

Abstract
Objective
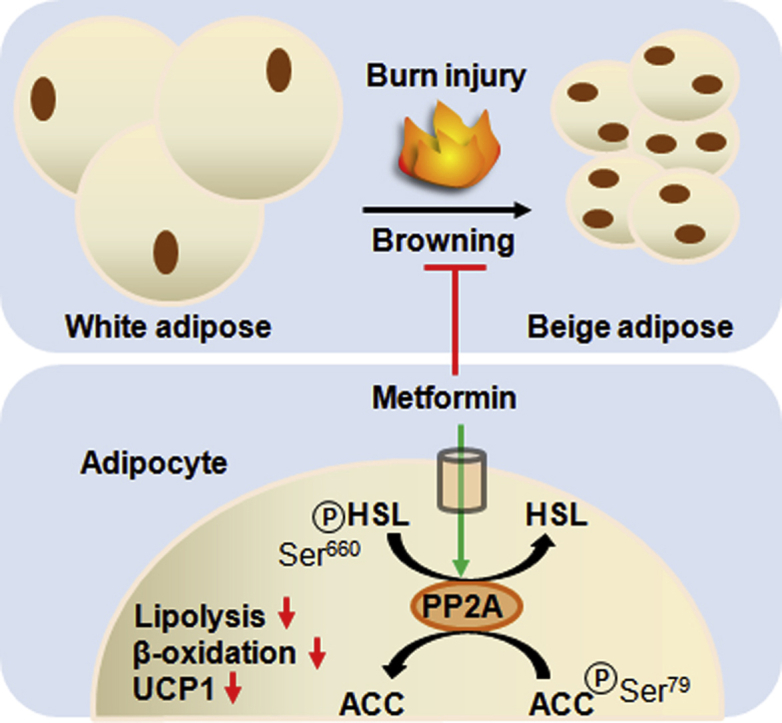
Browning, the conversion of white adipose tissue (WAT) to a beige phenotype, has gained interest as a strategy to induce weight loss and improve insulin resistance in metabolic disorders. However, for hypermetabolic conditions stemming from burn trauma or cancer cachexia, browning is thought to contribute to energy wasting and supraphysiological nutritional requirements. Metformin's impact on this phenomenon and underlying mechanisms have not been explored.
Methods
We used both a murine burn model and human ex vivo adipose explants to assess metformin and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR)'s effects on the development of subcutaneous beige adipose. Enzymes involved in fat homeostasis and browning, as well as mitochondrial dynamics, were assessed to determine metformin's effects.
Results
Treatment with the biguanide metformin lowers lipolysis in beige fat by inducing protein phosphatase 2A (PP2A) independently of adenosine monophosphate kinase (AMPK) activation. Increased PP2A activity catalyzes the dephosphorylation of acetyl-CoA carboxylase (Ser 79) and hormone sensitive lipase (Ser 660), thus promoting fat storage and the “whitening” of otherwise lipolytic beige adipocytes. Moreover, co-incubation of metformin with the PP2A inhibitor okadaic acid countered the anti-lipolytic effects of this biguanide in human adipose. Additionally, we show that metformin does not activate this pathway in the WAT of control mice and that AICAR sustains the browning of white adipose, offering further evidence that metformin acts independently of this cellular energy sensor.
Conclusions
This work provides novel insights into the mechanistic underpinnings of metformin's therapeutic benefits and potential as an agent to reduce the lipotoxicity associated with hypermetabolism and adipose browning.
Keywords: Trauma, Oxidative phosphorylation, Metformin, Burns, Mitochondria, Uncoupling
Graphical abstract
Highlights
-
•
Metformin prevents the catabolism of murine iWAT tissue post-burn injury.
-
•
Mitochondrial respiration and uncoupling in adipose are decreased by metformin.
-
•
Metformin, independently of AMPK, reduces adipose lipolysis and β-oxidation via PP2A.
-
•
AICAR treatment activates AMPK in peripheral adipose leading to sustained browning.
-
•
PP2A is directly induced by metformin in scWAT, lowering ACC/HSL phosphorylation.
1. Introduction
Hypermetabolism is a chronic physiological response to severe trauma such as burns and is associated with cachexia that occurs in advanced cancers and infectious diseases such as tuberculosis and the human immunodeficiency virus (HIV) [1], [2]. Trademarks of this phenomenon include increases in resting energy expenditure (REE), supraphysiological nutritional requirements, and the systemic wasting of adipose tissue reserves and muscle tissue [1]. Although not understood in its entirety, the hypermetabolic response is thought to be driven primarily by catecholamines, corticosteroids and pro-inflammatory cytokines [3]. Fortunately, interventions exist which lower these mediators and curb the degree of hypermetabolism. These include insulin to restore euglycemia, beta blockers such as propranolol, and anabolic steroids such as oxandrolone to prevent muscle catabolism [4]. However, while these therapeutic agents can mitigate the damage stemming from a hypermetabolic state, an absolute cure for this condition has eluded researchers and clinicians. This is of utmost importance as the increase in REE seen in burn patients brings with it higher incidences of infection, sepsis, and death, and cancer cachexia has been shown to contribute to 20% of cancer-related deaths [4], [5].
The preservation of white adipose tissue (WAT) is seen as a valuable strategy to improve patient outcomes in cachexic/hypermetabolic states. Not only does the loss of WAT lead to a decrease in body mass and higher susceptibility to infections and sepsis, but the increased circulation of free fatty acids (FFAs) due to WAT lipolysis leads to the ectopic deposition of fats in organs such as the liver and skeletal muscle, further exacerbating the damage to these tissues and promoting organ dysfunction [6]. This increased fat catabolism is further compounded due to WAT undergoing a browning process following severe burns and during cancer-associated cachexia [7]. This phenotypic switch, whereby adipocytes increase mitochondrial biogenesis and the expression of uncoupling protein 1 (UCP-1), is associated with augmented lipolytic activity and increased systemic energy expenditure [8]. While browning is often regarded as a positive biological phenomenon with the potential to improve outcomes for metabolic disorders such as obesity and diabetes, it is postulated that the conversion of WAT to a beige phenotype during hypermetabolic/cachexic states worsens outcomes and impedes the conservation of body mass [9]. To that effect, therapeutic agents that preserve fat should also ameliorate patient wellbeing.
Metformin, a biguanide with a high safety profile, has long been marketed as a treatment for type 2 diabetes due to its ability to normalize glucose levels [10]. However, in recent decades, the application of this compound has been expanded to fields such as cancer, cardiovascular disease, and aging, prompting an investigation into the molecular mechanisms underlying its benefits [11], [12], [13]. Metformin's cellular actions are often attributed to this drug inhibiting complex I of the electron transport chain (ETC), subsequently increasing the AMP:ATP ratio and activating adenosine monophosphate kinase (AMPK) [14]. Downstream targets of the latter include acetyl-CoA carboxylase-1 (ACC-1), which becomes phosphorylated and inactivated, the outcome of which is decreased fat synthesis and increased β-oxidation [15]. In the context of hypermetabolic/cachexic states, AMPK activation and increased β-oxidation in tissues such as the liver and muscle may be beneficial, as ectopic fat accumulation impedes organ function [16]. However, metformin has also been shown to have AMPK-independent mechanisms of action, particularly in peripheral tissues where the concentrations of this therapeutic agent are not expected to accumulate at levels sufficient to activate this nutritional sensor. For instance, there is evidence that metformin's activity is dependent on Rag GTPases, protein phosphatases or redox state [17], [18], [19]. Moreover, genetic ablation of AMPK or liver kinase B1 does not diminish metformin's ability to reduce hepatic glucose output, further suggesting that our knowledge of metformin's underlying mechanism is incomplete [20].
The aim of this study was to determine if metformin can be used as a pharmacological strategy to maintain body mass and adipose tissue in a murine model of burn injury as well as burn patients. Furthermore, we sought to determine metformin's mechanism of action in beige adipocytes from inguinal WAT (iWAT), including its role in regulating mitochondrial dynamics and fat homeostasis. To accomplish this, C57BL/6 mice were subjected to a 30% total body surface area (TBSA) burn, a traumatic injury which induces severe and prolonged adrenergic stress, the browning of iWAT and a hypermetabolic state accompanied by the loss of body mass [8]. Metformin or the AMPK activator 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) were administered and iWAT collected to study mitochondrial dynamics, AMPK signaling, and fat homeostasis (Figure 1A). The findings from this study were simultaneously replicated in subcutaneous white adipose tissue (scWAT) samples collected from burn patients, the implications of which are discussed herein.
Figure 1.

The biguanide metformin preserves mass while remodeling inguinal white adipose tissue (iWAT). (A) Timeline and study design. Adult C57BL/6 mice were subjected to a 30% total body surface area (TBSA) scald burn or designated as controls. Select cohorts received metformin (100 mg/kg), 5-aminoimidazole-4-carboxamide ribonucleotide (500 mg/kg; AICAR) or saline. (B) Kaplan–Meier curves demonstrating mortality in experimental cohorts. (C) Percentage change in murine body mass from day 0 to day 7, taken prior to euthanasia. (D) Percentage of body mass occupied by iWAT at day 7. (E) Concentration of circulating free fatty acids (FFAs) measured in serum. (F) Percentage of body mass occupied by the liver at day 7. (G) Circulating activity levels of alanine transaminase (ALT) measured in plasma. (H) Representative transmission electron microscopy images taken of control (i, ii), burn (iii, iv), and burn + metformin (v, vi) iWAT samples. Arrows are used to designate mitochondria at 500 nm magnification.  : control (n = 10);
: control (n = 10);  : burn (n = 10);
: burn (n = 10);  : burn + metformin treatment (n = 10). Values are presented as mean ± standard error. *p ≤ 0.05; **p ≤ 0.01.
: burn + metformin treatment (n = 10). Values are presented as mean ± standard error. *p ≤ 0.05; **p ≤ 0.01.
2. Methods
2.1. Murine model
C57BL/6 mice (8 weeks of age; Jackson Laboratory) were housed at ambient temperature and cared for in accordance with the Guide for the Care and Use of Laboratory Animals. All procedures performed in this study were approved by the Sunnybrook Research Institute Animal Care Committee. Mice were anesthetized by inhalation of 3–5% isoflurane and given a 0.1 mg/kg intraperitoneal (i.p.) injection of buprenorphine. Approximately 40% of the dorsum and ventral regions were shaved with electrical clippers and ∼ 1cc of lactated ringers solution was injected under the skin along the spinal column as well as i.p. Full thickness third degree scald burns covering 30% of the TBSA were achieved by immersing the back of the mice at 98 °C for 10 s and the ventral region for 2 s to avoid organ damage. Burned mice were subsequently housed individually in sterile cages and fed ad libitum until sacrifice. Food consumption was monitored daily until euthanasia. Select mice were treated with a daily i.p. injection of metformin (100 mg/kg) or AICAR (500 mg/kg). All injured rodents were scored by both certified veterinarians and laboratory staff daily to minimize animal pain and distress, and the analgesic buprenorphine was administered to mitigate pain when indicated. Mice were euthanized at 7 days post-thermal injury. Organs were harvested for further analysis and blood collected following cardiac puncture. Sham mice (control) underwent identical experimental procedures, with the exception of the burn injury. Mice not receiving metformin or AICAR were given vehicle (saline) injections.
2.2. Blood glucose measurements
Following the excision of 1–2 mm from the tail-tip, blood glucose levels (mmol/L) were measured via a glucometer (OneTouch UltraMini) after gentle massaging of the tail to obtain a drop of blood.
2.3. Patients and explant preparation
Patients admitted to the Ross Tilley Burn Centre at Sunnybrook Hospital (Toronto, Canada) and requiring surgery were consented preoperatively for tissue collection. These procedures were approved by the Research Ethics Board at Sunnybrook Hospital under study #194–2010. In agreement with previous studies, samples were collected from adult patients, between 20 and 40 years of age, undergoing surgery at 10 days to 3 weeks post-burn, when browning of WAT has been demonstrated [21]. Subcutaneous WAT obtained post-operatively was immediately transferred to the laboratory for the preparation of adipose tissue-conditioned media [22]. For the latter, tissue was washed 3X in ice-cold phosphate buffered saline (PBS) and cut into small pieces, with caution exercised to exclude connective tissue and vessels. Twelve g of diced adipose tissue were incubated in a culture plate with 50 mL of Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in an atmosphere of 5% CO2 at 37 °C. Select samples contained varying concentrations of metformin or the PP2A inhibitor okadaic acid. After 24 h (wash out period), media was removed and replaced with fresh DMEM (±metformin/okadaic acid) for another 24 h. This was subsequently removed from the culture plate, filter-sterilized, and stored at −80 °C. Adipose was washed in PBS and stored at −80 °C until further use.
2.4. Mitochondrial isolation and respirometry
Freshly-excised iWAT was minced in 10 volumes of mitochondrial isolation buffer (MHSE + BSA; 210 mM mannitol, 70 mM sucrose, 5 mM HEPES, 1 mM EGTA, 0.5% (w/v) fatty acid-free BSA, pH 7.2) as described [23]. The tissue was then homogenized using a Teflon glass homogenizer with 6 strokes and filtered through three layers of cheesecloth. Mitochondria were isolated via differential centrifugation. Briefly, the homogenate was centrifuged at 600 g for 10 min and the supernatant decanted into a new tube. This fraction was centrifuged at 9000 g for 10 min to afford a mitochondrial pellet. A clear lipid layer surrounding the mitochondria was carefully aspirated and the pellet re-suspended in 50 uL of MHSE + BSA. A second centrifugation to purify mitochondria was performed as per the protocol by Rogers et al. [23]. BCA assays were performed to gauge protein concentrations. Mitochondria were plated in a 96-well plate via centrifugation at 2000 g for 20 min at 4 °C and bioenergetics assessed using a Seahorse XF96 analyzer (Agilent Technologies). Mitochondrial respiration in a coupled state (10 μg/well) was measured in mitochondrial assay solution (MAS; 220 mM mannitol, 70 mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA and 0.2% (w/v) fatty acid-free BSA, pH 7.2 at 37 °C) containing 10 mM succinate as a substrate with 2 μM rotenone (complex II-driven respiration) or 40 μM palmitoyl carnitine with 1 mM malate (β-oxidation-driven respiration). State 3 respiration (phosphorylating respiration) was triggered via the injection of 4 mM ADP. State 4° respiration was assessed by the addition of 2.5 μg/mL oligomycin, while maximal uncoupler-stimulated respiration was observed following the injection of 4 μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP). Antimycin A (4 μM), a complex III inhibitor, was added at the end of the experiment to inhibit mitochondrial respiration, as described [23]. Mitochondria from at least 6 mice were included in each Seahorse analysis, and every sample analyzed in triplicate. The Seahorse XF Wave software was used to group the respiration data from separate mice into a single representative curve and data was normalized to mitochondrial protein content.
2.5. Transmission electron microscopy of iWAT
Murine iWAT was fixed, sectioned and imaged at the University of Toronto Microscopy Imaging Laboratory, as described previously [24].
2.6. Alanine transaminase (ALT) determination
Circulating ALT activity levels were analyzed by colorimetric assay as per the manufacturer's instructions (Biovision Incorporated; catalog # K752).
2.7. PP2A precipitation and activity measurements
Immunoprecipitation of PP2A from iWAT was performed as per the manufacturer's protocol. Subsequent activity measurements were obtained with malachite green following dephosphorylation of the phosphopeptide K-R-pT-I-R-R (Millipore Sigma; catalog # 17-313).
2.8. Plasma cortisol and adrenaline determination
Levels of circulating cortisol (Cayman Chemical; catalog # 500360) and adrenaline (Elabscience; catalog # E-EL-0045) following euthanasia were measured via ELISA kits as per the manufacturer's protocol.
2.9. FFA measurements
Concentrations of FFAs in circulating plasma and the adipose secretome were determined using FFA colorimetric assay kits according to the manufacturer's instructions (Abcam; catalog # ab65341).
2.10. Electrophoresis and in-gel activity assays
Blue native (BN) polyacrylamide gel electrophoresis (PAGE) was performed as described [25], [26]. Mitochondrial fractions were isolated and prepared in a non-denaturing buffer (50 mM Bis-Tris, 500 mM ε-aminocaproic acid, pH 7.0, 4 °C) at a concentration of 4 μg/μL. Ten percent n-dodecyl β-D-maltoside was added to mitochondrial fractions to solubilize membrane bound proteins. A 2–10% or 4–16% gradient gel was prepared (Bio-Rad MiniProteanTM 2 system) using 1 mm spacers to ensure optimal protein separation. Sixty μg of protein were loaded into each well and electrophoresed under native conditions in anode buffer (50 mM Bis-Tris, pH 7 at 4 °C) at 80 V to ensure proper stacking, then at 300 V for proper migration through the gel, ensuring the current does not surpass 25 mA. The blue cathode buffer (50 mM Tricine, 15 min Bis-Tris, 0.02% (w/v) Coomassie G-250, pH 7 at 4 °C) was utilized to help visualize the running front and was exchanged to a colorless cathode buffer (50 mM Tricine, 15 mM Bis-Tris, pH 7 at 4 °C) when the running front was halfway through the gel. Upon completion, the gel was equilibrated in reaction buffer (25 mM Tris-HCl, 5 mM MgCl2, pH 7.4) for 15–30 min. Equal loading was assured by Coomassie blue staining, as described [27]. Complex I was detected by the addition of 5 mM KCN, 1 mM NADH and 0.4 mg/mL iodonitrotetrazolium chloride. Analysis of complex II was achieved with a reaction mixture containing 20 mM succinate, 0.2 mM phenazine methosulfate and 25 mg nitrotetrazolium blue in 10 mL of 5 mM Tris/HCl, pH 7.4. Complex III activity was assessed with 5 mg diaminobenzidine (DAB) in 10 mL of 50 mM sodium phosphate, pH 7.2. Complex IV was assayed by the addition of 10 mg/mL of DAB, 10 mg/mL cytochrome C, and 562.5 mg/mL of sucrose. A reaction mixture containing 35 mM Tris, 270 mM glycine, 14 mM MgCl2, 0.2% Pb(NO3)2 and 8 mM ATP permitted the visualization of ATP synthase activity. Negative controls for the activity of ETC complexes I-IV and ATP synthase lacked NADH, succinate, DAB, cytochrome C and ATP, respectively. Inhibition of their respective activities was accomplished via the addition of 10 μM rotenone, 5 mM malonate, 40 μM antimycin A, 5 mM KCN or 5 μg/mL oligomycin to the reaction mixtures. Reactions were stopped using a destaining solution (40% methanol, 10% glacial acetic acid) once the activity bands had reached the desired intensity. Acetic acid was omitted for the ATP synthase reaction, as the acid would dissolve the lead phosphate precipitate. Gels were imaged colorometrically using a Bio-Rad Chemidoc Imaging System with the exception of ATP synthase which required fluorescent imaging settings to capture the lead phosphate precipitate.
2.11. Immunoblot experiments
To characterize protein expression, western blots were performed. Following SDS-PAGE of iWAT or scWAT lysate (30 μg), proteins were transferred using a Trans-Blot Turbo Transfer System (Bio-Rad) [27]. Upon completion, the nonspecific binding sites on the membrane were blocked using 5% (w/v) non-fat skim milk for 1 h. The membrane was then washed twice in TTBS (12.1 g Tris, 40 g NaCl, 2% Tween 20; pH 7.6 at 4 °C) for 5 min. Blots were incubated overnight with the primary antibody [1:1000 ACC (Cell Signaling Technology; #3662); 1:1000 pACC (Ser79; Cell Signaling Technology; #3661); 1:1000 AMPK (Cell Signaling Technology; #2532S); 1:1000 pAMPK (Thr172; Cell Signaling Technology; #2531S); 1:1000 Cox IV (Cell Signaling Technology; #4850); 1:1000 DRP1 (Abcam; ab56788); 1:1000 FASN (Cell Signaling Technology; #3661); 1:5000 GAPDH (Abcam; ab8245); 1:1000 HSL (Cell Signaling Technology; #4107); 1:1000 pHSL (Ser660; Cell Signaling Technology; #4126; Ser565; Cell Signaling Technology; #4137); 1:1000 Mfn2 (Cell Signaling Technology; #9482); 1:1000 mTORC1 (Cell Signaling Technology; #2972); 1:1000 PGC-1α (Abcam; ab54481); 1:1000 TFAM (Cell Signaling Technology; #8076); 1:1000 UCP1 (Abcam; ab23841); The membrane was washed 3 X in TTBS for 5 min, followed by incubation with secondary antibody, which consisted of horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibodies (1:3000) for 1 h. Following three additional TTBS washes, detection of the desired protein was achieved with a BioRad ChemiDoc Imaging System. SuperSignal West Pico Chemiluminescent Substrate was a product of Thermo Scientific Inc. Densitometry was performed using Image J for Windows.
2.12. Statistical analysis
Data were expressed as mean +/− standard error. Statistical correlations of the data were checked for significance using the Student t test or one-way ANOVA followed by Tukey post-hoc (p ≤ 0.05). Six control mice and at minimum six thermally injured mice were included in each analysis.
3. Results
3.1. Metformin treatment decreases lipolysis and remodels iWAT
While 100% of control and treated animals survived the duration of the study, there was a 20% mortality rate in the injured untreated cohort (Figure 1B). As burn trauma induces a hypermetabolic phenotype, animals were weighed prior to the injury and at the time of euthanasia. Indeed, at 7 days post-burn, ∼5% of body mass was lost in the burn cohort (despite an increase in food consumption; Supplementary Figure 1A), and this outcome was significantly reduced by metformin treatment (Figure 1C). While not significant (p = 0.06), levels of plasma cortisol at day 7 appear reduced with metformin treatment versus burn alone while no change was measured in adrenaline concentrations (Supplementary Figure 1B, C). Moreover, no differences were observed in blood glucose over the duration of the study (Supplementary Figure 1D). The iWAT was collected and weighed and metformin treatment was found to have increased the mass of this organ (Figure 1D), while also lowering the circulation of FFAs in plasma, indicating a preservative effect on adipose (Figure 1E) relative to burn alone. Consistent with the lower circulation of FFAs, liver mass was decreased with metformin treatment (Figure 1F), and circulating levels of the liver damage biomarker alanine transaminase (ALT) were also significantly reduced by metformin (Figure 1G). To elucidate the upstream source of these changes, iWAT was visualized by TEM to observe cellular features. While control mice displayed larger lipid stores with elongated mitochondria (Figure 1H i, ii), injured rodents had multilocular lipid droplets and an abundance of punctate mitochondria, consistent with the browning phenomenon (Figure 1H, iii, iv). Metformin treatment appeared to reverse these effects, enlarging the lipid stores and the shape of select mitochondria, more closely resembling the iWAT from uninjured rodents (Figure 1H, v, vi).
3.2. Metformin treatment increases mitochondrial coupling control
Although there is a lack of consensus on metformin's intracellular mechanism, it is generally thought to act on mitochondria. To that effect, iWAT was collected following euthanasia and immediately homogenized for assays of mitochondrial activity via Seahorse XF96 flux assays. Indeed, metformin treatment reduces mitochondrial activity at 7 days post-burn (Figure 2A), while also increasing coupling efficiency relative to burn alone (Figure 2B). This reduction in oxygen consumption rates (OCR) is evident at all states of respiration measured (Figure 2C–E). To analyze the contribution of β-oxidation to respiratory rates, respirometric assays were repeated using palmitoyl carnitine and malate as substrates, with a similar effect noted in the metformin-treated cohort (Figure 2F–J). Given the time-consuming nature of these extracellular flux assays, we could not eliminate the possibility that mitochondrial integrity was compromised. As such, iWAT fat pads that were immediately transferred to liquid nitrogen were homogenized and mitochondria subsequently isolated for blue native polyacrylamide gel electrophoresis (BN-PAGE) in-gel activity assays. Consistent with the Seahorse analyses, all complexes of the electron transport chain as well as ATP synthase were up-regulated in the burn cohort (Figure 3A–E). Treatment with metformin decreased the activity of complexes I, II, and IV while having no effect on ATP synthase, contributing to the lowered OCR and increased coupling observed (Figure 2A,B). Furthermore, protein expression levels of UCP-1, a key component of the browning response, are decreased by metformin treatment relative to the burn group (Figure 3F).
Figure 2.

Metformin decreases inguinal white adipose tissue (iWAT) mitochondrial respiration while increasing coupling control. (A) Seahorse XF96 respiration profiles of iWAT mitochondria from control mice (grey), mice subjected to thermal injury (black) and injured mice treated with metformin (100 mg/kg; dotted grey), using 10 mM succinate and 2 μM rotenone as substrates. (B) Percentage of the electron transport chain coupled to ATP synthesis as measured by the Seahorse XF stress test report generator. Basal (C), state 3 (D), and state 3u (E) respiration parameters in isolated mitochondria as measured via Seahorse XF96 extracellular flux assays. (F–J) The same parameters outlined above were measured with 40 μM palmitoyl carnitine and 1 mM malate to simulate the contribution of β-oxidation to respiration. : control (n = 6); : burn (n = 6); : burn + metformin treatment (n = 6). Values are presented as mean ± standard error. *p ≤ 0.05; **p ≤ 0.01.
Figure 3.

Metformin acts post-translationally to alter mitochondrial dynamics in inguinal white adipose tissue (iWAT). (A–E) Mitochondrial protein was extracted from iWAT and run in a gradient gel via blue-native polyacrylamide gel electrophoresis, then subjected to activity assays for complexes I-IV and ATP synthase. (F–J) Representative western blots of uncoupling protein-1 (UCP1), dynamin-1-like protein (DRP1), mitofusin 2 (Mfn2), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and mitochondrial transcription factor A (TFAM) in iWAT. Densitometric measurements of in-gel activity assay bands and western blots were performed using ImageJ. : control (n = 8); : burn (n = 8); : burn + metformin treatment (n = 8). Values are presented as mean ± standard error. *p ≤ 0.05; **p ≤ 0.01.
Metformin's ability to suppress OCR and increase coupling prompted us to analyze proteins involved in biogenesis and dynamics of this organelle. DRP1 and Mfn2, key players in mitochondrial fission and fusion respectively, are both significantly increased in the iWAT at 7 days post-burn. Relative to burn alone, metformin does not modulate the expression of these proteins (Figure 3G,H). Moreover, the burn without treatment group had increased expression of the mitochondrial biogenesis markers PGC-1α and TFAM, neither of which was altered significantly by metformin treatment (Figure 3I,J). As such, the ability of this biguanide to alter mitochondrial respiration is seemingly contained to the post-translational level.
3.3. Suppression of lipolysis by metformin is achieved via induction of PP2A
Having established that metformin exerts effects on mitochondrial respiration in iWAT, we sought to determine the mechanism by which it decreases lipolysis in this fat depot post-burn. As AMPK activation is the canonical signaling pathway of metformin, we initially analyzed the phosphorylation of this protein and found that it is not activated by metformin treatment post-burn in iWAT (Figure 4A). Importantly, this is in direct opposition to the results obtained in the livers from the same mice, where metformin suppressed respiration (Supplementary Figure 2A) but did not alter coupling percentages (Supplementary Figure 2B), inducing a stark increase in AMPK phosphorylation (Supplementary Figure 2C). As mTORC1 has been shown to be down-regulated by metformin independently of AMPK activation, we analyzed the expression levels of this protein and found the expression to be lowered in iWAT with metformin treatment (Figure 4B) relative to burn alone. We next focused our efforts on determining metformin's anti-lipolytic mechanism. Fatty acid synthesis as catalyzed by FASN and ACC were first analyzed and while the former was not altered in burn groups relative to the control (Figure 4B), the inhibitory phosphorylation of ACC at serine 79 was diminished in the metformin group, promoting a shift towards fat synthesis and away from β-oxidation (Figure 4C).
Figure 4.

Independently of AMPK, metformin activates protein phosphatase 2A (PP2A) in inguinal white adipose tissue (iWAT). (A-D) Representative western blots of phosphorylated AMP-activated protein kinase (Thr 172; p-AMPK/t-AMPK), mammalian target of rapamycin complex 1 (mTORC1), fatty acid synthase (FASN), phosphorylated acetyl-CoA carboxylase (Ser 79; p-ACC/t-ACC) and phosphorylated hormone sensitive lipase (Ser 660; p-HSL/t-HSL) in iWAT. (E) Ten mg of iWAT was homogenized and PP2A immunoprecipitated for activity measurements using malachite green. Densitometric measurements of western blots were performed using ImageJ. : control (n = 8);  : control + metformin (n = 8) : burn (n = 8); : burn + metformin treatment (n = 8). Values are presented as mean ± standard error. *p ≤ 0.05; **p ≤ 0.01.
: control + metformin (n = 8) : burn (n = 8); : burn + metformin treatment (n = 8). Values are presented as mean ± standard error. *p ≤ 0.05; **p ≤ 0.01.
As ACC is a primary target of AMPK activation, this further suggests that metformin acts independently of this energy sensor in iWAT. Moreover, the activating phosphorylation of hormone sensitive lipase at serine 660 was diminished in the metformin-treated cohort (Figure 4D), indicative of decreased lipolysis underlying the lowered circulation of FFAs observed (Figure 1E). As dephosphorylation of ACC and HSL at these sites is catalyzed by PP2A, the latter was immunoprecipitated from iWAT for activity measurements. Interestingly, metformin increased PP2A activity in iWAT at 7 days post-burn, but did not significantly affect this enzyme in control animals on the same metformin regimen, suggesting a beige fat-specific mechanism for this drug (Figure 4E). To that effect, no significant changes were observed in AMPK activation, mTORC1 or ACC inhibition (Supplementary Figure 3A–C) in the control + metformin cohort, while HSL phosphorylation (Ser 660) was increased (Supplementary Figure 3D). These data suggest that metformin may have opposing effects in WAT (control) and beige adipose (burn), activating lipolysis in the former but not the latter.
3.4. AMPK activation via AICAR treatment sustains iWAT browning
To further establish metformin acting independently of AMPK signaling, select mice were treated with the AMPK activator AICAR to determine the effects of this energy sensing pathway in beige iWAT. Unlike metformin, AICAR did not significantly alter post-burn whole body mass (Figure 5A) nor the mass of iWAT (Figure 5B). Circulating FFAs, however, were decreased by AICAR relative to burn alone (Figure 5C). Mitochondrial respiration as well as thermogenesis as measured by Seahorse XF96 analysis remained elevated in this cohort (Figure 5D; Supplementary Figure 4A–D). These findings are supported by the sustained increases in the protein expression of complex IV (Figure 5E) as well as UCP-1 (Figure 5F). To confirm the signaling pathway of AICAR, AMPK phosphorylation was analyzed and indeed increased by this pharmacological intervention (Figure 5G). While not significantly increased relative to burn alone, downstream target of AMPK PGC-1α (Figure 5H) maintained high expression levels. However, the ratio of phospho-ACC to total ACC was increased, indicative of elevated β-oxidation (Figure 5I). To that effect, we also measured the phosphorylation of HSL at serine 660 and found that it was unaffected by AICAR treatment (Figure 5J), while no significant changes were observed in the inhibitory phosphorylation state (Ser 565; Figure 5K). Thus, while metformin induces the ‘whitening’ of beige adipose, AICAR has an opposing effect by sustaining and possibly potentiating the browning of iWAT.
Figure 5.

AMPK activation with 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) sustains inguinal white adipose tissue (iWAT) browning. (A) Percentage change in murine body mass from day 0 to day 7, taken prior to euthanasia. (B) Percentage of body mass occupied by iWAT at day 7. (C) Concentration of circulating free fatty acids (FFAs) measured in plasma. (D) Seahorse XF96 respiration profiles of iWAT mitochondria from control mice (grey), mice subjected to thermal injury (black) and injured mice treated with AICAR (500 mg/kg; dotted grey). (E–K) Cropped representative western blots of complex IV (COX IV), uncoupling protein-1 (UCP1), phosphorylated AMP-activated protein kinase (Thr 172; p-AMPK/t-AMPK), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), phosphorylated acetyl-CoA carboxylase (Ser 79; p-ACC/t-ACC) and phosphorylated hormone sensitive lipase (Ser 565 and 660; p-HSL/t-HSL) in iWAT. Densitometric measurements of western blots were performed using ImageJ. : control (n = 8); : burn (n = 8); : burn + AICAR treatment (n = 8). Values are presented as mean ± standard error. *p ≤ 0.05; **p ≤ 0.01.
3.5. Metformin directly induces PP2A in human scWAT
As metformin has been shown to improve insulin sensitivity in select conditions, we elected to remove these systemic effects via the direct treatment of human adipose in an ex vivo explant model. Here, freshly excised human burn adipose was incubated in media +/− metformin to determine effects on the signaling pathways regulating lipolysis. As observed by eye, concentrations of this biguanide as low as 10 μM were sufficient to reduce the catabolism of adipose over the course of 48 h (Figure 6A). The media was subsequently analyzed to determine the concentration of FFAs. Indeed, 10 μM metformin diminished the release of FFAs into explant media, a decrease that was inhibited when fat was co-incubated with metformin and 10 nM of the PP2A inhibitor, okadaic acid (Figure 6B). Following the incubation, adipose tissue was collected and prepared for western blots. As observed in the murine assays, the low dose of metformin does not increase phosphorylation and thus activation of AMPK (Figure 6C). However, 10 μM of this biguanide did decrease the phosphorylation of both ACC (Ser 79; Figure 6D) and HSL (Ser 660; Figure 6E) while activating PP2A (Figure 6F). Consistent with metformin's mechanism of action proceeding via PP2A, these events were significantly diminished in the presence of 10 nM okadaic acid. Moreover, as with the murine model, metformin (10 μM) significantly lowered the expression of UCP-1, suggesting that the impairment of lipolysis and β-oxidation reduces the thermogenesis associated with beige adipose at physiological concentrations (Figure 6G).
Figure 6.

Metformin directly activates protein phosphatase 2A (PP2A) in human subcutaneous white adipose tissue (scWAT). (A) Representative image of ex vivo explant cultures from a burn patient ± metformin added to the media. (B) Concentration of circulating free fatty acids (FFAs) released by adipose into explant media. (C–E) Representative western blots of phosphorylated AMP-activated protein kinase (Thr 172; p-AMPK/t-AMPK), phosphorylated acetyl-CoA carboxylase (Ser 79; p-ACC/t-ACC) and phosphorylated hormone sensitive lipase (Ser 660; p-HSL/t-HSL) in scWAT. (F) Ten mg of scWAT was homogenized and PP2A immunoprecipitated for activity measurements using malachite green. (G) Representative western blot of UCP1. Densitometric measurements of western blots were performed using ImageJ. : burn alone (n = 6); : burn + 10 μM metformin (n = 6); : burn + 10 μM metformin + 10 nM okadaic acid (n = 6); : burn + 100 μM metformin (n = 6). Values are presented as mean ± standard error. *p ≤ 0.05; **p ≤ 0.01.
4. Discussion
Often regarded as a positive phenomenon with potential benefits in metabolic disorders, the browning of WAT following severe burns or during cachexia as seen in the end stage of cancer appears to be detrimental [28]. Indeed, the atrophying of adipose tissue upstream of other metabolic organs such as the liver and muscle exacerbates cellular dysfunction via the ectopic deposition of FFAs in these tissues [29]. These patients often have supraphysiological nutritional requirements, rendering it difficult, if not impossible, to reverse the systemic catabolism, thus leading to poor outcomes and increased morbidity [30], [31]. To that end, therapeutic interventions that quell the browning response should preserve tissue and body mass while limiting the systemic dysfunction of cachectic states. The biguanide metformin has been shown to reduce the circulation of FFAs in patients with burn injury, as well as limit cancer cachexia in rat models while maintaining a good safety profile [32], [33]. However, despite widespread use and benefits to various extents for a plethora of disorders, the underlying mechanisms of metformin are hotly debated, and the potential role of this drug in mitigating hypercatabolic responses has not been adequately explored.
The data presented herein demonstrate that metformin exerts a ‘whitening’ effect on the iWAT of mice following severe burn injury, limiting mitochondrial activity, thermogenesis, and lipolysis, thus preserving both whole body and adipose mass. While this drug activates AMPK in the liver tissue, where concentrations are expected to be elevated, its actions proceed independently of this energy sensor in peripheral adipose (iWAT) [34]. This is of key importance in hypermetabolic conditions as AMPK-mediated phosphorylation of ACC decreases fat synthesis while activating β-oxidation. Theoretically, while the enhanced ability of the liver and muscle tissues to break down fat for energy is beneficial in cachexia/hypermetabolism, ACC phosphorylation in the adipose is detrimental, fueling the thermogenic and lipolytic response in this tissue. The decreased phosphorylation of ACC following metformin treatment supports an AMPK-independent role of this drug in adipose, while the induction of PP2A permits a shift from lipolysis towards fat storage via activation of ACC and deactivation of HSL [35]. Indeed, it has been reported that metformin interferes with the MID1-α4 protein complex, which regulates the degradation of the catalytic subunit PP2Ac, thus suggesting metformin directly affects this phosphatase independently of AMPK/TOR signaling [19]. One potential drawback of our study is that the day 7 time point alone only provides a snapshot of metformin's effects on browning, and may not fully recapitulate the plethora of changes to nutrient and energy signaling observed in iWAT post-burn. For instance, a clear increase in phospho-ACC is observed in the burn alone group, despite no increase in AMPK activation or decrease in PP2A activity in the burn cohort. To fully capture the temporal changes to adipose post-burn, a comprehensive time course study which examines the transient changes to adipose signaling pathways, morphology and function is currently in progress.
It is of interest to note that metformin does not activate PP2A nor does it decrease lipolysis in the iWAT of control mice. As biguanides carry a positive charge, we hypothesize that the increased mitochondrial density in beige iWAT allows the accumulation of these moieties at higher levels than in white iWAT from control mice [36]. Moreover, metformin's activation of HSL at serine 660 in control animals lends further credence to the ability of this drug to suppress obesity in a healthy state [37]. Interestingly, burn alone or metformin treatment did not alter blood glucose levels. An important limitation of the murine burn model is that mice retain full mobility, whereas human patients are confined to bed rest for weeks to months following thermal trauma [2], [3]. Therefore, the extent of muscle catabolism and atrophy in the murine model does not mimic the human experience, and we propose that maintenance of skeletal muscle glucose uptake via GLUT4 underlies the absence of hyperglycemia in murine studies despite the elevated glucose levels and insulin resistance observed in burn patients [38]. This may be corrected with a hindlimb unloading model, which more accurately captures the magnitude of muscle atrophy post-burn [39].
The observation that metformin fails to activate AMPK in peripheral iWAT prompted the study of this pathway using AICAR, a well characterized activator of this energy sensor. As observed in other conditions, AMPK activation sustains the increased mitochondrial activity, thermogenesis, and β-oxidation seen post-burn [40]. This is not surprising, as signaling an energy deficit induces the activation of PGC-1α, the master regulator of mitochondrial biogenesis [41]. Interestingly, although not significant, AICAR appears to reduce whole body mass loss and does significantly decrease circulating FFAs. Studies have shown that AICAR improves insulin sensitivity as well as glucose uptake and fat oxidation in skeletal muscle while suppressing glucose production in the liver [42], [43]. As such, while this pharmacological agent does not affect the browning of adipose, it may exert downstream benefits via modulation of hyperglycemia/insulin resistance, hallmarks of the hypermetabolic response to severe burn injury [2]. To that end, cachexia/hypermetabolism is characterized by increases in REE, defined by increased oxygen usage. As approximately 90% of cellular O2 is consumed in mitochondria, AICAR's benefits may be limited by its ability to potentiate AMPK signaling and mitochondrial biogenesis, further fueling the hypermetabolic state, though this remains to be explored systematically. Despite the activation of AMPK by AICAR, we did not observe an increase in the phosphorylation of HSL at serine 565, as others have previously [44]. It is possible that the magnitude of PKA-mediated lipolysis in burns opposes the inhibition of HSL or that currently unknown systemic mechanisms prevent this occurrence in vivo.
The fact that adipose tissue expresses organic cation transporters (OCT1) renders it possible for hydrophilic positively-charged biguanides to enter this organ and induce biological effects [45]. To determine if metformin can directly influence molecular pathways in adipose, fat explants were prepared ex vivo and incubated with this compound at varying concentrations. As in the murine model, metformin decreased phosphorylation of ACC and HSL at serines 79 and 660, respectively, via the activation of PP2A. Furthermore, this was observed at concentrations (10 μM) that were insufficient for AMPK activation. Co-incubation of metformin with okadaic acid, a potent inhibitor of PP2A, significantly reduced the anti-lipolytic activity of metformin. However, PP2A inhibition alone was not sufficient in restoring p-HSL/HSL and p-ACC/ACC to the burn alone state. While this may be rectified with longer incubation times, it cannot be ruled out that metformin is acting via alternative mechanisms. For instance, mTORC1 suppresses lipolysis, and if metformin is acting independently of AMPK to inhibit mTORC1 signaling, it is possible for this biguanide to increase lipolysis and counter PP2A activation, creating a unique dynamic within adipocytes, likely depending on the concentration of metformin used [18], [34]. It's of interest to note the dichotomy in metformin's ability to activate both AMPK and PP2A, as these exert opposing effects on ACC activation. At higher concentrations (100 μM), metformin does induce a slight yet insignificant increase in AMPK/ACC phosphorylation, suggesting PP2A signaling dominates only at lower concentrations of this biguanide. Further, owing to the increased bioenergetics and ATP production in catabolic beige adipose, we propose that higher concentrations of metformin would be required to increase the AMP:ATP ratio sufficiently for AMPK activation.
5. Conclusion
Collectively, these data demonstrate that metformin directly affects adipose, independently of systemic effects, and that activation of PP2A in burns, owing to the anti-lipolytic action of this enzyme, appears to be a viable strategy for adipose preservation. We suggest that metformin's ability to induce the activity of this phosphatase may exert benefits in other conditions, such as cancer cachexia, wherein the hyperactivation of oncogenic kinases could be countered by targeting PP2A as a tumor suppressor [46]. While AMPK activation is often acknowledged as the primary signaling pathway of metformin, this work demonstrates that this biguanide's intracellular effects can proceed independently of AMPK in peripheral adipose. Moreover, metformin appears to have tissue-specific mechanisms, activating AMPK in the liver and PP2A in beige but not white adipose. Overall, our results support the use of metformin in countering the hypermetabolic phenotype brought about by severe thermal trauma by inducing the phenotypic switch of catabolic beige adipose to a fat-storing white state. Future studies should focus on burn patients themselves, rather than an explant model, to determine if a regimen of metformin can prevent adipose browning and its associated lipotoxicity. As more evidence arises supporting the use of this anti-diabetic agent for other disorders, a comprehensive examination of metformin's tissue and concentration-dependent mechanisms will be required.
Author contributions
CA conceived, designed, and performed experiments as well as wrote the manuscript; CMK, AA, OS, and AP performed experiments and analyzed data; MGJ directed the studies and wrote portions of the manuscript.
Acknowledgements
Abdikarim Abdullahi is a recipient of the Vanier Canada Graduate Scholarship from the Canadian Institutes of Health Research. This work was supported by grants from the Canadian Institutes of Health Research (#123336), the Canada Foundation for Innovation Leader's Opportunity Fund (#25407) and National Institutes of Health (2R01GM087285-05A1).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2019.08.011.
Conflict of interest
The authors declare they have no conflict of interest.
Appendix A. Supplementary data
The following is the supplementary data to this article:
References
- 1.Argiles J.M., Lopez-Soriano F.J., Busquets S. Novel approaches to the treatment of cachexia. Drug Discovery Today. 2008;13:73–78. doi: 10.1016/j.drudis.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Stanojcic M., Abdullahi A., Rehou S., Parousis A., Jeschke M.G. Pathophysiological response to burn injury in adults. Annals of Surgery. 2018;267:576–584. doi: 10.1097/SLA.0000000000002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jeschke M.G., Gauglitz G.G., Kulp G.A., Finnerty C.C., Williams F.N., Kraft R. Long-term persistence of the pathophysiologic response to severe burn injury. PLoS One. 2011;6:e21245. doi: 10.1371/journal.pone.0021245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Auger C., Samadi O., Jeschke M.G. The biochemical alterations underlying post-burn hypermetabolism. Biochimica et Biophysica Acta Molecular Basis of Disease. 2017;1863:2633–2644. doi: 10.1016/j.bbadis.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aoyagi T., Terracina K.P., Raza A., Matsubara H., Takabe K. Cancer cachexia, mechanism and treatment. World Journal of Gastrointestinal Oncology. 2015;7:17–29. doi: 10.4251/wjgo.v7.i4.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snel M., Jonker J.T., Schoones J., Lamb H., de Roos A., Pijl H. Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. International Journal of Endocrinology. 2012;2012:983814. doi: 10.1155/2012/983814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tamucci K.A., Namwanje M., Fan L., Qiang L. The dark side of browning. Protein Cell. 2018;9:152–163. doi: 10.1007/s13238-017-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abdullahi A., Auger C., Stanojcic M., Patsouris D., Parousis A., Epelman S. Alternatively activated macrophages drive browning of white adipose tissue in burns. Annals of Surgery. 2019;269:554–563. doi: 10.1097/SLA.0000000000002465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abdullahi A., Jeschke M.G. White adipose tissue browning: a double-edged sword. Trends in Endocrinology and Metabolism. 2016;27:542–552. doi: 10.1016/j.tem.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li M., Li X., Zhang H., Lu Y. Molecular mechanisms of metformin for diabetes and cancer treatment. Frontiers in Physiology. 2018;9:1039. doi: 10.3389/fphys.2018.01039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vancura A., Bu P., Bhagwat M., Zeng J., Vancurova I. Metformin as an anticancer agent. Trends in Pharmacoligical Sciences. 2018;39:867–878. doi: 10.1016/j.tips.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barzilai N., Crandall J.P., Kritchevsky S.B., Espeland M.A. Metformin as a tool to target aging. Cell Metabolism. 2016;23:1060–1065. doi: 10.1016/j.cmet.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madeo F., Carmona-Gutierrez D., Hofer S.J., Kroemer G. Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential. Cell Metabolism. 2019;29:592–610. doi: 10.1016/j.cmet.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 14.Howell J.J., Hellberg K., Turner M., Talbott G., Kolar M.J., Ross D.S. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metabolism. 2017;25:463–471. doi: 10.1016/j.cmet.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee M., Katerelos M., Gleich K., Galic S., Kemp B.E., Mount P.F. Phosphorylation of acetyl-CoA carboxylase by AMPK reduces renal fibrosis and is essential for the anti-fibrotic effect of metformin. Journal of the American Society of Nephrology. 2018;29:2326–2336. doi: 10.1681/ASN.2018010050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanchez A.M., Candau R.B., Csibi A., Pagano A.F., Raibon A., Bernardi H. The role of AMP-activated protein kinase in the coordination of skeletal muscle turnover and energy homeostasis. American Journal of Physiology Cell Physiology. 2012;303:C475–C485. doi: 10.1152/ajpcell.00125.2012. [DOI] [PubMed] [Google Scholar]
- 17.Madiraju A.K., Qiu Y., Perry R.J., Rahimi Y., Zhang X.M., Zhang D. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nature Medicine. 2018;24:1384–1394. doi: 10.1038/s41591-018-0125-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalender A., Selvaraj A., Kim S.Y., Gulati P., Brule S., Viollet B. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metabolism. 2010;11:390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kickstein E., Krauss S., Thornhill P., Rutschow D., Zeller R., Sharkey J. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proceedings of the National Academy of Sciences. 2010;107:21830–21835. doi: 10.1073/pnas.0912793107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foretz M., Hebrard S., Leclerc J., Zarrinpashneh E., Soty M., Mithieux G. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. Journal of Clinical Investigation. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patsouris D., Qi P., Abdullahi A., Stanojcic M., Chen P., Parousis A. Burn induces browning of the subcutaneous white adipose tissue in mice and humans. Cell Reports. 2015;13:1538–1544. doi: 10.1016/j.celrep.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hendijani F. Explant culture: an advantageous method for isolation of mesenchymal stem cells from human tissues. Cell Proliferation. 2017;50 doi: 10.1111/cpr.12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogers G.W., Brand M.D., Petrosyan S., Ashok D., Elorza A.A., Ferrick D.A. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diao L., Auger C., Konoeda H., Sadri A.R., Amini-Nik S., Jeschke M.G. Hepatic steatosis associated with decreased beta-oxidation and mitochondrial function contributes to cell damage in obese mice after thermal injury. Cell Death & Disease. 2018;9:530. doi: 10.1038/s41419-018-0531-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Auger C., Sivayoganathan T., Abdullahi A., Parousis A., Jeschke M.G. Hepatic mitochondrial bioenergetics in aged C57BL/6 mice exhibit delayed recovery from severe burn injury. Biochimica et Biophysica Acta Molecular Basis of Disease. 2017;1863:2705–2714. doi: 10.1016/j.bbadis.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han S., Auger C., Appanna V.P., Lemire J., Castonguay Z., Akbarov E. A blue native polyacrylamide gel electrophoretic technology to probe the functional proteomics mediating nitrogen homeostasis in Pseudomonas fluorescens. Journal of Microbiological Methods. 2012;90:206–210. doi: 10.1016/j.mimet.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Auger C., Sivayoganathan T., Abdullahi A., Parousis A., Pang B.W., Jeschke M.G. Metformin adapts its cellular effects to bioenergetic status in a model of metabolic dysfunction. Scientific Reports. 2018;8:5646. doi: 10.1038/s41598-018-24017-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abdullahi A., Jeschke M.G. Taming the flames: targeting white adipose tissue browning in hypermetabolic conditions. Endocrine Reviews. 2017;38:538–549. doi: 10.1210/er.2017-00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaitkus J.A., Celi F.S. The role of adipose tissue in cancer-associated cachexia. Experimental Biology and Medicine. 2017;242:473–481. doi: 10.1177/1535370216683282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gullett N.P., Mazurak V.C., Hebbar G., Ziegler T.R. Nutritional interventions for cancer-induced cachexia. Current Problems in Cancer. 2011;35:58–90. doi: 10.1016/j.currproblcancer.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall K.L., Shahrokhi S., Jeschke M.G. Enteral nutrition support in burn care: a review of current recommendations as instituted in the Ross Tilley Burn Centre. Nutrients. 2012;4:1554–1565. doi: 10.3390/nu4111554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeschke M.G., Abdullahi A., Burnett M., Rehou S., Stanojcic M. Glucose control in severely burned patients using metformin: an interim safety and efficacy analysis of a phase II randomized controlled trial. Annals of Surgery. 2016;264:518–527. doi: 10.1097/SLA.0000000000001845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oliveira A.G., Gomes-Marcondes M.C. Metformin treatment modulates the tumour-induced wasting effects in muscle protein metabolism minimising the cachexia in tumour-bearing rats. BMC Cancer. 2016;16:418. doi: 10.1186/s12885-016-2424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He L., Wondisford F.E. Metformin action: concentrations matter. Cell Metabolism. 2015;21:159–162. doi: 10.1016/j.cmet.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Kinney B.P., Qiao L., Levaugh J.M., Shao J. B56alpha/protein phosphatase 2A inhibits adipose lipolysis in high-fat diet-induced obese mice. Endocrinology. 2010;151:3624–3632. doi: 10.1210/en.2010-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bridges H.R., Jones A.J., Pollak M.N., Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochemical Journal. 2014;462:475–487. doi: 10.1042/BJ20140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Aa M.P., Hoving V., van de Garde E.M., de Boer A., Knibbe C.A., van der Vorst M.M. The effect of eighteen-month metformin treatment in obese adolescents: comparison of results obtained in daily practice with results from a clinical trial. Journal of Obesity. 2016;2016:7852648. doi: 10.1155/2016/7852648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richter E.A., Hargreaves M. Exercise, GLUT4 and skeletal muscle glucose uptake. Physiological Reviews. 2013;93:993–1017. doi: 10.1152/physrev.00038.2012. [DOI] [PubMed] [Google Scholar]
- 39.Song J., Baer L.A., Threlkeld M.R.S., Geng C., Wade C.E., Wolf S.E. Insulin and exercise improved muscle function in rats with severe burn and hindlimb unloading. Physiological Reports. 2019;7:e14158. doi: 10.14814/phy2.14158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu L., Zhang L., Li B., Jiang H., Duan Y., Xie Z. AMP-activated protein kinase (AMPK) regulates energy metabolism through modulating thermogenesis in adipose tissue. Frontiers in Physiology. 2018;9:122. doi: 10.3389/fphys.2018.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaidhu M.P., Fediuc S., Anthony N.M., So M., Mirpourian M., Perry R.L. Prolonged AICAR-induced AMP-kinase activation promotes energy dissipation in white adipocytes: novel mechanisms integrating HSL and ATGL. Journal of Lipid Reseasrch. 2009;50:704–715. doi: 10.1194/jlr.M800480-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith A.C., Bruce C.R., Dyck D.J. AMP kinase activation with AICAR simultaneously increases fatty acid and glucose oxidation in resting rat soleus muscle. Journal of Physiology. 2005;565:537–546. doi: 10.1113/jphysiol.2004.081679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Logie L., Lees Z., Allwood J.W., McDougall G., Beall C., Rena G. Regulation of hepatic glucose production and AMPK by AICAR but not by metformin depends on drug uptake through the equilibrative nucleoside transporter 1 (ENT1) Diabetes, Obesity and Metabolism. 2018;20:2748–2758. doi: 10.1111/dom.13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daval M., Diot-Dupuy F., Bazin R., Hainault I., Viollet B., Vaulont S. Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. Journal of Biological Chemistry. 2005;280:25250–25257. doi: 10.1074/jbc.M414222200. [DOI] [PubMed] [Google Scholar]
- 45.Moreno-Navarrete J.M., Ortega F.J., Rodriguez-Hermosa J.I., Sabater M., Pardo G., Ricart W. OCT1 Expression in adipocytes could contribute to increased metformin action in obese subjects. Diabetes. 2011;60:168–176. doi: 10.2337/db10-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perrotti D., Neviani P. Protein phosphatase 2A: a target for anticancer therapy. The Lancet Oncology. 2013;14:e229–e238. doi: 10.1016/S1470-2045(12)70558-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.