer cells by a histone deacetylase inhibitor. J Cell Physiol 187: 392–401 [DOI] [PubMed] [Google Scholar]
- Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ , Lowe SW (2003) Rb‐mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703–716 [DOI] [PubMed] [Google Scholar]
- Oh M, Choi IK, Kwon HJ (2008) Inhibition of histone deacetylase1 induces autophagy. Biochem Biophys Res Commun 369: 1179–1183 [DOI] [PubMed] [Google Scholar]
- Ooi JYY, Tuano NK, Rafehi H, Gao XM, Ziemann M, Du XJ, El‐Osta A (2015) HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics 10: 418–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasyukova EG, Vaiserman AM (2017) HDAC inhibitors: a new promising drug class in anti‐aging research. Mech Ageing Dev 166: 6–15 [DOI] [PubMed] [Google Scholar]
- Peleg S, Feller C, Ladurner AG, Imhof A (2016) The metabolic impact on histone acetylation and transcription in ageing. Trends Biochem Sci 41: 700–711 [DOI] [PubMed] [Google Scholar]
- Qing H, Aono J, Findeisen HM, Jones KL, Heywood EB, Bruemmer D (2016) Differential regulation of telomerase reverse transcriptase promoter activation and protein degradation by histone deacetylase inhibition. J Cell Physiol 231: 1276–1282 [DOI] [PubMed] [Google Scholar]
- Rebelo AP, Dillon LM, Moraes CT (2011) Mitochondrial DNA transcription regulation and nucleoid organization. J Inherit Metab Dis 34: 941–951 [DOI] [PubMed] [Google Scholar]
- Ricobaraza A, Cuadrado‐Tejedor M, Pérez‐Mediavilla A, Frechilla D, Del Río J, García‐Osta A (2009) Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology 34: 1721–1732 [DOI] [PubMed] [Google Scholar]
- Robert C, Rassool FV (2012) HDAC inhibitors: roles of DNA damage and repair. Adv Cancer Res 116: 87–129 [DOI] [PubMed] [Google Scholar]
- Roos WP, Krumm A (2016) Survey and summary: the multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res 44: 10017–10030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scognamiglio A, Nebbioso A, Manzo F, Valente S, Mai A, Altucci L (2008) HDAC‐class II specific inhibition involves HDAC proteasome‐dependent degradation mediated by RANBP2. Biochim Biophys Acta 1783: 2030–2038 [DOI] [PubMed] [Google Scholar]
- Sharma S, Taliyan R (2016) Epigenetic modifications by inhibiting histone deacetylases reverse memory impairment in insulin resistance induced cognitive deficit in mice. Neuropharmacology 105: 285–297 [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE (2011) Role of telomeres and telomerase in cancer. Semin Cancer Biol 349–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD et al (2013) Suppression of oxidative stress by β‐hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339: 211–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW (2010) Nonhistone protein acetylation as cancer therapy targets. Expert Rev Anticancer Ther 10: 935–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smerdon MJ, Lan SY, Calza RE, Reeves R (1982) Sodium butyrate stimulates DNA repair in UV‐irradiated normal and xeroderma pigmentosum human fibroblasts. J Biol Chem 257: 13441–13447 [PubMed] [Google Scholar]
- Soliman MA, Berardi P, Pastyryeva S, Bonnefin P, Feng X, Colina A, Young D, Riabowol K (2008) ING1a expression increases during replicative senescence and induces a senescent phenotype. Aging Cell 7: 783–794 [DOI] [PubMed] [Google Scholar]
- Stephens AD, Liu PZ, Banigan EJ, Almassalha LM, Backman V, Adam SA, Goldman RD, Marko JF (2017) Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol Biol Cell 29: 220–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout GJ, Stigter ECA, Essers PB, Mulder KW, Kolkman A, Snijders DS, Van Den Broek NJF, Betist MC, Korswagen HC, MacInnes AW et al (2013) Insulin/IGF‐1‐mediated longevity is marked by reduced protein metabolism. Mol Syst Biol 9: 679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs MC, Kim W, Bariteau M, Davis T, Vempati S, Minehart J, Witkin M, Qi J, Krivtsov AV, Bradner JE et al (2015) Selective inhibition of HDAC1 and HDAC2 as a potential therapeutic option for B‐ALL. Clin Cancer Res 21: 2348–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Youle RJ, Finkel T (2016) The mitochondrial basis of aging. Mol Cell 61: 654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura M, Kyo S, Sowa Y, Wang Z, Yatabe N, Maida Y, Tanaka M, Inoue M (2001) Telomerase activation by histone deacetylase inhibitor in normal cells. Nucleic Acids Res 29: 3006–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Dean B, Thomas EA (2011) Disease‐ and age‐related changes in histone acetylation at gene promoters in psychiatric disorders. Transl Psychiatry 1: e64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao D, Lu J, Sun H, Zhao Y‐M, Yuan Z‐G, Li X‐X, Huang B‐Q (2004) Trichostatin A extends the lifespan of Drosophila melanogaster by elevating hsp22 expression. Acta Biochim Biophys Sin (Shanghai) 36: 618–622 [DOI] [PubMed] [Google Scholar]
- Trüe O, Matthias P (2012) Interplay between histone deacetylases and autophagy – from cancer therapy to neurodegeneration. Immunol Cell Biol 90: 78–84 [DOI] [PubMed] [Google Scholar]
- Vaiserman AM (2011) Hormesis and epigenetics: is there a link?. Ageing Res Rev 10: 413–421 [DOI] [PubMed] [Google Scholar]
- Vargas JE, Filippi‐Chiela EC, Suhre T, Kipper FC, Bonatto D, Lenz G (2014) Inhibition of HDAC increases the senescence induced by natural polyphenols in glioma cells. Biochem Cell Biol 92: 297–304 [DOI] [PubMed] [Google Scholar]
- Venkatesh R, Ramaiah MJ, Gaikwad HK, Janardhan S, Bantu R, Nagarapu L, Sastry GN, Ganesh AR, Bhadra M (2015) Luotonin‐A based quinazolinones cause apoptosis and senescence via HDAC inhibition and activation of tumor suppressor proteins in HeLa cells. Eur J Med Chem 94: 87–101 [DOI] [PubMed] [Google Scholar]
- Walsh ME, Bhattacharya A, Sataranatarajan K, Qaisar R, Sloane L, Rahman MM, Kinter M, Van Remmen H (2015) The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell 14: 957–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh ME, Van Remmen H (2016) Emerging roles for histone deacetylases in age‐related muscle atrophy. Nutr Healthy Aging 4: 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Salisbury E, Shi X, Timchenko L, Medrano EE, Timchenko NA (2008) HDAC1 cooperates with C/EBPα in the inhibition of liver proliferation in old mice. J Biol Chem 283: 26169–26178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen T, Yan H, Qi H, Deng C, Ye T, Zhou S, Li FR (2013) Role of histone deacetylase inhibitors in the aging of human umbilical cord mesenchymal stem cells. J Cell Biochem 114: 2231–2239 [DOI] [PubMed] [Google Scholar]
- Ware CB, Wang L, Mecham BH, Shen L, Nelson AM, Bar M, Lamba DA, Dauphin DS, Buckingham B, Askari B et al (2009) Histone deacetylase inhibition elicits an evolutionarily conserved self‐renewal program in embryonic stem cells. Cell Stem Cell 4: 359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley JC, Pettan‐Brewer C, Ladiges WC (2011) Phenylbutyric acid reduces amyloid plaques and rescues cognitive behavior in AD transgenic mice. Aging Cell 10: 418–428 [DOI] [PubMed] [Google Scholar]
- Willis‐Martinez D, Richards HW, Timchenko NA, Medrano EE (2010) Role of HDAC1 in senescence, aging, and cancer. Exp Gerontol 45: 279–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson‐Edell KA, Yevtushenko MA, Rothschild DE, Rogers AN, Benz CC (2014) mTORC1/C2 and pan‐HDAC inhibitors synergistically impair breast cancer growth by convergent AKT and polysome inhibiting mechanisms. Breast Cancer Res Treat 144: 287–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S, Zhang X, Zheng S, Khanabdali R, Kalionis B, Wu J, Wan W, Tai X (2016) An update on inflamm‐aging: mechanisms, prevention, and treatment. J Immunol Res 2016: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying M, Xu R, Wu X, Zhu H, Zhuang Y, Han M, Xu T (2006) Sodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLA. J Biol Chem 281: 12580–12586 [DOI] [PubMed] [Google Scholar]
- Yoo YE, Ko CP (2011) Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp Neurol 231: 147–159 [DOI] [PubMed] [Google Scholar]
- Zhao Y (2005) Lifespan extension and elevated HSP gene expression in Drosophila caused by histone deacetylase inhibitors. J Exp Biol 208: 697–705 [DOI] [PubMed] [Google Scholar]