

Abstract
Objective
Roux-en-Y gastric bypass surgery (RYGB) improves the first phase of glucose-stimulated insulin secretion (GSIS) in patients with type 2 diabetes. How it does so remains unclear. Farnesoid X receptor (FXR), the nuclear receptor of bile acids (BAs), is implicated in bariatric surgery. Moreover, the transient receptor potential ankyrin 1 (TRPA1) channel is expressed in pancreatic β-cells and involved in insulin secretion. We aimed to explore the role of BAs/FXR and TRPA1 in improved GSIS in diabetic rats after RYGB.
Methods
RYGB or sham surgery was conducted in spontaneous diabetic Goto-Kakizaki (GK) rats, or FXR or TRPA1 transgenic mice. Gene and protein expression of islets were assessed by qPCR and western blotting. Electrophysiological properties of single β-cells were studied using patch-clamp technique. Binding of FXR and histone acetyltransferase steroid receptor coactivator-1 (SRC1) to the TRPA1 promoter, acetylated histone H3 (ACH3) levels at the TRPA1 promoter were determined using ChIP assays. GSIS was measured using enzyme-linked immunosorbent assays or intravenous glucose tolerance test (IVGTT).
Results
RYGB increases GSIS, particularly the first-phase of GSIS in both intact islets and GK rats in vivo, and ameliorates hyperglycemia of GK rats. Importantly, the effects of RYGB were attenuated in TRPA1-deficient mice. Moreover, GK β-cells displayed significantly decreased TRPA1 expression and current. Patch-clamp recording revealed that TRPA1−/− β-cells displayed a marked hyperpolarization and decreased glucose-evoked action potential firing, which was associated with impaired GSIS. RYGB restored TRPA1 expression and current in GK β-cells. This was accompanied by improved glucose-evoked electrical activity and insulin secretion. Additionally, RYGB-induced TRPA1 expression involved BAs/FXR-mediated recruitment of SRC1, promoting ACH3 at the promoter of TRPA1.
Conclusions
The BAs/FXR/SRC1 axis-mediated restoration of TRPA1 expression plays a critical role in the enhanced GSIS and remission of diabetes in GK rats after RYGB.
Keywords: RYGB, TRPA1, FXR, Insulin secretion, Diabetes
Highlights
-
•
RYGB-restored TRPA1 expression in diabetic β-cells.
-
•
RYGB-induced TRPA1 is mediated by FXR-SRC1-increased ACH3 at TRPA1 promoter.
-
•
TRPA1 is required for β-cell stimulus-secretion coupling.
-
•
Modulation of TRPA1 expression in β-cells may constitute a novel therapeutic strategy to improve β-cell function in T2D.
1. Introduction
The prevalence of type 2 diabetes (T2D) originates from β-cell failure to compensate insulin resistance and secrete the necessary amount of insulin to maintain glucose homeostasis [1]. RYGB has been recognized the choice for treatment of T2D patients because it leads to normalization of hyperglycemia within days after surgery, an effect is thought independent of weight loss [2], [3]. How it does so remain unclear, although an improved secretory capacity of pancreatic β-cells has been postulated [4]. Particularly, the first phase of GSIS, which is severely impaired in T2D, has been restored shortly after RGYB in T2D patients [4], [5] and animal models [6].
The first phase of GSIS is attributable to ATP-driven closure of ATP-sensitive potassium channels (KATP), depolarization of the plasma membrane, and opening of the voltage-gated Ca2+ channels with subsequent increase in cytoplasmic Ca2+ concentration. Importantly, this process requires an additional small inward current that can bring the membrane potential away from the equilibrium potential of K+, which results in initiation of action potential firing and insulin secretion [7], [8]. The transient receptor potential (TRP) channels have been hypothesized to be responsible for this inward cation current [9], [10]. As a member of the transient receptor potential (TRP) channel superfamily, TRP ankyrin 1 (TRPA1) is abundantly expressed in pancreatic β-cells, but not α-cells [11]. TRPA1 is a Ca2+ permeable non-selective cation channel that responds to noxious cold, divalent cations and polyunsaturated fatty acids [12]. Activation of TRPA1 by its agonist induces inward current, which leads to membrane depolarization and Ca2+ influx and potentiates insulin secretion [11], [13]. Although these pharmacological data suggest that TRPA1 may be involved in stimulus-secretion coupling in β-cells, the physiological significance of this channel in β-cell function remains to be confirmed in TRPA1-deficient mice. Furthermore, the alterations of TRPA1 in β-cells in diabetes, particularly its association with impaired GSIS have not been explored.
RYGB leads to elevation of serum bile acids (BAs) in T2D patients [14] and diabetic rats [15]. As an important metabolic regulator, BA signaling plays a critical role in improved glucose homeostasis and remission of diabetes after RYGB [3], [16]. Emerging evidences demonstrate that BAs enhance GSIS, an effect that is attributed to activation of its nuclear farnesoid X receptor (FXR) [17], [18], since BAs fail to potentiate GSIS in FXR−/- islets [18]. FXR, a ligand-activated transcription factor belonging to the nuclear receptor superfamily [19], has been identified in human and rodent β-cells [17], [18]. Activation of FXR by BAs or FXR agonist GW4064 potentiates GSIS [17], [18]. However, the precise mechanism by which this is achieved remains largely unknown. In this study, we found that RYGB restores TRPA1 expression in β-cells from diabetic GK rats, via increasing BAs with subsequent stimulating FXR-mediated recruitment of histone acetyltransferase steroid receptor coactivator-1 (SRC1), and promoting the acetylation of histone H3 (ACH3) at the promoter of TRPA1. We also demonstrate that TRPA1 is required for the ability of glucose to evoke electrical activity and stimulate insulin secretion in β-cells. Thus, increased TRPA1 in β-cells would account for improved GSIS and glycemic control in diabetes after RYGB.
2. Materials and methods
2.1. Experimental animals
Male Wistar and GK rats aged 10 wk were purchased from SLRC Laboratory Animal company (Shanghai, China). FXR knockout mice (C57Bl/6) were kindly provided by Prof. Youfei Guan at Dalian University, China [20]. TRPA1 knockout mice (C57BL/6J) were purchased from The Jackson Laboratory (Bar Harbor, USA). All animals were maintained on a normal diet and kept in the conventional vivarium with a 12-h day/night cycle (lights on at 7:00 and off at 19:00). The animals were housed at room temperature (22–25 °C) and were allowed to adapt to the new environment for 1 wk before the surgery. GK rats were randomized to RYGB or sham operation. Male FXR or TRPA1 transgenic mice aged 18–20 wk with body weight >25 g were used for RYGB or sham operation. Survival rates after surgery were 100% for RYGB and sham GK rats; and 75% for RYGB mice and 100% for sham mice. The animal procedures were performed according to the Principles of Laboratory Animal Care and approved by the Shenzhen University Animal Care Committee.
2.2. RYGB on GK rats
The RYGB procedure was performed as described previously [6]. Rats were fasted overnight and anesthetized with isoflurane (3% for induction, 2% for maintenance). Under sterile conditions, a midline laparotomy was performed. The stomach was divided into two by a suture along the white line between the forestomach and glandular stomach. A biliopancreatic limb extending 16 cm from the ligament of Treitz was transected. The distal segment was anastomosed to the gastric remnant, and the proximal segment was drained into 30 cm of the Roux limb by side-to-side anastomosis. For the GK-PF-sham-operated animals, following laparotomy, an incision was made at a spot 16 cm from the ligament of Treitz. The intestine was then reconnected by side-to-side anastomosis, without intestinal rearrangement, and the incision was closed.
2.3. RYGB on FXR or TRPA1 transgenic mice
The RYGB procedure was conducted using a modification of the approach described by Nestoridi et al. [21]. The stomach was ligated between the glandular portion and the gastric fundus (forestomach). The jejunum was transected at 4 cm from the ligament of Treitz and 6 cm from the site of gastroenterostomy. The distal segment of jejunum was anastomosed to the forestomach. The sham procedure involved mobilization of the forestomach and proximal and distal jejunum and ileum without any transection.
After surgery, animals were left to recover in a warm box and then returned to the animal facility. They were injected with buprenorphine and ampicillin for 3 days. All surgical GK rats were given purified water for 12 h before started on the normal chow; whereas the surgical mice were maintained on a liquid diet (Novartis, NY) for 10 days until weaned back to solid normal chow.
2.4. Intravenous glucose tolerance test
Rats were fasted overnight and anesthetized with isoflurane. The rats were given glucose at a dose of 1 g/kg through a jugular vein catheter. Blood samples were then collected at 0, 2, 5, 10, and 30 min after glucose administration to measure insulin.
2.5. Measurement of plasma bile acids
GK-sham and GK-RYGB rats were fasted 12 h, and blood samples were collected from the tail vein. Total bile acids (TBA) were determined using TBA test kit from Nanjing Jiancheng Bioengineering Institute (China) (E003) according to the manufacturer's instructions. Chenodeoxycholic acid (CDCA) levels were examined with CDCA ELISA kit (MET-5008) from Cell Biolabs (USA) in accordance with the instructions.
2.6. Isolation of islets and cell culture
Pancreatic islets were isolated by collagenase digestion, as described previously [22]. For preparation of single β-cells for electrophysiology, the islets were dissociated into single cells using a Ca2+-free solution.
2.7. Retroviral infection and RNAi transfection
INS-1 832/13 cells were used in these experiments. For FXR overexpression, plasmid pMX-puro-FXR was generated by PCR using primers listed in electronic Supplemental Table 1. The product was cleaved with BamH I and Not I and cloned into those sites of pMX-puro vector. INS-1 832/13 cells were retrovirally transduced with either pMX-puro or pMX-puro-FXR. For knockdown of FXR or SRC1, cells were lentivirally transduced with either scramble or shRNA (Cat. RMM3981-201756319) targeted against FXR mRNA or shRNA (Cat. RMM3981-201813658) targeted against SRC1 mRNA from GE Dharmacon.
2.8. Electrophysiological recordings
Electrical activity was recorded from single β-cells freshly isolated from islets using the perforated patch whole-cell configuration. β-cells were functionally identified by the absence of a voltage-gated Na+-currents when using a holding potential of −70mV, and a larger whole-cell membrane capacitance (≥7 pF) [23]. The pipette resistance ranged between 2 and 5 MΩ when the pipettes were filled with the intracellular solutions (mM): 128 K-gluconate, 10 KCl, 10 NaCl, 1 MgCl2, 5 HEPES (pH 7.3, adjusted with KOH). The extracellular medium consisted of (in mM): 140 NaCl, 5 NaHCO3, 3.6 KCl, 0.5 NaH2PO4, 1.5 CaCl2, 0.5 mM MgSO4, 10 HEPES and 5 mM d-glucose (pH 7.4, adjusted with NaOH). All experiments were conducted using an EPC-10 patch-clamp amplifier and the PULSE software (HEKA Electronics). Data were filtered and sampled with Pulse/Pulsefit and software (Heka Electronik). All electrophysiological experiments were performed at 20–25 °C.
2.9. Western blot analysis
These experiments were performed as previously described [22]. The antibodies were used: primary antibodies against TRPA1 (1:1,000, #ABN1009; Millipore, Billerica, MA), FXR (1:1,000, #417200; Thermo Fisher Scientific, Waltham, MA), GAPDH (1:3,000, #5174; Cell signaling Technology, Danvers, MA) and β-actin (1:10,000, #A5441; Sigma–Aldrich).
2.10. Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using a ChIP assay kit (Millipore) according to the manufacturer's instructions. Soluble chromatin was prepared from INS-1 832/13 cells, followed by immunoprecipitated with antibodies (2 μg) against FXR (#417200, Thermo Fisher Scientific) or acetylated histone 3 (#06–599, Millipore) or SRC1 (#ab2859, Abcam, Cambridge, MA), respectively. DNA extractions were sequenced from −1717 bp to −1605 bp in TRPA1 promoter by using the primers listed in Supplemental Table 2.
2.11. Construction of TRPA1-Luc plasmid and promoter reporter assay
Rat TRPA1 promoter fragment between −1979 bp and −59 bp was amplified by PCR using the primers listed in Supplemental Table 3 and cloned into pGL3-basic luciferase reporter vector (Promega, Madison, WI). Site-directed mutagenesis of the putative FXR binding site from −1657bp to −1655bp was accomplished using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technology, Santa Clara, CA) by using the mutagenic primers listed in Supplemental Table 3. 293 T cells were transfected with TRPA1-Luc plasmid and Renilla luciferase plasmid, followed by stimulation with 5 μM GW4064 for 24 h. TRPA1 promoter activity was determined by using the Dual-Luciferase Reporter Assay kit (Promega) according to the manufacturer's instructions. The plasmid expressing Renilla luciferase was used for normalization of luciferase activity.
2.12. RNA purification and real time PCR
Total RNA was extracted from INS-1 832/13 cells or islets using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Real time PCR was performed using qTOWER 2.2 real time PCR System (Germany). The primer sequences are listed in Supplemental Table 4. TRPA1 expression was normalized to GAPDH or β-actin.
2.13. Insulin measurements
Insulin secretion was assayed as described previously [22].
2.14. Statistical analyses
Data are presented as mean ± S.E.M. for the indicated number of experiments (n). Statistical significance was evaluated using the independent t-test or One-way ANOVA. Data were considered significant when p < 0.05.
3. Results
3.1. RYGB improves GSIS and reverses hyperglycemia in GK rats
RYGB and sham surgeries were successfully conducted in GK rats. Intravenous glucose tolerance test (IVGTT) was performed to evaluate plasma insulin and glucose levels two months after surgeries. GK-RYGB rats had significant improvement of the first phase of GSIS (GK-RYGB vs. GK-sham, 3.77 ± 0.33 vs. 1.74 ± 0.32, 2.43 ± 0.30 vs. 1.18 ± 0.09, and 2.07 ± 0.33 vs. 1.15 ± 0.18 ng/ml/kg at 2, 5 and 10 min, respectively, after glucose administration), whereas lower than the values (thus 7.77 ± 0.27, 6.6 ± 0.6 and 6.01 ± 0.63 ng/ml/kg at 2, 5 and 10 min, respectively) of the Wistar controls (Figure 1A), consistent with the previous observations [6]. In accordance with these findings in vivo, treatment of GK-RYGB islets with 16.8 mM glucose for 10 min led to a ∼7-fold increase of insulin secretion (p < 0.05), in contrast to only a ∼1.4-fold stimulation in GK-sham islets (Figure 1B). This was paralleled by improved glycemic controls (Figure 1C) and a sustained reduction in glucose levels (Figure 1D) in GK-RYGB rats, consistent with remission of diabetes. Note that there was no significant difference of the body weights between GK-RYGB and GK-sham groups (Supplemental Fig. 1), as reported previously [6].
Figure 1.

Effects of RYGB on insulin secretion and plasma glucose level. (A) Plasma insulin of Wistar (symbols), GK-Sham (triangles) and GK-RYGB (squares) rats. The insulin values were normalized against body weight of rats in each group. Data are means ± S.E.M. n = 5–6. *, p < 0.05, **, p < 0.01 vs. GK-Sham. (B) Insulin secretion was stimulated with 2.8 and 16.8 mM glucose for 10 min in islets isolated from Wistar, GK-Sham, and GK-RYGB rats. Values represent the amount of secretion per islet. Data are means ± S.E.M. of 3–8 independent experiments. *, p < 0.05. (C) Plasma glucose levels after intravenous glucose injection (1 g/kg) in Wistar (symbols, n = 12), GK-Sham (triangles, n = 8), and GK-RYGB rats (squares, n = 6). Data are means ± S.E.M. **, p < 0.01 vs. GK-Sham. (D) Plasma glucose in Wistar (symbols), GK-Sham (triangles) and GK-RYGB (squares) rats. Data are means ± S.E.M. n = 5–10. *, p < 0.05; **, p < 0.01 vs. GK-Sham. Statistics in A–D: One-way ANOVA with least significant difference (LSD) post hoc test.
3.2. RYGB restores TRPA1 expression and current in GK β-cells
Analysis of TRPA1 expression revealed that GK-sham islets had ∼47% (p < 0.01) and ∼46% (p < 0.01) lower TRPA1 mRNA and protein levels, respectively, than the Wistar controls (Figure 2A, B). Notably, RYGB led to an increase of TRPA1 expression: GK-RYGB islets had ∼34% (p < 0.01) and ∼39% (p < 0.01) higher in TRPA1 mRNA and protein levels, respectively, than the sham controls (Figure 2A, B). The increase in TRPA1 expression was consistent with an enhanced TRPA1 current recorded in single β-cells from these rats (RYGB vs. sham, −5.25 ± 0.8 pA/pF vs. −2.29 ± 0.37 pA/pF, p < 0.01) (Figure 2C–F), albeit still lesser than the value of Wistar controls (−8.91 ± 1.36 pA/pF) (Figure 2C, E and F).
Figure 2.

Effects of RYGB on TRPA1 expression and currents in β-cells. (A, B) TRPA1 mRNA (A) and protein (B) in islets isolated from Wistar, GK-Sham, and GK-RYGB rats. A representative immunoblot is shown on the top (B). β-actin was used as internal and loading control. Intensities were quantified and normalized against the level of actin and expressed as the percentage of protein abundance of Wistar islets. Data are means ± S.E.M. of 5 experiments. *, p < 0.05; **, p < 0.01. (C–E) Representative recording of AITC (100 μM)-induced currents in β-cells isolated from Wistar (C), GK-Sham (D), and GK-RYGB (E) rats. (F) Mean (±S.E.M.) of AITC-sensitive currents of β-cells from Wistar (white bar, n = 10), GK -Sham (grey bar, n = 10), and GK-RYGB (dark bar, n = 13) rats. **, p < 0.01. Statistics in A, B and F: One-way ANOVA with LSD post hoc test.
We next examined whether the observed reduction of TRPA1 in GK rats was due to hyperglycemia. We conducted the experiments by culturing INS-1 832/13 cells in 5.5 mM glucose (G5.5) or 30 mM glucose (G30) medium for 3 days. Western blotting revealed that TRPA1 protein levels were similar in the two groups (Supplemental Fig. 2). Thus, reduced TRPA1 expression in GK rats is not attributed to extracellular high glucose levels.
3.3. RYGB-potentiated GSIS is impaired in TRPA1−/− mice
To explore whether TRPA1 is involved in RYGB-potentiated GSIS, we performed RYGB and sham surgeries on TRPA1+/+ and TRPA1−/− mice, and determined GSIS in islets isolated from these mice (Figure 3A) or IVGTT in vivo (Figure 3B). Thus, treatment with 16.8 mM glucose for 10 min led to a ∼3.4-fold increase of insulin secretion as compared to 2.8 mM glucose in TRPA1+/+ sham islets, in contrast to ∼1.8-fold increase in TRPA1−/− sham islets (Figure 3A). Importantly, glucose stimulation produced a further ∼80% greater (p < 0.01) of insulin secretion in TRPA1+/+ RYGB islets, but not in TRPA1−/− RYGB islets (Figure 3A). In agreement with the observation made in islets, IVGTT results revealed that TRPA1+/+ RYGB mice displayed a marked increase (p < 0.01) of GSIS than sham controls, especially insulin secretion at 2 min after glucose administration (Figure 3B). Strikingly, however, TRPA1−/− RYGB mice responded with an attenuated increase (∼2.5-fold lower peak; p < 0.01) in insulin levels (Figure 3B). This was paralleled by a diminished effect of RYGB on glucose control in TRPA1−/− mice (Figure 3C). Notably, GSIS assayed either in islets (Supplemental Fig. 3) or IVGTT (Supplemental Fig. 4) in vivo was comparable between sham and non-surgery groups, suggesting that surgery did not affect secretory capacity of β-cells in these TRPA1 mice.
Figure 3.

GSIS and glucose levels in TRPA1+/+ and TRPA1−/− mice after RYGB and sham surgery. (A) Insulin secretion in islets isolated from TRPA1+/+ and TRPA1−/− mice after RYGB or sham surgeries. The islets were treated with 2.8 or 16.8 mM glucose for 10 min. Values represent the amount of secretion per islet. Data are means ± S.E.M. of 5 independent experiments per group. **, p < 0.01. (B, C) Plasma insulin (B) or glucose (C) levels in TRPA1+/+-sham (open symbols), TRPA1+/+-RYGB (filled symbols), TRPA1−/−-sham (open triangles) and TRPA1−/−-RYGB (filled triangles) mice. Data are means ± S.E.M. n = 5 (A) or 4–6 (B) mice per group. Data were analyzed by One-way ANOVA with LSD post hoc test. *, p < 0.05 vs. TRPA1+/+-sham, ##, p < 0.01 vs. TRPA1−/−-RYGB.
These findings prompted us to investigate the role of TRPA1 in β-cell electrical activity and insulin secretion, by analyzing the β-cell electrophysiological properties and insulin secretion in TRPA1+/+ and TRPA1−/− mice. As shown in Figure 4A, B, exposure to 2.8 mM glucose, TRPA1−/− β-cells had a more hyperpolarized membrane potential (TRPA1−/− vs. TRPA1+/+, −77.2 ± 1.1 mV (n = 66) vs. −70.2 ± 0.9 mV (n = 25), p < 0.001). Upon stimulation with 16.8 mM glucose, TRPA1−/− β-cells displayed lower firing rate (TRPA1−/− vs. TRPA1+/+, 0.54 ± 0.09 Hz (n = 54) vs. 1.24 ± 0.31 Hz (n = 25), p < 0.01), as well as a lesser extent of GSIS (∼2-fold vs. ∼3.6-fold increase in TRPA1−/− and TRPA1+/+ islets, respectively; p < 0.01) (Figure 4C). This is consistent with an attenuated increase of GSIS in TRPA1−/− mice in vivo (Figure 4D).
Figure 4.

β-cell electrical activity and insulin secretion in TRPA1+/+ and TRPA1−/− mice. (A, B) Representative membrane potential recordings in single β-cells isolated from TRPA1+/+ (A) and TRPA1−/− (B) mice. Resting potentials were determined from slow time-scale recordings on which a clear baseline was evident. (C) Insulin secretion in islets isolated from TRPA1+/+ and TRPA1−/− mice. The islets were treated with 2.8 or 16.8 mM glucose for 10 min. Values represent the amount of secretion per islet. Data are means ± S.E.M. of 7–9 independent experiments. **, p < 0.01. (D) Plasma insulin levels in TRPA1+/+ and TRPA1−/− mice. Data are means ± S.E.M. n = 6–10 mice. *, p < 0.05. (E, F) Representative membrane potential recordings in β-cells isolated from Wistar rats. AITC (100 μM) (E) or HC-030031 (100 μM) (F) was applied as indicated. No holding current was applied. (G, H) Insulin secretion was stimulated with 2.8 or 16.8 mM glucose for 30 min in islets from Wistar rats, with DMSO or AITC (100 μM) (G) or HC-030031 (100 μM) (H). Data are means ± S.E.M. of 7–13 (G) or 8–13 (H) independent experiments. **, p < 0.01. Statistics in C, D, G and H: One-way ANOVA with LSD post hoc test.
Moreover, we confirmed that, as previously reported [11], TRPA1 agonist AITC stimulated whereas its antagonist HC-030031 suppressed glucose-elicited firing (Figure 4E, F) and GSIS (Figure 4G, H) in β-cells from normal Wistar rats.
3.4. Lack of RYGB-induced TRPA1 expression in islets from FXR−/- mice
It has been reported that RYGB causes significant increase in BA levels in Zucker diabetic rats [15]. We determined whether RYGB surgery also exerts similar effects on BA levels in non-obese GK rats. Plasma BA levels were assessed in GK rats 2 weeks after RYGB or sham surgery. This revealed ∼1.5-fold higher levels of BAs in GK-RYGB rats (p < 0.05) (Figure 5A). As CDCA is one of the major primary bile acids in rodents [24], [25] and in humans [26], [27], and also the most potent endogenous FXR agonist [19], [27], we sought to determine whether RYGB also increases CDCA levels. The results indicate that GK-RYGB rats had ∼1.9-fold higher level of CDCA (p < 0.05) as compared with the sham controls (Figure 5B). Since FXR activation leads to suppression of expression of phosphoenolpyruvate carboxykinase (PEPCK), the well-known target gene regulated by FXR [28], we next examined whether RYGB influences expression of PEPCK. This revealed that PEPCK mRNA level was decreased by ∼60% (p < 0.05) in GK-RYGB islets as compared with the sham controls (Supplemental Fig. 5), confirming FXR activation after RYGB.
Figure 5.

Lack of RYGB-induced TRPA1 expression in islets from FXR−/- mice. (A) Plasma total bile acid in GK-Sham (white bar) and GK-RYGB (grey bar) rats. Data are means ± S.E.M. n = 3. Two-sided unpaired t-test was used for statistical analysis. *, p < 0.05. (B) Plasma CDCA in GK-Sham (white bar) and GK-RYGB (grey bar) rats. Data are means ± S.E.M. n = 3. Data were analyzed by two-sided unpaired t-test. *, p < 0.05. (C) A representative immunoblot of TRPA1 protein expression in islets from FXR+/+ and FXR−/- mice after RYGB or sham surgery. β-actin was used as internal and loading control. (D) TRPA1 protein was quantified and normalized against the level of β-actin and expressed as the percentage of protein abundance of FXR+/+ islets. Data are means ± S.E.M. of 4 independent experiments. Data were analyzed by One-way ANOVA (LSD). *, p < 0.05.
Given that FXR is responsible for the beneficial effects of BAs in remission of T2D after bariatric surgery [29], that it mediates the effects of BAs on GSIS in β-cells [18], and the crucial role of TRPA1 in RYGB-enhanced GSIS (Figure 3A, B), we reasoned whether FXR is involved in RYGB-induced increase in TRPA1 expression in β-cells. To this end, we performed RYGB and sham surgeries on FXR+/+ and FXR−/- mice, and determined TRPA1 protein expression in islets from these mice. Western blot results revealed that FXR+/+-RYGB islets had a more abundance of TRPA1 protein levels than the sham controls (Figure 5C, lane 2 vs. 1; Figure 5D, column 2 vs. 1). Whereas, TRPA1 protein abundance in FXR−/--RYGB islets was identical to that in FXR−/--sham controls (Figure 5C, lane 4 vs. 3; Figure 5D, column 4 vs. 3). These results indicate that RYGB-induced TRPA1 expression requires the participation of FXR.
To further determine whether FXR affects TRPA1 expression in β-cells, we examined TRPA1 expression in FXR knockdown and overexpressing INS-1 832/13 cells. FXR knockdown led to reduced TRPA1 mRNA (Figure 6A) and protein (Figure 6B) levels. In agreement, FXR−/- islets had ∼55% (p < 0.05) and ∼37% (p = 0.01) lower TRPA1 mRNA and protein levels, respectively, than FXR+/+ islets (Figure 6C, D). On the other hand, FXR overexpressing INS-1 832/13 cells (Supplemental Fig. 6) displayed an increased TRPA1 mRNA (Figure 6E) and protein (Figure 6F) levels. The same observations were also made in INS-1 832/13 cells chronically treated with either GW4064 (Figure 6G, H) or CDCA (Figure 6I).
Figure 6.

FXR-dependent TRPA1 expression. (A, B) qPCR (A) and western blot (B) analysis of TRPA1 mRNA (A) and protein (B) expression in FXR knockdown INS-1 832/13 cells. A representative immunoblot is shown on the top, and GAPDH was used as internal and loading control (B). Data are means ± S.E.M. of 3 (A) and 5 (B) independent experiments. *, p < 0.05. (C, D) As in (A, B) but TRPA1 mRNA (C) and protein (D) expression in islets from FXR+/+ and FXR−/- mice. Data are means ± S.E.M. of 4 independent experiments. *, p < 0.05. (E, F) As in (A, B) but TRPA1 mRNA (E) and protein (F) levels in FXR overexpressed INS-1 832/13 cells. Data are means ± S.E.M. of 4 independent experiments. *, p < 0.05. (G–I) TRPA1 mRNA (H) and protein (G, I) levels were determined by treatment with 5 μM GW4064 for 2 h (G) or 12 h (H) or 50 μM CDCA for 15 h (I) in INS-1 832/13 cells, respectively. Data are means ± S.E.M. of 4 (G, H) or 3 (I) independent experiments. *, p < 0.05. Data were analyzed by two-sided unpaired t-test.
Finally, we confirmed the FXR-dependent effect of BAs on GSIS. Thus, treatment with GW4064 (Supplemental Fig. 7) or CDCA (Supplemental Fig. 8) led to a significant increase of GSIS in islets from FXR+/+ mice, but not in islets from FXR−/- mice, consistent with previous reports [18].
3.5. FXR activation induces direct binding of FXR to the promoter of TRPA1 to promote TRPA1 expression
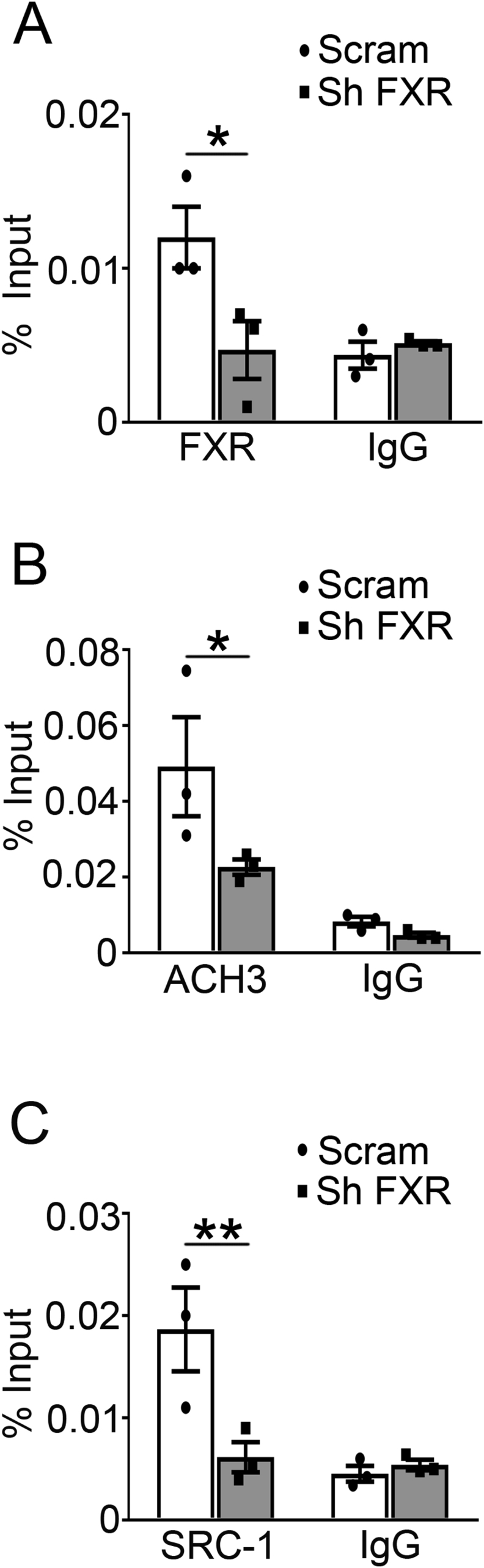
To get insight into the mechanisms underlying FXR-regulated TRPA1 expression, we surveyed DNA sequence in the TRPA1 locus, which revealed a consensus “AGGTCA” sequence of the FXR binding site (FXRE) in the TRPA1 promoter that is conserved across a varies of species (Figure 7A). Chromatin immunoprecipitation (ChIP) assay revealed that stimulation of FXR with GW4064 for 2 h (Figure 7B) increased whereas shRNA knockdown of FXR suppressed (Supplemental Fig. 9A) binding of FXR to the TRPA1 promoter. To investigate whether mutation of the FXR binding site would abolish FXR-induced TRPA1 expression, we constructed a luciferase reporter driven by either the wild-type FXRE or its mutant form “AGGCAG” (Figure 7C), and transfected them into 293 T cells, followed by GW4064 treatment and detection of the luciferase activity of the reporter gene. We found that GW4064 increased expression of the reporter driven by the wide-type FXRE, but not by the mutant FXRE (Figure 7D), demonstrating that FXRE in the TRPA1 promoter is essential for FXR-mediated TRPA1 expression. Our ChIP results also showed that GW4064 increased (Figure 7E) whereas FXR knockdown decreased (Supplemental Fig. 9B) ACH3 marker at the TRPA1 promoter. Since the gene expressing histone acetyltransferase SRC1 is one of the key histone modifiers generating ACH3 [30], we next determined whether SRC1 mediates ACH3 of TRPA1 promoter in INS-1 832/13 cells. ChIP assay revealed that GW4064 markedly increased (Figure 7F) whereas FXR knockdown decreased (Supplemental Fig. 9C) SRC1 binding to the TRPA1 promoter. To further determine the role of SRC1 in mediating FXR-dependent stimulation of the TRPA1 gene expression, we analyzed TRPA1 expression in INS-1 832/13 cells transfected with a scramble or SRC1 shRNA. We found that SRC1 knockdown abolished the stimulatory effect of GW4064 on TRPA1 expression, albeit the FXR agonist caused a significant increase (p < 0.01) of TRPA1 expression in scramble control cells (Figure 7G). These results suggest that FXR activation recruits SRC1 to increase ACH3 markers at the promoter of TRPA1 and, subsequently, promotes TRPA1 expression and GSIS in β-cells. The essential role of SRC1 in mediating FXR-dependent regulation of GSIS is further evidenced by experiments in SRC1 knockdown cells (Figure 7H). Thus, treatment with GW4064 led to ∼1.8-fold (p < 0.01) increase of GSIS in control INS-1 832/13 cells, but not in SRC1 knockdown cells. Furthermore, the FXR agonist failed to enhance GSIS in islets from TRPA1−/− mice (Figure 7I), again indicating that stimulatory effect of FXR on GSIS is mediated by TRPA1.
Figure 7.

Binding of FXR, recruitment of the histone modifier SRC1, and histone acetylation at the promoter of TRPA1. (A) A conserved FXR binding element (FXRE) at the promoter of TRPA1 gene in various species. (B) ChIP assays were performed using anti-FXR antibody in INS-1 832/13 cells treated with 5 μM GW4064 for 2 h. Control IgG served as a negative control. Data are means ± S.E.M. n = 3. *, P < 0.05. (C) Site-directed mutagenesis of FXRE at the promoter of TRPA1. (D) 293 T cells transfected with a luciferase reporter driven by either wild-type or mutant FXRE followed by treatment with 5 μM GW4064 for 24 h prior to luciferase activity analysis for the reporter expression. Data are means ± S.E.M. n = 3. *, P < 0.05. (E) ChIP assays were performed to determine histone acetylation at the promoter of TRPA1 in INS-1 832/13 cells treated with 5 μM GW4064 for 2 h. Control IgG served as a negative control. Data are means ± S.E.M. n = 3. **, P < 0.01. (F) As in (E) but ChIP assays were performed using anti-SRC1 antibody. Data are means ± S.E.M. n = 3. **, P < 0.01. (G) qPCR analysis of TRPA1 mRNA expression in scrambled or shSRC1 INS-1 832/13 cells treated with DMSO or 5 μM GW4064 for 2 h. Data are means ± S.E.M. of 3 independent experiments. **, P < 0.01. (H) Insulin secretion in shSRC1 INS-1 832/13 cells at 16.8 mM glucose with DMSO or 5 μM GW4064 for 30 min. Data are means ± S.E.M. of 6 independent experiments. **, p < 0.01. (I) Insulin secretion in islets isolated from TRPA1+/+ and TRPA1−/− mice. The islets were treated with 16.8 mM glucose with of DMSO or 5 μM GW4064 for 30 min. Values are expressed as percentage of secretion of islets from TRPA1+/+ mice at 16.8 mM glucose and DMSO. Data are means ± S.E.M. n = 6–8. *, p < 0.05. Data in B, D, E, F, G, H, and I were analyzed by One-way ANOVA with LSD post hoc test. (J) Schematic representation of the molecular mechanism of RYGB-induced TRPA1 expression, and the role of TRPA1 in stimulus-secretion coupling in β-cells. See text for details.
4. Discussion
In this study, we demonstrate that RYGB restores TRPA1 expression in β-cells from diabetic GK rats, which significantly improves the ability of diabetic β-cells to respond to glucose, thereby enhancing the first phase of GSIS and ameliorating hyperglycemia in GK rats. For the first time to our knowledge, we show that RYGB increases TRPA1 expression via BAs/FXR-mediated increase of ACH3 at the promoter of TRPA1. Our data also revealed that TRPA1 plays a crucial role in glucose-evoked electrical activity and insulin secretion in β-cells. Of note, we find that TRPA1 participates in RYGB-improved GSIS and glycemic controls in diabetic GK rats. These findings therefore assign FXR-dependent regulation of TRPA1 a critical role in improvement of β-cell function and glucose homeostasis in diabetes after RYGB surgery.
Bile Acids have been implicated as key mediators of the weight-independent effects of bariatric surgery with respect to glucose homeostasis. Our data show that RYGB results in a significant increase of the plasma total bile acid, particularly CDCA in non-obese GK rats (Figure 5A, B). Interestingly, this effect of RYGB appears to be more pronounced in high-fat-high-cholesterol diet Wistar rats [31] and obese diabetic Zucker rats [32]. Given that CDCA has the highest affinity for FXR among BA species and the most potent endogenous FXR agonist [19], [27], elevated CDCA would leads to FXR activation in GK-RYGB rats. In agreement, expression of PEPCK, the target gene that is negatively regulated by FXR activation [28], was markedly suppressed in GK-RYGB rats (Supplemental Fig. 5), again confirming FXR activation by RYGB.
Studies in rodent β-cells and β-cell lines have revealed that BAs potentiates GSIS, an effect dependent on FXR activation [17]. However, the precise molecular mechanism underlying this FXR-mediated effect remains unclear. We now demonstrate that FXR activation leads to increased TRPA1 expression in β-cells, which mediate the stimulatory effect of FXR activation on GSIS. This concept is corroborated by five pieces of evidence. First, there is existence of the FXR binding site (FXRE) at the TRPA1 promoter (Figure 7A). Stimulation with FXR agonist GW4064 led to an increase of FXR binding to the TRPA1 promoter (Figure 7B). Second, activation of FXR by CDCA or GW4064 increased TRPA1 expression at mRNA (Figure 6G) and protein (Figure 6H, I) levels. Third, knockdown FXR by shRNA resulted in reduced TRPA1 expression (Figure 6A, B). Fourth, TRPA1 expression was substantially decreased in FXR−/- islets (Figure 6C, D). Fifth, GW4064 failed to potentiate GSIS in TRPA1−/− mice, whereas it caused ∼1.6-fold enhancement in control mice (Figure 7I). Importantly, our findings demonstrate that FXR activation promoted the recruitment of the epigenetic regulator SRC1 (Figure 7F), and subsequently enhancing histone acetylation at the TRPA1 locus (Figure 7E). This would lead to increased expression of TRPA1 gene, given that acetylation of nucleosomal histones increases the accessibility of DNA to transcription factors and leads to increased transcription at the target DNA locus [33]. Consistently, the lack of effect of GW4064 on TRPA1 gene transcription (Figure 7G) and GSIS (Figure 7H) in shSRC1 cells again indicates that SRC1 is responsible for these effects of FXR. Thus, our results unravel a novel molecular mechanism of regulation of TRPA1 expression in pancreatic β-cells: FXR-mediated increase of TRPA1 expression via SRC1 and histone acetylation (Figure 7J).
Glucose-induced insulin secretion is attributed to the membrane depolarization upon closure of KATP channels as a result of glucose metabolism and ATP production, which results in activation of voltage-dependent Ca2+ channels and initiation of insulin secretion [8]. Importantly, however, closure of KATP channels alone is not sufficient to cause membrane depolarization to reach the threshold for regenerative electrical activity (−60 mV) [34]. Hence, β-cells must be equipped with an inward current that may be tonically active, to bring the membrane potential away from the K+-equilibrium potential. It has been reported previously that activation of TRPA1 results in an increase of cytosolic Ca2+ and insulin secretion in β-cells [13]. We now confirm and extend these observations. We found that ablation of TRPA1 resulted in decreased glucose-evoked electrical activity (Figure 4B) and insulin secretion (Figure 4C, D). This observation is further supported by the experiments performed in rat β-cells treated with TRPA1 agonist AITC (Figure 4E, G) or antagonist HC-030031 (Figure 4F, H), which resulted in enhanced or suppressed response to glucose. Based on these observations and those published [11], [13], it is justifiable to conclude that the TRPA1 channel contributes, at least partly, to the background inward current in β-cells. Moreover, our data also suggest that TRPA1 involves in the development of resting membrane potential in β-cells, given that TRPA1-deficient β-cells possessed a more hyperpolarized membrane potential. It is worth noting that, in addition to TRPA1, other TRP members such as TRPM2 may also contribute to the glucose-induced increase in background current [35]. Nevertheless, our current findings suggest a more prominent role for TRPA1 in β-cell stimulus-secretion coupling.
It is interesting to note that diabetic GK β-cells displayed a marked reduction in TRPA1 expression (Figure 2). This is likely to impair the secretory response to glucose stimulation, given that TRPA1 is required for the ability of glucose to evoke β-cell electrical activity (Figure 4A, B) and insulin secretion (Figure 4C, D). Thus, reduced TRPA1 expression would account, at least partly, for the diminished GSIS in GK rats (Figure 1A, B). Importantly, our data also suggest that restored TRPA1 expression would contribute to improved GSIS after RYGB. Three pieces of evidence corroborate this notion. First, GK-RYGB islets displayed higher TRPA1 expression than GK-sham controls (Figure 2A, B). Increase in TRPA1 expression was in agreement with a greater TRPA1 agonist-induced current in β-cells from GK-RYGB rats (Figure 2E). Second, increased TRPA1 expression is paralleled by improved GSIS in GK-RYGB rats (Figure 1A, B). Third, the effect of RYGB on GSIS was significantly attenuated in TRPA1−/− mice (Figure 3A, B). This led to decreased efficacy of RYGB in improving glycemic controls (Figure 3C).
Collectively, our findings suggest that the BA/FXR/SRC1 axis plays a crucial role in improved GSIS and glycemic control in diabetic GK rats following RYGB (Figure 7J). These are important findings as previously little was known as to whether and how BA/FXR signaling can enhance the secretory capacity of β-cells after RYGB, and whether and how alterations in TRPA1 in β-cells would interfere with the ability of glucose to evoke β-cell electrical activity and stimulate insulin secretion, in particular in diabetes. Thus, our studies unravel the BA/FXR/SRC1 axis-mediated regulation of TRPA1 expression critical for improved GSIS in GK rats after RYGB. It is also worth pointing out that the significance of FXR-dependent regulation of TRPA1 may not only be in non-obese GK rats. It can be speculated that it may also contribute to RYGB-induced improvement of the first phase of insulin secretion in obese diabetes, given the evident effects of RYGB on elevating CDCA and the total BAs [32], as well as TRPA1 expression (unpublished data) observed in obese Zucker rats. Thus, further exploration of this pathway would help to define novel approaches to treatment of T2D.
Author contributions
X.K., Y.T., B.L., L.Z., L.F., L.W., and L.Z. performed the experiments. X.K., Y.T., and X.M. analyzed the data. H. Z. provided expertise of RYGB surgery and GK rats. X.H. contributed to the discussion and assisted with the editing of the manuscript. X.M. designed the project and wrote the manuscript.
Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 81670708 and No. 81370914 to XM; No.81400801 to XK). Shenzhen Basic Research Project (JCYJ20160307155501522 to XK). New teacher natural science research project of Shenzhen University (2016093 to XK).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2019.08.009.
Conflict of interest
The authors declare no competing financial interests.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Fig s1.
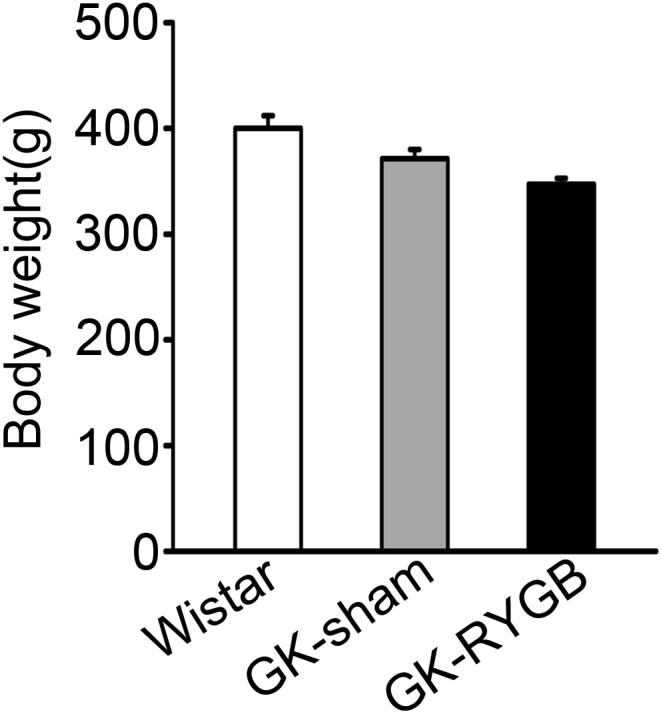
Fig s2.
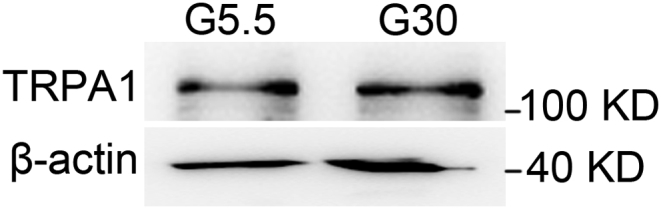
Fig s3.

Fig s4.

Fig s5.
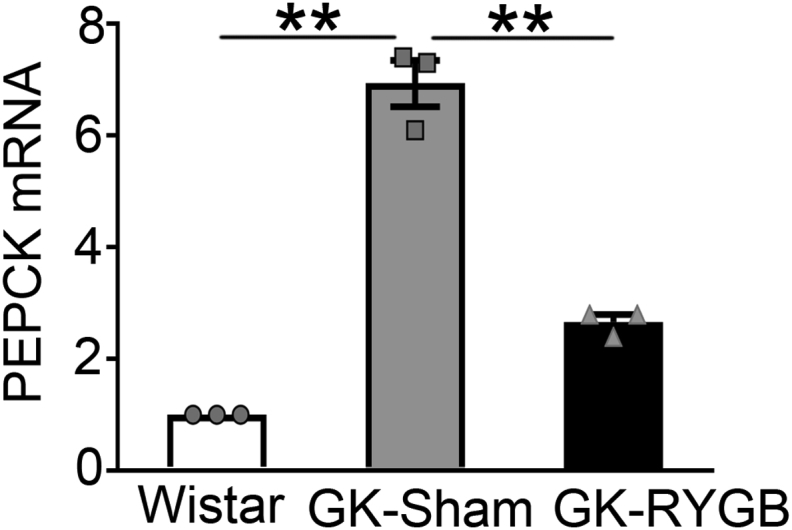
Fig s6.
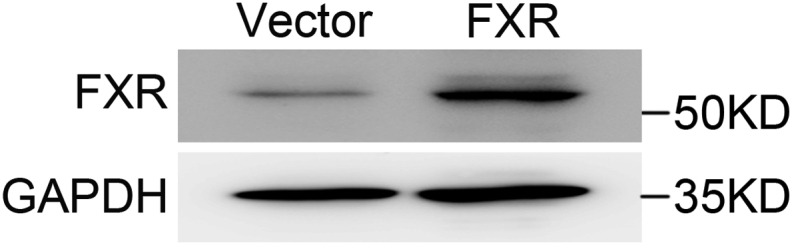
Fig s7.
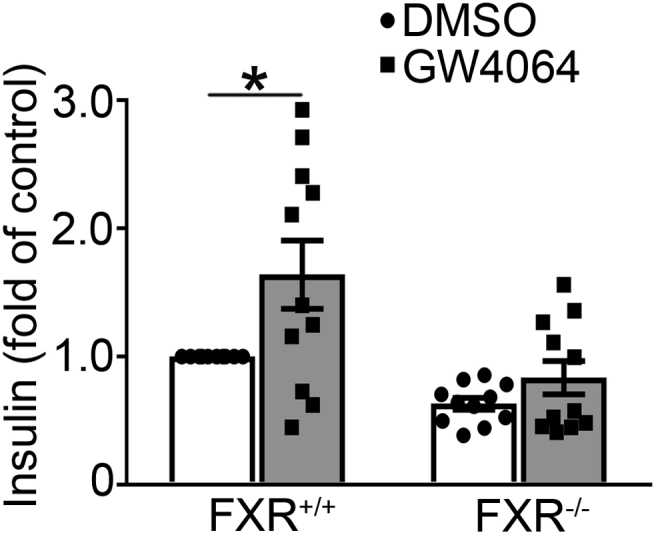
Fig s8.
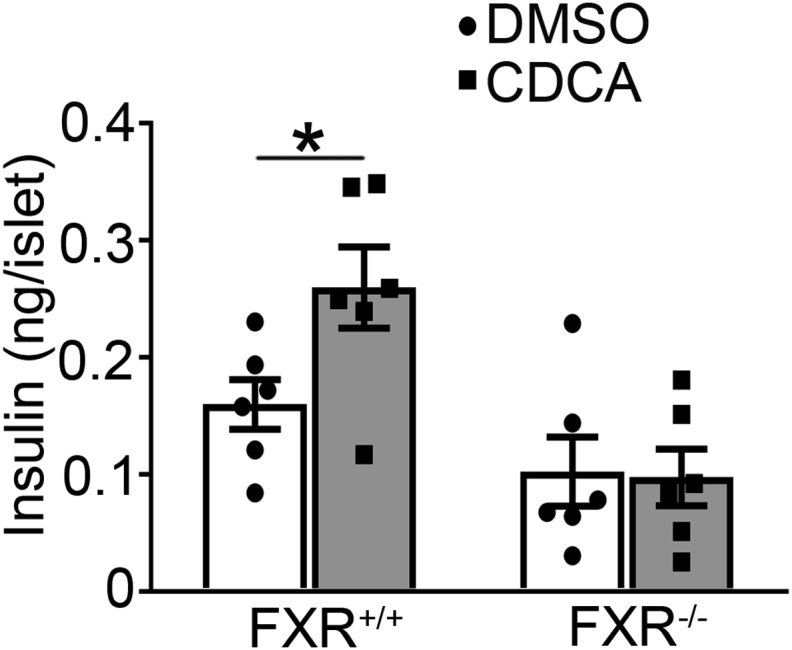
Fig s9.
References
- 1.Turner R., Cull C., Holman R. United Kingdom Prospective Diabetes Study 17: a 9-year update of a randomized, controlled trial on the effect of improved metabolic control on complications in non-insulin-dependent diabetes mellitus. Annals of Internal Medicine. 1996;124:136–145. doi: 10.7326/0003-4819-124-1_part_2-199601011-00011. [DOI] [PubMed] [Google Scholar]
- 2.Schauer P.R., Burguera B., Ikramuddin S., Cottam D., Gourash W., Hamad G. Effect of laparoscopic Roux-en Y gastric bypass on type 2 diabetes mellitus. Annals of Surgery. 2003;238:467–484. doi: 10.1097/01.sla.0000089851.41115.1b. discussion 484-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fried M., Ribaric G., Buchwald J.N., Svacina S., Dolezalova K., Scopinaro N. Metabolic surgery for the treatment of type 2 diabetes in patients with BMI <35 kg/m2: an integrative review of early studies. Obesity Surgery. 2010;20:776–790. doi: 10.1007/s11695-010-0113-3. [DOI] [PubMed] [Google Scholar]
- 4.Salinari S., Bertuzzi A., Guidone C., Previti E., Rubino F., Mingrone G. Insulin sensitivity and secretion changes after gastric bypass in normotolerant and diabetic obese subjects. Annals of Surgery. 2013;257:462–468. doi: 10.1097/SLA.0b013e318269cf5c. [DOI] [PubMed] [Google Scholar]
- 5.Martinussen C., Bojsen-Moller K.N., Dirksen C., Jacobsen S.H., Jorgensen N.B., Kristiansen V.B. Immediate enhancement of first-phase insulin secretion and unchanged glucose effectiveness in patients with type 2 diabetes after Roux-en-Y gastric bypass. American Journal of Physiology, Endocrinology and Metabolism. 2015;308:E535–E544. doi: 10.1152/ajpendo.00506.2014. [DOI] [PubMed] [Google Scholar]
- 6.Qian B., Zhou X., Li B., Li B., Liu Z., Wu J. Reduction of pancreatic beta-cell dedifferentiation after gastric bypass surgery in diabetic rats. Journal of Molecular Cell Biology. 2014;6:531–534. doi: 10.1093/jmcb/mju042. [DOI] [PubMed] [Google Scholar]
- 7.Henquin J.C. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52:739–751. doi: 10.1007/s00125-009-1314-y. [DOI] [PubMed] [Google Scholar]
- 8.Rorsman P., Eliasson L., Renstrom E., Gromada J., Barg S., Gopel S. The cell physiology of biphasic insulin secretion. News in Physiological Sciences. 2000;15:72–77. doi: 10.1152/physiologyonline.2000.15.2.72. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson D.A., Philipson L.H. TRP channels of the pancreatic beta cell. Handbook of Experimental Pharmacology. 2007:409–424. doi: 10.1007/978-3-540-34891-7_24. [DOI] [PubMed] [Google Scholar]
- 10.Drews G., Krippeit-Drews P., Dufer M. Electrophysiology of islet cells. Advances in Experimental Medicine & Biology. 2010;654:115–163. doi: 10.1007/978-90-481-3271-3_7. [DOI] [PubMed] [Google Scholar]
- 11.Cao D.S., Zhong L., Hsieh T.H., Abooj M., Bishnoi M., Hughes L. Expression of transient receptor potential ankyrin 1 (TRPA1) and its role in insulin release from rat pancreatic beta cells. PLoS One. 2012;7 doi: 10.1371/journal.pone.0038005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Motter A.L., Ahern G.P. TRPA1 is a polyunsaturated fatty acid sensor in mammals. PLoS One. 2012;7 doi: 10.1371/journal.pone.0038439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Numazawa S., Takase M., Ahiko T., Ishii M., Shimizu S., Yoshida T. Possible involvement of transient receptor potential channels in electrophile-induced insulin secretion from RINm5F cells. Biological & Pharmaceutical Bulletin. 2012;35:346–354. doi: 10.1248/bpb.35.346. [DOI] [PubMed] [Google Scholar]
- 14.Ferrannini E., Camastra S., Astiarraga B., Nannipieri M., Castro-Perez J., Xie D. Increased bile acid synthesis and deconjugation after biliopancreatic diversion. Diabetes. 2015;64:3377–3385. doi: 10.2337/db15-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar S., Lau R., Hall C., Palaia T., Brathwaite C.E., Ragolia L. Bile acid elevation after Roux-en-Y gastric bypass is associated with cardio-protective effect in Zucker Diabetic Fatty rats. International Journal of Surgery. 2015;24:70–74. doi: 10.1016/j.ijsu.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Patti M.E., Houten S.M., Bianco A.C., Bernier R., Larsen P.R., Holst J.J. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obesity (Silver Spring) 2009;17:1671–1677. doi: 10.1038/oby.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dufer M., Horth K., Wagner R., Schittenhelm B., Prowald S., Wagner T.F. Bile acids acutely stimulate insulin secretion of mouse beta-cells via farnesoid X receptor activation and K(ATP) channel inhibition. Diabetes. 2012;61:1479–1489. doi: 10.2337/db11-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Renga B., Mencarelli A., Vavassori P., Brancaleone V., Fiorucci S. The bile acid sensor FXR regulates insulin transcription and secretion. Biochimica et Biophysica Acta. 2010;1802:363–372. doi: 10.1016/j.bbadis.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Modica S., Gadaleta R.M., Moschetta A. Deciphering the nuclear bile acid receptor FXR paradigm. Nuclear Receptor Signaling. 2010;8:e005. doi: 10.1621/nrs.08005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X., Huang S., Gao M., Liu J., Jia X., Han Q. Farnesoid X receptor (FXR) gene deficiency impairs urine concentration in mice. Proceedings of the National Academy of Sciences of the U S A. 2014;111:2277–2282. doi: 10.1073/pnas.1323977111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nestoridi E., Kvas S., Kucharczyk J., Stylopoulos N. Resting energy expenditure and energetic cost of feeding are augmented after Roux-en-Y gastric bypass in obese mice. Endocrinology. 2012;153:2234–2244. doi: 10.1210/en.2011-2041. [DOI] [PubMed] [Google Scholar]
- 22.Kong X., Yan D., Sun J., Wu X., Mulder H., Hua X. Glucagon-like peptide 1 stimulates insulin secretion via inhibiting RhoA/ROCK signaling and disassembling glucotoxicity-induced stress fibers. Endocrinology. 2014;155:4676–4685. doi: 10.1210/en.2014-1314. [DOI] [PubMed] [Google Scholar]
- 23.Barg S., Ma X., Eliasson L., Galvanovskis J., Gopel S.O., Obermuller S. Fast exocytosis with few Ca(2+) channels in insulin-secreting mouse pancreatic B cells. Biophysical Journal. 2001;81:3308–3323. doi: 10.1016/S0006-3495(01)75964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Aguiar Vallim T.Q., Tarling E.J., Edwards P.A. Pleiotropic roles of bile acids in metabolism. Cell Metabolism. 2013;17:657–669. doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwarz M., Russell D.W., Dietschy J.M., Turley S.D. Alternate pathways of bile acid synthesis in the cholesterol 7alpha-hydroxylase knockout mouse are not upregulated by either cholesterol or cholestyramine feeding. The Journal of Lipid Research. 2001;42:1594–1603. [PubMed] [Google Scholar]
- 26.Chiang J.Y. Bile acid metabolism and signaling. Comparative Physiology. 2013;3:1191–1212. doi: 10.1002/cphy.c120023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khurana S., Raufman J.P., Pallone T.L. Bile acids regulate cardiovascular function. Clin Transl Sci. 2011;4:210–218. doi: 10.1111/j.1752-8062.2011.00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y.D., Chen W.D., Moore D.D., Huang W. FXR: a metabolic regulator and cell protector. Cell Research. 2008;18:1087–1095. doi: 10.1038/cr.2008.289. [DOI] [PubMed] [Google Scholar]
- 29.Ryan K.K., Tremaroli V., Clemmensen C., Kovatcheva-Datchary P., Myronovych A., Karns R. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature. 2014;509:183–188. doi: 10.1038/nature13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sterner D.E., Berger S.L. Acetylation of histones and transcription-related factors. Microbiology and Molecular Biology Reviews. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spinelli V., Lalloyer F., Baud G., Osto E., Kouach M., Daoudi M. Influence of Roux-en-Y gastric bypass on plasma bile acid profiles: a comparative study between rats, pigs and humans. International Journal of Obesity. 2016;40:1260–1267. doi: 10.1038/ijo.2016.46. [DOI] [PubMed] [Google Scholar]
- 32.Bhutta H.Y., Rajpal N., White W., Freudenberg J.M., Liu Y., Way J. Effect of Roux-en-Y gastric bypass surgery on bile acid metabolism in normal and obese diabetic rats. PLoS One. 2015;10 doi: 10.1371/journal.pone.0122273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haery L., Thompson R.C., Gilmore T.D. Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes Cancer. 2015;6:184–213. doi: 10.18632/genesandcancer.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rorsman P., Ashcroft F.M. Pancreatic beta-cell electrical activity and insulin secretion: of mice and men. Physiological Reviews. 2018;98:117–214. doi: 10.1152/physrev.00008.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yosida M., Dezaki K., Uchida K., Kodera S., Lam N.V., Ito K. Involvement of cAMP/EPAC/TRPM2 activation in glucose- and incretin-induced insulin secretion. Diabetes. 2014;63:3394–3403. doi: 10.2337/db13-1868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.