

Abstract
Lung cancer, the leading cause of cancer-related mortality in the US, occurs primarily due to prolonged exposure to an array of carcinogenic compounds in cigarette smoke. These carcinogens create bulky DNA adducts, inducing alterations including missense mutations in the tumor suppressor gene TP53. TP53 is the most commonly mutated gene in many human cancers, and a specific set of these variants are enriched in lung cancer (at amino acid residues V157, R158, and A159). This perspective postulates that lung-enriched mutations can be explained, in part, by biological selection for oncogenic gain-of-function (GOF) mutant p53 alleles at V157, R158, and A159. This hypothesis explaining tissue-specific TP53 mutations, is further supported by mouse model studies of the canonical TP53 hotspots showing that tumor spectra and GOF activities are altered with mutation type. Therefore, while smoking-related lung cancer unequivocally arises due to the mutagenic environment induced by tobacco carcinogens, this perspective provides a rationale for the preferential selection of lung-enriched V157, R158, and A159 mutant p53.
Keywords: lung cancer, mutant p53, gain of function, tumor suppressor gene, carcinogenesis/tobacco
Introduction
Although the p53 tumor suppressor, as the “guardian of the genome,” has been studied extensively, the availability of more complete sequencing data across the entire genome has allowed the current era of human genetic studies to be more comprehensive than ever.(1,2) One of the most active areas of research is in mutant p53 gain of oncogenic function (GOF), whereby single nucleotide missense mutations exhibit not only loss of tumor suppressor function but also acquisition of novel activities such as increased invasion, chemoresistance, and metabolic reprogramming.(3,4) Identification of novel tumorigenic functions may open new avenues for p53-targeted therapies.
Somatic alterations in the TP53 gene occur primarily at one of six major canonical hotspots in the DNA-binding domain: R175, G245, R248, R249, R273, and R282. Specifically, the eight most common mutants individually range in frequency from 2.9 to 7% and together account for 27.7% of all TP53 mutations in a database of 11,145 sequenced tumors.(5) Notably, when lung cancer cases are reviewed in isolation, the frequency of mutations at amino acid residues V157 and R158 increases 5- to 6-fold (0.9% to 4.6% and 1.1% to 5.9%, respectively) and surpasses that of many of the traditional hotspots. This phenomenon has been reported extensively and attributed to preferential DNA adduct formation at these codons by carcinogenic polycyclic aromatic hydrocarbons (PAH) found in cigarette smoke.(6–10)
The PAH benzo[a]pyrene, which is metabolically activated to benzo[a]pyrene-diol-epoxide (BPDE), preferentially binds the N2 position of guanine at TP53 codons 157, 248, and 273 in normal human bronchial epithelial cells and normal human fibroblasts.(6) These bulky DNA adducts generate a COSMIC smoking signature described as G:C > T:A transversion mutations with a bias for the nontranscribed DNA strand, where DNA is less efficiently repaired.(11) Moreover, DNA adduct formation by BPDE is enhanced at methylated CpG sites in both lung-enriched V157F and R158 codons and the canonical hotspots G245, R248, and R273.(6) While the precise mechanism is not yet understood, it has been proposed that cytosine methylation increases the electron density of the amine group of the paired guanine. This enhances its affinity with BPDE and acrolein, a mutagenic aldehyde also found in cigarette smoke.(12–14) Alternative hypotheses have been put forth as well, with the methyl group of 5-methylcytosine allowing increased intercalation of BPDE with subsequent increase in covalent interaction; or increased probability of base flipping of the paired guanine into the major groove, allowing it to interact with BPDE. (12,13) A variety of in vitro and in vivo studies to examine mutations induced by exposure of p53 cDNA, bronchial epithelial cells, lung fibroblasts, and human TP53 knock-in mouse embryo fibroblasts to BPDE have confirmed a high frequency of G>T transversions at codons V157 and R158 in addition to the canonical p53 hotspots. (12–18)
TP53 mutational spectra in lung tumors
To investigate the clinical significance of V157, R158, and A159 mutations in lung cancer, we evaluated lung adenocarcinoma and lung squamous cell lung cancer sequencing data from the Pan-Lung Cancer Study of The Cancer Genome Atlas (TCGA). (19) As noted above, the frequency of R158 and V157 mutations in lung cancers is markedly elevated. This finding is not observed in head and neck squamous cell carcinomas (HNSCC), for which cigarette smoking is also the primary risk factor (Figure 1). In the TCGA database which includes 528 patient samples, TP53 is altered in 367 cases of HNSCC (69.5%), and 253 (68.9%) of these are missense mutations.(20) These frequencies are nearly identical to the TCGA Pan-Lung Cancer Study, where 1,144 patient samples exhibit TP53 alterations in 776 cases (67.8%), and 505 (65.1%) of these are missense mutations.(19) However, in sharp contrast to lung cancer, only five samples harbor V157 mutations (5/5 V157F, 2.0% of total), three have R158 mutations (2/3 R158L, 1.2% of total), and two have A159 mutations (1/2 A159P, 0.8%). In bladder urothelial carcinoma where cigarette smoking is also a major risk factor, there is a much lower rate of TP53 mutations.(21) Among 412 patient samples, 74 (18.0%) exhibit alterations in TP53, and 57 (77.0%) of these are missense mutations. No V157 mutations and only one R158 mutations (R158H) exist in this cohort, and although there are four A159 mutations, the overall lower frequency of TP53 mutations suggests that changes in the p53 tumor suppressor do not play a major role in bladder tumorigenesis. These data show that the V157, R158, and A159 TP53 mutations occur preferentially in lung cancer over HNSCC and bladder cancer despite the critical role that cigarette smoke exposure plays in all three tumor types. Hence, it is possible that these TP53 mutations are not only induced by tobacco carcinogens but exhibit a relative abundance in the lung driven by selective biological pressure which may be tissue-specific.
Figure 1. Patterns in TP53 mutations vary by tumor type.
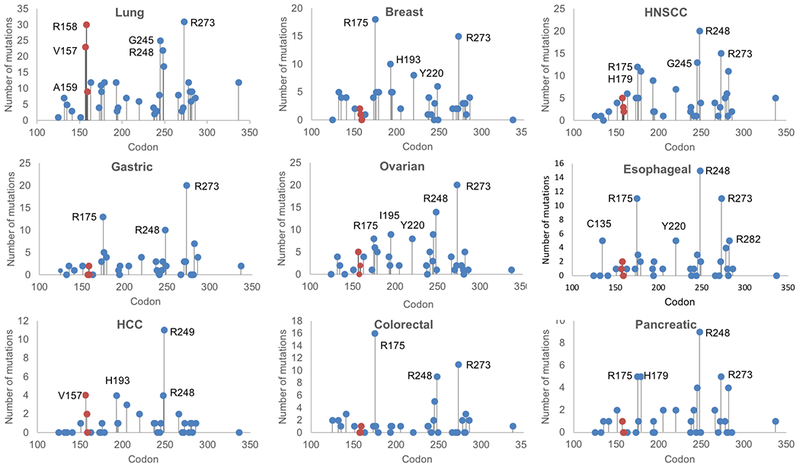
Number of somatic missense mutations are plotted for the most frequently mutated amino acid residues in the TP53 gene, by solid tumor type. Data are from The Cancer Genome Atlas (TCGA). (19) Total numbers of samples with TP53 missense mutations are: Lung adenocarcinoma and squamous cell carcinoma, 505; Breast adenocarcinoma, 179; Head and neck squamous cell carcinoma (HNSCC), 253; Gastric adenocarcinoma, 131; Ovarian serous cystadenocarcinoma, 179; Esophageal adenocarcinoma, 110; Hepatocellular carcinoma (HCC), 71; Colorectal adenocarcinoma, 83; Pancreatic adenocarcinoma, 72.
Our analysis of the larger and more recently curated AACR Project GENIE cohort revealed a similar enrichment for mutations at V157, R158, and A159 among non-small cell lung cancer samples (Table 1). Specifically, in lung adenocarcinoma (n = 5,060 samples), the overall prevalence of TP53 alterations was 46.1%, or 2,333 samples, and R158 was the third most frequent amino acid residue at which mutations occurred (increasing 4-fold in frequency to 4.2%, from 1.0% when compared to non-lung cancers). Alterations in TP53 in general were more common in squamous cell lung cancer (n = 559 samples) than in adenocarcinomas, with an overall prevalence of 78.4%, or 438 samples. Mutations at the traditional hotspot R248 and the lung-enriched hotspot R158 occurred most commonly, each with a frequency of 7.4%, or 22 of 297 missense mutations. The lung-enriched hotspots V157 and A159 also occurred with higher frequency than in non-lung cancers. In small cell lung cancer (SCLC, n = 236 samples) TP53 alterations occurred in 78.0%, or 184 samples. This is lower than in other published cohorts, where alterations were seen in 98% of samples.(22) While the general distribution of mutations is starkly different from that in lung adenocarcinoma and squamous cell lung cancers, with mutations seen at a variety of amino acid residues including H179, Y220, and P278, the lung-enriched loci V157 and R158 continue to be hotspots in SCLC. Together, these findings show that while minor differences exist among histologic groups – primarily, high R158 frequencies in squamous cell lung cancer in particular – mutations at the lung-enriched hotspots have a high relative abundance in all subtypes of lung cancer.
Table 1.
TP53 mutations in AACR Project GENIE registry v3.0.0.
| Non-lung cancers n = 33,011 samples | Adenocarcinoma n = 5,060 samples | Squamous cell lung cancer n = 559 samples | Small cell lung cancer (SCLC) n = 236 samples | |
|---|---|---|---|---|
| TP53 mutations# | 13,080 mutations in11,983 samples (36.3%) | 2,513 mutations in 2,333 samples (46.1%) | 479 mutations in 438 samples (78.4%) | 190 mutations in 184 samples (78.0%) |
| TP53 missense mutations | 8,813 | 1,750 | 297 | 112 |
| Traditional TP53 hotspot missense mutations* | ||||
| R273 | 951 (10.8%) | 117 (6.7%) | 16 (5.4%) | 8 (7.1%) |
| R248 | 847 (9.6%) | 106 (6.1%) | 22 (7.4%) | 4 (3.6%) |
| R175 | 736 (8.4%) | 44 (2.5%) | 7 (2.4%) | 4 (3.6%) |
| G245 | 324 (3.7%) | 68 (3.9%) | 10 (3.4%) | 3 (2.7%) |
| Lung-enriched TP53 hotspot missense mutations* | ||||
| R158 | 87 (1.0%) | 73** (4.2%) | 22 (7.4%) | 6 (5.4%) |
| V157 | 67 (0.8%) | 49** (2.8%) | 9 (3.0%) | 5 (4.5%) |
| A159 | 48 (0.5%) | 25** (1.4%) | 4 (1.3%) | 0 (0%) |
Frequency is represented in percent of samples with TP53 mutations (including missense, truncating, inframe, and other mutations) out of total number of samples for each histologic subtype.
Frequency is represented in percent of missense mutations out of total number of missense mutations for each histologic subtype.
One case had V157F and A159 mutations (counted separately), and one case had R158L and R158C mutations (counted separately).
Recent studies have defined subsets of lung adenocarcinoma based upon frequently co-occurring mutations, namely KRAS and TP53 or STK11, with impacts on treatment response and survival.(23–25) We sought to determine whether co-mutational events differ among lung cancers harboring the lung-enriched mutations in the Project GENIE cohort. Similar to all TP53-altered lung adenocarcinomas, the most frequently co-occurring mutations in V157-, R158-, and A159-mutated lung adenocarcinomas were found in KRAS and EGFR (which are mutually exclusive). Co-mutated TP53 and KRAS were slightly more common among lung-enriched p53-mutated samples (35.6%, or 52 of 146 samples vs. 26.0%, or 434 of 1,667 samples; Figure 2A and Table 2A). Co-occurrence of KRAS and STK11 in p53-mutant tumors was rare in both groups (2.7%, or 4 of 146 samples, vs 3.1%, or 52 of 1,667 samples), consistent with existing literature.(26) Among lung squamous cell cancers, the most commonly co-mutated genes with TP53 were KMT2D and CDKN2A. (Table 2B) In the lung-enriched mutant p53 cohort, the frequency of co-occurring CDKN2A mutations was higher (31.4%, or 11 of 35 samples) than among squamous cell lung cancers with any TP53 mutation (20.6%, or 59 of 287 samples; Figure 2B). Together, these findings show that common co-occurring mutations (KRAS in lung adenocarcinoma and CDKN2A in squamous cell lung cancer) are even more frequent in tumors harboring lung-enriched p53 mutations.
Figure 2. Co-occurring mutations with TP53 in lung adenocarcinoma and lung squamous cell samples from AACR Project GENIE v3.0.0.
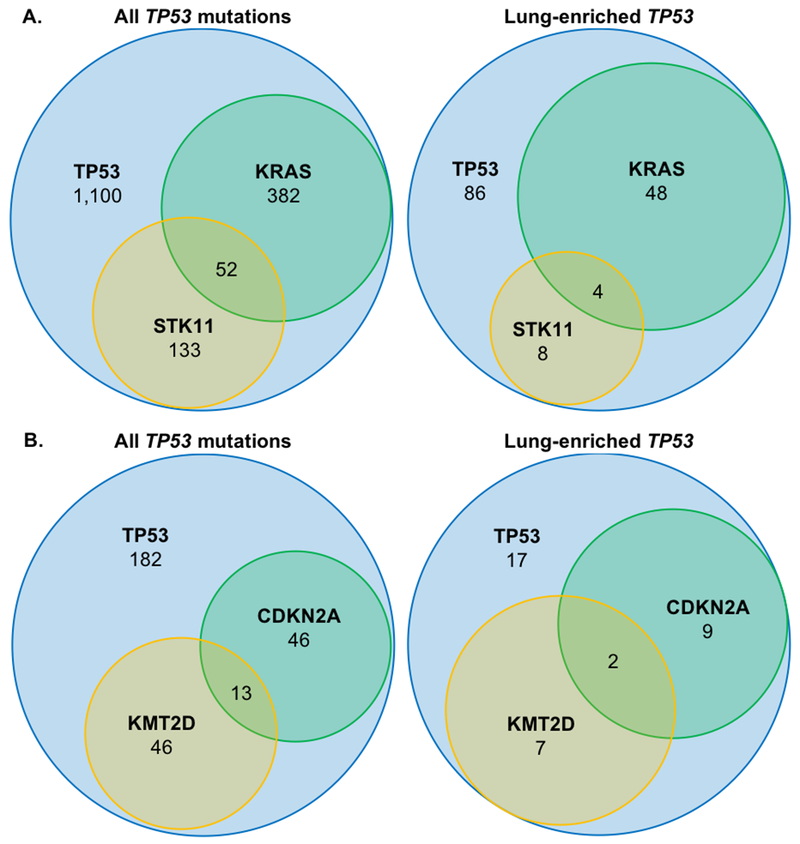
Prevalence of common co-occurring mutations in A. lung adenocarcinoma, and B. squamous cell lung cancers is shown for tumors harboring any mutation in TP53 vs. the cohort of lung-enriched mutations.
Table 2A.
AACR GENIE Lung Adenocarcinoma Co-Occurring Mutations
| All lung adenocarcinomas (n = 5,060 samples) | ||
|---|---|---|
| TP53 mutation* | 2,333 (46.1%) | |
| KRAS mutation** | 1,649 (32.6%) | |
| STK11 mutation*** | 665 (13.4%) | |
| Mutations# | All TP53 mutations n = 1,667 samples (1,750 missense mutations) | Lung-enriched TP53 mutations (V157, R158, A159) n = 146 samples (147 missense mutations) |
| TP53 without KRAS or STK11 | 1,100 (66.0%) | 86 (58.9%) |
| TP53 and KRAS | 434 (26.0%) | 52 (35.6%) |
| TP53 and STK11 | 185 (11.1%) | 12 (8.2%) |
| TP53, KRAS, and STK11 | 52 (3.1%) | 4 (2.7%) |
TP53 mutations include 2,325 samples with mutations, 3 samples with fusions, and 5 samples with multiple alterations.
KRAS mutations include 1,625 samples with mutations and 24 samples with multiple alterations.
STK11 mutations include 652 samples with mutations, 12 samples with fusions, and 1 sample with multiple alterations.
Amplifications and deep deletions are excluded.
Frequency is represented in percent of samples with missense mutations out of total number of samples with TP53 missense mutations.
Table 2B.
AACR GENIE Squamous Cell Lung Cancer Co-Occurring Mutations
| All squamous cell lung cancer (n = 559 samples) | ||
|---|---|---|
| TP53 mutation* | 438 (78.4%) | |
| KMT2D mutation** | 96 (17.2%) | |
| CDKN2A mutation*** | 88 (15.7%) | |
| Mutations# | All TP53 mutations n = 287 samples (297 missense mutations) | Lung-enriched TP53 mutations (V157, R158, A159) n = 35 samples (35 missense mutations) |
| TP53 without KMT2D or CDKN2A | 182 (63.4%) | 17 (48.6%) |
| TP53 and KMT2D | 59 (20.6%) | 9 (25.7%) |
| TP53 and CDKN2A | 59 (20.6%) | 11 (31.4%) |
| TP53, CDKN2A, and KMT2D | 13 (4.5%) | 2 (5.7%) |
TP53 mutations include 435 samples with mutations, 2 samples with fusions, and 1 sample with multiple alterations.
KMT2D mutations include 94 samples with mutations, 1 sample with fusions, and 1 sample with multiple alterations.
CDKN2A mutations include 86 samples with mutations and 2 samples with fusions.
Amplifications and deep deletions are excluded.
Frequency is represented in percent of samples with missense mutations out of total number of samples with TP53 missense mutations.
While a comparison of lung tumors occurring in current or former smokers vs. never smokers would better elucidate the relationship between tobacco carcinogens and the lung-enriched p53 mutations, 64 of the 65 samples with missense mutations at V157, R158, or A159 in the TCGA Pan-Lung Cancer study are from patients with a history of cigarette smoke exposure. (19) However, this delineation has been addressed in older datasets, with the conclusion that the p53 mutation spectra are highly different between smokers and non-smokers. (7,9) Newer datasets such as the AACR Project GENIE, which is not yet fully clinically annotated, may provide more detailed knowledge as smoking rates continue to decline in the US. (27)
In addition to the enriched loci at which TP53 mutations occur in lung cancer, the frequency of specific nucleotide changes is also altered in these tumors. Smoking-related lung cancer arises in a highly mutagenic environment and is associated with a specific G>T mutational signature. This G>T signature is found frequently in p53 mutations in lung cancer – in contrast to the G>A transitions that predominate p53 mutations in non-lung cancers – and supports the important role of smoking in oncogenic p53 mutations in the pathogenesis of lung cancer. Given this, one would expect that if G>T transversions at the canonical hotspots are possible and sufficient for lung tumorigenesis, the presence of non-classical nucleotide changes at non-canonical codons might be unnecessary. Notably, while G>T transversions are also the most common nucleotide substitution in V157 and R158 mutations, a minority of tumors show a G>C transversion.(19) Furthermore, TCGA data reveals that A159, despite including a guanine, exclusively participates in G>C transversions (A159P) or C>T transitions (A159V) rather than the canonical G>T smoking-related signature. Finally, it has been noted that the G>T smoking signature dominates the nucleotide changes at the canonical hotspots R273 (51.6% of R273 mutations in the Pan-Lung Cancer Study), G245 (84%), and R248 (32%) which still occur with moderate frequency, albeit reduced. In contrast, at R175 there are no G>T transversions in the Pan-Lung Cancer Study but rather an increase in C>G transversions Furthermore, there is an overall decrease in frequency of mutations at this traditional hotspot which is usually among the most commonly altered amino acid residues in other solid tumors.(19) This diversity of nucleotide changes suggests that factors beyond cigarette smoke exposure may play a role in selection for p53 mutation and cancer progression in the lung.
Additional support for preferential biological selection of the V157 mutant is illustrated by a case report of a 22-year old patient with Li Fraumeni-like syndrome due to germline V157D p53 and PMS2 mutations who developed lung cancer with loss of heterozygosity (LOH) restricted to lung tumor cells.(28) Notably, the proband’s father also carried a germline p53 V157D mutation with heterozygous mutation noted in a sample of the colon cancer from which he died at age 31. While one interpretation would be that selective pressure exists specifically in the lung for mutations at V157F p53, it is also possible that LOH in the lung adenocarcinoma resulted from its inability to tolerate the presence of even a single allele of wildtype p53 tumor suppressive capacity. Larger scale studies of Li Fraumeni patients harboring different p53 mutations have demonstrated a pattern of variable tumor type and age at onset depending on the mutant allele.(29,30)
Together, this evidence supports our hypothesis that while mutations at codons V157, R158, and A159 may occur as a result of tobacco carcinogen exposure, they may be preferentially selected in lung cancer for additional reasons such as GOF activities with inherent benefit to lung tumors.
Biochemical and molecular rationale for selection of p53 mutations
It is plausible that the lung-enriched mutations are selected for structural alterations resulting in a highly debilitated mutant protein. Fersht and colleagues characterized V157F as a severely destabilizing mutation, with a large number of neighboring side chains affected by the substitution of a larger, hydrophobic phenylalanine for valine, which eliminates function at physiological temperature.(31) Located in the β-sandwich strand S4, V157F mutations cause global effects on the DNA-binding core domain and loop-sheet helix motif, which directly contacts DNA.(32) Additional structural studies, however, have reported that the most common p53 mutant proteins exhibit a wide range of structural debility, suggesting that p53 mutations are selected for more than just a disrupted structure.(5,33)
Early in vivo studies of GOF effects by mutant p53 demonstrated allele-specific differences in tumor type and invasiveness.(34–36) Specifically, Olive and colleagues found, in genetically engineered murine models, that p53R270H/− and p53R172H/− mice exhibited similar survival to that of p53−/− mice, but the heterozygous mutant p53 mice exhibited an increased incidence of epithelial tumors with evidence of invasion and/or metastasis while none of the null mice in the study developed carcinomas.(34) Similarly, p53R270H/+ mice had an increased incidence of carcinomas, most commonly lung adenocarcinomas. In contrast, p53R172H/+ mice were found to develop osteosarcomas at a rate twice that of p53R270H/+ mice. These results together demonstrated GOF effects (invasion and metastasis) due to structural (R175H) and conformational (R273H) mutations in p53 and the emergence of distinct tumor spectra depending on the specific allele present. More recently, Hanel and colleagues demonstrated differential gain of function in tumorigenesis, with accelerated tumor onset of all tumor types and shortened survival in humanized p53 knock-in (HUPKI) p53hupkiR248Q/− mice, in contrast to p53hupkiG245S/− mice which were similar to p53 null mice in tumor latency and survival.(37) To examine the effect of p53 alterations on K-ras-induced lung cancer, Jackson and colleagues generated a compound conditional mouse harboring the LSL-K-rasG12D allele and combinations of p53LSL.R270H, p53LSL.R172H, and p53Flox alleles.(38) This work demonstrated that mice with endogenous expression of the contact mutant p53R270H develop significantly greater tumor burden compared with mice expressing the structural mutant p53R172H. The authors postulated that this difference in tumorigenic potential may explain the relative underrepresentation of mutations at the conventional hotspot codon R175 in human non-small cell lung cancers. Using a similar model, Turrell and colleagues found genotype-specific changes in transcriptional GOF, with increased dependency on mevalonate pathway gene expression in R270H but not R172H lung tumor cells.(39) Furthermore, p53R270H mutant KrasG12D-driven lung tumors exhibited a robust decrease in cell proliferation and survival upon mevalonate pathway inhibition by statin treatment, while p53-null and R172H lung tumors displayed no antiproliferative or apoptotic effect with statin therapy. Collectively, these data demonstrate that mutant p53 proteins exhibit GOF activities during tumor progression in vivo, and also that different hotspot mutations in p53 manifest GOF properties of varying magnitudes and tumor spectra. As yet, no murine studies have examined the lung-enriched p53 mutations at V157, R158, and A159.
Conclusions
We have proposed a paradigm for tissue-specificity in the selection of p53 mutations, which translates into varying frequencies of hotspot mutations unique to specific tumor types. Additionally, we hypothesize that while the unique pattern of mutations at V157, R158, and A159 in lung cancer is in part due to the mutagenic effect of tobacco carcinogens, these specific codons may also impart an oncogenic GOF leading to selection for V157, R158, or A159 mutations. Notably, the clustered nature of the lung-enriched loci suggests structurally or functionally related mutant p53 proteins, although the innate hypersensitivity of this genomic region to DNA adducts cannot be ruled out. From a translational standpoint, elucidating the biological activities associated with the lung-enriched p53 mutations will add to the growing understanding of mutant p53 gain of function and may allow for specific therapies targeted against GOF pathways. Existing treatment strategies against mutant p53 include restoration of wildtype p53 function or degradation of mutant p53 proteins. The GOF pathways induced or repressed by lung-enriched mutant p53 may identify actionable targets for precision therapy in certain subsets of lung cancer. As an example, statins hold promise in targeting the mevalonate pathway, which is responsible for disordered and invasive morphologic GOF exhibited by p53 R273H mutant breast cancers.(40) Many additional questions remain, including the impact of tissue- and microenvironment context on GOF activities and whether different mutant p53 alleles function through different molecular mechanisms.
Acknowledgments
Financial support:
NCI R01 CA164834 (SM)
American Cancer Society 1300042-IRG-16-244-10 (JB)
Footnotes
Conflict of interest disclosure statement:
The authors have no conflicts of interest to disclose.
References
- 1.Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979;278:261–3 [DOI] [PubMed] [Google Scholar]
- 2.Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979;17:43–52 [DOI] [PubMed] [Google Scholar]
- 3.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes & Development 2012;26:1268–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller Patricia AJ, Vousden Karen H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014;25:304–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS. Why are there hotspot mutations in the TP53 gene in human cancers[quest]. Cell Death Differ 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denissenko MF, Pao A, Tang M-s, Pfeifer GP. Preferential Formation of Benzo[a]pyrene Adducts at Lung Cancer Mutational Hotspots in P53. Science 1996;274:430–2 [DOI] [PubMed] [Google Scholar]
- 7.Hainaut P, Pfeifer GP. Patterns of p53 G→T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis 2001;22:367–74 [DOI] [PubMed] [Google Scholar]
- 8.Hussain SP, Amstad P, Raja K, Sawyer M, Hofseth L, Shields PG, et al. Mutability of p53 Hotspot Codons to Benzo(a)pyrene Diol Epoxide (BPDE) and the Frequency of p53 Mutations in Nontumorous Human Lung. Cancer Research 2001;61:6350–5 [PubMed] [Google Scholar]
- 9.Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002;21:7435–51 [DOI] [PubMed] [Google Scholar]
- 10.Rodin SN, Rodin AS. Human lung cancer and p53: The interplay between mutagenesis and selection. Proceedings of the National Academy of Sciences 2000;97:12244–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Z, Muehlbauer K-R, Schmeiser HH, Hergenhahn M, Belharazem D, Hollstein MC. p53 Mutations in Benzo(a)Pyrene-Exposed Human p53 Knock-in Murine Fibroblasts Correlate with p53 Mutations in Human Lung Tumors. Cancer Research 2005;65:2583–7 [DOI] [PubMed] [Google Scholar]
- 12.Kucab JE, van Steeg H, Luijten M, Schmeiser HH, White PA, Phillips DH, et al. TP53 mutations induced by BPDE in Xpa-WT and Xpa-Null human TP53 knock-in (Hupki) mouse embryo fibroblasts. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 2015;773:48–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H-T, Weng M-w, Chen W-c, Yobin M, Pan J, Chung F-L , et al. Effect of CpG methylation at different sequence context on acrolein- and BPDE-DNA binding and mutagenesis. Carcinogenesis 2013;34:220–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng Z, Hu W, Hu Y, Tang M-s. Acrolein is a major cigarette-related lung cancer agent: Preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proceedings of the National Academy of Sciences 2006;103:15404–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menzies GE, Reed SH, Brancale A, Lewis PD. Base damage, local sequence context and TP53 mutation hotspots: a molecular dynamics study of benzo[a]pyrene induced DNA distortion and mutability. Nucleic Acids Research 2015;43:9133–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen Y-M, Troxel AB, Vedantam S, Penning TM, Field J. Comparison of p53 Mutations Induced by PAH o-Quinones with Those Caused by anti-Benzo[a]pyrene Diol Epoxide in Vitro: Role of Reactive Oxygen and Biological Selection. Chemical Research in Toxicology 2006;19:1441–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matter B, Wang G, Jones R, Tretyakova N. Formation of Diastereomeric Benzo[a]pyrene Diol Epoxide-Guanine Adducts in p53 Gene-Derived DNA Sequences. Chemical Research in Toxicology 2004;17:731–41 [DOI] [PubMed] [Google Scholar]
- 18.Glick J, Xiong W, Lin Y, Noronha AM, Wilds CJ, Vouros P. The influence of cytosine methylation on the chemoselectivity of benzo[a]pyrene diol epoxide-oligonucleotide adducts determined using nanoLC/MS/MS. J Mass Spectrom 2009;44:1241–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 2016;48:607–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014;507:315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non–Small Cell Lung Cancer. Clinical Cancer Research 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS - mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer discovery 2015;5:860–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biton J, Mansuet-Lupo A, Pécuchet N, Alifano M, Ouakrim H, Arrondeau J, et al. TP53, STK11 and EGFR Mutations Predict Tumor Immune Profile and the Response to anti-PD-1 in Lung Adenocarcinoma. Clinical Cancer Research 2018 [DOI] [PubMed] [Google Scholar]
- 26.Schabath MB, Welsh EA, Fulp WJ, Chen L, Teer JK, Thompson ZJ, et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene 2015;35:3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discovery 2017;7:818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Sun Y, Gao B, Lu Y, Fang R, Gao Y, et al. Two co-existing germline mutations P53 V157D and PMS2 R20Q promote tumorigenesis in a familial cancer syndrome. Cancer letters 2014;342:36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J, Qian J, Hu Y, Wang J, Zhou X, Chen H, et al. Heterogeneity of Li-Fraumeni Syndrome links to unequal gain-of-function effects of p53 mutations. Scientific Reports 2014;4:4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J, Wang J, Hu Y, Qian J, Xu B, Chen H, et al. Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death &Amp; Disease 2014;5:e1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. [DOI] [PubMed]
- 32.Calhoun S, Daggett V. Structural effects of the L145Q, V157F, and R282W cancer-associated mutations in the p53 DNA-binding core domain. Biochemistry 2011;50:5345–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baugh EH, Simmons-Edler R, Muller CL, Alford RF, Volfovsky N, Lash AE, et al. Robust classification of protein variation using structural modelling and large-scale data integration. Nucleic Acids Res 2016;44:2501–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 Gain of Function in Two Mouse Models of Li-Fraumeni Syndrome. Cell 2004;119:847–60 [DOI] [PubMed] [Google Scholar]
- 35.Lang GA, Iwakuma T, Suh Y-A, Liu G, Rao VA, Parant JM, et al. Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell 2004;119:861–72 [DOI] [PubMed] [Google Scholar]
- 36.Kim MP, Lozano G. Mutant p53 partners in crime. Cell Death Differ 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanel W, Marchenko N, Xu S, Xiaofeng Yu S, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ 2013;20:898–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The Differential Effects of Mutant p53 Alleles on Advanced Murine Lung Cancer. Cancer Research 2005;65:10280. [DOI] [PubMed] [Google Scholar]
- 39.Turrell FK, Kerr EM, Gao M, Thorpe H, Doherty GJ, Cridge J, et al. Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes & Development 2017;31:1339–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freed-Pastor William A, Mizuno H, Zhao X, Langerød A, Moon S-H, Rodriguez-Barrueco R, et al. Mutant p53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell 2012;148:244–58 [DOI] [PMC free article] [PubMed] [Google Scholar]