

Abstract
Within the materials science community, proteins with cage-like architectures are being developed as versatile nanoscale platforms for use in protein nanotechnology. Much effort has been focused on the functionalization of protein cages with biological and non-biological moieties to bring about new properties of not only individual protein cages, but collective bulk-scale assemblies of protein cages. In this review, we report on the current understanding of protein cage assembly, both of the cages themselves from individual subunits, and the assembly of the individual protein cages into higher order structures. We start by discussing the key properties of natural protein cages (for example: size, shape and structure) followed by a review of some of the mechanisms of protein cage assembly and the factors that influence it. We then explore the current approaches for functionalizing protein cages, on the interior or exterior surfaces of the capsids. Lastly, we explore the emerging area of higher order assemblies created from individual protein cages and their potential for new and exciting collective properties.
1. Introduction
Protein cages are self-assembled, monodispersed, three dimensional structures. Many of these cages are spherical in shape, but other shapes such as rods, rings and more complex geometries are also common. The protein cage family consists of virus-like particles (VLPs),1,2 ferritins,3,4 chaperonins,5 and heat shock proteins,6 to name a few. They self-assemble from multiple copies of a single, or just a few, protein monomers into intricate supramolecular structures and possess well-defined interior, and exterior surfaces, as well as interfaces between subunit building blocks. The interior cavity is typically used to confine cargo molecules, and it is physically segregated from the external environment. In the case of viruses, nucleic acids (required for host infectivity and virus replication) are sequestered on the interior of the cage.2,7 Similarly, ferritin uses its interior cavity to store iron as a nanoparticle of ferric oxyhydroxide.8,9 The exterior surface of viruses is responsible for cellular interaction both for the purposes of infection and immunological defense responses.7 The interface between sub-units also plays a critical role in protein cage assembly and disassembly, affecting both the pathways of assembly and resulting particle morphology and also the stability of the final cage structures formed.2
The vast number of protein cages and their mechanisms of assembly have intrigued biologists and virologists for decades, while simultaneously inspiring and capturing the imagination of scientists and engineers far beyond the fields of biology and virology. Viruses are by far the most abundant biological entities on earth with estimated numbers of individual particles that are difficult to imagine (e.g. an estimated 4 × 1030 viruses in ocean water alone),10 and yet just a tiny fraction of these have been well-characterized or have known structures. Because of this fact, virology is still a widely unexplored area that continues to expand. We do know, however, that the formation of virus capsids from protein subunits is an intriguing example of molecular self-assembly11 and that the process has been perfected resulting in a fine balance between structural stability and functional dynamics over millions of years of evolution. The sophisticated mechanisms of protein cage self-assembly and their delicate functionalities are beyond what current materials science can replicate, and scientists have increasingly been inspired by these protein cages for new materials. Biomimetic/bioinspired materials chemistry has used the principles behind the natural assembly of functional protein cage architectures that have been discovered to date to design and create new protein based materials.12,13 In addition to using individual protein based nanoparticle structures, protein cages have been exploited as building blocks for construction of higher order bulk materials because of their size homogeneity and ability to impart a wide range of functionalities.14–17
Protein cages serve as a unique platform for synthetic chemistry not only because the diverse reactive groups available on naturally occurring amino acid side chains (e.g. amines, carboxylates, thiols, amides, phenols), but also the genetic capability to introduce non-native amino acids with a wide range of functionality in a site specific manner.18,19 Most of protein cages mentioned in this review have near-atomic resolution structure models (either X-ray crystal structures or high-resolution cryo-EM structural models) and based on this structural information, one can predict and select the location on the protein cage (e.g. interior, interface, or exterior) where the targeted functionality is to be introduced.
Here we focus on the assembly of protein cages across multiple length scales (i.e. from individual capsid assembly to assembly of individual capsids into higher order structures). We also cover the wide range of techniques for biofunctionalization of the cages to explore new uses and properties of the capsids and hierarchically assembled capsids. We begin with the assembly of protein cages from individual protein monomer building blocks into a cage-like structure, and the effects of solution conditions on the final protein cage structure. A reasonably well understood mechanism of protein cage self assembly from individual sub-units has been exploited to incorporate cargo molecules into the cage either in vivo20–24 or in vitro.25–27 This has proven to be a powerful approach for the development of functional protein cage nanoparticles. Additionally we review recent development of 3D bulk materials constructed via assembly of individual protein cages.14,28 The ability to incorporate wide range of functionality in conjunction with the excellent homogeneity and monodispersity of protein cages make them unique building blocks to construct higher order materials. Self-assembly of individual protein cages themselves into hierarchical structures has led to new materials with the potential for emergent or collective properties.29,30 Other recent reviews have covered a number of applications in which functionalized protein cages, particularly virus particles, have been used.31–34 These include medicine (vaccines, imaging, gene therapy/delivery, drug delivery, tissue engineering), biotechnology (phage display, sensing, nanoreactors), and energy (catalysts, devices, battery electrodes, data storage).31
2. Morphology, size, symmetry and structure of protein cages
In nature, protein cages are found in a wide range of related morphologies, sizes, and symmetries and in turn adopt many different functions. Some examples of the most familiar and widely used protein cages for materials synthesis are shown in Fig. 1. Many (most) are very homogeneous in both size and shape and are highly symmetrical. This is a result of the fairly limited number of protein monomers that assemble to form morphologically distinct and precise protein cage assemblies. Typical sizes of these protein cages range from a few nanometers up to ~500 nm. VLPs are by far the largest and most diverse class of protein cages. Simplistically, viruses consist of an assembled protein shell (capsid) with their nucleic acid cargo sequestered on the inside. VLPs assemble to form protein shells nearly identical to their native virus structures but lacking the viral genome (and other components such as membranes and accessory proteins), thus they are non-infectious. The majority of the structurally characterized viruses consist of roughly spherical protein cages usually built on an icosahedral lattice. Rod shaped viruses are another common shape found among viruses as well as more exotic shapes discovered recently among the archaeal viruses.35 Of course there are many viruses, particularly enveloped viruses, that adopt less regular morphologies.36–38
Fig. 1.

Space filling models of some protein cages discussed in this review. P22 procapsid (56 nm diameter, T = 7) PDBI: 3IYI, CCMV (28 nm, T = 3) PDB: 1CWP, CPMV (30 nm, pseudo T = 3) PDB: 1NYZ, MS2 (27 nm, T = 3) PDB: 2MS2, Qβ (30 nm, T = 3) PDB: 1QBE, ferritin (12 nm) PDB: 2FHA, sHsp (12 nm) PDB: 1SHS, LS (15 nm, T = 1) PDB: 1RVV, and Dps (9 nm) PDB: 1QGH. These images were reproduced using UCSF Chimera (http://www.cgl.ucsf.edu/chimera) from the Resource for Biocomputing, Visualization, and Informatics at the University of California (supported by NIH RR-01081).
An underlying icosahedral symmetry is common among the spherical viruses. A true icosahedral virus will contain 12 5-fold, 20 3-fold, and 30 2-fold rotation axes. The smallest number of subunits needed to make a cage of this symmetry is 60, arranged so that there are 12 pentameric capsomers (capsomers are the basic building blocks from which the capsid is assembled) and no hexamers. Descriptions of viruses often invoke Triangulation number (T),39 which is an allowable integer used to describe the number of nonequivalent positions in the asymmetric unit of the capsid, based on a description by Caspar and Klug.39 Detailed descriptions of triangulation number can be found in the literature.39–41 Briefly, a capsid with more than 60 subunits does not form a strict icosahedron with symmetrically equivalent subunits, but can be formed with minimal distortions using different numbers of equivalent subunits. The triangular faces on these capsids are enlarged and further subdivided into smaller triangles. The triangulation number arises from geometrical constraints where only certain numbers are allowed (T = 1, 3, 4, 7, 12, 13, …, etc.). For an icosahedral capsid, the number of pentamers is always 12, and the number of hexamers can be calculated using the formula 10 × (T – 1). For example, T = 1 viruses such as Adeno-associated virus (AAV) are composed of only 12 pentamers and no hexamers.42 Bacteriophage P22 forms a T = 7 capsid with 12 pentamers and 60 hexamers, for a total of 420 subunits (although in the infectious P22 phage, one pentamer is replaced with a portal complex which plays critical roles in DNA packaging as well as entry and ejection of the viral genome).43
2.1. Virus and virus-like particle protein cages
2.1.1. Spherical protein cages.
Naturally occurring viruses provide a vast library of possible architectures that can be envisioned as starting materials for nanomaterials design and synthesis. The viruses most often encountered and used for materials applications are those derived from plants and bacteria, which are well characterized in terms of structure and biophysical characteristics, and importantly can be produced reproducibly in high yield using their native host plants or heterologous expression system (typically E. coli). Here we briefly cover some of the key properties of the most commonly used VLPs encountered in the biomimetic and bioinspired approaches to materials chemistry.
CPMV.
Cowpea mosaic virus (CPMV) is a 30 nm icosahedral plant virus that infects a number of legume species that was first reported in 1959.44 The genome consists of 2 separate positive sense RNA strands. The outer shell is made up of 60 copies of a small (S) subunit protein and 60 copies of a large(L) subunit that consists of two distinct domains.45 The crystal structure was reported in 1999 to 2.8 A resolution.46 It has become a popular platform for materials design in part because it can be produced with high yield in plants. Until recently a limitation of the CPMV system was that there was no easy method for making the CPMV cage from individual subunit building blocks and that it was difficult to reproducibly make empty virus like particles devoid of infections RNA genome.47,48
CCMV.
Cowpea chlorotic mottle virus (CCMV) is an icosahedral plant virus that infects the cowpea plant creating yellow spots, hence the term “chlorotic”. The CCMV capsid assembles into a simple T = 3 icosahedral capsid comprising 180 identical 20 kDa subunits.49 It has an approximately 28 nm outer diameter and an 18 nm inner diameter. It was first isolated and characterized in 1960s.50 The virus has a tripartite genome (i.e. its genome is in 3 parts, RNA1 [3171 nt], RNA2 [2774 nt], and RNA 3 [2173 nt]) and requires 3 morphologically identical particles to pack the entire genome, RNA1 and RNA2 are packaged alone, and RNA 3 is co-packaged with a fourth RNA, RNA4 (824 nt), which is subgenomic, meaning it is synthesized from the (−) RNA3 strand.51–53 Thus each capsid particle contains ~3000 nt.53 Structural analysis of CCMV has demonstrated that the highly basic N-termini (with 6 Arg, 3 Lys residues) are not required for capsid assembly and project into the interior of the capsid. These 180 N-termini (a total of 1620 basic amino acids) are required to package and condense the anionic RNA viral genome through complementary electrostatic interactions. The N-terminal region is disordered and therefore not observed in the crystal structure. Using the techniques of limited proteolysis combined with mass spectrometry revealed that the N-terminal region is susceptible to proteolytic digestion and is the first part of the capsid to be cleaved despite the fact that it interacts with RNA on the inside of the capsid, suggesting that this region is highly dynamic and is transiently exposed to the outside of the capsid.54 The crystal structure was reported in 1995 to 3.2 angstrom resolution.49 Importantly, CCMV has well studied in vitro assembly system53,55,56 and undergoes a fascinating structural transition in which pores in the structure open and close in response to changes in pH and [Ca2+], going from a closed form to a swollen form49 (Fig. 2). This virus can also be produced with high yield in plant and bacterial expression systems making it a very useful platform for synthetic materials applications.57,58
Fig. 2.

Cryo EM reconstruction images of the swollen and closed forms of CCMV. The swollen condition is triggered upon raising the pH to above ~6.5 and lowering the Ca2+ concentration. The swelling occurs at the quasi 3-fold axes to form ~2 nm pores and increases the particle size by ~10%. Figure reproduced from ref. 59 with permission from Wiley, copyright 2007.
BMV.
Brome mosaic virus is a positive sense RNA icosahedral virus that is 28 nm is size with T = 3 triangulation number. BMV is very similar to CCMV, sharing 70% amino acid sequence identity.49,52 The crystal structure was reported in 2002.60 Like CCMV, the genome consists of 3 separate RNA strands packaged in three separate virions. RNA1 and RNA2 encode proteins 1a and 2a which are necessary for genome replication and transcription of a fourth RNA (a single subgenomic RNA or sgRNA4) from the minus strand of RNA3. RNA3 also encodes for a protein necessary for cell-to-cell movement that direct infection in the host. Coat protein is expressed from the RNA4 strand.61 The CP sequence has a highly positively charged amino acid segment that interacts with the negatively charged genome. Also, like CCMV, it undergoes a structural transition depending on solution pH and ionic strength. The native state is observed at low pH and ionic strength. At neutral pH, it undergoes a conformational change that can be stabilized by added Mg2+. At higher salt and neutral pH, the capsid disassembles and the RNA genome precipitates. The capsid can be reassembled into empty T = 3 particles and T = 1 particles, among others, upon lowering the pH and ionic strength.60,62
Bacteriophage P22.
Bacteriophage P22 is a 56 nm double stranded DNA phage that infects Salmonella typhimurium. It has icosahedral symmetry and forms a T = 7 capsid, with 415 coat proteins (CP, product of gene 5, gp5,) 60–300 molecules of scaffold protein (SP, gp8), minor proteins (gp7, 16, and 20) and a portal protein (gp1).43,63–66 The infectious P22 virion also contains other gene products known as the tail machinery, necessary for assembly and infection shown in Fig. 3. The P22 system has developed into a robust platform based in part on the ease with which it self-assembles in vitro from purified scaffolding and coat protein,67,68 and it has served as a model virus for understanding dsDNA phage assembly mechanism and structures. The assembly process for the VLP, which lacks the portal complex and is comprised of 420 CP and 100–300 copies of the SP, will be described below in Section 3.2. Reports of the structure of P22 have emerged over the years,66,69–72 with the most recent cryo-EM structure model of P22 reported by Hyrc and coworkers to 3.3 angstrom resolution.73
Fig. 3.

(A) A surface volume reconstruction image of the infectious P22 virion. The T = 7 organization is indicated by the yellow lattice cage. The portal complex is located at one of the 5-fold vertices. CP (gp5) is in dark blue. (B) A cutaway interior view of the infections P22 virion. The gene products of the tail machinery are shown in different colors: gp1 (red), gp4, (magenta), gp7, 16, and 20 (purple), gp9 (orange), gp10 (light blue), and gp26 (yellow). Figure reproduced from ref. 43 with permission from AAAS, copyright 2006.
Bacteriophage MS2.
Bacteriophage MS2 is another bacteriophage widely used as a platform for nanomaterials synthesis and VLP-based vaccine development.27,74,75 It is an icosahedral virus that is ~27 nm in diameter composed of 180 subunits (T = 3). The structure was solved to 2.8 Å resolution in the early 1990s.76–78 The genome is a positive sense single-stranded RNA that is 3569 nucleotides in length that codes for 4 proteins: the major coat protein, a maturation protein known as protein A which mediates phage attachment to the bacterial pili, a replicase, and a lysis protein.79 Interestingly, the fully assembled capsid consists of 3 quasi-equivalent CP structures: 30 copies of a symmetric dimer (known as a “C/C” dimer) and 60 copies of an asymmetric dimer (A/B). The primary difference for these structures is in the FG loop region. RNA hairpin structures direct the conformational change in the FG loop region to form the A/B dimers.79–81
Bacteriophage Qβ.
Bacteriophage Qβ is a 25 nm diameter icosahedral capsid that forms a T = 3 structure.82,83 The crystal structure was reported in 1996.82 The genome is made up of a single strand of positive sense RNA that is ~4200 bases long.82 A 29 nt hairpin in the genome has a high affinity for the CP and initiates assembly.84–86 Like MS2, its CP adopts three different conformations with 3 different dimer types. Disulfides link the individual CPs into pentamers and hexamers and result in a highly stable structure (Fig. 4).87 The dissociation temperature (thermal stability) decreases from 85–100 °C to ~40° upon removal of the disulfide bonds.88
Fig. 4.

(A) Chimera reconstruction of bacteriophage Qβ with A chains colored red, B chains colored blue, and C chains colored green. The Cysteine residues C74 and C80 that form disulfide bonds are labeled in yellow. PDB: 1QBE (B) the locations of the disulfide bonds along the FG loop are highlighted (red arrows), both along the 5-fold and 3 fold axes of symmetry. Part B reproduced from ref. 88 with permission from Elsevier, copyright 2011.
2.1.2. Tubular protein structures
TMV.
Perhaps the quintessential example of a tubular rod-shaped protein structure in Nature is the tobacco mosaic virus (TMV). TMV was the first virus to be discovered over a century ago and it has been extensively studied since then.89 TMV was also the first virus to be crystalized and the structure was reported in 1989 to 2.9 angstrom resolution.90 Cryo-EM reconstructions91 are shown in Fig. 5. TMV consists of a helical coat protein structure surrounding a single RNA strand cargo that is bound to the hollow inner channel. There are 2130 identical coat proteins formed in the right handed helix that is 300 nm in length, 18 nm in diameter and has a 4 nm inner channel with 17 coat proteins per turn.92,93 TMV can be reconstituted using a double disk of the coat protein comprising 34 CPs, known as the 20S aggregate, acting as a scaffold. A specific 9 nt RNA sequence acts as a recognition sequence by the TMV CPs and facilitates hydrogen bonding interactions between the RNA and the RNA binding sites on the 20S aggregates. More 20S particles are assembled in a cooperative process and the 5′ end of the RNA is pulled through the central cavity.89,92,94
Fig. 5.
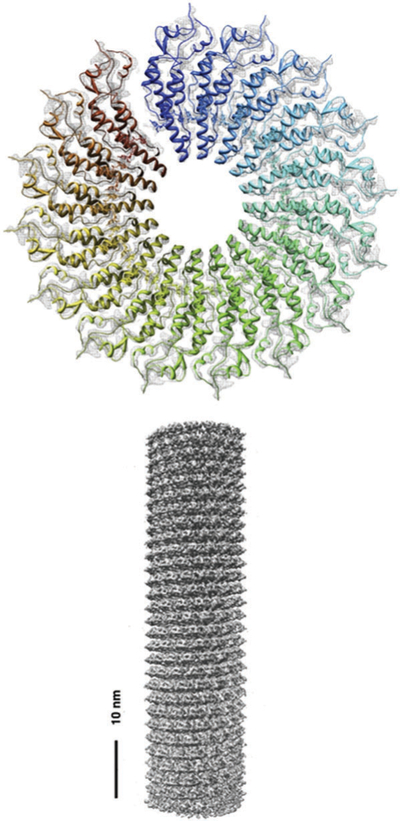
Density map of the TMV structure obtained by cryoEM. (a) Density of a single turn composed of 16 subunits (b) 3D construction image of a TMV rod. Figure adapted from ref. 91 with permission from Elsevier, copyright 2007.
Bacteriophage M13.
Bacteriophage M13 is another well-characterized rod shaped filamentous virus. It is a cylindrical capsid of 880 nm in length with approximately 2700 copies of a pVIII protein along the cylinder with a width of 6.5 nm. There are 5 copies each of pIX and pVII at one end, and 5 copies of proteins pIII and pVI at the other end, which forms the tail of the phage that infects bacteria. The genome is circular single stranded DNA, and dictates the length of the capsid.95–97 One of the most notable applications of M13 in biotechnology is its use for phage display. Here, a library of random peptide sequences is displayed at the end of M13 coat protein (typically pIII) and used for high-throughput screening to investigate protein–protein, protein–peptide, peptide–DNA, and peptide–materials interactions (see Section 5.1).
2.2. Non-viral protein cages
A number of non-viral protein cages have also been explored for materials design and synthesis. While the number of non-viral protein cages are far fewer than the number of viral protein cages, they have wide diversity in size and inherent function-alities, hence they have great utility as platforms for nano-materials synthesis. A few of the most commonly encountered are discussed here.
2.2.1. Ferritin.
Ferritin protein cages are found in nearly all forms of life3,98 and function in vivo as iron storage containers where they encapsulate and sequester iron, usually in the form of a poorly crystalline iron oxyhydroxide.8,9 The general structure consists of 24 subunits with octahedral (4-fold, 3-fold, 2-fold) symmetry (Fig. 6). The subunits consist of a very robust four helix bundle fold with a left-hand twist arranged in 12 antiparallel pairs. The ferritin cage has an outer diameter of ~12 nm and an inner cavity of 7–8 nm.99 Ferritins can be further subdivided into maxi-ferritins (24 subunits), of which classical ferritins and bacterio-ferritins are members, and mini-ferritins (12 subunits), of which DNA-binding protein from starved cells (Dps) proteins are members.100,101 While almost all the true ferritins have octahedral symmetry, mini-ferritins show tetrahedral symmetry (i.e. 3-fold, 2-fold).4,102 Despite the quite significant differences in primary sequence among ferritins, the secondary, tertiary and quaternary structures are highly conserved.92,99 One notable exception to this is the ferritin isolated from the hyperthermophilic Archaeon Archaeoglobus fulgidus (AfFn). Despite the fact that AfFn is assembled from 24 subunits and the subunit structure has a high degree of structural similarity with other ferritins, it assembles into a cage structure not having octahedral symmetry but rather tetrahedral symmetry with four large pores (B4.5 nm) (Fig. 6).103,104 Interestingly, a double mutant of AfFn, K150A/R151A, assembles into a closed cage with octahedral symmetry, resembling the archetypal ferritin structure, suggesting a critical role for these two residues in inter-subunit interactions to form the quaternary structure.105 The assembly mechanism of ferritin from its constituent subunits is highly concentration dependent and starts with the unfolded monomers becoming properly folded. It is followed by rapid formation of dimers that accumulate since the dimerization is fast compared to the subsequent associations. The dimers go on to make trimers, hexamers, and dodecamers and finally forms the 24-mer fully assembled cage. The purported hexamer intermediate is the most transient of the intermediates and only detected in small amounts.92,106 On the other hand, the assembly mechanism for mini-ferritins remains poorly understood.
Fig. 6.

Ribbon diagrams of exterior surface of view of (A) human heavy-chain ferritin (PDB: 2FHA) looking down from 4 fold axis (left) and 3 fold axis,(B) ferritin from Archaeoglobus fulgidus (PDB: 1S3Q) looking down from 3 fold axis, and (C) Dps from Sulfolobus sofataricus (PDB: 2CLB) looking down from 3 fold axis. These images were reproduced using UCSF Chimera (http://www.cgl.ucsf.edu/chimera) from the Resource for Biocomputing, Visualization, and Informatics at the University of California (supported by NIH RR-01081).
2.2.2. Other non-viral protein cages.
Lumizine synthase is an enzyme involved with riboflavin biosynthesis that can form icosahedral capsids with 60 subunits, i.e. it structurally resembles a T = 1 virus capsid.107–109 Other structurally characterized icosahedral protein cages include encapsulin,93 clatherin,110 and dihydrolipoyl acetyltransferase (E2) from Bacillus stearothermophilus.111 Small heat shock protein (sHsp) from M. jannaschii is a 12 nm protein cage with octahedral symmetry composed of 24 subunits,6 meaning the overall morphology and size resemble ferritin. However, the subunit structure of sHsp consists of β-sheets instead of the α-helix bundles of the ferritin subunit. Hsp has large (3 nm) pores at the 3-fold symmetry axis and smaller(1.6 nm) pores at the 4-fold symmetry axis. Other protein cages with non-spherical morphologies have also been discovered, such as chaperonins and vault particles. The chaperonins, which are hollow cylinder-like protein-assemblies, function to encapsulate non-native state proteins within the cylindrical cavity to mediate their proper folding to the native states.5 The chaperonins use their ATP-dependent conformational changes to assist folding of the guest proteins. The chaperonins are categorized into two types; group I and group II based on their structure and origin. Group I chaperonins are composed of two stacked rings of supramolecular proteins each with 7-fold rotational symmetry (e.g. GroEL in bacteria, HSP60 in mitochondria) which is capped by a smaller protein (GroES in bacteria, HSP10 in mitochondria) on the top of the rings. Group II chaperonins (e.g. Thermosome in archaea) have a built-in protrusion that functions as a “lid” structure. Vault particles adopt a barrel-like shape, with a 41 nm diameter and 73 nm length. They are termed vault particles of their structural resemblance to vaulted ceilings in Gothic cathedrals.112 To date, they have been found in number of eukaryotes, however their function remains poorly understood.113
3. Assembly of monomers into protein cages
3.1. Assembled structures have common protein folds
Since their discovery at the end of the nineteenth century, viruses have provoked discussion among scientists regarding the question of whether viruses should be considered living organisms.114 The origin of viruses from an evolutionary perspective also remains an active area of investigation.115,116 With a number of viral structures and viral protein structures now known (>6900 entries in the PDB with the keyword virus), a few families with common protein folds have been identified and structural relationships to these have been proposed as a method for classification of viruses.117 Interestingly, capsids with similar architecture/folds infect hosts of all domains of life, which suggests a common ancient origin for virus families.117–120 Four major classes have been proposed for icosahedral viruses:(1) PRD1/adenovirus-like, (2) picornavirus-like (3) HK97-like, and(4) BTV-like.117 Here, we focus on the well-established PRD1/adenovirus and HK97-like viruses because their members are the most commonly encountered in the protein cages used for materials synthesis. The others are reviewed elsewhere.117
The capsids of the PRD1/adenovirus family consist of trimeric major capsid protein with a double β-barrel structure. The major capsid proteins form trimers, that become hexagonal capsomers that eventually go on to make an icosahedral structure.120 Bacteriophage PRD1 serves as the naming member of this line-age. The structure of the infectious virus consists of the protein capsid surrounding a protein-rich lipid membrane. The dsDNA genome is contained within the lipid membrane. The structure of the major capsid protein was first reported in 1999,121 followed by the entire virion structure solved by X-ray crystallography in 2004.122,123 Since then, other capsid types with similar double β-barrel fold have been identified and classified as part of the lineage, some of which are given in Fig. 7.
Fig. 7.

Structures of member capsid proteins of the PRD1-adenovirus lineage. (a) The structures are STIV (Sulfolobus turreted icosahedral virus, cyan), human adenovirus (purple), bacteriophage PRD1 (green), bacteriophage PM2 (blue), and Paramecium bursaria chlorella virus type 1 (PBCV-1, red). (b) The structure of the PRD1 virion, which forms a T = 25 capsid. The inset shows the trimeric capsomer with the individual monomers indicated. Figure adapted from ref. 120 with permission from Nature Publishing Group, copyright 2008.
Another group that has been well characterized is the HK97 protein fold, named because HK97 was the first virus structure solved in this group. Over 40 different structures have been identified having HK97-like protein folds (Fig. 8). Interestingly, only about 10–15% of the amino acid sequence is shared among the members. The structure of this protein fold is identified by a number of features that include an N-terminal region arm (N-arm) sometimes containing alpha helical portions, an extended loop of varying lengths (E-loop) that is a two-stranded anti parallel beta sheet.124 It is also characterized by a peripheral domain (P domain) containing a “spine helix” and an unusually long beta sheet, and an axial domain (A domain) with a central beta sheet surrounded by short helices and loops.124 The family of viruses having this protein fold structure are also characterized by the presence of an internal scaffolding protein that directs capsid assembly. Additional domains are also observed among other family members.
Fig. 8.

HK97 and related protein structures with common features high-lighted. (A) HK97 in the immature state (PDB: 3E8K), (B) P22, (C) T4, (PDB: 1YUE), (D) BPP-1 (PDB: 3J4U), and (E) P-SSP7 (PDB: 2XD8). The common features are N-arm (red), E-loop (yellow), P-domain (green), A-domain (cyan), β-hinge (orange). (F) An electron density map of the HK97 capsid with capsomers (both the hexamer [6-asymmetric subunits, multicolored] and the pentamer [red]) indicated. Parts A–E adapted from ref. 124 with permission from Elsevier, copyright 2015. Part F adapted from ref. 125 with permission from Nature Publishing Group, copyright 2009.
3.2. What drives the assembly process?
The assembly process is driven primarily by favorable interactions, typically a number of individual, weak contacts are made among capsid subunits and cargos that add up to give a globally stable structure. The primary interactions to be considered are subunit–subunit, subunit–cargo, and cargo–cargo interactions.126,127 Assembly of capsids can be divided into two categories: those that can assemble without the need for a cargo molecule (empty capsid assembly) and those in which interactions between capsid subunit and cargo direct the capsid assembly. The second categories can be further subdivided into two sub-class based on type of cargo molecules: either proteinaceous molecules (called scaffold protein) or nucleic acids (typically virus genome, but can be non-genomic nucleic acid).
3.2.1. Empty capsid assembly.
Empty capsid assembly can be understood from a model of nucleation and growth.127,128 Viruses need to encapsulate their genomes inside of the capsids and the assembly of empty capsids can be modeled and under-stood from relatively simple models, which nevertheless provide useful insights into how subunit–subunit interactions lead to the self-assembly of the capsid architecture. In this mechanism, an assembly nucleus or seed is formed from just a few protein subunits. These clusters are transient and relatively unstable because of the low number of favorable intersubunit inter-actions. A critical nucleus is formed when a sufficient number of subunits form in the correct geometry that allows for further growth. Growth is rapid in comparison to the formation of nuclei. The assembly process kinetics are sigmoidal in which an initial lag phase, where the nuclei are formed, is followed by rapid addition of subunits to complete the capsid (growth), followed by an asymptotic approach to equilibrium due to depletion of subunits (saturation). In general, increasing the subunit concentration or strengthening inter-subunit inter-actions will lead to more rapid assembly and the possibility increases for the formation of an excess of partially formed capsids, due to too many nucleation sites formed or kinetically trapped assembles, that might not lead to the correct close shell capsid structure. Lowering the subunit concentration leads to large nucleation barriers, due to fewer collisions between subunits, and therefore fewer chances of forming the critical nuclei.127,129
CCMV is model virus that has been studied for decades in order to elucidate the self-assembly process of capsid formation. It has been demonstrated that under appropriate conditions, subunits of CCMV can self-assemble to form empty capsid in the absence of RNA.56,130 For empty capsids, it follows the nucleation and growth model where CCMV subunit dimers associate to form a pentamer of dimers that serves as the critical nucleus for subsequent growth.130 When the protein concentration is low, assembly continues with the addition of dimers and the formation of T = 3 capsids is favored. When the subunit protein concentration is high, many nucleation sites are formed which causes incomplete formation of capsids and leads to formation of pseudo T = 2 particles.130 As we will discuss later, CCMV capsid can also self-assemble around RNAs,53 non-viral anionic polymers,131 and negatively charged gold nanoparticles.132 In vitro reconstruction of VLPs directed by negatively charged non-viral cargo molecules have been demonstrated with other viruses such as BMV and red clover necrotic mosaic virus (RCNMV).133–135
3.2.2. Assembly mechanisms involving scaffold proteins.
Virus particle formation may also require the use of scaffold protein (SP) to direct the self-assembly of subunit components into the correct capsid structure. One well known example of this is the bacteriophage P22, whose assembly involves association of both scaffolding proteins (SP) and coat proteins (CP) via specific interactions,136 which results in co-assembly of 415 subunits of CP (gp5) with about 60–300 subunits of SP (gp8) (Fig. 9) and CP is added along the growing edge of the shell until the capsid is formed. SP stabilizes and directs the CP into the correct geometry.124 Intriguingly, scaffold protein can form oligomers (dimers and tetramers) as well as monomers, with the dimer being the dominant, active form in assembly.137 This suggests a mechanism for striking a proper balance between nucleation and growth by changes in the oligomerization state of the SP.138 The interaction between coat and scaffold protein is largely electrostatic in nature because about 30% of the C-terminal helix–turn–helix of the SP is positively charged, with R293 on the SP and D14 on the CP being essential for proper assembly.124,139,140 Additionally, interactions between K296 (SP) and E15 (CP) are also likely to be important.140 Scaffold protein is also critical for formation of T = 7 capsids, rather than either smaller or larger capsids. Without scaffolding protein, aberrantly assembled particles, including small shells and spirals that cannot package DNA are formed at a slower rate.141 Also during in vivo assembly, a dodecameric portal complex and ejection proteins, which are required for formation of the infectious virion, are incorporated into the procapsid at this stage (one pentameric vertex of icosahedron is occupied by the portal complex43). The portal complex has been proposed to be part of the nucleation complex, which would ensure that only one portal complex is incorporated per virion.142 In the infectious virus, DNA is packaged through this portal complex after assembly of the P22 procapsid and the scaffold protein is removed after procapsid assembly during the morphological transformation of the capsid through packaging of nucleic acid (Fig. 9). Upon DNA packaging and removal of the scaffold proteins, the spherical procapsid transforms into the mature polyhedral capsid – resulting in a change in conformation of the coat protein and an approximately 10–15% increase in radius.124,143 The exact exit location of the SP is not known, but it is presumed to be through large 2.5 nm pores at the center of the hexamers.143 The scaffold protein can be recycled to take part in assembling more capsids as shown schematically in Fig. 9. Most importantly and usefully for materials synthesis is the fact that assembly of the bacteriophage P22 capsid can be reproduced by expressing only CP and SP in heterologous protein expression system using E. coli, or mixing CP and SP under a controlled manner in vitro.67,135,144 In this case, the portal complexes of the infectious P22 virions are replaced with 5 CP subunits, thus the capsid is composed of 420 copies of CP with approximately 60–300 copies of SP. Removal of SP form the capsid and morphological transformation from the procapsid form to the matured polyhedral capsid can also be reproduced in vitro by treating the procapsid form of P22 with 0.5 M guanidine hydrochloride67,145 and heating it at 65 °C, respectively.146,147
Fig. 9.

Scheme of P22 assembly. Figure adapted from ref. 65 with permission from Elsevier, copyright 2010.
3.2.3. Assembly around a nucleic acid cargo.
Other assembly mechanisms involve assembly directly around the nucleic acid cargo as demonstrated experimentally53,148–151 and suggested by simulations.127 This mechanism generally includes most ssRNA viruses. The primary driving force for this process is electrostatic interactions between the positively charged capsid proteins and the negatively charged nucleic acid cargo, but recent work has shown that specific RNA sequences act as a signal for packaging and play a key role in the assembly of some capsids.151,152 The RNA of tobacco mosaic virus initiates its own packaging and the coat protein subunits co-assemble with RNA to form the characteristic rod-shaped particle in which the nucleic acid is sequestered on the interior of the 300 nm long rod.153,154 In another example, based on recent cryo-EM structures of empty VLP and fully formed CPMV, a model for assembly both with and without the RNA cargo (Fig. 10) has been established. The C-terminal extension of the small subunit (S) plays an important role by not only forming favorable hydrophobic and electrostatic contacts with neighboring subunits to stabilize penton formation, but also promotes formation of an RNA binding site, and therefore can be considered a scaffolding protein or molecular chaperone.155 The C-terminal extension is highly positively charged and it is cleaved during virion assembly, but stay intact longer when no RNA is present. In the virion, the interface between the two pentamers (two fold axis) has the strongest interaction with the RNA cargo and begins the encapsulation process. Many other members of the Comoviridae family (of which CPMV is a member) have a similar C-terminal extension that is cleaved during assembly, however the length and sequence are highly variable.
Fig. 10.

A model for CPMV virion and empty VLP assembly. Pentamers are made up of large (L, green) and small (S, blue) subunits. The C-terminal extension is represented by the violet circle. RNA is the orange rectangle. In RNA free assembly (upper panel) C-terminal cleavage is slow. In the presence of RNA (bottom panel), the C-terminal extension is cleaved fast and RNA binds at the 2-fold axis. Figure reproduced from ref. 155 with permission from Nature Publishing Group, copyright 2015.
From these experimental observations and simulations for assembly around a polyelectrolyte (i.e. a nucleic acid cargo), two proposed mechanisms have emerged (Fig. 11).127,156,157 In one mechanism the CPs assemble “en masse” in a disordered orientation and cooperatively rearrange to form the ordered capsid.158 The other mechanism is similar to the nucleation and growth mechanism described above, in which the poly-electrolyte helps stabilize CP–CP interactions of an ordered nucleus, and growth continues with sequential addition of CPs. Simulations suggest that ionic strength plays a role in determining which mechanism is followed. At low ionic strength, CP–polyelectrolyte cargo interactions dominate and the en masse mechanism is observed, whereas at higher ionic strength, CP–CP interactions dominate and nucleation and growth is observed.156 These observations point to the critical role pH and ionic strength play in capsid assembly, as has been observed experimentally.
Fig. 11.

Assembly of a capsid around a polyelectrolyte cargo (e.g. a nucleic acid). Red is the nucleic acid cargo and blue is the coat protein. At low ionic strength (stronger coat–cargo interactions) and weak cargo–cargo interactions, an en masse mechanism is favored (A). The nucleation and growth mechanism is favored when coat–cargo interactions are weaker (higher ionic strength) and stronger coat–coat interactions. Figure adapted from ref. 156 with permission from Elsevier, copyright 2014.
3.2.4. Assembly of other protein cages.
A few studies have investigated the assembly mechanism for non-viral protein cages. For example, a mechanism for the assembly of apoferritin was first put forth by Gerl and coworkers.106,159 The assembly is proposed to proceed first by unstructured monomers (mi) adopting a structured confirmation (M1), which quickly form stable dimers. Dimers, trimers, hexamers and dodecamers intermediates were all detected. In the final step, two dodecamers assemble to make a fully formed 24-mer99 (eqn (1)).
| (1) |
Recent work has used time-resolved small angle X-ray scattering (TR-SAXS) to investigate this mechanism further. The observed TR-SAXS profiles were explained by a slightly modified simple scheme given in Fig. 12.160 The main difference being monomers and trimers are not included as intermediates ofhe reaction. Like the other assembly mechanisms described previously, pH, ionic strength, and subunit concentration are important considerations. In the case of ferritin, these factors may influence not only the kinetics of the reaction, but which intermediates are observed.160,161
Fig. 12.

Proposed model for the assembly of ferritin by Sato et al. Each dimer unit is depicted as a different color. In this mechanism, hexamers (a trimer of dimers) can form directly from dimers, but also from a tetramer and a dimer. In the final step, 2 dodecamers form the 24-mer cage. Figure reproduced from ref. 160 with permission from the American Chemical Society, copyright 2016.
Another important example of non-viral protein cage assembly are the bacterial microcompartments, in which multiple copies of enzymes are enclosed inside of a single proteinaceous shell. Hagan and coworkers have adapted their previous model156 for encapsulation of a polyelectrolyte cargo to study encapsulation of multiple cargos,162 like that observed with a bacterial micro-compartment. Their simulation showed that assembly can occur via two distinct pathways: (1) a single-step assembly in which cargo molecules and shell subunits assemble simultaneously, or (2) a multi-step assembly in which the cargo molecules assemble first to form a dense globules, followed by adsorption and assembly of shell subunits around the globules. Relatively weak cargo–cargo interactions lead to single-step pathway, whereas stronger cargo interaction tend to follow the multi-step pathway. Their simulation study also indicates that encapsulation of high density cargo requires a balance between cargo–cargo, cargo–shell and shell–shell interactions (in the range of 5–10kBT),162 much like virus particle assembly described earlier. These observations indicate that a delicate balance of molecular interactions (a “sweet spot”) between all the subunits of a protein cage must be struck in order to form functional particles.
3.3. Factors influencing in vitro assembly
3.3.1. Changes in pH and ionic strength can lead to different assembled structures.
CCMV is the first icosahedral virus that was reassembled in vitro to form an infectious particle.55,163–166 Since then many studies have investigated the factors influencing assembly, such as changes in pH and salt concentration, coat protein concentration, and CP ratio to nucleic acid.130,167–169 The CP interacts with the RNA primarily through highly basic, N-terminal arginine-rich motifs (ARM). Under appropriate conditions, CCMV capsids can self-assemble in the absence of RNA. Assembled structures range from not only single-walled capsids, but multishell and tubular structures, depending on the pH and salt concentration (Fig. 13). Multishell formation is favored at low pH (and low ionic strength) starting near pH 3.7 (the pI for CCMV CP) and up to pH 5. As pH increases, the capsid exterior becomes increasingly negatively charged, and electrostatic interactions between the exterior surface and the positively charged interior surface dominate assembly. The number of shells formed is limited to ~3 due to the unfavorable changes in the shell curvature as the layers grow. Multishell formation decreases and a single shell regular capsid with 28 nm becomes dominant as ionic strength increases, due to charge screening.56
Fig. 13.

The range of particle morphologies possible of CCMV assembly as a function of pH and ionic strength. (A) Phase diagram of protein assembly outcome over a range of pH and ionic strength. Samples were buffered with sodium cacodylate (red) or sodium citrate (black). The blue area represents conditions of assembly from ref. 168 and 169. (B) Electron microscopy images of different observed morphologies. (a) Multishelled structures are dominant at pH 4.8 and I = 0.01. (b) Tubular structure observed at pH 6.0 and I = 0.01 (c) Dumbbell shaped particles at pH 7.5, 0.001 M cacodylate buffer.(d) Single walled shells at pH 4.67 and 0.1 M acetate buffer and I = 0.1 Figure reproduced from ref. 56 with permission from the American Chemical Society, copyright, 2009.
The crossover to tubular rather than spherical structures occurs near pH 6 and above. This arises due to favorable hexamer–hexamer interactions rather than hexamer–pentamer interactions. The hexagonal sheets fold to form tubular structures some of which are capped with hemispheres of hexamers and pentamers. Smaller diameter tubes are favored at higher ionic strength due screening of the inner and outer surface mentioned above.56
pH also plays a critical role in formation of CCMV capsids around an RNA cargo. Near neutral pH, strong interactions between RNA and CP (containing the highly-positively charged arginine containing N-terminus) result in partially formed protocapsids that condense the RNA. Lowering the pH increases the strength of CP–CP interactions due to protonation of Glu81 and results in procapsid formation.53 The assembly can be described as two separate steps required to obtain the properly assembled structure. First, at low ionic strength and neutral pH, the CPs undergo disordered absorption on the RNA. When the pH is reduced, CP–CP interactions are strengthened, and the ordered capsid is formed. Fine-tuning these interactions is critical for proper in vitro assembly; if the attractions are too weak, the capsid is not formed. If they are too strong, the assembly falls into kinetic traps.149 For complete packaging of the native RNA, an excess of CP is needed – specifically when there is a matched amount of charge among CP and the RNA.53
3.3.2. Formation of non-native viral cages.
Non-native, aberrantly assembled, virus particles that form tubular structures are known as polyheads. These have been observed in HIV,170 bacteriophage T4,171 bacteriophage T7,172 and bacteriophage P22.173,174 Generally these structures are formed as a result of mutations in the CP sequence. Remarkably, a single mutation of the F170 residue in the CP of P22 results in formation of polyheads up to 2 mm in length.173,174 F170 is located in the β-hinge region of the CP, and causes decreased flexibility in the rest of the CP (especially in the A domain) and consequently favors assembly of hexamers over pentamers, suggesting the importance of CP flexibility to direct assembly of CPs into a proper capsid structure. The polyheads structures consist mostly of hexamers, but pentamers can be incorporated at the ends, which leads to termination of the growing structure.173
3.4. Synthetic/computational approaches to designing protein cages
Inspired by the intricate self-assembly of native proteins into larger closed shell architectures, researchers have applied the principles learned from nature to computationally design proteins to assemble into a range of synthetic protein cages. A key concept to designing these protein cages is the underlying symmetry of the assembled cages. Highly symmetrical structures assembled from a defined number of subunit building blocks allow for a minimum number of subunit interactions to be designed, as each new interface increases the complexity of the design.175 One such approach is to create fusions of known oligomeric proteins that already have intrinsic protein–protein interaction interfaces.12,13,175–179 For example, the design of a protein cage with cubic symmetry (432) has been successfully demonstrated by fusing two natural protein oligomers, a dimeric and a trimeric protein (Fig. 14). This defines two different symmetries, two-fold and three-fold, that are necessary for assembly into the correct structure. A computationally designed linker molecule provided the correct angle needed for self-assembly of the cubic structure, but other geometries such as tetrahedral and triangular prism structures are also observed.13 The other approach is to design new protein– protein interfaces.175,176,180,181 This is a new, very promising approach for precisely designing protein cages with desired size, shape, and amino acid composition for specific applications. While very specific interfaces can be designed, it comes at a cost of requiring sophisticated algorithms for design.175 This can be minimized however, by using natural oligomerization motifs as a model and then begin building new interfaces by sequence mutations.176 King et al. were able to engineer a number of two component protein cages using a library of dimers and trimers.180 Computationally this is accomplished by first choosing the desired architecture and symmetry docking the components (regions with large areas of contact) followed by re-designing the contact interface by changing the amino acid composition at the interface. The design feature makes use of hydrophobic protein cores surrounded by polar rims. More recent work by Baker et al. showed that a 60 subunit protein cage could be computationally designed from trimeric protein building blocks.182 In another example, Jerala et al. showed tetrahedral protein cages, four-sided pyramids, and a triangular prism could be assembled from protein coiled-coil dimers.183 De novo designed cages offer advantages over natural protein cages, such as customizable geometry, functionality, and increased stability. However, one disadvantage of this approach is that when computationally designed proteins are expressed in vivo, many sequences often result in insoluble inclusion bodies, which cannot yet be accurately predicted a priori.
Fig. 14.

Model of the engineered fusion protein and the resulting octahedral structure formed. (a) The fusions are composed of a trimeric protein (KDPGal aldolase, green) and a dimeric protein (FkpA, orange) with a four residue helical linker (blue). The lines represent the three-fold (cyan) and two-fold (magenta) axes. (b) The 24-mer cage with octahedral symmetry, with the symmetry axes shown. (c) 2D class averages of the 24-mer and 12-mer obtained after aligning, clustering, and average particles from several particles (left) which are consistent with calculated projections (center). The 3D atomic models (right) are also included. Figure reproduced from ref. 13 with permission from Nature Publishing Group, copyright 2014.
4. Encapsulation within protein cages
Although the biological function of the previously mentioned protein cages are different from each other, they have a relatively simple common task, i.e. sequester and protect the encapsulated cargo. The closed shell architecture of protein cages clearly defines a unique interior environment that is physically separated from the bulk, exterior, environment. Millions of years of evolution has taken advantage of this for the sequestration and protection of viral genomes within viral capsids, for the sequestration of an inert and non-toxic form of iron as iron oxide in the protein cage of ferritin and in some bacterial microcompartments the shell may act to localize enzymes capable of transforming harmful inter-mediates. There are many ways to achieve sequestration of cargos, and the synthetic community has paid attention to the structural and mechanistic understanding of how biological systems have achieved this, which has provided inspiration for biomimetic approaches to materials design and synthesis.2 In this section, we discuss the approaches and the inspiration behind using protein cage architectures as size constrained containers for the encapsulation and sequestration of a wide range of cargos.
Conceptually the encapsulation of a cargo on the interior of any one of these protein cages can usefully be approached in two ways: either through the initial formation of the protein cage which can then subsequently be filled with cargo or alternatively, through the entrapment of the cargo on the interior of the cage during the process of assembly from individual components into a cage. As we discussed above, examples of each of these approaches can be found in the packaging of nucleic acids within viral capsids, i.e. genomic DNA packing into a preformed procapsid as observed in bacteriophage P22,138 or subunit proteins interacting with nucleic acids and packaging it during capsid assembly, which is observed in CCMV.53
Using the preformed capsid as a reaction vessel requires that the cargo (or a cargo precursor) can traverse the shell. This usually implies the use of a small molecule or at least a cargo of size commensurate with the pore size of the capsid or that the capsid has dynamic behavior allowing the pore size to fluctuate, which could allow larger molecules to pass across the shell. Alternatively the cargo can be synthesized on the interior of the preformed capsid from small molecule precursors that can freely diffuse across the capsid shell. This is akin to a ‘ship-in-a-bottle’ synthesis/assembly and is the approach seen biologically in the biomineralization of an iron oxide particle on the interior of the iron storage protein ferritin where iron ions can diffuse through the shell, but the inorganic polymer of iron oxide is too large to leave and is therefore sequestered on the interior.3,184
Co-assembly of capsid and cargo on the other hand suggests an interaction between the capsid building blocks and the cargo. The type and strength of this interaction can vary widely from direct covalent fusion of the cargo and the capsid protein to weaker non-covalent interactions. Multivalent interactions and templating of the capsid assembly through interactions between the capsid and the cargo have biological relevance and have provided a conceptual framework that has been effectively exploited for synthetic applications.
4.1. Ship-in-a-bottle like synthesis in preformed capsids
As we pointed out previously, the ferritin family of iron storage proteins are widespread in almost all domains of life and function largely to catalyze the oxidation of ferrous ions and nucleation of a ferric oxyhydroxide nanoparticle sequestered within the cage.4 The protein subunits that assemble into these cage-like architectures contain catalytically active sites for oxidation of Fe(ii) ion to Fe(iii) (ferroxidase sites) and iron oxide nucleation which is largely through electrostatic interactions between the capsid and the accumulated ions. The cage has small well-defined pores of roughly 3 Å in diameter along the 3-fold and 4-fold symmetry axes.185 Electrostatic potential calculation of ferritin suggests that the 3-fold channel is likely to be a major entrance that enables iron ions to traverse the shell and reach the interior. This indicates that ferritin has efficiently evolved this design of taking up iron from external environment into its cavity. The overall oxidation and mineralization reactions are catalyzed on the interior of the cage.185–187 This mineralization of the small diffusible precursors is an excellent example of the ship-in-a-bottle synthesis on the interior that results in the formation of a nanoparticle product that is sequestered on the interior of the cage. In the case of ferritin this mineralization has the additional effect of continually removing soluble ions from solution thus creating a concentration gradient across the shell, which effectively drives the transport of ions into the cage and results in further accumulation on the interior. These components are all programmed into relatively simple protein subunits that can act both as the building blocks for capsid assembly and the active catalyst for the biomineralization.
Many studies have shown that a wide range of ionic and metallic nanoparticles, with no direct biological relevance, can be selectively grown inside the pre-formed protein cages derived from ferritin and related Dps proteins (Fig. 15A).4,188–192 The substrate flexibility in ferritins and Dps is largely due to the facile transport of cations across the capsid shell in response to an electrostatic gradient, and facilitated nucleation on the interior of the cage driven by ion accumulation at patches of high negative charge density on the interior protein interface. In addition, the protein cages themselves provide a well-defined volume and thus control and limit the size of the inorganic nanoparticles grown.
Fig. 15.

(A) Schematic illustration of magnetite (or maghemite) nanoparticle synthesis in ferritin. Magnetite nanoparticles can be formed and sequestered inside of ferritin even though temperature and pH used for this synthesis are far from physiological condition. TEM image on the right shows magnetite nanoparticles with approximately 6 nm in diameter synthesized inside of ferritin. (B) TEM image of a TMV containing an approximately 250 nm long nickel nanowire in the central channel. Part B reproduced from ref. 196 with permission from Wiley, copyright 2004.
The strategy of using electrostatic interaction between protein cages and cargo molecules for cargo entrapment has been extended beyond ferritin and Dps. The observation that CCMV packages its RNA cargo primarily through electrostatic interactions between the RNA and the capsid is reminiscent of the ion–capsid interactions in ferritin, despite the fact that the charge relation between cage interior and cargo is opposite, i.e. interaction between highly positively charged N-terminus domain of capsid protein and negatively charged nucleic acid cargo. Thus the CCMV viral protein cage was initially explored as a constrained environment for a range of nanomaterial synthesis and encapsulation from negatively charged precursor molecules. The viral capsid has pores at subunit–subunit interfaces allowing free access to small molecules through the capsid shell. The native CCMV, with its highly positively charged interior, was used to nucleate the growth of polyoxometalate crystals comprising the highly anionic H2W12O4210− ions with NH4+ counterions.193 The positively charged N-terminus of the CCMV was postulated to act as a charged interface to accumulate the anions, which after nucleation could grow into the final particle filling the capsid. Using an anionic Ti ion precursor, this approach was subsequently also used to nucleate and grow TiO2 within the CCMV capsids.194
The importance of interfacial electrostatics and its utility in approaches for size constrained inorganic materials synthesis was further demonstrated in the CCMV system by means of a dramatic alteration of the charge on the interior of the capsid from positive to negative. Genetic modification of the N-terminus of the CCMV coat protein, in which all the basic residues (i.e. lysine and arginine) in the N-terminus were replaced with acidic, glutamic acid residues195 did not affect the capsid assembly. However, the altered electrostatic character of the interior of the protein cage favored strong interaction with cations, which in conjunction with free molecular passage of ions across the capsid acts to promote oxidative hydrolysis leading to the formation of size-constrained iron oxide particles exclusively on the inside of the CCMV capsid – a functional mimic of the ferritin protein.
Other protein cages such as chaperonins have been exploited to encapsulate or synthesize inorganic nanoparticles.197–199 Interestingly, ATP-dependent release of re-folded guest protein from chaperonins was successfully mimicked using preformed CdS nanoparticles and GroEL or chaperonin from Thermus thermophilus HB8. The CdS encapsulated inside of the chaperonin was released from the cage upon conformational changes induced by ATP hydrolysis.197
The controlled synthesis of nanowires is practically important and protein cage assemblies have been used to direct these syntheses. The rod-shaped tobacco mosaic virus (TMV) has been demonstrated as a constrained template to grow inorganic nanowires (Fig. 15B),196,200–207 and one dimensionally aligned nanoparticle arrays.208,209 The 300 nm long and 18 nm wide TMV has a 4 nm diameter interior pore in which metal nanowires such as Ni,202 Co,202 Cu,206 and Co–Fe alloy203,206,208 have been selectively grown. Although nanowires with the length of 100–200 nm are typically formed, fiber lengths up to 600 nm were observed, longer than a single TMV particle, suggesting some end-to-end assembly of individual capsids.202
4.2. Polymers inside protein cages
All viruses are comprised of a capsid that surrounds and protects a viral genome that is a charged polymer of RNA or DNA. As pointed out above, in some viruses, the genome packaging takes place after the capsid has already formed. In phages, this process is energetically costly and the DNA genome is packaged at very high density with large internal pressure via ATP-driven packing process but with little or no attractive interactions between DNA and the capsid. Other viruses package their genome at much lower density and exhibit strong electrostatic interaction between the capsid and the packaged genome. As described earlier, the plant virus CCMV can be produced as an empty, nucleic acid-free capsid. Initial synthetic studies with empty CCMV demonstrated that incubation with an anionic polymer (poly(anetholesulfonic acid)) resulted in efficient encapsulation within the CCMV capsid.193 Here, poly(anetholesulfonic acid) was incubated with the CCMV at pH 7.5 in which the CCMV took the swollen form (i.e. had larger inter-subunits pores) as discussed above, followed by lowering the pH to 4.5, well below the transition of the CCMV to the closed form. The polymer which passed across the CCMV capsid and accumulated was trapped inside of the capsid at the low pH condition. This strongly suggests that preformed capsids can be significantly more porous at the swollen form, although the size limit for free diffusion across the capsid has not been quantified.
The permeability and porous nature of the capsids suggested that site-specific extension of polymer strands anchored on the interior of the capsid could be achieved by transport of the monomers to the growing end of the polymer inside that capsid. Thus introduction of reactive amino acids, by site-directed mutagenesis, selectively on the interior of a capsid allows modified at these positions to create polymer-initiation sites from which the polymer strand can grow. Initiated polymer growth on the capsid interior enables not only the introduction of new chemical moieties beyond what can easily be incorporated using genetic methods, but it does so at high density making efficient use of the entire capsid volume.
Organic polymer synthesis constrained inside of protein cages was first demonstrated using the copper(i)-catalyzed azide alkyne cycloaddition (CuAAC) coupling of azide and alkyne bearing monomers. A series of polymers were synthesized through a stepwise molecular synthesis approach. In the small heat shock protein cage (sHsp) from Methanocaldococcus jannaschii, a branched polymer with pendant amines was synthesized using alternate addition of azide and alkyne monomers.210 This resulted in an 8-fold increase in the number of functionalizable amine sites in comparison with wild-type sHsp, which could then be chemically labeled.211 Thus high density labeling of sHsp at amines with Gd–DTPA, a T1 enhanced MRI contrast agent, was achieved. A similar approach was taken using azide or alkyne containing metal containing compounds as the monomers to create a coordination polymer connected through the CuAAC coupling.212 Alternatively, a coordinatively unsaturated metal–ligand was attached selectively to the interior of the capsid lumen and served as a site for coordination polymer extension through iterative addition of a ditopic ligand and metal ion.213
By far the most successful approach for high density polymer incorporation using functionalizable monomers has been with the use of “living” polymerization approaches,214,215 which are fast and accommodate a wide range of monomers. Atom-transfer radical polymerization (ATRP) inside of a protein cage was first demonstrated in the bacteriophage P22 using meth-acrylate monomers.216 An initiation site was introduced on the interior of the capsid through the site selective attachment of a tertiary bromide moiety on each of the 420 coat proteins. The polymerization of 2-aminoethyl methacrylate (AEMA) resulted in polymerized particles having 12 000 ± 3000 AEMA monomers per particle constrained within the P22 capsid. The AEMA polymer served as a scaffold for the conjugation of a number of functional small molecules with high loading density via the pendant amine groups (Fig. 16), including Gd–DTPA,216 fluorescein isothiocyanate (FITC),216 an isothiocyanate derivative (CoCl(dmgH)2Pyr) and eosin-Y isothiocynate.216,217 For example, chemical attachment of up to 10 000 Gd–DTPA to the polymer inside the P22 cavity led to development of a novel MRI contrast agent with a significantly high r1 relaxivity. This approach was also amenable to co-polymer formation with a range of other monomers.218 Notably, polymer formation with ATRP has also been demonstrated on the interior (and exterior) of the Qβ capsid where an azide-containing group was introduced and subsequently reacted with an ATRP initiating tertiary bromide via a CuAAC reaction.219,220
Fig. 16.

Atom-transfer radical polymerization (ATRP) can be used for high loading of Gd–DTPA within the P22 VLP. In the first step, an internal cysteine is labeled with a radical initiator (2-bromoisobutyryl maleimide, 1). Next poly-2-aminoethyl methacryalate (AEMA) is formed via ATRP. In the last step, the amino groups can be modified with FITC dye (2) or GD-DTPA-NCS (3). Figure reproduced from ref. 216 with permission from Nature Publishing Group, copyright 2012.
Some protein cages have pores, which are large enough to allow macromolecules to pass through the protein shells to reach the interior cavity.221–224 For example, a Group II chaperonin, thermosome,225 has large pores of approximately 5.4 nm diameter.221 Generation four Poly(amidoamine) dendrimer, which has about 4.5 nm diameter, has successfully encapsulated inside of thermosome to use the protein cage for siRNA delivery vehicle and gold nanoparticles have been synthesized on the interior.222,223 Similarly, a semiconducting polymer poly(2-methoxy-5-propyloxy sulfonate phenylene vinylene) (MPS-PPV) has been encapsulated inside of a vault cage, while the MPS-PPV could not enter the cavity if vault subunit proteins were covalently cross-linked together prior to the polymer loading. This result indicates that the MPS-PPV can diffuse into the cage through inter subunit pores.226
4.3. Encapsulation via co-assembly into cage-like structures
Assembly of some virus capsids is often driven through specific directed interactions between the capsids and their endogenous cargos that facilitate the capsid assembly. As an approach it is thus amenable to synthetic manipulation and has been demonstrated in many different capsid assemblies and with many different types of cargo. This highlights the power and versatility of a bioinspired approach which harnesses sophisticated self-assembly mechanisms observed in nature for smart materials design and synthesis.
Electrostatic interactions between capsid and cargo is perhaps the simplest example of this approach. As mentioned earlier, CCMV uses electrostatic interactions between negatively charged RNA and positively charged regions of the capsid to package the genome. This strategy of self assembly of capsid subunits around nanoparticles has been applied for encapsulating pre-formed inorganic nanoparticles (e.g. gold, iron oxides and quantum dots) inside of capsids derived from many viruses (e.g. BMV, red clover necrotic mosaic virus (RCNMV), CCMV, HIV-1 Gag protein and hepatitis B virus (HBV)).132–134,227–235 In a notable example demonstrating a high yield of encapsulation (>95%), gold nanoparticles modified with carboxylic acid terminated poly(ethyleneglycol) (PEG) directed the assembly of BMV subunits (Fig. 17A). Complementary electrostatics provided the interactions between the carboxyl groups and the capsid protein subunits while the PEG moiety served as a mimic for a dis-ordered hydration layer in the RNA packaged in BMV.134 BMV VLPs with discrete sizes, each adopting a specific T number, were obtained upon varying the size of the gold nanoparticle template, with the number of subunits in the final assembled capsid increasing with increasing template size (Fig. 17B–D).231 The co-assembly process of capsid and guest molecules through electrostatic interaction has also been exploited to encapsulate other cargo molecules, such as proteins and polymers.25,236–238
Fig. 17.

(A) Proposed mechanism of BMV virus-like particle (VLP) assembly around a gold nanoparticle (NP). First, electrostatic interaction leads to the formation of a disordered capsid protein (CP)–gold NP complex. The second step is a crystallization phase in which the protein–protein interactions result in the formation of a regular capsid. (B–D) Three-dimensional reconstruction (using negative stain data) of BMV VLP self-assembled around gold NPs with (B) 6 nm, (C) 9 nm, and (D) 12 nm diameter gold NPs. Structure and size of reconstituted models of (B), (C), and (D) resemble to a T = 1, a pseudo T = 2, and T = 3 models of BMV capsids, respectively. Part A reproduced from ref. 134 with permission from the American Chemical Society, copyright 2006. Part B reproduced from ref. 231 with permission from the National Academy of Sciences, copyright 2007.
A more elaborate level of directed cargo encapsulation has been demonstrated via an approach inspired by the assembly mechanism of some viruses in which specific interactions between capsid coat protein and cargo (protein or nucleic acid) play a critical role. For example, bacteriophages MS2, Qβ and RCNMV all use hairpin structures in their ssRNA to bind to the capsid proteins and thereby direct both capsid assembly and RNA encapsulation.85,239–241 Similarly, as we discussed previously, bacteriophage P22 assembles initially into a procapsid form, templated by a scaffolding protein (SP), which interacts with the coat protein to direct assembly and during that process the SP is encapsulated within the procapsid.67 Co-opting these natural specific interactions has allowed for the selective sequestration of cargo with high encapsulation efficiency. Representative examples of cargo co-assembly by using specific interaction of capsids and cargos are discussed below.
4.4. Encapsulation of enzymes
Among the many cargo molecules that have been encapsulated inside of protein cages, the encapsulation of enzymes has drawn particular interest recently. Because the interior of a viral capsid is a privileged environment, spatially separated from the bulk exterior environment by the capsid shell, an enzyme can be sequestered inside the capsid to create well-defined nanoreactors capable of catalyzing a wide range of chemical transformations that conceptually resemble bacterial microcompartments. Encapsulation of enzymes within protein cages is a promising approach towards creating efficient catalytic materials and understanding the effects of enzyme crowding as well as multi-enzyme transformations.
Initial work in the encapsulation of active enzymes inside VLPs used the CCMV system, which undergoes reversible in vitro disassembly and reassembly in response to changes in solution pH as we discussed earlier. Reassembly of the capsid in the presence of the enzyme horse radish peroxidase (HRP) resulted in some fortuitous encapsulation of the enzyme within the reassembled CCMV capsid.26 This passive encapsulation of an active enzyme, while not particularly efficient, paved the way for more directed approaches towards cargo sequestration and demonstrated key concepts: the capsid shell was shown to be porous towards small molecule substrates and the enzymes encapsulated within the CCMV were active but displayed some different kinetic behavior as compared to enzymes in the bulk. This observation suggests that the local environment of the capsid might affect enzyme function.
The in vitro assembly approach in CCMV has been refined to incorporate molecular recognition tags between the cargo and the capsid protein to achieve more efficient cargo encapsulation. Thus complementary peptides that form a heterodimeric coiled-coil were fused to the CCMV CP and the cargo protein236 allowing non-covalent coiled-coil interactions to occur prior to capsid assembly. This approach was also used to encapsulate the enzyme lipase B (PalB) from Pseudozyma antarctica inside CCMV and resulted in packaging of an average of 1–4 PalB per capsid.242 The initial velocity of singly encapsulated PalB was reportedly five times faster than the velocity observed for PalB free in solution. This strategy demonstrated successful encapsulation, but the method required incorporating unmodified CCMV CPs into the in vitro assembly system, together with the fusion proteins, to achieve assembly of the CCMV capsid with the PalB cargo. This approach naturally results in a distribution of species, ranging from assembled capsids containing no enzyme cargo to fully packed capsids.
There is a growing desire to directly harness the synthetic capabilities of biology and let biological systems direct the synthesis and assembly of complex materials directed only through altered genetics. Thus a goal is to establish robust methods for in vivo nanoreactor assembly. This requires producing all the components for assembly in the cell and introducing specific molecular recognition between the capsid protein and the cargo, with no interference from the other biomolecules within the cell. This genetically programmed assembly would take advantage of molecular level design and self-assembly, as well as metabolic engineering on the organismal level.
Directed enzyme cargo encapsulation has been shown for a number of different enzymes and capsids. Using a highly directed approach for enhanced molecular recognition between capsid and cargo, Hilvert et al. used the protein cage derived from lumazine synthase (LS) and created mutants with an increased negative charge density on the interior of the capsid through introduction of glutamic acid residues. The fusion of a deca-arginine tag to green fluorescent protein (GFP) cargo ensured the directed encapsulation through complementary electrostatic interactions between the cargo and capsid interior.237 This approach was then demonstrated in vivo where the directed evolution of Aquifex aeolicus (AaLS) variants were screened for enhanced binding and encapsulation of HIV protease within the LS capsid. Encapsulation of the protease rendered it inaccessible to proteins in the cell and resulted in enhanced cell survival.238 The negatively charged LS mutants have been shown to be highly effective at the directed encapsulation of a wide range of positively charge cargo proteins including positively “supercharged” GFP and ferritin variants.243,244 Additionally, various enzymes such as RuBisCO and carbonic anhydrase were tethered with the supercharged proteins to direct encapsulation of the enzymes in the negatively charged LS mutants, which lead to construction of a functional carboxysome mimic.245,246 Interestingly the loading of cargo in the LS mutant did not require a co-assembly but could be achieved by incubation of the assembled LS with the charged cargo. Since the cargo proteins themselves are larger than the pores of the LS cage (ferritin has a molecular weight of 5 × 106 Da) it was uncertain how the large cargos could be transported across the already formed LS cages. One suggested possibility was that the LS capsid assembly could be dynamic. However, a recent cryo-EM structural model of two LS variants was determined revealing a highly unusual, cage structures with large pores (each variant exhibits tetrahedral and icosahedral symmetry, respectively) that might accommodate transport of the cargo proteins across the capsid to the interior.247
Directed enzyme encapsulation has also been achieved using specific interactions, through molecular recognition. Finn and coworkers have developed a unique method for directed enzyme encapsulation in the virus Qβ. They used a chimeric single stranded RNA composed of 3 parts: (1) a hairpin that associates with the internal surface of the Qβ CP, (2) an RNA aptamer that binds to an arginine-rich peptide (Rev), and (3) the mRNA sequence that codes for the Qβ CP that also acts as spacer between the other components. Cargo proteins tagged with the Rev peptide are thus primed for directed encapsulation into the Qβ capsid through the interaction between the Rev peptide and the RNA aptamer (Fig. 18).24 The transcribed mRNA thus serves as both as the transcript for CP expression and as the recognition element for directed encapsulation. This approach, using the co-expression of the CP/RNA mediator together with the tagged protein cargo, has been effectively used for the directed encapsulation of other protein cargoes within the Qβ system.248 In another example, MS2 shows sequence specific interaction between its CP and a short RNA stem-loop to trigger capsid assembly,81 hence the RNA sequence has been utilized to encapsulate cargo molecules inside of the MS2 VLPs.249,250 Furthermore, encapsulation of cargo proteins has also been demonstrated by using DNA oligomers and negatively charged peptides as tags to induce capsid assembly and mediate cargo-capsid interaction.27 Intriguingly, the presence of a protein stabilizing osmolyte, trimethylamine-N-oxide, has been reported to increase yield of capsid reassembly.27
Fig. 18.

An example of a direct encapsulation of enzyme into a capsid by molecular recognition. A Rev-tagged enzyme is bound to the interior of the capsid via an RNA with a Rev aptamer and a Qβ genome packaging hairpin that that is bound to the interior of the capsid. Figure reproduced from ref. 24 with permission from Wiley, copyright 2010.
Yet another approach for directed encapsulation involves the genetic fusion of a cargo of interest to a scaffolding protein. Of the protein cages used for directed encapsulation, the P22 bacteriophage has emerged as an extremely versatile platform for the programmed and directed encapsulation of a wide range of gene product cargos within the E. coli cells used for heterologous expression. Recall that the P22 SP templates capsid assembly and that SP bound to the interior of the capsid is incorporated into the assembled P22 VLP. The wild type SP is a 303 residue long protein and it has been shown that it can be truncated to an essential C-terminal scaffolding domain without disrupting its ability to direct assembly.251,252 This truncation has been used to simultaneously direct capsid assembly and cargo encapsulation as genetic fusions to either the N-20,21,23,253–257 or C-termini22 of the SP. Remarkably, the small helix–turn–helix domain, that is essential for directing capsid assembly, can be fused to protein cargo of up to 180 kDa without affecting the fidelity of capsid assembly.21 By co-expressing a genetic fusion of a protein cargo and the truncated SP together with CP, a number of functional enzyme and protein cargoes have been successfully encapsulated inside the P22 VLP, which maintain their functional activity. Directed assembly of an enzyme cargo has also been demonstrated with other cages. For example, Handa and co-workers used the minor coat proteins (VP2) of the SV40 virus to direct encapsulation by genetic fusion of an enzyme cargo, cytosine deaminase within the VLP. Co-expression of the modified VP2 together with the major coat protein VP1 and the other minor coat protein VP3 resulted in assembly of SV40 VLPs with active enzymes entrapped on the interior of the capsid.258
The capsid morphology can have a profound effect on the enzyme cargo in terms of its local concentration, crowding effects, and access between the interior and exterior. The P22 VLP capsid undergoes a natural morphogenesis which mimics the native maturation of the infectious virus upon DNA packaging. The initially formed procapsid (PC) form of the P22 VLP expands, upon heating at 65 °C, from the spherical PC to the expanded (EX) form, which nearly doubles the interior volume of the capsid.71 Upon further heating, to 75 °C, the 12 pentameric vertices of the capsid are irreversibly lost leaving 12 pores 10 nm in size in the capsid that forms a structure affectionately referred to as wiffleball (WB).259 In some cases, where the cargo is thermally stable, the EX and WB morphologies of P22 can be accessed by heating the capsid and this has allowed an investigation of the kinetic behavior of encapsulated enzymes as a function of both local enzyme concentration (crowding) and capsid porosity (substrate diffusion across the shell).255,256 In the P22 encapsulation, the enzyme cargo is initially constrained to the interior capsid lumen in the PC form, but upon expansion to the EX the SP binding site is lost due to the conformational change of CP, and the cargo is free to occupy the entire volume of the capsid (Fig. 19).260
Fig. 19.

The morphologies of P22 and the mapping of cargo location within them. (A) P22 procapsid (PC) undergoes a morphological change to a more angular expanded (EX) from upon heating, mimicking the structure of the native virion. The internal volume increases approximately 35% and porosity of the capsid decreases. Heating further forms the wiffleball (WB) morphology, where pentamers are removed leaving large holes in the capsid wall. (B) The central sections of three-dimensional reconstructions of the empty P22 and CelB-P22. The darker region on the interior lumen of the capsid indicates higher density, caused by the presence of the bound cargos. The arrows indicate the increased density from the SP-C terminal region of the SP and the CelB-SP binding to the capsid interior. (C) Reconstructions of the EX forms with cargo. Decreased electron density reveals that the cargos are no longer bound to the capsid and can freely move within the entire capsid interior volume. Part A adapted from ref. 261 with permission from The Royal Society of Chemistry. Part B and C reproduced from ref. 260 with permission from The Royal Society of Chemistry.
While a number of enzymes that have been successfully encapsulated within the P22 VLP show slightly lower kcat values compared to the enzymes free in solution (likely due to the effects of molecular crowding and restricted dynamics during enzyme turnover),255,256 encapsulation of the heterodimeric NiFe hydrogenase from E. coli, Hyd-1, within the P22 capsid shows a dramatic enhancement of the activity as compared to the free enzyme (Fig. 20).23 The encapsulated Hyd-1 activity increases nearly 150 fold over the free enzyme and follows classic Michaelis–Menten kinetics. The enhancement is likely due to the entrapment of the enzyme at high concentration within the VLP thus shifting its equilibrium toward the more active hetero-dimeric form of the HyaA and HyaB subunits from the much less active monomers in bulk solution. It is also possible that the dimeric Hyd-1, presenting two copies of the SP from each subunit is favored for capsid assembly over a single copy as has been demonstrated in early studies on the effects of SP oligomerization on capsid assembly.137 Therefore, capsid assembly itself might drive the Hyd-1 equilibrium towards the more active dimeric form and simultaneously entrap that form at high concentration within the capsid. Indeed, recent in vitro P22 VLP assembly study indicated that cargo proteins displaying multivalent SP preferentially encapsulated over monovalent wild type SP while assembling into VLP.262
Fig. 20.

Encapsulation of an active hydrogenase within a P22 VLP (P22-Hyd-1). (A) Scheme showing expression of both subunits of the hydrogenase as fusions with truncated scaffold protein (hyaA-SP and hyaB-SP) under IPTG control with coat protein (CP) in a separate plasmid under L-arabinose control. The result is P22 capsid with both subunits packaged within it. Structures are PDB: 3USE (E. coli hydrogenase-1), PDB: 2XYY (P22 procapsid), PDB: 2GP8 (scaffold protein binding domain). (B) Hydrogen is assayed via gas chromatography using methyl viologen (MV+•) as the electron transfer mediator and an electron source (dithionite or electrochemical reduction). (C) TEM image (negative staining) of P22-Hyd particles. (D) Specific activity of P22-Hyd at varying concentrations of methyl viologen. The enzyme activity follows Michealis–Menten kinetics. (E) Specific activity traces of P22-Hyd-1 (black triangles) and free Hyd-1 (gray squares) shows a marked increase in activity of the P22 encapsulated Hyd-1 compared to Hyd-1 in solution. Figure adapted from ref. 23 with permission from Nature Publishing Group, copyright 2016.
Unlike many other protein cages, chaperonins have relatively large pores likely due to their native function of assisting folding of guest proteins to it native state. These large pores of chaperonins allow the encapsulation of enzymes without disassembling the cage structure into subunits. For example, horseradish peroxidase was encapsulated into thermosome and used for polymerization (via ATRP) of an acrylate which occurred selectively inside of the cavity.221
5. Functionalization of the exterior of viruses and VLPs
In nature, the exterior of protein cages are in contact with the surrounding environment and must interact with the surroundings in order to function and protect the valuable cargo. The exterior surfaces of protein cages can also be rationally designed to conduct specific tasks and properties. For a virus, presentation of different functionalities on the exterior is necessary for targeting the virus to a particular location in vivo. Also, the exterior surfaces of ferritin has evolved to create electrostatic gradients around the three-fold channels, which provides a guidance mechanism for cations entering the protein cavity through the channels.185 Because all the protein cages described here possess a highly symmetric polyvalent surface, the exterior functionalities are typically presented in a multivalent and highly symmetrical manner. For viruses, the polyvalent presentation of targeting and cell fusion moieties can enhance affinity to their host cells and infection.263 These examples found in nature inspire us to explore the exterior surfaces of protein cages to impart functionality by design. In this section, we present an overview of the variety of methods for functionalizing the exterior of a protein cage (summarized in Table 1).
Table 1.
Commonly used methods of exterior (and in some cases, interior) modification of protein cages discussed in this section
| Method of modification | Examples |
|---|---|
| Genetic |
|
| Covalent (bioconjugation) |
|
| Non-covalent genetic |
|
5.1. Genetic functionalization
Genetic modification of the protein cage is perhaps the most widely used method for exterior functionalization. Modifications of the exterior can be achieved using molecular biology techniques to add, delete, or replace amino acid residues. Typically the protein cages are quite robust, and remain intact after minor genetic modifications.
Phage display is a powerful and widely used technique for high throughput screening of not only protein–protein and protein–peptide interactions but also interaction between peptides and abiotic materials such as inorganic materials. In phage display system, infected bacteriophage have a peptide or protein of interest “displayed” on the exterior of capsid. Some notable examples include using phage display to present a library of short peptide sequences.264–266 A foreign gene is inserted within the coat protein gene and it is expressed along with the CP. In this technique, viruses that have affinity for a particular target can be selectively amplified and characterized. Modifications to the protein cage are typically done at termini or exposed loops so that the coat protein can still fold properly.190,267–272 The gene (nucleic acid) is packaged within the capsid, which allows for linkage of the phenotype (the displayed peptide) and the genotype (the encoded DNA). In a process termed biopanning, large libraries of random sequences can be screened against individual targets such as proteins, peptides, DNA, and abiotic materials. The binding phage can be isolated and its sequence determined. The filamentous phage M13 is one of the most commonly used platforms for phage display technology because of the ease of replication and commercial availability.265 There are a wide range of applications in molecular biology where phage display has proven useful, such as in vitro protein evolution, drug discovery, enzyme inhibitors, and finding protein partners.265,273 This technique has also proven useful in the material science community. Certain peptides can be selected that show preferential binding for different inorganic surfaces/nanoparticles, based on the composition and crystallographic orientation of the material.274–277 In one early example, M13 bacteriophage was engineered with peptide sequences to act as a template for nucleation of ZnS or CdS nanocrystals to form nanowires.278
Whole proteins and protein domains of interest can also be genetically fused to capsid coat proteins.279–281 For example, Gleiter and Lilie displayed a 6.8 kDa domain from protein Z on surface exposed loops of polyoma VLPs.282 Protein Z is an antibody binding protein that was still capable of binding antibodies when attached to the VLP. In another example, Nabel and coworkers displayed trimeric antigen cargo (the ectodomain of the influenza hemagglutinin protein) at the 3-fold axes on a ferritin cage by creating a fusion at the N-terminus of the ferritin subunit to effectively create a nanoparticle based vaccine.283 Here, symmetry matching, i.e. the trimeric antigen cargo was fused at a trimeric symmetry site of the platform protein cage, is an important design consideration and it lead to multivalent antigen display (eight trimeric antigen per ferritin as ferritin is composed of 24 subunit). The nano-particle based vaccine produced an increased immunogenic response to many viral subtypes compared to the commercially available vaccine. Cell targeting peptides have also successfully displayed on the exterior of protein cages in a multi-valent manner by genetically introducing the peptides either at the end of N- or C-terminus of an individual subunit,190,269,284 or inserting into a surface exposed loop region.285,286 For example, Kang and coworkers demonstrated that a non-viral protein cages, encapsulin, can also be genetically modified to display a targeting peptide ligand (Fig. 21),285 and an encapsulated drug molecule can then be delivered to the targeted cell.
Fig. 21.

Functionalization and targeting of encapsulin. Encapsulin is genetically modified to contain the targeting ligand (SP94) and is assembled to form a protein cage. An anticancer drug aldoxorubicin was delivered to the HepG3 cells, resulting in cytotoxicity to the cells. Figure reproduced from ref. 285 with permission from the American Chemical Society, copyright 2014.
Despite the successes described above the genetic fusion of functional proteins and peptides to protein cages can often result in undesired outcomes, such as the formation of insoluble fusion proteins or disruption of subunit assembly into a proper cage-like structure. To overcome this drawback, a bioconjugation technique using the enzyme sortase287 to mediate site specific attachment has recently been applied for functionalization of protein cages because this approach possesses great flexibility and versatility.288,289 For example, GFP and influenza hemag-glutinin head were successfully conjugated to the exterior surface of P22 VLP (Fig. 22).288 The SpyTag/SpyCatcher protein ligation system290 shows great potential for bioconjugation to protein cages.291 Thus, if a sortase recognized motif or a SpyTag/SpyCatcher motif is successfully introduced onto a protein cage, nearly any protein cargo could potentially be conjugated to the protein cage in a “plug and play” modular manner using this strategy.
Fig. 22.

A modular approach to presentation of cargo on the exterior of a protein cage. The CP of P22 was genetically engineered to contain a LPETG amino acid sequence at the C-terminal that can be covalently bound to a poly glycine sequence an N-terminal of cargo (here, GFP or hemagglutinin head) via the enzyme sortase. This modular approach allows for many different cargo types to be presented without the need for redesigning the modified CP sequence. Figure reproduced from ref. 288 with permission from the American Chemical Society, copyright 2017.
5.2. Covalent modification
Chemical conjugation is another widely used functionalization/bioconjugation approach that allows for incorporation of many different desired functional groups, both biological and chemical, into protein cages to change or enhance their function.292 Because many of the protein cages utilized for nanomaterials syntheses have near-atomic resolution structure models, the number and location of functionalizable sites in a protein cage can be precisely known with knowledge of the protein sequence and structure. Covalent modification of protein cages initially utilized natural amino acids with reactive functional groups such as the sulfhydryl group (cysteine), the amino group (lysine) and the carboxyl group (glutamate and aspartate). For example, the sulfhydryl group can serve as reactive sites for thiol selective chemistry,293,294 or for binding to a gold surface.295 High density labeling can also be achieved by labeling at amines using activated N-esters, and carboxylates can be activated into reactive esters. One major drawback of this conjugation approach is the poor selectivity of sites with the same functional group. Typically there are multiple cysteine, lysine, glutamate or aspartate residues in a protein subunit, and it is difficult to conjugate a target molecule to a specific site over others, which results in heterogeneous mixtures of modified protein cages.
In order to overcome the sometimes undesired non-selectivity of the reactions, there has been a continuing interest to develop new bioconjugation reactions with high level of site- and chemo-selectivity under mild conditions in aqueous environments.292,296,297 For instance, the α-amine of the N-terminus is chemically unique and shows distinctive reactivity over lysines. Thus an increasing number of chemical conjugation strategies have developed to utilize the N-terminus amine as site-specific reactive group.297 For example, the N-terminus amine shows lower pKa (6–8) than other aliphatic amine (pKa ~ 10.5) as a consequence of the inductive effects of the nearby carbonyl group.297,298 Hence selective acylation and alkylation of the N-terminus amine can be carried out at low to neutral pH.297 In an application relevant to protein cages, the TMV virus was covalently modified at both the N-terminus and at an introduced cysteine residue with two different chromophores that undergo FRET to produce a “light harvesting rod.”299 Another emerging bioconjugation strategy is the incorporation of unnatural amino acids with bio-orthogonal reactive groups such as azido, alkyne, alanine and norbornene into a protein cage.18,19 These functional groups can be uniquely modified over other natural amino acids in the protein. For example, Finn and coworkers have demonstrated functionalization of Qβ mutants with azide- or alkyne-containing unnatural amino acids using Cu(i)-catalyzed [3+2] cycloaddition (known as “click chemistry”).19
Pioneering work by Francis and coworkers has developed a number of oxidative coupling reactions with electron rich aromatic species with both natural and unnatural amino acids that can be performed on proteins in aqueous solution under mild conditions.296 In one example, they utilize this strategy to dually functionalize both the interior and exterior of bacteriophage MS2.300 The interior was functionalized with a porphyrin maleimide that was introduced at a mutated C87 residue. The exterior was functionalized at the unnatural amino acid p-amino-phenylalanine via oxidative coupling with a DNA aptamer that targets protein kinase 7 receptors on Jurkat leukemia cells. After incubation of the cells with capsids and upon light illumination, the porphyrin generated a cytotoxic singlet oxygen species. The capsids selectively targeted and killed 76% of the Jurkat cells in the presence of erythrocytes (Fig. 23).
Fig. 23.

Overall scheme for the construction of a dually functionalized recombinant bacteriophage MS2. Interior cysteines were modified with porphyrin (purple); exterior p-aminophenylalanine were modified with DNA aptamers (~20 per particle). (A) Live control cells showing the difference in size and shape between Jurkat cells and red blood cells (RBCs). (B) Exposure of the cells to the functionalized MS2 capsids to light for 20 minutes results in selective killing of Jurkat cells as indicated by trypan blue staining. RBCs are left unharmed. (C) Positive control showing death of both cell types induced by 30% ethanol. Scale bars are 100 0μm. Figure adapted from ref. 300 with permission from the American Chemical Society, copyright 2010.
Atom transfer radical polymerization (ATRP) has also been used to decorate the exterior of capsids with polymers, which serve as a scaffold to conjugate imaging agents and drug molecules as demonstrated by Finn and coworkers.219,220 In this study, approximately 180 ATRP initiating bromides were introduced on the exterior surface of Qβ by using azide–alkyne click chemistry. Oligo(ethyleneglycol)-methacrylate with functional groups were selectively polymerized on the surface of the decorated Qβ via ATRP. Direct attachment of polymer molecules instead of conducting a polymerization reaction on the capsids has also been demonstrated. The most notable example is the bioconjugation of polyethylene glycol (PEG) to proteins (so called “PEGylation”) because it is a common technique for functionalizing the proteins in an attempt to reduce renal clearance and immunogenicity, and improve pharmacokinetics. PEG with various chain lengths have successfully been conjugated on the exterior surface of protein cages such as CPMV, MS2 and ferritin.75,301–303 The PEGylated protein cages exhibited a reduced interaction with a cell and an antibody.75,303
5.3. Noncovalent-genetic attachment
Another approach that has gained traction is the fusion of a cargo to secondary structural proteins (sometimes called auxiliary proteins) with a natural affinity for a capsid exterior. Secondary proteins that bind to the exterior of some viruses are collectively known as decoration proteins. This approach offers the advantage of using genetic expression of the necessary proteins, and removes the cleanup steps typically needed for after bioconjugation to the capsid. Decoration protein functions range from being actively involved in viral infection to providing structural support to the capsid. In fact, some decoration proteins are part of the conserved structure of the virus.304–306 Decoration proteins were initially used as means to display proteins or protein fragments as an alternative display platform to the M13 phage.307,308 In another example, the bacteriophage T4 structure includes two secondary proteins known as Soc, which binds to local threefold interfaces between capsomers, and Hoc, which binds to the center of each capsomer. Whole proteins were functionally displayed on Soc and Hoc, and the fusions have similar binding affinities as the unmodified proteins.309
While P22 does not have a native decoration protein, the decoration protein “Dec” found in the highly similar bacterio-phage L binds to the EX form of P22.310 The Dec protein binds tightly as a trimer at the 60 quasi 3-fold axes and with a much lower affinity at the 20 true 3-fold sites. The N-terminus remains close to the capsid shell, while the C-terminus is projected away from the capsid,311 which provides an ideal environment for presenting proteins on the exterior of the P22 capsid. For example, Schwarz et al. showed presentation of both a monomeric small peptide (“self peptide” derived from CD47, which inhibits phagocytosis by macrophages312) and a trimeric protein cargo (the soluble region of CD40L, which is a key signal in adaptive immunity) on the exterior of P22 with Dec fusions and each of the cargo remained biologically active (Fig. 24).313 Dissociation constants, KD, of the two Dec mutants to the tight binding sites on P22 were measured as 18–30 nM, which is nearly identical to KD of wild type Dec (9 nM). The half-life of the tight binding site was at least 60 hours, which suggests that Dec bound P22 is a good candidate for in vivo cargo delivery.
Fig. 24.

Presentation of a target protein trimer CD40L (PDB 1ALY) on a P22 VLP exterior. The Dec trimer is pictured in green. A polyhistidine tag (purple) is used for purification. The CD40L is genetically fused to the C-terminus of Dec and presented as a trimer. Dec binds to P22 at the quasi 3-fold axes to present the CD40L on the exterior. Figure reproduced from ref. 313 with permission from the American Chemical Society, copyright 2015.
In other examples, entire proteins as well as small molecules can be non-covalently conjugated to the surface of platform protein cages via a well-characterized biotin–streptavidin interaction. For example, papillomavirus VLPs have been functionalized with protein antigens conjugated with streptavidin.314,315
6. Higher order assembly of protein cages
Construction of higher order structure from colloidal nanoparticles in a controlled manner has drawn considerable interest in the field of materials chemistry because the assembled materials could display collective properties that are different from individual nanoparticles.29,30 Exploiting the self-assembly of nanoparticle building blocks into higher order structures is a powerful, versatile and low-cost approach to designing such materials. Biological building blocks including VLPs and related cage-like proteins have several unique advantages over synthetic colloidal particles as will be discussed below.316 In this part, we will review recent progress towards the design of higher order structures built from protein cage building blocks via self-assembly processes.
6.1. Protein cages as building blocks for higher order assembly
Self-assembly of biomolecular components into higher order structure with hierarchically organized arrangement over multiple length scales is one of the hallmarks of biological systems.317 The highly organized structural arrangements of building block components could endow collective and orchestrated functionalities to the assembled structures beyond those of individual building blocks. For example, the excellent mechanical property of bone is a consequence of the highly regulated hierarchical structure of the hydroxyapatite and collagen assembly across nano to macro scales.318 The vertebrate striated muscle is another elegant example of organized assembly of multiple biomolecular building blocks.319 The functional properties of muscle emerge as a consequence of the hierarchical structure composed of multiple proteins including actin, myosin and titin.
Viruses are also excellent examples of nature’s self-assembled architectures which are constructed from a limited number of biomolecular building blocks. They exhibit complex functionalities through their life-cycle including host-entry, replication and shedding. From a materials viewpoint, cage-like proteins are excellent building block components to construct higher order structures across multiple length scales for a number of reasons. First, protein are genetically encoded materials, hence they are produced with near perfect uniformity. The homogeneity of a building block is critical to construct higher order structures particularly if long-range order is desired. Secondly, as we discussed earlier, a wide range of functional protein cage building blocks can be produced by encapsulation of various functional cargo molecules. Third, the established chemical and genetic modification strategies can also be used to modify the exterior surface of the protein cages, which can lead to modulation of inter-particle interactions and control the assembly of individual building blocks into higher order structures. Fourth, numerous types of protein cages, which share the same spherical morphology but with a wide range of sizes and chemical and physical properties, are available, either naturally occurring or by synthetic design. Therefore, similar construction strategies could be applied to different cages, resulting in a new class of protein-based super-lattice materials. Lastly, VLPs and related cage-like proteins are in general chemically and mechanically robust, perhaps as an evolutionary consequence of protecting their genomic nucleic acids cargos from the external environment. Thus the protein shells can be used as protective coatings for encapsulated cargos including fragile enzymes.
6.2. One- and two-dimensional assembly of protein cages
One-dimensional (1D) assembly of protein cages into tube like structures has been demonstrated through a head-to-head assembly of individual cages. Aida et al. have demonstrated fabrication of a micrometer long nanotube self-assembled from a chemically modified GroEL chaperonin.320–322 The apical domain of GroEL was modified with merocyanine (MC). The MC decorated GroEL “polymerized” into a tube structure in the presence of Mg2+ through a head-to-head assembly of GroEL due to coordination between MC and Mg2+. A 1D tube of GroEL encapsulating superparamagnetic iron oxide nanoparticles within the cavity was shown to form a bundle of tubes under the influence of an external magnetic field.323
Ordered two-dimensional (2D) arrays of proteins have been studied for decades as they are of interest in electron crystallography and nanotechnology. Such arrays can be developed at flat surfaces including air–liquid and liquid–liquid interfaces as substrate to support the growth of the arrays.324–331 Some of the earliest examples of self-assembled 2D arrays used the protein cage ferritin, and were prepared on lipid monolayers at the air–water interface,324–326 where interaction of protein cages with polar head groups of lipids was key to manipulating the array formation. This approach is versatile and close-packed 2D arrays have also been developed from larger protein cages including CPMV,327 TYMV,328 and P22.329 It is important to note that arrangement of protein cages into ordered arrays is primarily dependent on the properties of the exterior surfaces of protein cages. This allows for the development of the same array structures regardless of the cargo molecules encapsulated within protein cages. For example, ferritin proteins encapsulating iron oxide or indium oxide nanoparticles within the interior cavity have been successfully assembled into near perfect hexagonally packed 2D arrays under the same conditions that lead to the empty apoferritin array assembly.332,333
Assembly of protein cages at the air–liquid and liquid–liquid interfaces is typically a dynamic process, thus the arrays formed at the interfaces are usually transferred to solid substrates, or an inter-particle chemical cross-linkage is introduced to obtain stable 2D monolayers.330,331 Alternatively, direct deposition of protein cages on the surfaces of solid substrates has also been investigated. The deposition can be achieved either through the formation of covalent bonds between the protein cage and the substrate or through weak physical interactions between them.334,335 For example, an engineered CCMV Janus particle, which possessed a reactive thiol group only on one side of its exterior surface, was shown to bind selectively to a gold substrate via a covalent linkage and formed a 2D array of the CCMV.295 Controlled deposition of CCMV was also achieved by manipulating electrostatic interactions between the overall negatively charged CCMV and the positively charged substrate.336
The bottom-up approaches for constructing 2D arrays of protein cages can be readily combined with top-down techniques such as lithography, which lead to controlled placement of protein cages into a designed pattern. Specific interaction between protein cages and substrates are required to accomplish site-specific immobilization of the cages. For example, De Yoreo et al. demonstrated alignment of virus capsids in a line (one capsid wide) through a combination of exterior surface engineering of CPMV and a dip-pen lithography patterning of a substrate.334,335 They genetically engineered a CPMV mutant decorated with six contiguous histidine residues (His-CPMV). The His-CPMV was successfully placed as lines with 30 nm width on nitrilotriacetic acid (NTA)-terminated patterned on a gold substrate (Fig. 25) via a bond formation with Ni-NTA. The line width corresponds to the 1D alignment of single capsids.
Fig. 25.

(A) Schematic of nanografting and virus deposition process. (A) A Ni-NTA terminated surface is created at specific locations on a gold substrate (red lines) to allow for deposition of His-tagged CPMV (yellow spheres) via Ni(ii) chelation. (B–E) AFM images showing the coverage of the CPMV on the substrate as well as the increasing order observed by increases in virus concentration (flux) and increasing inter-viral attractive interaction (increasing PEG concentration). (B) At a condition of low flux and weak inter-viral attractive interaction (0 wt% PEG), His-CPMV attaches almost exclusively to the Ni-NTA line, but the coverage is low. (C) At higher flux with weak interaction, His-CPMV still exclusively attaches to the line, but with increased coverage. (D and E) As the inter-viral interaction is increased (by addition of 1 wt% PEG), virus assembly spreads outward from the lines, and clusters of viruses lie between the lines.(F) Higher resolution AFM image of condition (C), showing His-CPMV alignment at a single capsid width. (G) Higher resolution image of condition (E), showing ordered packing of virus capsid on the Ni-NTA line. Figure reproduced from ref. 335 with permission from the American Chemical Society, copyright 2006.
Modification of the exterior surface of a protein cage with an additional peptide sequence is another approach that has been used to immobilize a protein cage in site-specific manner. Peptide sequences which exhibit high affinity to specific metal, inorganic and polymer materials have been identified by several techniques such as phage display library277 and de novo design.337 These peptides can be readily introduced on the exterior surfaces of protein cages via genetic or chemical modification and are utilized to immobilize protein cages on a targeted substrate.338,339 For example, a titanium binding peptide (RKLPDA) was introduced onto the exterior surface of ferritin allowing the ferritin to be localized to titanium patterns on a silica substrate.340 This result illustrates that engineering of protein cages in combination with top-down patterning techniques will lead to further development of fabricating protein cage based devices.
6.3. Three-dimensional assembly of protein cages
Construction of three-dimensional (3D) superlattice from functional nanoparticle building blocks in a controlled manner is of significant interest because they have the potential for making a new class of materials that exploit collective behavior and properties arising from those of the individual particles.341–344 Although synthetic particles such as metallic, inorganic and polymer nanoparticles have been extensively studied as building blocks, protein cages are appealing multifunctional nanoparticles as has been described in the previous sections. In the assembly of nanoparticles, the formation of amorphous aggregates is common28,345–347 but the successful formation of long-range ordered assemblies of nanoparticles, i.e. superlattices, has been demonstrated by optimizing interactions between building block components.16,343,348–350 A range of methods have been explored to construct 3D assemblies of protein cages and in many cases linker molecules have been utilized to direct and control assembly. Electrostatic interaction is one of the most well explored interactions to mediate assembly of protein cages into bulk materials.14,15,17,351–353 For example, Kostiainen et al. have demonstrated the formation of protein cage based superlattice materials through electrostatic interactions between the protein cages and oppositely charged mediator particles.14,15 Chemically modified gold nanoparticles bearing positive surface charge were used to mediate 3D assembly of CCMV, which has a negatively charged exterior surface. Interparticle interactions were manipulated by changing the ionic strength and pH of the solution (Fig. 26A). When the interparticle interactions were too strong, an amorphous aggregate was formed, whereas an ordered array with a face-centered-cubic (FCC) arrangement of CCMV was obtained under an optimal range of interaction (Fig. 26B).14 They have further explored the approach and demonstrated that superlattices with not only FCC structures, but also hexagonal-close-packed (HCP) structures, and body-centered-cubic (BCC) structures have been realized by changing the type and size of the mediator (Fig. 26C).15,353
Fig. 26.

(A) Superlattice formation of positively charged gold nanoparticles (AuNP) and negatively charged CCMV at different Debye screening lengths, k−1, (adjusted by NaCl concentration), AuNP/CCMV mass ratios (top) and pH (bottom) determined by small angle X-ray scattering (SAXS). At a given pH, the superlattice is formed only in a narrow NaCl concentration window (Region 2), which is shifted to smaller k−1 (i.e. higher NaCl concentration) when the pH is increased. Superlattice formation is possible only when the pH is higher than pI of CCMV (pH = 3.8). High NaCl concentration screens the electrostatic interaction between CCMV and AuNP, resulting in non-assembly of the particles (Region 3). (B) Two-dimensional (inset) and integrated one-dimensional SAXS data obtained from a CCMV superlattice (sample 2 in (A)) (top). The experimental scattering pattern (black trace) matches well with the respective theoretical scattering pattern of an AB8FCC-type structure (red and green traces). Crystalline superlattices are also observed with cryo-TEM (bottom). The particles arrange into an AB8FCC type lattice, where CCMV (blue) adopts an FCC structure and the voids between the CCMVs are filled with eight AuNPs (yellow) as shown in inset. (C) Crystal structure models and lattice parameters of CCMV–avidin, CCMV–G6 PAMAM dendrimer and CCMV–Au nanoparticle superlattice. Superlattices with different structures are yielded depending on linker molecules, which mediate assembly of CCMV. Parts A and B reproduced from ref. 14 with permission from Nature Publishing Group, copyright 2013. Part C adapted from ref. 15 with permission from Nature Publishing Group, copyright 2014.
Catalytically active VLP superlattices were recently developed using a similar assembly approach combined with genetic modification of VLP exterior surface to modulate interparticle interactions.17 In this study, two enzymes, which catalyze a sequential two-step reaction to form isobutanol, were encapsulated individually into the P22 VLPs. The superlattices constructed from the two populations of P22 VLPs retained the coupled two-step catalytic activity, despite the fact that this was now a bulk material with the enzymes immobilized within it. This result indicates that the assembled lattice is porous and the diffusion of small molecule substrates and products through the lattice is not the rate-limiting step in the overall reaction under the conditions it was investigated. As it is a bulk material, the catalytically active superlattice is readily condensed, recycled and reused.
Higher order assembly mediated by electrostatic interaction has been applied for developing protein cage templated binary arrays of encapsulated inorganic nanoparticles. Beck et al., used two types of ferritin cages, one with a positively charged exterior surface and the other having a negatively charged exterior surface.354 The oppositely charged ferritin mutants were co-crystalized with a tetragonal lattice structure. By synthesizing cerium oxide and cobalt oxide inside of negatively charged and positively charged ferritins independently prior to the co-crystallization process, the protein cages served as a template to construct a binary crystalline array of cerium oxide and cobalt oxide nanoparticles.
Three-dimensional assembly of protein cages do occur naturally in vivo. Iridovirus such as Wiseana iridescent virus (WIV) is known to display a “rainbow-like” opalescent hue in heavily infected host insects.355,356 This is because paracrystal-line arrays of viral particles are formed within the cell cytoplasm of the infected hosts and a weak optical interference effect due to Bragg scattering from the assembled arrays is observed.357 Vaia et al., have successfully reproduced the assembly of WIV particles in vitro by various sedimentation or thin-film assembly techniques and demonstrated that optical iridescence of the in vitro assembled materials is much stronger than that commonly observed in vivo.357 The inter-particle interaction is characterized by weak long-range electrostatic repulsion and short-range attractive interaction due to depletion and van der Waals forces.
Complementary pairs of oligonucleotides have been another widely used linker molecule to mediate assembly of nanoparticles into higher order structures.342–345,348,349 Various physical parameters of the oligonucleotide linkers including strength and specificity of the complementary strand interactions, and the length and shapes of the linkers can be finely tuned by rationally designed sequence of the linkers. Thus a wide range of ordered arrays of not only close packed structures, but also loosely packed structures such as a diamond lattice have been realized via this approach.16,343,350 Finn et al., applied oligonucleotide linkers for directing assembly of protein cages.346 They demonstrated temperature dependent assembly and disassembly of two populations of virus capsids, each conjugated with one of the complementary pair of oligonucleotide linkers. They have also reported construction of a binary superlattice with NaTl (B32)-type crystalline structures assembled from oligonucleotide decorated virus capsid and gold nanoparticle.16 In principle, complementary pairs of peptide motifs such as coiled-coil peptides have the potential to mediate assembly of protein cages in a sequences specific manner similar to the oligonucleotide-based linker.358 Although they have not yet been explored as assembly mediators to nearly the same extent that oligonucleotide-based linkers have, polypeptide linkers could be significantly more diverse that oligonucleotide linkers and have great potential to direct and control assembly of protein cage building blocks into complex structures.359–362
Two-dimensional arrays of protein cages discussed earlier are readily expanded to construction of 3D architectures via layer-by-layer deposition.28,338,363–371 For instance, Sano et al. fabricated 3D layer of a ferritin mutant (T1-LF) which possesses a titanium binding peptide on its exterior surface.338 The T1-LF exhibit two unique functions, specific binding to Ti and capability to induce titania mineralization around the cage. In the fabrication process, the first layer of the T1-LF was selectively adsorbed on a Ti pattern formed on a Pt substrate due to specific affinity of the Ti binding peptide. Next, the substrate was soaked in a solution containing titanium bis(ammonium lacto)dihydroxide (TiBALDH), a precursor of titania and a thin titania layer was formed on the top of T1-LF layer. The titania layer worked as a binding target of the second T1-LF layer. Hence stacking layers of ferritin and titania were successfully fabricated by utilizing binding and mineralization capability of the T1-LF.
Anisotropic shaped filamentous and tubular cages such as TMV and bacteriophage M13 have also been utilized to fabricate 2D film and 3D assemblies of cages, because they could exhibit liquid-crystalline ordering under appropriate conditions.372–376 For example, Lee et al. demonstrated formation of a film of M13 filaments with long-range order on a substrate through simply dipping and pulling the substrate vertically from a suspension of the M13 phage at precisely controlled speeds.376 Importantly, the helical structure of individual M13 phage was mirrored in the hierarchical assembly and a helical twist to the organization of the assembly was observed when fabricated under appropriate conditions. TMV, whose surface is negatively charged, has been assembled into a bundle of superlattice wires mediated by an oppositely charged metal nanoparticle. Similar to the example of M13 film, the assembled bundle shows right-handed helical twisting because of the right-handed structure of the individual TMV building units.373
One of the unique features of protein cages over other nanoparticles is their ordered and highly symmetric arrangement of subunits (as discussed in previous sections), thus they present well-defined symmetry specific sites. By exploiting symmetry specific sites on the protein cages as binding sites for linker molecules, protein cages can serve as coordination nodes for the construction of three dimensional network structures, which conceptually resemble the metal ions in metal–organic frameworks (MOFs). For example, Tezcan et al. have exploited the 3-fold symmetry site of the ferritin cage to direct its assembly into a crystalline material.377 They substituted threonine 122, located at the 3-fold symmetry site of ferritin, with histidine. The three histidine residues at the 3-fold symmetry site of the cage (i.e. one from each subunit) coordinate with Zn2+ with tetrahedral coordination geometry, and an additional ditopic ligand molecule can occupy the last coordination site of Zn2+ and serve as a linker to mediate assembly between ferritin cages. As a consequence, the engineered ferritin formed a BCC type crystal in the presence of Zn2+ and a ditopic linker, instead of an FCC type crystal which wild type ferritin typically forms (Fig. 27). Each ferritin cage possesses eight 3-fold symmetry axes, hence the engineered ferritin crystalized into a BCC structure that has eight nearest neighbor positions.
Fig. 27.

Schematic illustration for metal/linker-directed self-assembly of ferritin into three-dimensional crystals. Surface exposed Zn2+ binding sites are engineered at 3-fold symmetry (C3) sites of ferritin by mutating threonine 122 to histidine (T122H). In the presence of ditopic organic linkers, the T122H ferritin mutant forms a BCC lattice through coordination of ferritins at the C3 sites. On the other hand, in the absence of such ditopic linkers, the mutant forms a FCC lattice, which ferritin typically crystalized into, through coordination at 2-fold symmetry (C2) sites. Figure reproduced from ref. 377 with permission from the American Chemical Society, copyright 2015.
An engineered protein can also serve as a linker molecule to direct self-assembly of protein cages into higher order structures. For example, a decoration protein (Dec), which binds at symmetry specific sites on the expanded (mature) form of P22 was used to construct protein-based linkers to mediate controlled assembly of the P22 VLP.28 A linear ditopic linker and a tetrahedral tetratopic linker were genetically engineered through either a point mutation of Dec or fusion of Dec to the exposed termini on the smaller Dps protein cage, respectively. Bulk assembly and layer-by-layer deposition of P22 VLP in solution was successfully achieved using both linker molecules (Fig. 28). Although the obtained bulk assemblies of P22 VLPs in this study did not show a long-range ordered structure, optimal manipulation of interaction between protein cages and linker proteins could lead to construction of protein only superlattice materials because Dec binds to symmetry specific sites of P22.310,311
Fig. 28.

(A) TEM images of P22 mixed with a ditopic linker derived from Dec (left) or wild type (wt) Dec (right). In the case of P22 with ditopic Dec linker, large clusters of micrometer to tens of micrometers in size, which composed of P22 VLP, were observed. In the case of P22 with wtDec, each VLP were well dispersed and no cluster of VLP was observed. (B) Quartz crystal microbalance (QCM) resonance frequency changes upon alternate injection of P22, ditopic Dec linker and wtDec on a gold coated QCM sensor. The decrease in resonance frequency corresponds to the increase in mass on the sensor due to the deposition of the proteins. Layer-by-layer deposition was demonstrated through the alternative addition of P22 and ditopic Dec linker. Addition of wtDec caps the deposited layer and passivates from further attachment of P22 because it has only one binding site for P22. The cartoon on the right depicts observed layer-by-layer deposition. Figure adapted from ref. 28 with permission from Wiley, copyright 2015.
7. Conclusion
It is evident that the natural world has a seemingly infinite number of protein cages, specifically viral protein cages waiting to be discovered and exploited. Even the small number of cages that have been thoroughly characterized to date have provided us with a vast library that can be used for further design and manipulation for materials synthesis. Fundamental studies into the mechanisms of viral capsid assembly and protein–protein interactions are being harnessed for development of novel nanomaterials and armed with the knowledge of Nature’s synthetic methods, novel designer cages can be constructed to have specific features (such as stability, size, rigidity, and porosity). With increasing synthetic precision a wide range of desired moieties can be presented on either the interior or exterior of these cages taking advantage of the high symmetry and associated multivalent presentation of these platforms. Thus these closed shell protein architectures provide interior and exterior surfaces for presentation and cargo encapsulation as well as intersubunit interfaces that allow for fine tuning of the structural properties and inspiration for de novo design. The diversity and synthetic versatility of these platforms has made them attractive materials for an increasing number of applications in materials science, chemistry, biology, and medicine. These applications have grown extensively in the last few decades, and will continue to grow as the library of cages, our foundational understanding of individual protein cages, and the generality of new approaches develops. New advances in directing the interactions between functionally active individual particles also holds the promise of future applications where the collective properties of an ensemble of particles can be harnessed. Viruses no longer need to be understood only as hostile pathogens but rather as a source of active nanomaterial inspiration and the raw materials for a new and exciting materials future.
Acknowledgements
The authors would like to acknowledge funding support from the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering (DE-SC0016155), the National Institutes of Health (R01 AI104905), and the National Science Foundation (NSF-BMAT DMR-1507282).
Biographies

William M. Aumiller Jr received a BS degree in chemistry from Juniata College in 2008. In 2015 he received his PhD in chemistry from The Pennsylvania State University under the direction of Prof. Christine D. Keating. He developed model systems based on aqueous phase separation of polymers for the purpose of mimicking intracellular compart-mentalization and phase separation. He is currently a postdoctoral research associate in Trevor Douglas’s group at Indiana University where he is studying higher order assembly of virus particles.

Masaki Uchida is an Associate Scientist in the Douglas Lab at Indiana University. His current research interests are in bioinspired materials synthesis and assembly of protein cages into higher order structures. He received his PhD degree in Materials Chemistry in 2002 from Kyoto University, Japan under the supervision of Prof. Tadashi Kokubo. He did his post-doctoral training at the National Institute of Advanced Science and Technology, Japan and at Montana State University.

Trevor Douglas is the Earl Blough Professor of Chemistry at Indiana University. His research has focused on the development of biomimetic approaches to construction of new functional materials. He received his PhD in 1991 from Cornell University working with Professor Klaus Theopold and did his post-doctoral training at Bath University with Professor Stephen Mann, in the area of biomineralization and biomimetic materials.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1.Johnson JE and Chiu W, Curr. Opin. Struct. Biol, 2000, 10, 229–235. [DOI] [PubMed] [Google Scholar]
- 2.Douglas T and Young M, Science, 2006, 312, 873–875. [DOI] [PubMed] [Google Scholar]
- 3.Harrison PM and Arosio P, Biochim. Biophys. Acta, Bioenerg, 1996, 1275, 161–203. [DOI] [PubMed] [Google Scholar]
- 4.Uchida M, Kang S, Reichhardt C, Harlen K and Douglas T, Biochim. Biophys. Acta, 2010, 1800, 834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horwich AL, Fenton WA, Chapman E and Farr GW, Annu. Rev. Cell Dev. Biol, 2007, 23, 115–145. [DOI] [PubMed] [Google Scholar]
- 6.Kim KK, Kim R and Kim SH, Nature, 1998, 394, 595–599. [DOI] [PubMed] [Google Scholar]
- 7.Lodish HF, Molecular cell biology WH Freeman-Macmillan Learning, New York, 8th edn, 2016. [Google Scholar]
- 8.Theil EC and Goss DJ, Chem. Rev, 2009,109, 4568–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arosio P and Levi S, Ferritin, iron homeostasis, and oxidative damage, Free Radical Biol. Med, 2002, 33, 457–463. [DOI] [PubMed] [Google Scholar]
- 10.Suttle CA, Nature, 2005, 437, 356–361. [DOI] [PubMed] [Google Scholar]
- 11.Mateu MG, Arch. Biochem. Biophys, 2013, 531, 65–79. [DOI] [PubMed] [Google Scholar]
- 12.Lai YT, Cascio D and Yeates TO, Science, 2012, 336, 1129. [DOI] [PubMed] [Google Scholar]
- 13.Lai YT, Reading E, Hura GL, Tsai KL, Laganowsky A, Asturias FJ, Tainer JA, Robinson CV and Yeates TO, Nat. Chem, 2014, 6, 1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kostiainen MA, Hiekkataipale P, Laiho A, Lemieux V, Seitsonen J, Ruokolainen J and Ceci P, Nat. Nanotechnol, 2013, 8, 52–56. [DOI] [PubMed] [Google Scholar]
- 15.Liljestrom V, Mikkila J and Kostiainen MA, Nat. Commun, 2014, 5, 4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cigler P, Lytton-Jean AKR, Anderson DG, Finn MG and Park SY, Nat. Mater, 2010, 9, 918–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uchida M, McCoy K, Fukuto M, Yang L, Yoshimura H, Miettinen HM, Lafrance B, Patterson DP, Schwarz B, Karty JA, Prevelige PE Jr, Lee B and Douglas T, ACS Nano, 2018, 12, 942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrico ZM, Romanini DW, Mehl RA and Francis MB, Chem. Commun, 2008, 1205–1207. [DOI] [PubMed] [Google Scholar]
- 19.Strable E, Prasuhn DE, Udit AK, Brown S, Link AJ, Ngo JT, Lander G, Quispe J, Potter CS, Carragher B, Tirrell DA and Finn MG, Bioconjugate Chem, 2008, 19, 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Neil A, Prevelige PE, Basu G and Douglas T, Biomacromolecules, 2012, 13, 3902–3907. [DOI] [PubMed] [Google Scholar]
- 21.Patterson DP, Schwarz B, Waters RS, Gedeon T and Douglas T, ACS Chem. Biol, 2014, 9, 359–365. [DOI] [PubMed] [Google Scholar]
- 22.Qazi S, Miettinen HM, Wilkinson RA, McCoy K, Douglas T and Wiedenheft B, Mol. Pharmaceutics, 2016, 13, 1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jordan PC, Patterson DP, Saboda KN, Edwards EJ, Miettinen HM, Basu G, Thielges MC and Douglas T, Nat. Chem, 2016, 8, 179–185. [DOI] [PubMed] [Google Scholar]
- 24.Fiedler JD, Brown SD, Lau JL and Finn MG, Angew. Chem., Int. Ed, 2010, 49, 9648–9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wörsdörfer B, Pianowski Z and Hilvert D, J. Am. Chem. Soc, 2012, 134, 909–911. [DOI] [PubMed] [Google Scholar]
- 26.Comellas-Aragones M, Engelkamp H, Claessen VI, Sommerdijk NAJM, Rowan AE, Christianen PCM, Maan JC, Verduin BJM, Cornelissen JJLM and Nolte RJM, Nat. Nanotechnol, 2007, 2, 635–639. [DOI] [PubMed] [Google Scholar]
- 27.Glasgow JE, Capehart SL, Francis MB and Tullman-Ercek D, ACS Nano, 2012, 6, 8658–8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uchida M, LaFrance B, Broomell CC, Prevelige PE Jr. and Douglas T, Small, 2015, 11, 1562–1570. [DOI] [PubMed] [Google Scholar]
- 29.Böker A, He J, Emrick T and Russell TP, Soft Matter, 2007, 3, 1231. [DOI] [PubMed] [Google Scholar]
- 30.Nie Z, Petukhova A and Kumacheva E, Nat. Nanotechnol, 2010, 5, 15–25. [DOI] [PubMed] [Google Scholar]
- 31.Wen AM and Steinmetz NF, Chem. Soc. Rev, 2016, 45, 4074–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Culver JN, Brown AD, Zang FH, Gnerlich M, Gerasopoulos K and Ghodssi R, Virology, 2015, 479, 200–212. [DOI] [PubMed] [Google Scholar]
- 33.Rother M, Nussbaumer MG, Renggli K and Bruns N, Chem. Soc. Rev, 2016, 45, 6213–6249. [DOI] [PubMed] [Google Scholar]
- 34.5w?>Schoonen L and van Hest JCM, Nanoscale, 2014, 6, 7124–7141. [DOI] [PubMed] [Google Scholar]
- 35.Prangishvili D, Annu. Rev. Microbiol, 2013, 67, 565–585. [DOI] [PubMed] [Google Scholar]
- 36.Noda T, Front. Microbiol, 2012, 3, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao GP, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning JY, Ahn J, Gronenborn AM, Schulten K, Aiken C and Zhang PJ, Nature, 2013, 497, 643–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ge P, Tsao J, Schein S, Green TJ, Luo M and Zhou ZH, Science, 2010, 327, 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caspar DLD and Klug A, Cold Spring Harbor Symp. Quant. Biol, 1962, 27, 1–24. [DOI] [PubMed] [Google Scholar]
- 40.Prasad BV and Schmid MF, Adv. Exp. Med. Biol, 2012, 726, 17–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carrillo-Tripp M, Shepherd CM, Borelli IA, Venkataraman S, Lander G, Natarajan P, Johnson JE, Brooks CL 3rd and Reddy VS, Nucleic Acids Res, 2009, 37, D436—D442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daya S and Berns KI, Clin. Microbiol. Rev, 2008, 21, 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B and Johnson JE, Science, 2006, 312, 1791–1795. [DOI] [PubMed] [Google Scholar]
- 44.Chant SR, Ann. Appl. Biol, 1959, 47, 565–572. [Google Scholar]
- 45.Sainsbury F, Canizares MC and Lomonossoff GP, Annu. Rev. Phytopathol, 2010, 48, 437–455. [DOI] [PubMed] [Google Scholar]
- 46.Lin TW, Chen ZG, Usha R, Stauffacher CV, Dai JB, Schmidt T and Johnson JE, Virology, 1999, 265, 20–34. [DOI] [PubMed] [Google Scholar]
- 47.Sainsbury F, Saxena P, Aljabali AAA, Saunders K, Evans DJ and Lomonossoff GP, Virus Hybrids as Nanomaterials: Methods and Protocols, 2014, vol. 1108, pp. 139–153. [DOI] [PubMed] [Google Scholar]
- 48.Saunders K, Sainsbury F and Lomonossoff GP, Virology, 2009, 393, 329–337. [DOI] [PubMed] [Google Scholar]
- 49.Speir JA, Munshi S, Wang G, Baker TS and Johnson JE, Structure, 1995, 3, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bancroft JB, Hills GJ and Markham R, Virology, 1967, 31, 354–379. [DOI] [PubMed] [Google Scholar]
- 51.Annamalai P and Rao ALN, Virology, 2005, 332, 650–658. [DOI] [PubMed] [Google Scholar]
- 52.Rao ALN, Annu. Rev. Phytopathol, 2006, 44, 61–87. [DOI] [PubMed] [Google Scholar]
- 53.Garmann RF, Comas-Garcia M, Knobler CM and Gelbart WM, Acc. Chem. Res, 2016, 49, 48–55. [DOI] [PubMed] [Google Scholar]
- 54.Speir JA, Bothner B, Qu C, Willits DA, Young MJ and Johnson JE, J. Virol, 2006, 80, 3582–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bancroft JB and Hiebert E, Virology, 1967, 32, 354–356. [DOI] [PubMed] [Google Scholar]
- 56.Lavelle L, Gingery M, Phillips M, Gelbart WM, Knobler CM, Cadena-Nava RD, Vega-Acosta JR, Pinedo-Torres LA and Ruiz-Garcia J, J. Phys. Chem. B, 2009, 113, 3813–3819. [DOI] [PubMed] [Google Scholar]
- 57.Lavelle L, Michel JP and Gingery M, J. Virol. Methods, 2007, 146, 311–316. [DOI] [PubMed] [Google Scholar]
- 58.Michel JP, Gingery M and Lavelle L, J. Virol. Methods, 2004, 122, 195–198. [DOI] [PubMed] [Google Scholar]
- 59.Uchida M, Klem MT, Allen M, Suci P, Flenniken M, Gillitzer E, Varpness Z, Liepold LO, Young M and Douglas T, Adv. Mater, 2007, 19, 1025–1042. [Google Scholar]
- 60.Lucas RW, Larson SB and McPherson A, J. Mol. Biol, 2002, 317, 95–108. [DOI] [PubMed] [Google Scholar]
- 61.Diez J, Ishikawa M, Kaido M and Ahlquist P, Proc. Natl. Acad. Sci. U. S. A, 2000, 97, 3913–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pfeiffer P and Hirth L, Virology, 1974, 61, 160–167. [DOI] [PubMed] [Google Scholar]
- 63.King J, Botstein D, Casjens S, Earnshaw W, Harrison S and Lenk E, Philos. Trans. R. Soc. London, Ser. B, 1976, 276, 37–49. [DOI] [PubMed] [Google Scholar]
- 64.Prevelige PE and King J, Prog. Med. Virol, 1993, 40, 206–221. [PubMed] [Google Scholar]
- 65.Teschke CM and Parent KN, Virology, 2010, 401, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lander GC, Khayat R, Li R, Prevelige PE, Potter CS, Carragher B and Johnson JE, Structure, 2009, 17, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prevelige PE, Thomas D and King J, J. Mol. Biol, 1988, 202, 743–757. [DOI] [PubMed] [Google Scholar]
- 68.Fuller MT and King J, J. Mol. Biol, 1982, 156, 633–665. [DOI] [PubMed] [Google Scholar]
- 69.ThumanCommike PA, Greene B, Jakana J, Prasad BVV, King J, Prevelige PE and Chiu W, J. Mol. Biol, 1996, 260, 85–98. [DOI] [PubMed] [Google Scholar]
- 70.Chang J, Weigele P, King J, Chiu W and Jiang W, Structure, 2006, 14, 1073–1082. [DOI] [PubMed] [Google Scholar]
- 71.Parent KN, Khayat R, Tu LH, Suhanovsky MM, Cortines JR, Teschke CM, Johnson JE and Baker TS, Structure, 2010, 18, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen DH, Baker ML, Hryc CF, DiMaio F, Jakana J, Wu W, Dougherty M, Haase-Pettingell C, Schmid MF, Jiang W, Baker D, King JA and Chiu W, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hryc CF, Chen DH, Afonine PV, Jakana J, Wang Z, Haase-Pettingell C, Jiang W, Adams PD, King JA, Schmid MF and Chiu W, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 3103–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hooker JM, Kovacs EW and Francis MB, J. Am. Chem. Soc, 2004, 126, 3718–3719. [DOI] [PubMed] [Google Scholar]
- 75.Kovacs EW, Hooker JM, Romanini DW, Holder PG, Berry KE and Francis MB, Bioconjugate Chem, 2007, 18, 1140–1147. [DOI] [PubMed] [Google Scholar]
- 76.Golmohammadi R, Valegard K, Fridborg K and Liljas L, J. Mol. Biol, 1993, 234, 620–639. [DOI] [PubMed] [Google Scholar]
- 77.Valegard K, Liljas L, Fridborg K and Unge T, Acta Crystallogr., Sect. B: Struct. Sci, 1991, 47, 949–960. [DOI] [PubMed] [Google Scholar]
- 78.Valegard K, Liljas L, Fridborg K and Unge T, Nature, 1990, 345, 36–41. [DOI] [PubMed] [Google Scholar]
- 79.Fu Y and Li JM, Virus Res, 2016, 211, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dykeman EC, Stockley PG and Twarock R, J. Mol. Biol, 2010, 395, 916–923. [DOI] [PubMed] [Google Scholar]
- 81.Stockley PG, Rolfsson O, Thompson GS, Basnak G, Francese S, Stonehouse NJ, Homans SW and Ashcroft AE, J. Mol. Biol, 2007, 369, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Golmohammadi R, Fridborg K, Bundule M, Valegard K and Liljas L, Structure, 1996, 4, 543–554. [DOI] [PubMed] [Google Scholar]
- 83.Brown SD, Fiedler JD and Finn MG, Biochemistry, 2009, 48, 11155–11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fang PY, Ramos LMG, Holguin SY, Hsiao C, Bowman JC, Yang HW and Williams LD, Nucleic Acids Res, 2017, 45, 3519–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Witherell GW and Uhlenbeck OC, Biochemistry, 1989, 28, 71–76. [DOI] [PubMed] [Google Scholar]
- 86.Ashcroft AE, Lago H, Macedo JMB, Horn WT, Stonehouse NJ and Stockley PG, J. Nanosci. Nanotechnol, 2005, 5, 2034–2041. [DOI] [PubMed] [Google Scholar]
- 87.Takamatsu H and Iso K, Nature, 1982, 298, 819–824. [DOI] [PubMed] [Google Scholar]
- 88.Bundy BC and Swartz JR, J. Biotechnol, 2011, 154, 230–239. [DOI] [PubMed] [Google Scholar]
- 89.Alonso JM, Gorzny ML and Bittner AM, Trends Biotechnol, 2013, 31, 530–538. [DOI] [PubMed] [Google Scholar]
- 90.Namba K, Pattanayek R and Stubbs G, J. Mol. Biol, 1989, 208, 307–325. [DOI] [PubMed] [Google Scholar]
- 91.Sachse C, Chen JZ, Coureux PD, Stroupe ME, Fandrich M and Grigorieff N, J. Mol. Biol, 2007, 371, 812–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pieters BJGE, van Eldijk MB, Nolte RJM and Mecinovic J, Chem. Soc. Rev, 2016, 45, 24–39. [DOI] [PubMed] [Google Scholar]
- 93.Sutter M, Boehringer D, Gutmann S, Gunther S, Prangishvili D, Loessner MJ, Stetter KO, Weber-Ban E and Ban N, Nat. Struct. Mol. Biol, 2008, 15, 939–947. [DOI] [PubMed] [Google Scholar]
- 94.Butler PJG, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee SY, Lim JS and Harris MT, Biotechnol. Bioeng, 2012, 109, 16–30. [DOI] [PubMed] [Google Scholar]
- 96.Kehoe JW and Kay BK, Chem. Rev, 2005, 105, 4056–4072. [DOI] [PubMed] [Google Scholar]
- 97.Makowski L, Curr. Opin. Struct. Biol, 1994, 4, 225–230. [Google Scholar]
- 98.Lewin A, Moore GR and Le Brun NE, Dalton Trans, 2005, 3597–3610. [DOI] [PubMed] [Google Scholar]
- 99.Zhang Y and Orner BP, Int. J. Mol. Sci, 2011, 12, 5406–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bevers LE and Theil EC, Prog. Mol. Subcell. Biol, 2011, 52, 29–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Almiron M, Link AJ, Furlong D and Kolter R, Genes Dev, 1992, 6, 2646–2654. [DOI] [PubMed] [Google Scholar]
- 102.Ilari A, Savino C, Stefanini S, Chiancone E and Tsernoglou D, Acta Crystallogr., Sect. D: Biol. Crystallogr, 1999, 55, 552–553. [DOI] [PubMed] [Google Scholar]
- 103.Swift J, Butts CA, Cheung-Lau J, Yerubandi V and Dmochowski IJ, Langmuir, 2009, 25, 5219–5225. [DOI] [PubMed] [Google Scholar]
- 104.Johnson E, Cascio D, Sawaya MR, Gingery M and Schröder I, Structure, 2005, 13, 637–648. [DOI] [PubMed] [Google Scholar]
- 105.Sana B, Johnson E, Le Magueres P, Criswell A, Cascio D and Lim S, J. Biol. Chem, 2013, 288, 32663–32672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gerl M and Jaenicke R, Eur. Biophys. J, 1987, 15, 103–109. [DOI] [PubMed] [Google Scholar]
- 107.Schott K, Ladenstein R, König A and Bacher A, J. Biol. Chem, 1990, 265, 12686–12689. [PubMed] [Google Scholar]
- 108.Zhang XF, Konarev PV, Petoukhov MV, Svergun DI, Xing L, Cheng RH, Haase I, Fischer M, Bacher A, Ladenstein R and Meining W, J. Mol. Biol, 2006, 362, 753–770. [DOI] [PubMed] [Google Scholar]
- 109.Ladenstein R, Fischer M and Bacher A, FEBS J, 2013, 280, 2537–2563. [DOI] [PubMed] [Google Scholar]
- 110.Cheng Y, Boll W, Kirchhausen T, Harrison SC and Walz T, J. Mol. Biol, 2007, 365, 892–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Milne JL, Wu X, Borgnia MJ, Lengyel JS, Brooks BR, Shi D, Perham RN and Subramaniam S, J. Biol. Chem, 2006, 281, 4364–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rome LH and Kickhoefer VA, ACS Nano, 2013, 7, 889–902. [DOI] [PubMed] [Google Scholar]
- 113.Tanaka H, Kato K, Yamashita E, Sumizawa T, Zhou Y, Yao M, Iwasaki K, Yoshimura M and Tsukihara T, Science, 2009, 323, 384–388. [DOI] [PubMed] [Google Scholar]
- 114.Moreira D and Lopez-Garcia P, Nat. Rev. Microbiol, 2009, 7, 306–311. [DOI] [PubMed] [Google Scholar]
- 115.Forterre P and Prangishvili D, Res. Microbiol, 2009, 160, 466–472. [DOI] [PubMed] [Google Scholar]
- 116.Koonin EV, Dolja VV and Krupovic M, Virology, 2015, 479, 2–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Abrescia NGA, Bamford DH, Grimes JM and Stuart DI, Annu. Rev. Biochem, 2012, 81, 795–822. [DOI] [PubMed] [Google Scholar]
- 118.Bamford DH, Grimes JM and Stuart DI, Curr. Opin. Struct. Biol, 2005, 15, 655–663. [DOI] [PubMed] [Google Scholar]
- 119.Benson SD, Bamford JKH, Bamford DH and Burnett RM, Mol. Cell, 2004, 16, 673–685. [DOI] [PubMed] [Google Scholar]
- 120.Krupovic M and Bamford DH, Nat. Rev. Microbiol, 2008, 6, 941–948. [DOI] [PubMed] [Google Scholar]
- 121.Benson SD, Bamford JKH, Bamford DH and Burnett RM, Cell, 1999, 98, 825–833. [DOI] [PubMed] [Google Scholar]
- 122.Cockburn JJB, Abrescia NGA, Grimes JM, Sutton GC, Diprose JM, Benevides JM, Thomas GJ, Bamford JKH, Bamford DH and Stuart DI, Nature, 2004, 432, 122–125. [DOI] [PubMed] [Google Scholar]
- 123.Abrescia NGA, Cockburn JJB, Grimes JM, Sutton GC, Diprose JM, Butcher SJ, Fuller SD, Martin CS, Burnett RM, Stuart DI, Bamford DH and Bamford JKH, Nature, 2004, 432, 68–74. [DOI] [PubMed] [Google Scholar]
- 124.Suhanovsky MM and Teschke CM, Virology, 2015, 479, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gertsman I, Gan L, Guttman M, Lee K, Speir JA, Duda RL, Hendrix RW, Komives EA and Johnson JE, Nature, 2009, 458, 646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kegel WK and van der Schoot P, Biophys. J, 2006, 91, 1501–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Perlmutter JD and Hagan MF, Annu. Rev. Phys. Chem, 2015, 66, 217–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Prevelige PE, Thomas D and King J, Biophys. J, 1993, 64, 824–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nguyen HD, Reddy VS and Brooks CL, Nano Lett, 2007, 7, 338–344. [DOI] [PubMed] [Google Scholar]
- 130.Zlotnick A, Aldrich R, Johnson JM, Ceres P and Young MJ, Virology, 2000, 277, 450–456. [DOI] [PubMed] [Google Scholar]
- 131.Hu YF, Zandi R, Anavitarte A, Knobler CM and Gelbart WM, Biophys. J, 2008, 94, 1428–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aniagyei SE, Kennedy CJ, Stein B, Willits DA, Douglas T, Young MJ, De M, Rotello VM, Srisathiyanarayanan D, Kao CC and Dragnea B, Nano Lett, 2009, 9, 393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huang X, Bronstein LM, Retrum J, Dufort C, Tsvetkova I, Aniagyei S, Stein B, Stucky G, McKenna B, Remmes N, Baxter D, Kao CC and Dragnea B, Nano Lett, 2007, 7, 2407–2416. [DOI] [PubMed] [Google Scholar]
- 134.Chen C, Daniel M-C, Quinkert ZT, De M, Stein B, Bowman VD, Chipman PR, Rotello VM, Kao CC and Dragnea B, Nano Lett, 2006, 6, 611–615. [DOI] [PubMed] [Google Scholar]
- 135.Parent KN, Zlotnick A and Teschke CM, J. Mol. Biol, 2006, 359, 1097–1106. [DOI] [PubMed] [Google Scholar]
- 136.Thuman-Commike PA, Greene B, Malinski JA, Burbea M, McGough A, Chiu W and Prevelige PE, Biophys. J, 1999, 76, 3267–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Parker MH, Stafford WF and Prevelige PE, J. Mol. Biol, 1997, 268, 655–665. [DOI] [PubMed] [Google Scholar]
- 138.Prevelige PE and Fane BA, Adv. Exp. Med. Biol, 2012, 726, 325–350. [DOI] [PubMed] [Google Scholar]
- 139.Cortines JR, Weigele PR, Gilcrease EB, Casjens SR and Teschke CM, Virology, 2011, 421, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cortines JR, Motwani T, Vyas AA and Teschke CM, J. Virol, 2014, 88, 5287–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Thuman-Commike PA, Greene B, Malinski JA, King J and Chiu W, Biophys. J, 1998, 74, 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Moore SD and Prevelige PE Jr., J. Mol. Biol, 2002, 315, 975–994. [DOI] [PubMed] [Google Scholar]
- 143.Prasad BV, Prevelige PE, Marietta E, Chen RO, Thomas D, King J and Chiu W, J. Mol. Biol, 1993, 231, 65–74. [DOI] [PubMed] [Google Scholar]
- 144.Fuller MT and King J, Biophys. J, 1980, 32, 381–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fuller MT and King J, Virology, 1981, 112, 529–547. [DOI] [PubMed] [Google Scholar]
- 146.Galisteo ML and King J, Biophys. J, 1993, 65, 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Teschke CM, McGough A and Thuman-Commike PA, Biophys. J, 2003, 84, 2585–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen C, Wang JCY and Zlotnick A, PLoS Pathog, 2011, 7, e1005802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Garmann RF, Comas-Garcia M, Gopal A, Knobler CM and Gelbart WM, J. Mol. Biol, 2014, 426, 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Garmann RF, Sportsman R, Beren C, Manoharan VN, Knobler CM and Gelbart WM, J. Am. Chem. Soc, 2015, 137, 7584–7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dykeman EC, Stockley PG and Twarock R, J. Mol. Biol, 2013, 425, 3235–3249. [DOI] [PubMed] [Google Scholar]
- 152.Stockley PG, Twarock R, Bakker SE, Barker AM, Borodavka A, Dykeman E, Ford RJ, Pearson AR, Phillips SEV, Ranson NA and Tuma R, J. Biol. Phys, 2013, 39, 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Stubbs G, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Klug A, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hesketh EL, Meshcheriakova Y, Dent KC, Saxena P, Thompson RF, Cockburn JJ, Lomonossoff GP and Ranson NA, Nat. Commun, 2015, 6, 10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Perlmutter JD, Perkett MR and Hagan MF, J. Mol. Biol, 2014, 426, 3148–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Elrad OM and Hagan MF, Phys. Biol, 2010, 7, 045003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.McPherson A, BioEssays, 2005, 27, 447–458. [DOI] [PubMed] [Google Scholar]
- 159.Gerl M, Jaenicke R, Smith JMA and Harrison PM, Biochemistry, 1988, 27, 4089–4096. [DOI] [PubMed] [Google Scholar]
- 160.Sato D, Ohtomo H, Yamada Y, Hikima T, Kurobe A, Fujiwara K and Ikeguchi M, Biochemistry, 2016, 55, 287–293. [DOI] [PubMed] [Google Scholar]
- 161.Sato D, Takebe S, Kurobe A, Ohtomo H, Fujiwara K and Ikeguchi M, Biochemistry, 2016, 55, 482–488. [DOI] [PubMed] [Google Scholar]
- 162.Perlmutter JD, Mohajerani F and Hagan MF, eLife, 2016, 5, e14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hiebert E, Bancroft JB and Bracker CE, Virology, 1968, 34, 492–508. [DOI] [PubMed] [Google Scholar]
- 164.Bancroft JB, Wagner GW and Bracker CE, Virology, 1968, 36, 146–149. [DOI] [PubMed] [Google Scholar]
- 165.Bancroft JB, Bracker CE and Wagner GW, Virology, 1969, 38, 324–335. [DOI] [PubMed] [Google Scholar]
- 166.Hiebert E and Bancroft JB, Virology, 1969, 39, 296–311. [DOI] [PubMed] [Google Scholar]
- 167.Zhao XX, Fox JM, Olson NH, Baker TS and Young MJ, Virology, 1995, 207, 486–494. [DOI] [PubMed] [Google Scholar]
- 168.Adolph KW and Butler PJ, J. Mol. Biol, 1974, 88, 327–341. [DOI] [PubMed] [Google Scholar]
- 169.Bancroft JB, Adv. Virus Res, 1970, 16, 99–134. [DOI] [PubMed] [Google Scholar]
- 170.Byeon IJL, Meng X, Jung JW, Zhao GP, Yang RF, Ahn JW, Shi J, Concel J, Aiken C, Zhang PJ and Gronenborn AM, Cell, 2009, 139, 780–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Steven AC, Couture E, Aebi U and Showe MK, J. Mol. Biol, 1976, 106, 187–221. [DOI] [PubMed] [Google Scholar]
- 172.Steven AC, Serwer P, Bisher ME and Trus BL, Virology, 1983, 124, 109–120. [DOI] [PubMed] [Google Scholar]
- 173.Suhanovsky MM, Parent KN, Dunn SE, Baker TS and Teschke CM, Mol. Microbiol, 2010, 77, 1568–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Parent KN, Sinkovits RS, Suhanovsky MM, Teschke CM, Egelman EH and Baker TS, Phys. Biol, 2010, 7, 045004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.King NP and Lai YT, Curr. Opin. Struct. Biol, 2013, 23, 632–638. [DOI] [PubMed] [Google Scholar]
- 176.Lai YT, King NP and Yeates TO, Trends Cell Biol, 2012, 22, 653–661. [DOI] [PubMed] [Google Scholar]
- 177.Padilla JE, Colovos C and Yeates TO, Proc. Natl. Acad. Sci. U. S. A, 2001, 98, 2217–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sinclair JC, Davies KM, Venien-Bryan C and Noble MEM, Nat. Nanotechnol, 2011, 6, 558–562. [DOI] [PubMed] [Google Scholar]
- 179.Lai YT, Tsai KL, Sawaya MR, Asturias FJ and Yeates TO, J. Am. Chem. Soc, 2013, 135, 7738–7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.King NP, Bale JB, Sheffler W, McNamara DE, Gonen S, Gonen T, Yeates TO and Baker D, Nature, 2014, 510, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bale JB, Gonen S, Liu Y, Sheffler W, Ellis D, Thomas C, Cascio D, Yeates TO, Gonen T, King NP and Baker D, Science, 2016, 353, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hsia Y, Bale JB, Gonen S, Shi D, Sheffler W, Fong KK, Nattermann U, Xu C, Huang PS, Ravichandran R, Yi S, Davis TN, Gonen T, King NP and Baker D, Nature, 2016, 535, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ljubetic A, Lapenta F, Gradisar H, Drobnak I, Aupic J, Strmsek Z, Lainscek D, Hafner-Bratkovic I, Majerle A, Krivec N, Bencina M, Pisanski T, Velickovic TC, Round A, Carazo JM, Melero R and Jerala R, Nat. Biotechnol, 2017, 35, 1094–1101. [DOI] [PubMed] [Google Scholar]
- 184.Theil EC, Curr. Opin. Chem. Biol, 2011, 15, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Douglas T and Ripoll DR, Protein Sci, 1998, 7, 1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tosha T, Ng H-L, Bhattasali O, Alber T and Theil EC, J. Am. Chem. Soc, 2010, 132, 14562–14569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tosha T, Hasan MR and Theil EC, Proc. Natl. Acad. Sci. U. S. A, 2008, 105, 18182–18187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Jutz G, van Rijn P, Santos Miranda B and Boker A, Chem. Rev, 2015, 115, 1653–1701. [DOI] [PubMed] [Google Scholar]
- 189.Meldrum FC, Wade VJ, Nimmo DL, Heywood BR and Mann S, Nature, 1991, 349, 684–687. [Google Scholar]
- 190.Uchida M, Flenniken ML, Allen M, Willits DA, Crowley BE, Brumfield S, Willis AF, Jackiw L, Jutila M, Young MJ and Douglas T, J. Am. Chem. Soc, 2006, 128, 16626–16633. [DOI] [PubMed] [Google Scholar]
- 191.Allen M, Willits D, Young M and Douglas T, Inorg. Chem, 2003, 42, 6300–6305. [DOI] [PubMed] [Google Scholar]
- 192.Kang S, Lucon J, Varpness ZB, Liepold L, Uchida M, Willits D, Young M and Douglas T, Angew. Chem., Int. Ed, 2008, 47, 7845–7848. [DOI] [PubMed] [Google Scholar]
- 193.Douglas T and Young M, Nature, 1998, 393, 152–155. [Google Scholar]
- 194.Jolley C, Klem M, Harrington R, Parise J and Douglas T, Nanoscale, 2011, 3, 1004–1007. [DOI] [PubMed] [Google Scholar]
- 195.Douglas T, Strable E, Willits D, Aitouchen A, Libera M and Young M, Adv. Mater, 2002, 14, 415–418. [Google Scholar]
- 196.Knez M, Sumser M, Bittner AM, Wege C, Jeske H, Martin TP and Kern K, Adv. Funct. Mater, 2004, 14, 116–124. [Google Scholar]
- 197.Ishii D, Kinbara K, Ishida Y, Ishii N, Okochi M, Yohda M and Aida T, Nature, 2003, 423, 628–632. [DOI] [PubMed] [Google Scholar]
- 198.McMillan RA, Paavola CD, Howard J, Chan SL, Zaluzec NJ and Trent JD, Nat. Mater, 2002, 1, 247–252. [DOI] [PubMed] [Google Scholar]
- 199.McMillan RA, Howard J, Zaluzec NJ, Kagawa HK, Mogul R, Li Y-F, Paavola CD and Trent JD, J. Am. Chem. Soc, 2005, 127, 2800–2801. [DOI] [PubMed] [Google Scholar]
- 200.Bromley KM, Patil AJ, Perriman AW, Stubbs G and Mann S, J. Mater. Chem, 2008, 18, 4796. [Google Scholar]
- 201.Zhou JC, Soto CM, Chen M-S, Bruckman MA, Moore MH, Barry E, Ratna BR, Pehrsson PE, Spies BR and Confer TS, J. Nanobiotechnol, 2012, 10, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Knez M, Bittner AM, Boes F, Wege C, Jeske H, Maib E and Kern K, Nano Lett, 2003, 3, 1079–1082. [Google Scholar]
- 203.Balci S, Hahn K, Kopold P, Kadri A, Wege C, Kern K and Bittner AM, Nanotechnology, 2012, 23, 045603. [DOI] [PubMed] [Google Scholar]
- 204.Kobayashi M, Onodera K, Watanabe Y and Yamashita I, Chem. Lett, 2010, 39, 616–618. [Google Scholar]
- 205.Fan XZ, Pomerantseva E, Gnerlich M, Brown A, Gerasopoulos K, McCarthy M, Culver J and Ghodssi R, J. Vac. Sci. Technol, 2013, 31, 050815. [Google Scholar]
- 206.Balci S, Bittner AM, Hahn K, Scheu C, Knez M, Kadri A, Wege C, Jeske H and Kern K, Electrochim. Acta, 2006, 51, 6251–6257. [Google Scholar]
- 207.Tsukamoto R, Muraoka M, Seki M, Tabata H and Yamashita I, Chem. Mater, 2007, 19, 2389–2391. [Google Scholar]
- 208.Kobayashi M, Seki M, Tabata H, Watanabe Y and Yamashita I, Nano Lett, 2010, 10, 773–776. [DOI] [PubMed] [Google Scholar]
- 209.Dujardin E, Peet C, Stubbs G, Culver JN and Mann S, Nano Lett, 2003, 3, 413–417. [Google Scholar]
- 210.Abedin MJ, Liepold L, Suci P, Young M and Douglas T, J. Am. Chem. Soc, 2009, 131, 4346–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Liepold LO, Abedin MJ, Buckhouse ED, Frank JA, Young MJ and Douglas T, Nano Lett, 2009, 9, 4520–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lucon J, Abedin MJ, Uchida M, Liepold L, Jolley CC, Young M and Douglas T, Chem. Commun, 2010, 46, 264–266. [DOI] [PubMed] [Google Scholar]
- 213.Uchida M, Morris DS, Kang S, Jolley CC, Lucon J, Liepold LO, Lafrance B, Prevelige PE and Douglas T, Langmuir, 2012, 28, 1998–2006. [DOI] [PubMed] [Google Scholar]
- 214.Wang J-S and Matyjaszewski K, J. Am. Chem. Soc, 1995, 117, 5614–5615. [Google Scholar]
- 215.Kato M, Kamigaito M, Sawamoto M and Higashimura T, Macromolecules, 1995, 28, 1721–1723. [Google Scholar]
- 216.Lucon J, Qazi S, Uchida M, Bedwell GJ, LaFrance B, Prevelige PE Jr and Douglas T, Nat. Chem, 2012, 4, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Edwards E, Roychoudhury R, Schwarz B, Jordan P, Lisher J, Uchida M and Douglas T, J. Mater. Chem. B, 2016, 4, 5375–5384. [DOI] [PubMed] [Google Scholar]
- 218.Lucon J, Edwards E, Qazi S, Uchida M and Douglas T, Eur. Polym. J, 2013, 49, 2976–2985. [Google Scholar]
- 219.Pokorski JK, Breitenkamp K, Liepold LO, Qazi S and Finn MG, J. Am. Chem. Soc, 2011, 133, 9242–9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hovlid ML, Lau JL, Breitenkamp K, Higginson CJ, Laufer B, Manchester M and Finn MG, ACS Nano, 2014, 8, 8003–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Renggli K, Sauter N, Rother M, Nussbaumer MG, Urbani R, Pfohl T and Bruns N, Polym. Chem, 2017, 8, 2133–2136. [Google Scholar]
- 222.Nussbaumer MG, Duskey JT, Rother M, Renggli K, Chami M and Bruns N, Adv. Sci, 2016, 3, 1600046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Nussbaumer MG, Bisig C and Bruns N, Chem. Commun, 2016, 52, 10537–10539. [DOI] [PubMed] [Google Scholar]
- 224.Wang X, Wang C, Pan M, Wei J, Jiang F, Lu R, Liu X, Huang Y and Huang F, ACS Appl. Mater. Interfaces, 2017, 9, 25387–25396. [DOI] [PubMed] [Google Scholar]
- 225.Ditzel L, Lowe J, Stock D, Stetter KO, Huber H, Huber R and Steinbacher S, Cell, 1998, 93, 125–138. [DOI] [PubMed] [Google Scholar]
- 226.Ng BC, Yu M, Gopal A, Rome LH, Monbouquette HG and Tolbert SH, Nano Lett, 2008, 8, 3503–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Dixit SK, Goicochea NL, Daniel M-C, Murali A, Bronstein L, De M, Stein B, Rotello VM, Kao CC and Dragnea B, Nano Lett, 2006, 6, 1993–1999. [DOI] [PubMed] [Google Scholar]
- 228.Aniagyei SE, DuFort C, Kao CC and Dragnea B, J. Mater. Chem, 2008, 18, 3762–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Malyutin AG, Cheng H, Sanchez-Felix OR, Carlson K, Stein BD, Konarev PV, Svergun DI, Dragnea B and Bronstein LM, ACS Appl. Mater. Interfaces, 2015, 7, 12089–12098. [DOI] [PubMed] [Google Scholar]
- 230.Tsvetkova I, Chen C, Rana S, Kao CC, Rotello VM and Dragnea B, Soft Matter, 2012, 8, 4571. [Google Scholar]
- 231.Sun J, DuFort C, Daniel M-C, Murali A, Chen C, Gopinath K, Stein B, De M, Rotello VM, Holzenburg A, Kao CC and Dragnea B, Proc. Natl. Acad. Sci. U. S. A, 2007, 104, 1354–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Huang X, Stein BD, Cheng H, Malyutin A, Tsvetkova IB, Baxter DV, Remmes NB, Verchot J, Kao C, Bronstein LM and Dragnea B, ACS Nano, 2011, 5, 4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Loo L, Guenther RH, Lommel SA and Franzen S, J. Am. Chem. Soc, 2007, 129, 11111–11117. [DOI] [PubMed] [Google Scholar]
- 234.Loo L, Guenther RH, Basnayake VR, Lommel SA and Franzen S, J. Am. Chem. Soc, 2006, 128, 4502–4503. [DOI] [PubMed] [Google Scholar]
- 235.Vieweger M, Goicochea N, Koh ES and Dragnea B, ACS Nano, 2011, 5, 7324–7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Minten IJ, Hendriks LJA, Nolte RJM and Cornelissen JJLM, J. Am. Chem. Soc, 2009, 131, 17771–17773. [DOI] [PubMed] [Google Scholar]
- 237.Seebeck FP, Woycechowsky KJ, Zhuang W, Rabe JP and Hilvert D, J. Am. Chem. Soc, 2006, 128, 4516–4517. [DOI] [PubMed] [Google Scholar]
- 238.Wörsdörfer B, Woycechowsky KJ and Hilvert D, Science, 2011, 331, 589–592. [DOI] [PubMed] [Google Scholar]
- 239.Peabody DS, EMBO J, 1993, 12, 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Lim F, Spingola M and Peabody DS,J. Biol. Chem, 1996, 271, 31839–31845. [DOI] [PubMed] [Google Scholar]
- 241.Parrott AM, Lago H, Adams CJ, Ashcroft AE, Stonehouse NJ and Stockley PG, Nucleic Acids Res, 2000, 28, 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Minten IJ, Claessen VI, Blank K, Rowan AE, Nolte RJM and Cornelissen JJLM, Chem. Sci, 2011, 2, 358–362. [Google Scholar]
- 243.Beck T, Tetter S, Kunzle M and Hilvert D, Angew. Chem., Int. Ed, 2015, 54, 937–940. [DOI] [PubMed] [Google Scholar]
- 244.Sasaki E and Hilvert D, J. Phys. Chem. B, 2016, 120, 6089–6095. [DOI] [PubMed] [Google Scholar]
- 245.Azuma Y, Zschoche R, Tinzl M and Hilvert D, Angew. Chem., Int. Ed, 2016, 55, 1531–1534. [DOI] [PubMed] [Google Scholar]
- 246.Frey R, Mantri S, Rocca M and Hilvert D, J. Am. Chem. Soc, 2016, 138, 10072–10075. [DOI] [PubMed] [Google Scholar]
- 247.Sasaki E, Bohringer D, van de Waterbeemd M, Leibundgut M, Zschoche R, Heck AJ, Ban N and Hilvert D, Nat. Commun, 2017, 8, 14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Rhee JK, Hovlid M, Fiedler JD, Brown SD, Manzenrieder F, Kitagishi H, Nycholat C, Paulson JC and Finn MG, Biomacromolecules, 2011, 12, 3977–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Wu M, Brown WL and Stockley PG, Bioconjugate Chem, 1995, 6, 587–595. [DOI] [PubMed] [Google Scholar]
- 250.Ashley CE, Carnes EC, Phillips GK, Durfee PN, Buley MD, Lino CA, Padilla DP, Phillips B, Carter MB, Willman CL, Brinker CJ, Caldeira J. d. C., Chackerian B, Wharton W and Peabody DS, ACS Nano, 2011, 5, 5729–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Parker MH, Casjens S and Prevelige PE Jr., J. Mol. Biol, 1998, 281, 69–79. [DOI] [PubMed] [Google Scholar]
- 252.Weigele PR, Sampson L, Winn-Stapley D and Casjens SR, J. Mol. Biol, 2005, 348, 831–844. [DOI] [PubMed] [Google Scholar]
- 253.O’Neil A, Reichhardt C, Johnson B, Prevelige PE and Douglas T, Angew. Chem., Int. Ed, 2011, 50, 7425–7428. [DOI] [PubMed] [Google Scholar]
- 254.Patterson DP, McCoy K, Fijen C and Douglas T, J. Mater. Chem. B, 2014, 2, 5948–5951. [DOI] [PubMed] [Google Scholar]
- 255.Patterson DP, LaFrance B and Douglas T, Chem. Commun, 2013, 49, 10412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Patterson DP, Prevelige PE and Douglas T, ACS Nano, 2012, 6, 5000–5009. [DOI] [PubMed] [Google Scholar]
- 257.O’Neil A, Prevelige PE and Douglas T, Biomater. Sci, 2013, 1, 881. [DOI] [PubMed] [Google Scholar]
- 258.Inoue T, Kawano M.-a., Takahashi R.-u., Tsukamoto H, Enomoto, Imai T, Kataoka Kand Handa H, J. Biotechnol, 2008, 134, 181–192. [DOI] [PubMed] [Google Scholar]
- 259.Teschke CM, McGough A and Thuman-Commike PA, Biophys. J, 2003, 84, 2585–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Llauro A, Luque D, Edwards E, Trus BL, Avera J, Reguera D, Douglas T, Pablo PJ and Caston JR, Nanoscale, 2016, 8, 9328–9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Patterson DP, Schwarz B, El-Boubbou K, van der Oost J, Prevelige PE and Douglas T, Soft Matter, 2012, 8, 10158. [Google Scholar]
- 262.Sharma J, Uchida M, Miettinen HM and Douglas T, Nanoscale, 2017, 9, 10420–10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Mammen M, Choi SK and Whitesides GM, Angew. Chem, 1998, 37, 2754–2794. [DOI] [PubMed] [Google Scholar]
- 264.Yang SH, Chung WJ, McFarland S and Lee SW, Chem. Rec, 2013, 13, 43–59. [DOI] [PubMed] [Google Scholar]
- 265.Gray BP and Brown KC, Chem. Rev, 2014, 114, 1020–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Smith GP and Petrenko VA, Chem. Rev, 1997, 97, 391–410. [DOI] [PubMed] [Google Scholar]
- 267.Uchida M, Willits AD, Muller K, Willis AF, Jackiw L, Jutila M, Young M, Porter AE and Douglas T, Adv. Mater, 2009, 21, 458–462. [Google Scholar]
- 268.Uchida M, Kosuge H, Terashima M, Willits DA, Liepold LO, Young MJ, Mcconnell MV and Douglas T, ACS Nano, 2011, 5, 2493–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Flenniken ML, Willits DA, Harmsen AL, Liepold LO, Harmsen AG, Young MJ and Douglas T, Chem. Biol, 2006, 13, 161–170. [DOI] [PubMed] [Google Scholar]
- 270.Servid A, Jordan P, O’Neil A, Prevelige P and Douglas T, Biomacromolecules, 2013, 14, 2989–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Phelps JP, Dang N and Rasochova L, J. Virol. Methods, 2007, 141, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Tissot AC, Renhofa R, Schmitz N, Cielens I, Meijerink E, Ose V, Jennings GT, Saudan P, Pumpens P and Bachmann MF, PLoS One, 2010, 5, e9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Wu CH, Liu IJ, Lu RM and Wu HC, J. Biomed. Sci, 2016, 23, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Whaley SR, English DS, Hu EL, Barbara PFand Belcher AM, Nature, 2000, 405, 665–668. [DOI] [PubMed] [Google Scholar]
- 275.Naik RR, Stringer SJ, Agarwal G, Jones SE and Stone MO, Nat. Mater, 2002, 1, 169–172. [DOI] [PubMed] [Google Scholar]
- 276.Chen HB, Su XD, Neoh KG and Choe WS, Anal. Chem, 2006, 78, 4872–4879. [DOI] [PubMed] [Google Scholar]
- 277.Sarikaya M, Tamerler C, Jen A, Schulten K and Baneyx F, Nat. Mater, 2003, 2, 577–585. [DOI] [PubMed] [Google Scholar]
- 278.Mao C, Flynn CE, Hayhurst A, Sweeney R, Qi J, Georgiou G, Iverson B and Belcher AM, Proc. Natl. Acad. Sci. U. S. A, 2003, 100, 6946–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Hoogenboom HR, Griffiths AD, Johnson KS, Chiswell DJ, Hudson P and Winter G, Nucleic Acids Res, 1991, 19, 4133–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Kratz PA, Böttcher B and Nassal M, Proc. Natl. Acad. Sci. U. S. A, 1999, 96, 1915–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Schneemann A, Speir JA, Tan GS, Khayat R, Ekiert DC, Matsuoka Y and Wilson IA, J. Virol, 2012, 86, 11686–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Gleiter S and Lilie H, Protein Sci, 2001, 10, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Kanekiyo M, Wei C-J, Yassine HM, McTamney PM, Boyington JC, Whittle JRR, Rao SS, Kong W-P, Wang L and Nabel GJ, Nature, 2013, 499, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Pokorski JK, Hovlid ML and Finn MG, ChemBioChem, 2011, 12, 2441–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Moon H, Lee J, Min J and Kang S, Biomacromolecules, 2014, 15, 3794–3801. [DOI] [PubMed] [Google Scholar]
- 286.Culpepper BK, Morris DS, Prevelige PE and Bellis SL, Biomaterials, 2013, 34, 2455–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Tsukiji S and Nagamune T, ChemBioChem, 2009, 10, 787–798. [DOI] [PubMed] [Google Scholar]
- 288.Patterson D, Schwarz B, Avera J, Western B, Hicks M, Krugler P, Terra M, Uchida M, McCoy K and Douglas T, Bioconjugate Chem, 2017, 28, 2114–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Schoonen L, Pille J, Borrmann A, Nolte RJ and van Hest JC, Bioconjugate Chem, 2015, 26, 2429–2434. [DOI] [PubMed] [Google Scholar]
- 290.Reddington SC and Howarth M, Curr. Opin. Chem. Biol, 2015, 29, 94–99. [DOI] [PubMed] [Google Scholar]
- 291.Moon H, Bae Y, Kim H and Kang S, Chem. Commun, 2016, 52, 14051–14054. [DOI] [PubMed] [Google Scholar]
- 292.Stephanopoulos N and Francis MB, Nat. Chem. Biol, 2011, 7, 876–884. [DOI] [PubMed] [Google Scholar]
- 293.Flenniken ML, Willits DA, Brumfield S, Young MJ and Douglas T, Nano Lett, 2003, 3, 1573–1576. [Google Scholar]
- 294.Miller RA, Presley AD and Francis MB, J. Am. Chem. Soc, 2007, 129, 3104–3109. [DOI] [PubMed] [Google Scholar]
- 295.Klem MT, Willits D, Young M and Douglas T, J. Am. Chem. Soc, 2003, 125, 10806–10807. [DOI] [PubMed] [Google Scholar]
- 296.ElSohly AM and Francis MB, Acc. Chem. Res, 2015, 48, 1971–1978. [DOI] [PubMed] [Google Scholar]
- 297.Rosen CB and Francis MB, Nat. Chem. Biol, 2017, 13, 697–705. [DOI] [PubMed] [Google Scholar]
- 298.Sereda TJ, Mant CT, Quinn AM and Hodges RS, J. Chromatogr, 1993, 646, 17–30. [DOI] [PubMed] [Google Scholar]
- 299.Dedeo MT, Duderstadt KE, Berger JM and Francis MB, Nano Lett, 2010, 10, 181–186. [DOI] [PubMed] [Google Scholar]
- 300.Stephanopoulos N, Tong GJ, Hsiao SC and Francis MB, ACS Nano, 2010, 4, 6014–6020. [DOI] [PubMed] [Google Scholar]
- 301.Kulmashiro Y, Ikezoe Y, Tamada K and Hara M, J. Phys. Chem. B, 2008, 112, 8291–8297. [DOI] [PubMed] [Google Scholar]
- 302.Raja KS, Wang Q, Gonzalez MJ, Manchester M, Johnson JE and Finn MG, Biomacromolecules, 2003, 4, 472–476. [DOI] [PubMed] [Google Scholar]
- 303.Steinmetz NF and Manchester M, Biomacromolecules, 2009, 10, 784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Lander GC, Evilevitch A, Jeembaeva M, Potter CS, Carragher B and Johnson JE, Structure, 2008, 16, 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Qin L, Fokine A, O’Donnell E, Rao VB and Rossmann MG, J. Mol. Biol, 2010, 395, 728–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Xiang Y and Rossmann MG, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 4806–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Gupta A, Onda M, Pastan I, Adhya S and Chaudhary VK, J. Mol. Biol, 2003, 334, 241–254. [DOI] [PubMed] [Google Scholar]
- 308.Yang F, Forrer P, Dauter Z, Conway JF, Cheng N, Cerritelli ME, Steven AC, Plückthun A and Wlodawer A, Nat. Struct. Mol. Biol, 2000, 7, 230–237. [DOI] [PubMed] [Google Scholar]
- 309.Ren Z.-j. and Black LW, Gene, 1998, 215, 439–444. [DOI] [PubMed] [Google Scholar]
- 310.Tang L, Gilcrease EB, Casjens SR and Johnson JE, Structure, 2006, 14, 837–845. [DOI] [PubMed] [Google Scholar]
- 311.Parent KN, Deedas CT, Egelman EH, Casjens SR, Baker TS and Teschke CM, Biomaterials, 2012, 33, 5628–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK and Discher DE, Science, 2013, 339, 971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Schwarz B, Madden P, Avera J, Gordon B, Larson K, Miettinen HM, Uchida M, LaFrance B, Basu G and Rynda-Apple A, ACS Nano, 2015, 9, 9134–9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Chackerian B, Lenz P, Lowy DR and Schiller JT, J. Immunol, 2002, 169, 6120–6126. [DOI] [PubMed] [Google Scholar]
- 315.Chackerian B, Lowy DR and Schiller JT, J. Clin. Invest, 2001, 108, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Liu Z, Qiao J, Niu Z and Wang Q, Chem. Soc. Rev, 2012, 41, 6178–6194. [DOI] [PubMed] [Google Scholar]
- 317.Whitesides GM and Grzybowski B, Science, 2002, 295, 2418–2421. [DOI] [PubMed] [Google Scholar]
- 318.Wegst UG, Bai H, Saiz E, Tomsia AP and Ritchie RO, Nat. Mater, 2015, 14, 23–36. [DOI] [PubMed] [Google Scholar]
- 319.Tskhovrebova L and Trinick J, Nat. Rev. Mol. Cell Biol, 2003, 4, 679–689. [DOI] [PubMed] [Google Scholar]
- 320.Biswas S, Kinbara K, Niwa T, Taguchi H, Ishii N, Watanabe S, Miyata K, Kataoka K and Aida T, Nat. Chem, 2013, 5, 613–620. [DOI] [PubMed] [Google Scholar]
- 321.Sendai T, Biswas S and Aida T, J. Am. Chem. Soc, 2013, 135, 11509–11512. [DOI] [PubMed] [Google Scholar]
- 322.Biswas S, Kinbara K, Oya N, Ishii N, Taguchi H and Aida T, J. Am. Chem. Soc, 2009, 131, 7556–7557. [DOI] [PubMed] [Google Scholar]
- 323.Sim S, Miyajima D, Niwa T, Taguchi H and Aida T, J. Am. Chem. Soc, 2015, 137, 4658–4661. [DOI] [PubMed] [Google Scholar]
- 324.Fromherz P, Nature, 1971, 231, 267–268. [DOI] [PubMed] [Google Scholar]
- 325.Uzgiris EE and Kornberg RD, Nature, 1983, 301, 125–129. [DOI] [PubMed] [Google Scholar]
- 326.Yoshimura H, Scheybani T, Baumeister W and Nagayama K, Langmuir, 1994, 10, 3290–3295. [Google Scholar]
- 327.Kewalramani S, Wang S, Lin Y, Nguyen HG, Wang Q, Fukuto M and Yang L, Soft Matter, 2011, 7, 939–945. [Google Scholar]
- 328.Fukuto M, Nguyen QL, Vasilyev O, Mank N, Washington-Hughes CL, Kuzmenko I, Checco A, Mao Y, Wang Q and Yang L, Soft Matter, 2013, 9, 9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Yoshimura H, Edwards E, Uchida M, McCoy K, Roychoudhury R, Schwarz B, Patterson D and Douglas T, J. Phys. Chem. B, 2016, 120, 5938–5944. [DOI] [PubMed] [Google Scholar]
- 330.Kaur G, He J, Xu J, Pingali S, Jutz G, Boker A, Niu Z, Li T, Rawlinson D, Emrick T, Lee B, Thiyagarajan P, Russell TP and Wang Q, Langmuir, 2009, 25, 5168–5176. [DOI] [PubMed] [Google Scholar]
- 331.Russell JT, Lin Y, Boker A, Su L, Carl P, Zettl H, He J, Sill K, Tangirala R, Emrick T, Littrell K, Thiyagarajan P, Cookson D, Fery A, Wang Q and Russell TP, Angew. Chem., Int. Ed, 2005, 44, 2420–2426. [DOI] [PubMed] [Google Scholar]
- 332.Yamashita I, Thin Solid Films, 2001, 393, 12–18. [Google Scholar]
- 333.Okuda M, Kobayashi Y, Suzuki K, Sonoda K, Kondoh T, Wagawa A, Kondo A and Yoshimura H, Nano Lett, 2005, 5, 991–993. [DOI] [PubMed] [Google Scholar]
- 334.Cheung CL, Camarero JA, Woods BW, Lin T, Johnson JE and De Yoreo JJ, J. Am. Chem. Soc, 2003, 125, 6848–6849. [DOI] [PubMed] [Google Scholar]
- 335.Cheung CL, Chung S-W, Chatterji A, Lin T, Johnson JE, Hok S, Perkins J and De Yoreo JJ, J. Am. Chem. Soc, 2006, 128, 10801–10807. [DOI] [PubMed] [Google Scholar]
- 336.Suci PA, Klem MT, Douglas T and Young M, Langmuir, 2005, 21, 8686–8693. [DOI] [PubMed] [Google Scholar]
- 337.Okochi M, Sugita T, Furusawa S, Umetsu M, Adschiri T and Honda H, Biotechnol. Bioeng, 2010, 106, 845–851. [DOI] [PubMed] [Google Scholar]
- 338.Sano K, Yoshii S, Yamashita I and Shiba K, Nano Lett, 2007, 7, 3200–3202. [DOI] [PubMed] [Google Scholar]
- 339.Hayashi T, Sano K, Shiba K, Kumashiro Y, Iwahori K, Yamashita I and Hara M, Nano Lett, 2006, 6, 515–519. [DOI] [PubMed] [Google Scholar]
- 340.Hashimoto T, Gamo K, Fukuta M, Zheng B, Zettsu N, Yamashita I, Uraoka Y and Watanabe H, Appl. Phys. Lett, 2011, 99, 263701. [Google Scholar]
- 341.Murray CB, Kagan CR and Bawendi MG, Annu. Rev. Mater. Sci, 2000, 30, 545–610. [Google Scholar]
- 342.Rogers WB, Shih WM and Manoharan VN, Nat. Rev. Mater, 2016, 1, 16008. [Google Scholar]
- 343.Macfarlane RJ, Lee B, Jones MR, Harris N, Schatz GC and Mirkin CA, Science, 2011, 334, 204–208. [DOI] [PubMed] [Google Scholar]
- 344.Zhang Y, Lu F, Yager KG, van der Lelie D and Gang O, Nat. Nanotechnol, 2013, 8, 865–872. [DOI] [PubMed] [Google Scholar]
- 345.Mirkin CA, Letsinger RL, Mucic RCand Storhoff JJ, Nature, 1996, 382, 607–609. [DOI] [PubMed] [Google Scholar]
- 346.Strable E, Johnson JE and Finn MG, Nano Lett, 2004, 4, 1385–1389. [Google Scholar]
- 347.Broomell CC, Birkedal H, Oliveira CLP, Pedersen JS, Gertenbach J-A, Young M and Douglas T, Soft Matter, 2010, 6, 3167. [Google Scholar]
- 348.Park SY, Lytton-Jean AK, Lee B, Weigand S, Schatz GC and Mirkin CA, Nature, 2008, 451, 553–556. [DOI] [PubMed] [Google Scholar]
- 349.Nykypanchuk D, Maye MM, van der Lelie D and Gang O, Nature, 2008, 451, 549–552. [DOI] [PubMed] [Google Scholar]
- 350.Liu W, Tagawa M, Xin HL, Wang T, Emamy H, Li H, Yager KG, Starr FW, Tkachenko AV and Gang O, Science, 2016, 351, 582–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Kostiainen MA, Kasyutich O, Cornelissen JJLM and Nolte RJM, Nat. Chem, 2010, 2, 394–399. [DOI] [PubMed] [Google Scholar]
- 352.Kostiainen MA, Ceci P, Fornara M, Hiekkataipale P, Kasyutich O, Nolte RJM, Cornelissen JJLM, Desautels RD and van Lierop J, ACS Nano, 2011, 5, 6394–6402. [DOI] [PubMed] [Google Scholar]
- 353.Liljestrom V, Seitsonen J and Kostiainen MA, ACS Nano, 2015, 9, 11278–11285. [DOI] [PubMed] [Google Scholar]
- 354.Kunzle M, Eckert T and Beck T, J. Am. Chem. Soc, 2016, 138, 12731–12734. [DOI] [PubMed] [Google Scholar]
- 355.Williams T, Neotrop. Entomol, 2008, 37, 615–632. [DOI] [PubMed] [Google Scholar]
- 356.Folwer M and Robertson JS, J. Invertebr. Pathol, 1972, 19, 154–155. [Google Scholar]
- 357.Juhl SB, Chan EP, Ha YH, Maldovan M, Brunton J, Ward V, Dokland T, Kalmakoff J, Farmer B, Thomas EL and Vaia RA, Adv. Funct. Mater, 2006, 16, 1086–1094. [Google Scholar]
- 358.Servid A, Jordan P, O’Neil A, Prevelige P and Douglas T, Biomacromolecules, 2013, 14, 2989–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Apostolovic B, Danial M and Klok HA, Chem. Soc. Rev, 2010, 39, 3541–3575. [DOI] [PubMed] [Google Scholar]
- 360.Fletcher JM, Harniman RL, Barnes FR, Boyle AL, Collins A, Mantell J, Sharp TH, Antognozzi M, Booth PJ, Linden N, Miles MJ, Sessions RB, Verkade P and Woolfson DN, Science, 2013, 340, 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Woolfson DN, Bartlett GJ, Bruning M and Thomson AR, Curr. Opin. Struct. Biol, 2012, 22, 432–441. [DOI] [PubMed] [Google Scholar]
- 362.Gradisar H, Bozic S, Doles T, Vengust D, Hafner-Bratkovic I, Mertelj A, Webb B, Sali A, Klavzar S and Jerala R, Nat. Chem. Biol, 2013, 9, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Kang S, Suci PA, Broomell CC, Iwahori K, Kobayashi M, Yamashita I, Young M and Douglas T, Nano Lett, 2009, 9, 2360–2366. [DOI] [PubMed] [Google Scholar]
- 364.Kang YJ, Uchida M, Shin H-H, Douglas T and Kang S, Soft Matter, 2011, 7, 11078. [Google Scholar]
- 365.Steinmetz NF, Bock E, Richter RP, Spatz JP, Lomonossoff GP and Evans DJ, Biomacromolecules, 2008, 9, 456–462. [DOI] [PubMed] [Google Scholar]
- 366.Steinmetz NF, Calder G, Lomonossoff GP and Evans DJ, Langmuir, 2006, 22, 10032–10037. [DOI] [PubMed] [Google Scholar]
- 367.Tiu BD, Kernan DL, Tiu SB, Wen AM, Zheng Y, Pokorski JK, Advincula RC and Steinmetz NF, Nanoscale, 2017, 9, 1580–1590. [DOI] [PubMed] [Google Scholar]
- 368.Suci PA, Klem MT, Arce FT, Douglas T and Young M, Langmuir, 2006, 22, 8891–8896. [DOI] [PubMed] [Google Scholar]
- 369.Suci P, Klem MT, Young M and Douglas T, Soft Matter, 2008, 4, 2519–2523. [Google Scholar]
- 370.Yoo PJ, Nam KT, Belchert AM and Hammond PT, Nano Lett, 2008, 8, 1081–1089. [DOI] [PubMed] [Google Scholar]
- 371.Yoo PJ, Nam KT, Qi JF, Lee SK, Park J, Belcher AM and Hammond PT, Nat. Mater, 2006, 5, 234–240. [DOI] [PubMed] [Google Scholar]
- 372.Li T, Winans RE and Lee B, Langmuir, 2011, 27, 10929–10937. [DOI] [PubMed] [Google Scholar]
- 373.Liljestrom V, Ora A, Hassinen J, Rekola HT, Nonappa, Heilala M, Hynninen V, Joensuu JJ, Ras RHA, Torma P, Ikkala Oand Kostiainen MA, Nat. Commun, 2017, 8, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Li T, Zan X, Winans RE, Wang Q and Lee B, Angew. Chem., Int. Ed, 2013, 52, 6638–6642. [DOI] [PubMed] [Google Scholar]
- 375.Yoo PJ, Nam KT, Qi JF, Lee SK, Park J, Belcher AM and Hammond PT, Nat. Mater, 2006, 5, 234–240. [DOI] [PubMed] [Google Scholar]
- 376.Chung WJ, Oh JW, Kwak K, Lee BY, Meyer J, Wang E, Hexemer A and Lee SW, Nature, 2011, 478, 364–368. [DOI] [PubMed] [Google Scholar]
- 377.Sontz PA, Bailey JB, Ahn S and Tezcan FA, J. Am. Chem. Soc, 2015, 137, 11598–11601. [DOI] [PubMed] [Google Scholar]