

Abstract
Background
In various biological processes and cell functions, Post Translational Modifications (PTMs) bear critical significance. Hydroxylation of proline residue is one kind of PTM, which occurs following protein synthesis. The experimental determination of hydroxyproline sites in an uncharacterized protein sequence requires extensive, time-consuming and expensive tests.
Methods
With the torrential slide of protein sequences produced in the post-genomic age, certain remarkable computational strategies are desired to overwhelm the issue. Keeping in view the composition and sequence order effect within polypeptide chains, an innovative in-silico> predictor via a mathematical model is proposed.
Results
Later, it was stringently verified using self-consistency, cross-validation and jackknife tests on benchmark datasets. It was established after a rigorous jackknife test that the new predictor values are superior to the values predicted by previous methodologies.
Conclusion
This new mathematical technique is the most appropriate and encouraging as compared with the existing models.
Keywords: PseAAC, Hydroxylation of proline, Post Translational Modifications (PTMs), Sequence-coupling model, Mammalian proteins, Hydroxyproline
1. INTRODUCTION
Collagens are profoundly plenteous mammalian proteins which possess abundant hydroxyproline [1] that plays a key role in its stability. The structure of collagen is stringy and long; nearly a quarter or even more of total protein content in mammals is comprised of collagen [2]. In medical applications, collagens work as a major constituent while contributing to wound healing [3], burns surgery [4] and cosmetic surgery [5]. Their asymmetrical behavior and irregular movements may contribute to stomach disease [6] and lung cancer [7]. The ability to predict hydroxyproline (HyP) sites as a result of post-translational modifications in proteins provides precious information useful for both biomedical research and medication evolution [8]. Hydroxyproline is a non-essential amino acid which means that it is mostly synthesized with other amino acids in the liver and need not to be obtained directly through systemic ingestion. Proline undergoes hydroxylation by the conversion of group in proline residue into group or a hydroxyl group [8] as shown in Fig. (1).
Fig. (1).
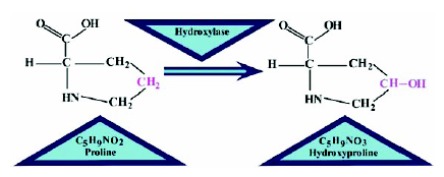
Figure shows how group is converted into group in the process of proline hydroxylation.
Owing to its significance for an in-depth understanding of the cellular biological process and discovering drug against cancers and other major diseases, many efforts have been made by other scientists in this regard [9-17]. Although, experimental techniques based on mass spectrometry exist that are used to determine hydroxylation sites of a given protein [18], however, this is laborious, tedious and high-priced. As a multitude of proteomic sequences are gathered into databanks each day, it is extremely desirable to devise an integrated and robust computational technique incorporating the composition and sequence order effect to determine potential hydroxylation sites with greater accuracy. Researchers have proposed a few methodologies for this purpose. However, the existing predictors lack the most pertinent details of features obscured within the primary sequences that prove crucial for reaching an accurate decision. Hydroxylation process had been of great interest to many researchers. Quantification of hydroxyproline was estimated by Colgrave, et al. [1] by using multiple-reaction-monitoring mass spectrometry. A mathematical modeling has been developed to understand the microbial behavior and their communities [19]. It was shown that the hydroxyproline and hydroxylysine in collagen were integrated by a clear extraordinary pathway, in which proline and lysine were hydroxylated after they were consolidated into a comprehensive polypeptide antecedent of collagen. Berg, et al. [20] defined a system that was set up to examine the inadequacy of collagen in connective tissues occurring due to lack of ascorbates to some extent.
The isolation and partial characterization of highly purified protocollagen proline hydroxylase and hydroxylation of proline in synthetic polypeptides with purified procollagen hydroxylase were elaborated by Halme et al. [21] and Kivirikko et al. [22]. Morgan, et al. [23] investigated, in terms of the distribution, the frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. Hydroxylation of lysine and crosslinking of collagens have been discussed in “Posttranslational Modifications of Proteins” [24]. Shi, Shao-Ping, et al. [25] presented a new method named as PredHydroxy to mechanize the forecast of the proline and lysine hydroxylation locales in term of position weight of 8 high-quality amino acid indices and support vector machines. The metabolism for the proline, hydroxyproline and a survey of activity of proline with the changing environment were also studied [26, 27]. Employing support vector machine and developing a tool for prediction of hydroxyproline sites were proposed by ZR Yang [28]. Hu, Le-Le, et al. [29] developed a sequence-based methodology for predicting hydroxylation of hydroxyproline and hydroxylysine. Xu, Yan, et al. [8] predicted hydroxyproline and hydroxylysine in proteins using dipeptide position and specific propensity into pseudo amino acid composition. An improved approach over this proficiency was proposed by Qiu, Wang-Ren, et al. [30] by integrating a sequence-coupled effect into general PseAAC.
2. RESULTS
To develop a worthwhile predictor for a biological phenomenon, one should observe the Chou's 5-step rule [31]. It is indeed good to present the new prediction method by observing the Chou's 5-step rule as many researchers followed this fundamental rule in their papers, published very recently [9, 32-38]. In the first step, benchmark dataset is accumulated for training and testing the predictor; in the next step, a mathematical model is formulated which sieves out the most momentous features of the polypeptide sequence. Later the feature vector is integrated into a prediction algorithm for training. Once the training is completed, the trained model is thoroughly tested and validated. Lastly, a web-server is developed for open use of the prediction model. In this study, the first four steps have been meticulously performed, however, the last step has been kept open for future work.
3. ACCURACY METRICS
In order to measure the predictive quality of the predictor, the following metrics are commonly used: is used to quantify the comprehensive accuracy of the predictor, is a stable measure of overall accuracy of the model, is used to estimate sensitivity, and is used for specificity [39]. To evaluate the prediction rate of the proposed model, this set of metrics is followed which are also employed by Ehsan et al. [40]. The formulation for the actual prediction of hydroxylated and non-hydroxylated site of proline is given below.
(1)
(2)
Where and represent the total number of peptides which was correctly predicted with proline hydroxylated site and the number of hydroxylated peptides which was incorrectly predicted as a non-hydroxylated proline site, respectively. Likewise and represent the total actual count of non-hydroxylated peptides and the number of wrongly predicted hydroxylated peptides, respectively.
(3)
It has been observed that when there are zero incorrectly predicted hydroxylated and non-hydroxylated proline peptides such that then equation (1) to (3) gives and signifying the highest possible accuracy rate. Subsequently, when then the prediction would be less than 1. There are a number of statistical equations which are used to measure the performance of the predictor given in eq (4).
(4)
Where , , and represent the true positive, true negative, false positive and false negative values, respectively. Expressions in equations (5) and (6) represent the symbols in terms of equation (1) to (3). It is also advantageous to use the intuitive metrics of Equations (5)-(6) to replace the traditional Equation (4). Either the set of traditional metrics copied from maths books or the intuitive metrics derived from the Chou's symbols [41-43] are valid only for the single-label systems (where each sample only belongs to one class). For the multi-label systems (where a sample may simultaneously belong to several classes), whose existence has become more frequent in system biology [32, 33, 36, 44], system medicine [45] and biomedicine [46], a completely different set of metrics as defined in the study represnted as reference [47] is absolutely needed.
(5)
(6)
It is relevant to discuss the following cases of the above equation (6), if then there is no incorrectly predicted hydroxylated proline peptides as non-hydroxylated proline peptides such that . Similarly, when it indicates that all hydroxylated proline peptides were incorrectly predicted as non-hydroxylated proline peptides, hence the sensitivity was computed as . Furthermore, yields specificity, represents that not even one non-hydroxylated proline peptide was incorrectly predicted as a hydroxylated proline peptide. Likewise yields specificity, and represents that all non-hydroxylated proline peptides were incorrectly predicted as hydroxylated proline peptides. Also, implies that all sequences of hydroxylated and non-hydroxylated proline peptides were predicted correctly such that . Further, the performance of binary classifications is often measured by Matthew correlative coefficient (MCC). There were three cases herein, indicates that no incorrectly predicted sequences were found both for hydroxylated and non-hydroxylated peptides yielding . In the second case, and generated indicating that this prediction was not more accurate than the random prediction. Lastly, with values of and , was obtained signifying a totally wrong binary classification and complete disagreement between the observed and predicted values.
4. VALIDATION METHOD
The metrics given in equation (6) are used to describe three frequently used test methods namely, independent dataset test, K-fold cross-validation test, and jackknife test. These tests are considered beneficial in validating the quality of the predictor. The jackknife test is considered the least arbitrary because it can agree to specific results for particularly obtained benchmark dataset as explained earlier in a study [31]. To study the statistical analysis of the new predictor, a comparison was made using the jackknife test with previous methodologies [8, 30]. In this study, all of these validation tests were employed to evaluate the quality of the proposed methodology. In addition, K-fold cross-validation test is based on sub-sampling to validate the classifier since several partitioning permutations exist therefore it cannot avoid ambiguity [8].
5. COMPARISON WITH PREVIOUS METHODS
Values given in Table 1 are the scores of the four metrics attained by the proposed predictor using the independent dataset test, 10-fold cross-validation test, and jackknife test on the dbptm benchmark dataset, while, Table 2 represents the scores of similar metrics using the most updated dataset obtained from UniProt. Furthermore, Table 3 shows a comparison with the existing techniques. Two existing predictors have been depicted, namely “iHyd-PseAAC” [8], and ” iHyd-PseCp” [30], for identifying the hydroxyproline sites. These methods also achieved the metrics scores using the jackknife test method. It can be observed from Table 3 that the accuracy (), stability (), sensitivity (), and specificity () scores evaluated by the newly proposed predictor are superior than those reported by the existing predictors. A comparison with previous methods was made using two benchmark datasets extracted from (a) dbptm and (b) uniprot database. To understand the complex biological systems, the graphical representation gives a valuable vision as represented by the list of earlier articles [48-50]. The same is depicted as a comparison in graphical representation showing the Receiver Operating Characteristic (ROC) [51] of the proposed predictor and previously existing predictors. In Fig. (2), the red curve represents the ROC curve for iHyd-PseAAC and green curve for iHyd-PseCp, while blue solid and dotted curves represent the ROC plotted by using the proposed predictor on dbptm and uniprot benchmark datasets. It is evident from the figure below that the area under the blue dotted and solid curves is extraordinarily larger than that under the red and green curves. Undoubtedly, the novel proposed predictor is certainly an improved approach over the existing predictors.
Table 1. Three tests result on set of metrics using proposed model on dbptm benchmark.
| Tests | Sn (%) | Sp (%) | Acc (%) | MCC |
|---|---|---|---|---|
| Independent dataset test |
98.30 | 98.02 | 98.77 | 0.96 |
| Cross- Validation |
98.73 | 94.87 | 96.85 | 0.93 |
| Jackknife test | 98.68 | 94.82 | 96.80 | 0.90 |
Table 2. Three tests result on four metrics using proposed model on recent uniprot benchmark.
| Tests | Sn (%) | Sp (%) | Acc (%) | MCC |
|---|---|---|---|---|
| Independent dataset test |
98.38 | 99.54 | 98.80 | 0.95 |
| Cross- Validation |
97.07 | 94.62 | 96.06 | 0.91 |
| Jackknife test | 97.02 | 94.57 | 96.01 | 0.88 |
Table 3. A comparison of the proposed model with the previous methods to identify hydroxylation of proline using jackknife test in the validation of benchmark datasets extracted from (a) dbptm and (b) uniprot.
| Predictors | Sn (%) | Sp (%) | Acc (%) | MCC |
|---|---|---|---|---|
| iHyd-PseAAC | 80.66 | 80.54 | 80.57 | 0.51 |
| iHyd-PseCp | 86.35 | 99.12 | 96.58 | 0.89 |
|
iHyd-PseAAC
(EPSV)a |
98.68 | 94.82 | 96.80 | 0.90 |
|
iHyd-PseAAC
(EPSV)b |
97.02 | 94.57 | 96.01 | 0.88 |
Fig. (2).
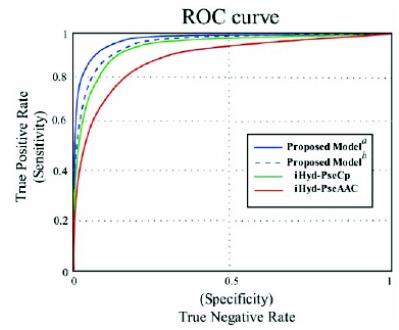
Comparison of the proposed model with the curves plotted with iHyd-PseAAC and iHyd-PseCp predictors.
The superior performance of the proposed system can be rationalized by a number of scientific and theoretical reasons. Some of these are discussed here. Firstly, the proposed model is a formulation based on the composition and sequence of primary structure which can conveniently handle diverse length sequences in a generous way without skipping any obscure information and form pairwise couplings in every possible permutation of amino acid residues. Secondly, it generates a fixed length vector, which imparts a non-variable size feature vector that equally separates proteins according to their attributes. This aspect enables the predictor to rigorously classify and conveniently recognize each sample. Thirdly, the correlation expression is the main mechanism that contributes towards the computation of a feature vector. It has been configured by incorporating each attribute group. Each expression deals with some specific metric and statistical expressions. For the sake of convenience, every property of amino acids was standardized numerically within a suitable range. Also, it has been observed that in comparison with previous methods proposed, the predictor outcomes are more superior and better than the former prediction rate.
6. WEB-SERVER
User-friendly and publicly accessible web-servers represent the current trend for developing various computational methods [52], as reflected by a series of recent publications [32, 33, 35, 36, 44]. Actually, they have significantly enhanced the impacts of computational biology in medical science [53], driving medicinal chemistry into an unprecedented revolution [54], here we shall do our best to provide a web-server for the predictor presented in this paper as soon as possible.
7. DISCUSSION
The proposed model is a new predictor to identify hydroxylation of proline. It can be analysed from Table 3 that the accuracy calculated for the proposed model is 96.80 and 96.01 which is higher than the accuracy calculated using previous predictors, that is 80.57 and 96.58. Also, MCC values were 0.90 and 0.88 which were superior to both the predictors i.e. iHyd-PseAAC and iHyd-PseCp. The proposed model was validated using benchmark datasets extracted from dbptm as well as from UniProt database.
8. METHODS
8.1. Benchmark Dataset
According to Chou's 5-step rule [31], the extraction of benchmark dataset is a crucial step that leads to the acquisition of a robust, diverse and updated dataset. In this study, a stringent benchmark dataset has been borrowed from two roots. One of the datasets is received from the resource http://www.uniprot.org/, and the other is leased from a post-translational modification database dbPTM 3.0 [55] that has also been utilized by Xu et al. [8]. The following two steps are used to select a stringent benchmark dataset.
Step-1: The data extracted from UniProt database, consists of positive and negative samples that represent the hydroxylated and non-hydroxylated polypeptide sequences at proline site. A query is generated to select protein sequences in the PTM/processing field as hydroxyproline. Entries annotated with any experimental assertion in Feature Table (FT) were exclusively selected.
Step-2: After a rigorous adoption of the above step, a first-rate benchmark dataset of hydroxyproline was collected. Total samples of 816 and 24,980 for positive and negative were extracted, respectively. After obtaining the duplicates, both were cut down to 782 and 24971 unique values. For the sake of convenience, and represent the positive and negative set of the hydroxylated polypeptides, respectively. Further, let be the total sum of these two. Also, it can be easily seen that there exist more negative peptides than positive peptides in nature. Thus, . Similarly, to extract another stringent benchmark dataset, the dbdtm 3.0 [55] was employed. The dataset was easily available in FASTA format and conveniently were downloaded for hydroxylation (positive and negative). There were found 226 positive sets and 3,865 negative sets. A demonstration in term of a Flowchart is given in Fig. (3), to understand the above steps. The primary structure of hydroxylated and non-hydroxylated proline sites can be found in Supplementary Tables S1, S2, S3 and S4 respectively.
Fig. (3).
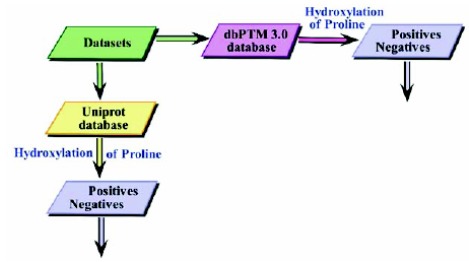
Flowchart is representing the database sources used to retrieve the datasets.
9. SAMPLE FORMULATION AND ALGORITHM DEVELOPMENT
According to the Chou's second and third step [31], a powerful mathematical formulation is proposed that can accurately reflect their indispensable correlation to arrange the sample in an effective way, also used by Ehsan et al. [40]. Considering a protein sample P, consisting of L amino acid residues.
(7)
Where is the first amino acid residue, is the second amino acid residue, and so on up to the last residue of protein sequence P, where indicates the length of the sequence (7). To identify the post translational modification in proline site, a computational methodology has been persuaded. This method upholds the sequence order effect and is adopted using the whole sequence data together with the occurrence of each amino acid residue of type (any one of the residues among twenty amino acid residues). Expression (8) to (11) describes the whole formulation strategy. The number of occurrences of residue and the possible number of correlated factors of with itself, such that is linked to expression (8). While, mean factors , and are connected with the deviation factors of at their respective positions and are represented by the expression (9), followed by condition (10). Whereas, runs over deviation factors and these factors are linked by a local mean. This deviation is denoted by , provided the positions of are labeled by p and q in , the polypeptide chain. While the subscript denotes the frequency of occurrence of deviation factors for similar amino acid residues discarding the occurrence at the first and last position residue , based on n total occurrences of ; similarly, is labeled for the difference, , and r represent the exact position of the residue appearing at of its occurrence in (7) while and denote the amino acid residues in the corresponding positions.
(8)
(9)
(10)
Combining expressions (8) and (9) and using constraint (10) yield the template for manipulating feature component related to , given in (11).
(11)
While and denote the number occurrences of before and after, with the remaining residues, respectively.
These are given in eq (12) and (13).
(12)
Where
(13)
Where shows the occurrence of binary function related to residue with any of the remaining nineteen residues and stands for none of its occurrence with others. , is defined as the pair function £h for all combinations of all residues, whereas the pair function in terms of is defined as , elaborated in a matrix (14). Equation (15) assigns all the possible pair factors concerning and together with (16). If a pair is found, then is labeled as 1 otherwise it will be assigned 0 value. Additionally, (15) admits to (14) with entries , and specifying lower triangular matrix for . Accordingly, the diagonal entries signify the combination among analogous residues and upper triangular matrix for .
(14)
(15)
Where
(16)
The manipulation of feature components in a matrix form, incorporating all the amino acid residues given in (17) can be viewed as an extension of (11).
(17)
Expression (11) together with equation (13) yields the component of feature vector that is , elaborated in eq (18) and (19).
(18)
Or
(19)
The structural scheme of the proposed formulation can be understood by considering term of a sequence (7), say, , which mirrors the amino acid residues say “A”. It must be noted that makes a pair with its adjacent residues before and after the residue in terms of and exemplified by blue and pink curvy lines and pairs with itself which is denoted by muddy green loops as shown in Fig. 4. The procedure must be followed till appears in place such that . Correspondingly, a similar procedure will be adopted for . The feature component agreeing to residue “A” is substituted in equation (20).
Fig. (4).
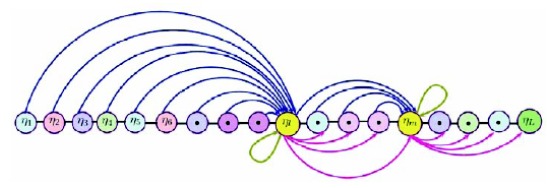
Graphical representation shows how to formulate the sequence for classification.
(20)
Where are the amino acid residues in ascending order. For simplicity, taking as the 20 amino acids in an alphabetical order for further generalization and onwards the 20 residues that periodically replicate themselves. Supposing ,,,..., are their associate feature components. These are given in equation (21).
(21)
The three main characteristics of amino acids, that is hydrophobicity, hydrophilicity and side chain mass of amino acids mainly take part in the above set of twenty feature components. Every characteristic relates 60 entries as coordinates,which contribute to 180 coordinates in total influenced by equation (22) to (24), identifies the characteristics.
(22)
(23)
(24)
Where ,, represent the normalized hydrophobicity, hydrophilicity and side-chain mass, respectively, and , , indicate the mean of the normalized values corresponding to the 20 amino acids related to attributes. The values used in (22) to (24) are normalized by using (25), and standardized in a range (-T, T), where T is the count for amino acids to be standardized. Entries for hydrophobicity are picked from Tanford C. [56], and for hydrophilicity, entries are taken from Hopp T.P., Woods K.R. [57], while the values of side-chain mass can be found in most of the books given in the bibliography.
(25)
The feature set is categorized into a vector with 220 components, of which, the first sixty are constructed by virtue of the hydrophobic nature of amino acids, the next sixty components depict their hydrophilic nature, the subsequent sixty components are related to side chain mass, whereas the last forty reflect the position and composition of each amino acid residue. The feature vectors hence obtained for the training data are clamped to a neural network for training. Once the training is completed, the trained network apparently gains the experience to categorize arbitrary input with an appreciable precision. While the process is carried on, the network normalizes its weights with a minimum slip.
Multilayer Perceptron (MLP) is an excellent model that can uncover and identify obscure patterns in diversified data sets. MLP is best suited for any classification problem as it can be fine tuned by changing the number of hidden layer neurons, training parameters and training algorithm to provide the best outcome. A Multi-Layer Perceptron (MLP) was trained using the extracted feature set for this purpose (Fig. 5). The feature vectors for the samples were assembled into a large array. Each row of the array represents the feature vector for a single sequence while each column represents a feature item extracted. Since 220 features were extracted for each sample; therefore each row had 220 columns while the total columns were 25796; out of which, 816 were positive samples.The weights of each layer were initialized randomly while a hidden layer with 75 neurons was used. Further, back propagation algorithm was used to adjust the weights after each epoch. Convergence was achieved after 2693 iterations while using gradient descent method for learning rate.
Fig. (5).
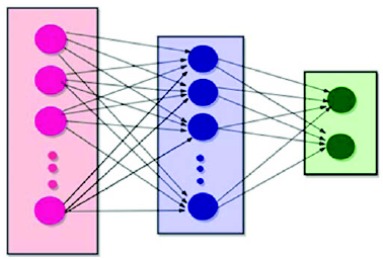
neural network.
The results were simulated on MATLAB R2017 version and were duplicated on python ver 3.6 platform along with Scikit Learn 0.20 for neural network training and simulation bearing identical results.
The algorithm which is developed by the following above method is called iHyd-PseAAC (EPSV), where “i” represents the first word of “identify”, Hyd is used for “hydroxylation” and Pse-AAC is the general term used for pseudo amino acid composition. Also the term “EPSV” stands for “enhanced position and sequence variant” technique which is used to construct an algorithm for polypeptide sequence.
ACKNOWLEDGEMENTS
Declared none.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s web site along with the published article.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AUTHORS’ CONTRIBUTIONS
A.E. proposed an algorithm and conducted experiments, K.M. supervised the results. Y.D. supervised the validation process. S.K. worked on the appropriateness of the suggested model. O.M. worked on the data collection and feature extraction. K.C. originally formulated the problem contributed equally to analyze and improve the results. All authors reviewed the manuscript.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article is available in the Kaggle at www.kaggle.com, reference number https://www.kaggle.com/ydkhan/starter-proline-hydroxylation-f4c3 c873-a”.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Colgrave M.L., Peter G.A., Jones A. Hydroxyproline quantification for the estimation of collagen in tissue using multiple reaction monitoring mass spectrometry. J. Chromatogr. A. 2008;1212(1-2):150–153. doi: 10.1016/j.chroma.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 2.Gelse K., Pöschl E., Aigner T. Collagens—structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003;55(12):1531–1546. doi: 10.1016/j.addr.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Ruszczak Z. Effect of collagen matrices on dermal wound healing. Adv. Drug Deliv. Rev. 2003;55(12):1595–1611. doi: 10.1016/j.addr.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Lee C.H., Singla A., Lee Y. Biomedical applications of collagen. Int. J. Pharm. 2001;221(1-2):1–22. doi: 10.1016/s0378-5173(01)00691-3. [DOI] [PubMed] [Google Scholar]
- 5.Becker G.D., Lawrence A.A., Hackett J. Collagen-assisted healing of facial wounds after mohs surgery. Laryngoscope. 1994;104(10):1267–1270. doi: 10.1288/00005537-199410000-00015. [DOI] [PubMed] [Google Scholar]
- 6.Guszczyn T., Soboleweki K. Deregulation of collagen metabolism in human stomach cancer. Pathobiology. 2004;71(6):308–313. doi: 10.1159/000081726. [DOI] [PubMed] [Google Scholar]
- 7.Sunila E.S., Kuttan G. A preliminary study on antimetastatic activity of Thuja occidentalis L. in mice model. Immunopharmacol. Immunotoxicol. 2006;28(2):269–280. doi: 10.1080/08923970600809017. [DOI] [PubMed] [Google Scholar]
- 8.Xu Y., Wen X., Shao X.J., Deng N.Y., Chou K.C. iHyd-PseAAC: Predicting hydroxyproline and hydroxylysine in proteins by incorporating dipeptide position-specific propensity into pseudo amino acid composition. Int. J. Mol. Sci. 2014;15(5):7594–7610. doi: 10.3390/ijms15057594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng P., Yang H., Ding H., Lin H., Chen W., Chou K.C. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics. 2019;111(1):96–102. doi: 10.1016/j.ygeno.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Xu Y., Ding J., Wu L.Y., Chou K.C. iSNO-PseAAC: Predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition. PLoS One. 2013;8(2):e55844. doi: 10.1371/journal.pone.0055844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Y., Shao X.J., Wu L.Y., Deng N.Y., Chou K.C. iSNO-AAPair: Incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. PeerJ. 2013;1:e171. doi: 10.7717/peerj.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia C., Lin X., Wang Z. Prediction of protein s-nitrosylation sites based on adapted normal distribution bi-profile bayes and chou’s pseudo amino acid composition. Int. J. Mol. Sci. 2014;15(1):10410–10423. doi: 10.3390/ijms150610410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia J., Liu Z., Xiao X., Liu B., Chou K.C. pSuc-Lys: Predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J. Theor. Biol. 2016;394(1):223–230. doi: 10.1016/j.jtbi.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 14.Jia J., Liu Z., Xiao X., Liu B., Chou K.C. iCar-PseCp: identify carbonylation sites in proteins by Monto Carlo sampling and incorporating sequence coupled effects into general PseAAC. Oncotarget. 2016;7(23):34558–34570. doi: 10.18632/oncotarget.9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia J., Zhang L., Liu Z., Xiao X., Chou K.C. pSumo-CD: Predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics. 2016;32(1):3133–3141. doi: 10.1093/bioinformatics/btw387. [DOI] [PubMed] [Google Scholar]
- 16.Khan Y.D., Rasool N., Hussain W., Khan S.A., Chou K.C. iPhosT-PseAAC: Identify phosphothreonine sites by incorporating sequence statistical moments into PseAAC. Anal. Biochem. 2018;550(1):109–116. doi: 10.1016/j.ab.2018.04.021. [DOI] [PubMed] [Google Scholar]
- 17.Khan Y.D., Rasool N., Hussain W., Khan S.A., Chou K.C. iPhosY-PseAAC: Identify phosphotyrosine sites by incorporating sequence statistical moments into PseAAC. Mol. Biol. Rep. 2018;550:109–116. doi: 10.1016/j.ab.2018.04.021. [DOI] [PubMed] [Google Scholar]
- 18.Cockman M.E., Webb J.D., Kramer H.B., Kessler B.M., Ratcliffe P.J. Proteomics-based identification of novel factor inhibiting Hypoxia-Inducible Factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol. Cell. Proteomics. 2009;8(3):535–546. doi: 10.1074/mcp.M800340-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ang K.S., Lakshmanan M., Lee N.R., Lee D.Y. Metabolic modeling of microbial community interactions for health, environmental and biotechnological applications. Curr. Genomics. 2018;19(8):712–722. doi: 10.2174/1389202919666180911144055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berg R.A., Steinmann B., Rennard S.I., Crystal R.G. Ascorbate deficiency results in decreased collagen production: under-hydroxylation of proline leads to increased intracellular degradation. Arch. Biochem. Biophys. 1983;226(2):681–686. doi: 10.1016/0003-9861(83)90338-7. [DOI] [PubMed] [Google Scholar]
- 21.Halme J., Kivirikko K.I., Simons K. Isolation and partial characterization of highly purified protocollagen proline hydroxylase. Biochim. Biophys. Acta. 1970;198(3):460–470. doi: 10.1016/0005-2744(70)90124-5. [DOI] [PubMed] [Google Scholar]
- 22.Kivirikko K.I., Prockop D.J. Hydroxylation of proline in synthetic polypeptides with purified protocollagen hydroxylase. J. Biol. Chem. 1967;242(18):4007–4012. [PubMed] [Google Scholar]
- 23.Morgan A.A., Rubenstein E. Proline: The distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS One. 2013;8(1):e53785. doi: 10.1371/journal.pone.0053785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamauchi M., Shiiba M. Posttranslational modifications of proteins. New York: Humana Press; 2002. Lysine hydroxylation and crosslinking of collagen. pp. 277–290. [Google Scholar]
- 25.Shi S.P., Chen X., Xu H.D., Qiu J.D. PredHydroxy: Computational prediction of protein hydroxylation site locations based on the primary structure. Mol. Biosyst. 2015;11(3):819–825. doi: 10.1039/c4mb00646a. [DOI] [PubMed] [Google Scholar]
- 26.Wu G., Bazer F.W., Burghardt R.C., Johnson G.A., Kim S.W., Knabe D.A., Li P., Li X., McKnight J.R., Satterfield M.C., Spencer T.E. Proline and hydroxyproline metabolism: Implications for animal and human nutrition. Amino Acids. 2011;40(4):1053–1063. doi: 10.1007/s00726-010-0715-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayat S., Hayat Q., Alyemeni M.N., Wani A.S., Pichtel J., Ahmad A. Role of proline under changing environments: A review. Plant Signal. Behav. 2012;7(11):1456–1466. doi: 10.4161/psb.21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Z.R. Predict collagen hydroxyproline sites using support vector machines. J. Comput. Biol. 2009;16(5):691–702. doi: 10.1089/cmb.2008.0167. [DOI] [PubMed] [Google Scholar]
- 29.Hu L.L., Niu S., Huang T., Wang K., Shi X.H., Cai Y.D. Prediction and analysis of protein hydroxyproline and hydroxylysine. PLoS One. 2010;5(12):e15917. doi: 10.1371/journal.pone.0015917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu W.R., Sun B.Q., Xiao X., Xu Z.C., Chou K.C. iHyd-PseCp: Identify hydroxyproline and hydroxylysine in proteins by incorporating sequence-coupled effects into general PseAAC. Oncotarget. 2016;7(28):44310. doi: 10.18632/oncotarget.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou K.C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011;273(1):236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng X., Xiao X., Chou K.C. pLoc-mPlant: Predict subcellular localization of multi-location plant proteins via incorporating the optimal GO information into general PseAAC. Mol. Biosyst. 2017;13(1):1722–1727. doi: 10.1039/c7mb00267j. [DOI] [PubMed] [Google Scholar]
- 33.Xiao X., Cheng X., Su S., Mao Q., Chou K.C. pLoc-mGpos: Incorporate key gene ontology information into general PseAAC for predicting subcellular localization of gram-positive bacterial proteins. Nat. Sci. 2017;9(1):331–349. [Google Scholar]
- 34.Wang J., Li J., Yang B., Xie R., Marquez-Lago T.T., Leier A., Hayashida M., Akutsu T., Zhang Y., Chou K.C., Selkrig J., Zhou T., Song J., Lithgow T. Bastion3: A two-layer approach for identifying type III secreted effectors using ensemble learning. Bioinformatics. 2018 doi: 10.1093/bioinformatics/xxxxx. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chou K.C., Cheng X., Xiao X. pLoc-bal-mHum: predict subcellular localization of human proteins by PseAAC and quasi-balancing training dataset. Genomics. 2018 doi: 10.1016/j.ygeno.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 36.Xiao X., Cheng X., Chen G., Mao Q. pLoc-bal-mGpos: Predict subcellular localization of Gram-positive bacterial proteins by quasi-balancing training dataset and PseAAC. Genomics. 2018 doi: 10.1016/j.ygeno.2018.05.017. [DOI] [PubMed] [Google Scholar]
- 37.Khan Y.D., Jamil M., Hussain W., Rasool N., Khan S.A., Chou K.C. pSSbond-PseAAC: Prediction of disulfide bonding sites by integration of PseAAC and statistical moments. J. Theor. Biol. 2019;463(1):47–55. doi: 10.1016/j.jtbi.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 38.Jia J., Li X., Qiu W., Xiao X., Chou K.C. iPPI-PseAAC(CGR): Identify protein-protein interactions by incorporating chaos game representation into PseAAC. J. Theor. Biol. 2019;460(1):195–203. doi: 10.1016/j.jtbi.2018.10.021. [DOI] [PubMed] [Google Scholar]
- 39.Chen J., Liu H., Yang J., Chou K.C. Prediction of linear b-cell epitopes using amino acid pair antigenicity scale. Amino Acids. 2007;33(1):423–428. doi: 10.1007/s00726-006-0485-9. [DOI] [PubMed] [Google Scholar]
- 40.Ehsan A., Mahmood K., Khan Y.D., Khan S.A., Chou K.C. A novel modeling in mathematical biology forclassification of signal peptides. Sci. Rep. 2018;8(1):1039. doi: 10.1038/s41598-018-19491-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chou K.C. Prediction of protein signal sequences and their cleavage sites. Proteins. 2001;42:136–139. doi: 10.1002/1097-0134(20010101)42:1<136::aid-prot130>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 42.Chou K.C. Using subsite coupling to predict signal peptides. Protein Eng. 2001;14(1):75–79. doi: 10.1093/protein/14.2.75. [DOI] [PubMed] [Google Scholar]
- 43.Chou K.C. Prediction of signal peptides using scaled window. Peptides. 2001;22(1):1973–1979. doi: 10.1016/s0196-9781(01)00540-x. [DOI] [PubMed] [Google Scholar]
- 44.Cheng X., Xiao X., Chou K.C. pLoc-mGneg: Predict subcellular localization of Gram-negative bacterial proteins by deep gene ontology learning via general PseAAC. Genomics. 2018;110(1):231–239. doi: 10.1016/j.ygeno.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 45.Cheng X., Zhao S.G., Xiao X., Chou K.C. iATC-mISF: A multi-label classifier for predicting the classes of anatomical therapeutic chemicals. Bioinformatics. 2017;33(3):341–346. doi: 10.1093/bioinformatics/btw644. [DOI] [PubMed] [Google Scholar]
- 46.Qiu W.R., Sun B.Q., Xiao X., Xu Z.C., Chou K.C. iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics. 2016;32(1):3116–3123. doi: 10.1093/bioinformatics/btw380. [DOI] [PubMed] [Google Scholar]
- 47.Chou K.C. Some remarks on predicting multi-label attributes in molecular biosystems. Mol. Biosyst. 2013;9:1092–1100. doi: 10.1039/c3mb25555g. [DOI] [PubMed] [Google Scholar]
- 48.Chou K.C. Graphic rule for drug metabolism systems. Curr. Drug Metab. 2010;11(1):369–378. doi: 10.2174/138920010791514261. [DOI] [PubMed] [Google Scholar]
- 49.Chou K.C., Lin W.Z., Xiao X. Wenxiang: A web-server for drawing wenxiang diagrams. Nat. Sci. 2011;3(1):862. [Google Scholar]
- 50.Wu Z.C., Xiao X., Chou K.C. 2d-mh: A web-server for generating graphic representation of protein sequences basedon the physicochemical properties of their constituent amino acids. J. Theor. Biol. 2010;267(1):29–34. doi: 10.1016/j.jtbi.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 51.Davis J., Goadrich M. Proceedings of the 23rd international conference on Machine learning. ACM; 2006. The relationship between precision-recall and roc curves. pp. 233–240. [Google Scholar]
- 52.Chou K.C., Shen H.B. Recent advances in developing web-servers for predicting protein attributes. Nat. Sci. 2009;1(1):63–92. [Google Scholar]
- 53.Chou K.C. Impacts of bioinformatics to medicinal chemistry. Med. Chem. 2015;11(1):218–234. doi: 10.2174/1573406411666141229162834. [DOI] [PubMed] [Google Scholar]
- 54.Chou K.C. An unprecedented revolution in medicinal chemistry driven by the progress of biological science. Curr. Top. Med. Chem. 2017;17(1):2337–2358. doi: 10.2174/1568026617666170414145508. [DOI] [PubMed] [Google Scholar]
- 55.Lu C.T., Huang K.Y., Su M.G., Lee T.Y., Bretana N.A., Chang W.C., Chen Y.J., Chen Y.J., Huang H.D. Dbptm 3.0: an informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res. 2012;41(1):295–305. doi: 10.1093/nar/gks1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanford C. Contribution of hydrophobic interactions to the stability of the globular conformation of proteins. J. Am. Chem. Soc. 1962;84(1):4240–4247. [Google Scholar]
- 57.Hopp T.P., Woods K.R. Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl. Acad. Sci. USA. 1981;78(1):3824–3828. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material is available on the publisher’s web site along with the published article.
Data Availability Statement
The data supporting the findings of the article is available in the Kaggle at www.kaggle.com, reference number https://www.kaggle.com/ydkhan/starter-proline-hydroxylation-f4c3 c873-a”.