

Abstract
Glutamate is the major excitatory neurotransmitter in the mammalian CNS. Here, we propose a new role for this neurotransmitter in the developing nervous system. We show that glutamate or the metabotropic class I agonist S-3,5-dihydroxyphenyl glycine, acting through the metabotropic glutamate receptor 1 (mGluR1), can reduce the activity of multiple axonal repellents in vitro. This effect is mediated by a pertussis toxin-sensitive activation of protein kinase A and the subsequent inactivation of Rho. This signaling pathway appears to be identical to the one we described previously for stromal derived factor-1-induced reduction of axonal repellent activities. Activation of mGluR1 can also promote increased survival of embryonic retinal ganglion cells in culture. We propose that neurotransmitter-induced modulation of repellent strength provides a novel mechanism by which activity can influence neuronal morphology.
Keywords: axon guidance, growth cone, metabotropic glutamate receptor, slit-2, semaphorin 3A, semaphorin 3C, cAMP, survival, neurotrophic
Introduction
Neurons interconnect in a mostly activity-independent manner early in development. Once basic neural circuitry is established and begins to be functionally active, however, interconnections are shaped through activity-dependent mechanisms. Considerable progress has been made in identifying the signaling molecules and their receptors that influence axonal and dendritic outgrowth during the activity-independent phase of neural development. For example, members of the semaphorin, netrin, ephrin, and slit families have been shown to act as either attractants or repellents that guide neuronal processes along specific pathways toward their targets (Tessier-Lavigne and Goodman, 1996; Yu and Bargmann, 2001). Less well understood, however, are the signals that control neurite growth and pruning during the activity-dependent phase of development. We hypothesize that the release of neurotransmitters from active neurons has a direct and profound influence on how axons and dendrites respond to these basic guidance signals.
Growth cone responses to guidance cues can be dramatically altered by manipulating cyclic nucleotide levels in extending neurites. For example, Xenopus spinal neurons are normally repelled by a gradient of MAG, however exposure to a cAMP agonist is reported to reverse their response to one of attraction (Song et al., 1998). Antagonizing cAMP converts an attractive response for netrin into one of repulsion (Song et al., 1997). Stimulation of adenosine A2b receptors can mimic the effect cAMP agonists and make retinal axons more responsive to netrin, whereas exposure to laminin-1 reduces cAMP and converts netrin from a retinal attractant into a repellent (Hopker et al., 1999; Shewan et al., 2002). Recently, our laboratory has found that SDF-1, a chemokine acting through the G-protein-coupled receptor CXCR4, can reduce growth cone responses to the repellents semaphorin 3A, semaphorin 3C, and Slit-2 (Chalasani et al., 2003a). Our pharmacological studies indicate that this modulatory effect is mediated by a pertussis toxin (PTX)-sensitive elevation of cAMP and activation of protein kinase A (PKA).
In an effort to identify other signaling molecules that influence axon responsiveness to repellent guidance cues, we focused on those known to elevate cAMP through a pertussis-sensitive mechanism. Prominent among these are the class I metabotropic receptors for the neurotransmitter glutamate. There are eight metabotropic glutamate receptors (mGluRs) that are divided into three classes based on their pharmacology and sequence homology (Pin and Duvoisin, 1995; Schoepp et al., 1999). Activation of class I mGluRs, composed of mGluR1 and mGluR5, has been shown to elevate cAMP levels via a PTX-sensitive signaling mechanism (Hermans and Challiss, 2001). The similarities between SDF-1/CXCR4 signaling and class I mGluR signaling pathways led us to hypothesize that glutamate could reduce the responsiveness of axonal growth cones to repellents.
We demonstrate here that glutamate, signaling through mGluR1, can reduce the effectiveness of Slit-2, semaphorin 3A, and semaphorin 3C on several different embryonic neural tissues. Our pharmacological studies indicate that this modulatory effect is cAMP dependent, induces the activation of PKA, and leads to the inactivation of Rho. We also found that mGluR1 activation promotes embryonic retinal ganglion cell (RGC) survival in minimal defined medium. These findings suggest a mechanism by which the release of neurotransmitters influence neuronal responses to traditional repellent cues.
Materials and Methods
Cultures and growth cone collapse assay. Retinal explants from chick embryonic day 6 (E6) animals, dorsal root ganglion (DRG) explants from E7 animals, and sympathetic explants from E8 animals were set up on laminin-coated coverslips in supplemented F-12 medium as described (Niclou et al., 2000). In the case of sympathetic explants, 5% FBS was added to the medium. Mouse embryos were dissected from timed pregnant Bl/6 females (day of plug was counted as 0.5). Dorsal root ganglia were dissected and cultured on laminin-coated coverslips in supplemented F-12 medium. Approximately 18-20 hr later, these explants sent out axons with growth cones at their tips. Repellents were added at the volumes indicated, and 30 min later, the cultures were fixed and the percentage of collapsed growth cones was identified. Glutamate (50 μm; Sigma, St. Louis, MO) or S-3,5 dihydroxy phenyl glycine (DHPG; 50 μm; Sigma) was added along with the repellents. Other agonists included 2 R,4 R-4-aminopyrrolidine-2,4-dicarboxylic acid (APDC; 50 μm; Tocris, Ellisville, MO) and l-AP-4 (5 μm; Tocris), which were also added along with the repellents. Antagonists such as (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385; 40 μm; Tocris), 2-methyl-6-(2-phenylethenyl) pyridine (SIB1893; 15 μm; Tocris), 7-(hydroxyimino) cyclopropa[b] chromen-1a-carboxylate ethyl ester (CPCCOEt; 20 μm; Tocris), and 2-methyl-6-(phenylethynyl) pyridine (MPEP; 20 μm; Tocris) were also added along with glutamate or DHPG. Various inhibitors were added to cultures at the same time as DHPG and the repellents. These included PKA inhibitor PKI-myr (200 nm; Calbiochem, La Jolla, CA), cAMP antagonist RpcAMPS (20 μm; Sigma), cGMP antagonist RpcGMPS (20 μm; Sigma), MAP kinase (MAPK) inhibitor PD98059 (20 μm; Calbiochem), phosphatidylinositol 3′-kinase (PI-3 kinase) inhibitor LY294002 (20 μm; Calbiochem), calmidazolium hydrochloride (CMZ; 20 μm; Calbiochem), PTX (100 ng/ml; Sigma), Y-27632 (10 μm; Tocris), and ML-7 (100 nm; Biomol, Plymouth Meeting, PA). GST-Tat-C3 (a gift from Dr. Michael Olson, University of Pennsylvania, Philadelphia, PA) was expressed in Escherichia coli using an isopropyl β-d-1-thiogalactopyranoside induction step and purified as described (Coleman et al., 2001). All the inhibitors and antagonists were used at concentrations that did not cause any collapse or detectable toxicity.
In situ hybridization and immunohistochemistry. An expressed-sequence tag clone (ID 5909116) was obtained from the research genetics covering a region of the mouse mGluR1 gene 3736-4140 bp. This clone was obtained in the pT7T3D-Pac (Pfizer, New York, NY). Antisense probes were generated from an EcoRI digest of the clone using a T3 primer. These probes were used to probe mouse embryonic sections at day 13.5 and were visualized with an anti-digoxygenin-alkaline phosphatase (Roche Applied Science, Indianapolis, IN). The same sections were reacted with anti-islet-1/2 (guinea pig polyclonal; a kind gift from Dr. Thomas Jessell, Columbia University, New York, NY) at 1:1000 and then visualized using anti-guinea pig Alexa Flour 546 (Molecular Probes, Eugene, OR).
cAMP response element-binding protein phosphorylation. Sympathetic chain ganglion from E8 chicks were dissociated and plated on laminin-coated glass coverslips in minimal F-12 medium with glucose, glutamine, penicillin, streptomycin, transferrin, and selenium. After 6 hr, these cultures were stimulated for 30 min with DHPG with and without 40 μm LY367385, 15 μm SIB1893, 100 ng/ml PTX, 20 μm CMZ, or 200 nm PKI. The cultures were then fixed for 20 min with 3.7% paraformaldehyde and stained with anti-islet-1 (1:200, 39.4D5; Hybridoma Bank, Iowa City, IA) and anti-phospho-specific cAMP response element-binding protein (CREB; Upstate Biochemicals, Charlottesville, VA). These antibodies were then detected by anti-mouse Alexa Flour 488 and anti-rabbit Alexa Flour 546 (Molecular Probes).
Survival assays. Dissociated RGCs were obtained from E6 chick retina and plated on poly-l-lysine-treated laminin-coated glass coverslips at a density of 1000 neurons per well (48-well plate; Costar, Acton, MA). The cells were grown in minimal F-12 medium supplemented with 6 mg/ml glucose, 2 mm glutamine, 100 U/ml penicillin, 100 U/ml streptomycin, 5 ng/ml transferrin, and 4 ng/ml selenium for 48 hr with or without DHPG (50 μm; Tocris co-op). Inhibitors of PKA (PKI, 200 nm; Calbiochem) and MAPK (PD98059, 20 μm; Calbiochem) were added for the entire culture period. These cultures were fixed for 20 min with a 3.7% paraformaldehyde, 20% sucrose in PBS solution and then stained with an anti-islet-1 antibody (39.4D5; Hybridoma Bank). Three coverslips were set up for each condition, and 10 random fields were counted for each coverslip. The average number of islet-positive cells was compared. Error bars on all graphs are SEM. p values were also obtained (two tailed, unequal variance heteroscedastic) using Microsoft Excel.
Results
The responsiveness of growth cones to multiple repellents is reduced by glutamate or the class I mGluR agonist DHPG
The growing tip of the axon, the growth cone, responds to the presence of repellents in the medium by withdrawing its filopodia, contracting its lamellipodia, and thereby displaying a “collapsed” morphology (Kapfhammer and Raper, 1987). The repellent semaphorin 3A was purified based on its ability to collapse DRG growth cones (Luo et al., 1993). Semaphorin 3C induces the collapse of sympathetic growth cones (Koppel et al., 1997), and slit-2 induces the collapse of RGC growth cones (Niclou et al., 2000). A dose-response curve can be constructed relating the percentage of collapsed RGC growth cones to the concentration of slit-2 (Fig. 1A, black diamonds). This curve is shifted to the right when 50 μm glutamate is added along with slit-2 (Fig. 1A, light gray squares). This rightward shift is indicative of a decreased responsiveness to the repellent activity of slit-2. The concentration of glutamate that reduces RGC responsiveness to slit-2 has previously been shown to be sufficient to induce the hydrolysis of phosphoinositides through the activation of metabotropic receptors (Pin and Duvoisin, 1995; Toms et al., 1995; Schoepp et al., 1999). Approximately 12-fold more slit-2 is required to obtain 50% growth cone collapse when glutamate is present in the medium (Fig. 1B). Agonists of various mGluR classes were tested to determine whether they could mimic this glutamatergic effect. A similar shift in the dose-response curve is seen when 50 μm DHPG, a class I mGluR agonist (Conn and Pin, 1997; Schoepp et al., 1999), is added to the medium (Fig. 1A, gray triangles). In contrast, neither the class II mGluR agonist APDC nor the class III mGluR agonist AP-4 has any effect on slit-2-mediated repellent responses (Fig. 1, open circles and open squares). The fold increase in the amount of slit-2 required for 50% collapse in various conditions is shown in Figure 1B. The relative potency of slit-2 by itself is set at 1 by definition. Approximately 12-fold more slit-2 is required to cause 50% collapse in cultures with either glutamate or the class I agonist DHPG added to the medium. However, the response of retinal growth cones to slit-2 is unaffected by the class II agonist APDC or class III agonist AP-4.
Figure 1.
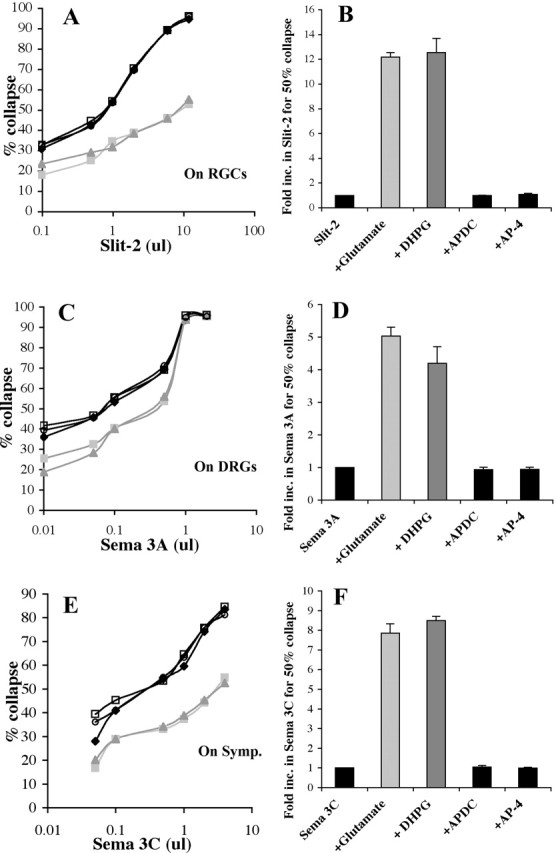
Glutamate or DHPG reduces the activity of Slit-2, Sema 3A, and Sema 3C. A, Dose-response curves showing the response of retinal growth cones to increasing concentrations of slit-2 (black diamonds). Glutamate (light gray squares) or DHPG (gray triangles) shifts the dose-response curve to right, indicating more slit is required to give the same response in their presence. Class II agonist APDC (open circles) and class III agonist AP-4 (open squares) do not change the dose-response curve of slit-2. B, Fold increase in the amount of slit-2 required to give 50% collapse is shown under various conditions. C, Increasing amounts of sema 3A (black diamonds) cause a greater percentage of DRG growth cones to collapse. More sema 3A is required, shifting the curves to the right, when glutamate (light gray squares) or DHPG (gray triangles) is present in the culture. D, The increase in the amount (fold) of sema 3A required to give 50% collapse in the presence of various metabotropic glutamate agonists is shown. E, Sema 3C (black diamonds) collapses an increasing percentage of sympathetic growth cones when more protein is added. This curve is shifted to the right when glutamate (light gray squares) or DHPG (gray triangles) is also present. On Symp., On sympathetic chain ganglia (symp). F, The fold increase in sema 3C to obtain 50% collapse is shown in the presence of various agonists.
To determine whether this effect of glutamate is specific to slit-2, the effectiveness of the repellent semaphorin 3A on DRG growth cones was tested in the presence or absence of glutamate and mGluR agonists. Sema 3A induces a dose-dependent increase in the percentage of collapsed DRG growth cones (Fig. 1C, black diamonds). Glutamate (Fig. 1C, light gray squares) and the class I mGluR agonist DHPG (Fig. 1C, gray triangles) shift the dose-response curve to the right, demonstrating that the activity of sema 3A is reduced. Class II and III mGluR agonists have no effect on the activity of sema 3A. Approximately fivefold more sema 3A is required to induce 50% collapse when glutamate or DHPG is present in the medium (Fig. 1D). The class II and III agonists APDC and AP-4 do not cause any significant change in the responsiveness of DRG growth cones to sema 3A. The same is true for semaphorin 3C effects on sympathetic axons. Both glutamate and the class I agonist DHPG reduce the responsiveness of sympathetic growth cones to sema 3C (Fig. 1E,F). Neither class II nor class III agonists affect sympathetic responses to sema 3C. These results demonstrate that glutamate or the class I mGluR agonist DHPG can reduce the effectiveness of slit-2 on retinal growth cones, sema 3A on DRG growth cones, or sema 3C on sympathetic growth cones.
The modulatory effect of glutamate on slit-2 responsiveness is mediated by mGluR I
The class I agonist DHPG can activate two different receptors: mGluR1 or mGluR5. Specific competitive and noncompetitive antagonists of these receptors were used to identify which of them mediates glutamate- and DHPG-induced modulation of slit-2 responsiveness. LY367385 is a competitive mGluR1 antagonist, and SIB1893 is a competitive mGluR5 antagonist (Conn and Pin, 1997; Schoepp et al., 1999). Glutamate and DHPG reduce the responsiveness of retinal growth cones to slit-2 (Fig. 2A, compare column 1 with columns 2, 3). These antagonists by themselves have no effect on the amount of slit-2 required to collapse 50% of retinal growth cones (Fig. 2A, compare column 1 with columns 4, 5). The mGluR1 antagonist LY367385 prevents both glutamate and DHPG from reducing the effectiveness of slit-2 (Fig. 2A, compare columns 2, 3 with columns 6, 8). The mGluR5 antagonist SIB1893 has no effect on either the glutamate- or DHPG-induced modulation of slit activity (Fig. 2A, compare columns 2, 3 with columns 7, 9). These results suggest that the ability of glutamate and DHPG to reduce slit-2 responses is specifically mediated by mGluR1 activation.
Figure 2.

Glutamate- or DHPG-induced modulation is mediated by mGluR1. A, Higher values (>1) indicate fold increases in the amount of slit-2 required to obtain 50% collapse in various conditions. All cultures with glutamate are shown in light gray, whereas those with DHPG are in dark gray. A competitive antagonist for mGluR1, LY367385, has no effect on the activity of slit-2 by itself (columns 1, 4) but can block both glutamate-induced (columns 2, 6) and DHPG-induced (columns 3, 8) modulation. A competitive mGluR5 antagonist, SIB1893, has no effecton activity of slit-2 (columns 1, 5). Also, together with glutamate (columns 2, 7) or DHPG (columns 3, 9), SIB1893 did not affect the modulation of slit-2. B, Noncompetitive antagonists for mGluR1 and mGluR5 were used to confirm the receptor being used by glutamate and DHPG. CPCCOEt, a noncompetitive mGluR1 antagonist, has no effect on slit-2 by itself (columns 1, 4) but can inhibit both glutamate-induced (columns 2, 6) and DHPG-induced (columns 3, 8) modulation. MPEP, a noncompetitive antagonist of mGluR5, has no effect on the activity of slit-2 by itself (columns 1, 5) or glutamate-induced (columns 2, 7) and DHPG-induced (columns 3, 9) modulation of slit-2.
Noncompetitive antagonists were also tested for their ability to block the glutamate- and DHPG-mediated modulation of retinal responses to slit-2 (Fig. 2B). CPCCOEt is a noncompetitive mGluR1 antagonist, whereas MPEP chloride is a noncompetitive mGluR5 antagonist (Conn and Pin, 1997; Schoepp et al., 1999). Glutamate and DHPG reduce the responsiveness of retinal growth cones to slit-2 (Fig. 2B, compare column 1 with columns 2, 3). Neither antagonist by itself has any effect on the activity of slit-2 (Fig. 2B, compare column 1 with columns 4, 5). The mGluR1 antagonist CPCCOEt prevents both glutamate and DHPG from reducing the effectiveness of slit-2 (Fig. 2B, compare columns 2, 3 with columns 6, 8). The mGluR5 antagonist MPEP chloride has no effect on either glutamate- or DHPG-induced reduction in slit activity (Fig. 2B, compare columns 2, 3 with columns 7, 9). The data from both the competitive and noncompetitive antagonists are in agreement and demonstrate that both glutamate and DHPG reduce retinal responsiveness to slit-2 through activation of mGluR1.
mGluR1 is expressed by embryonic mouse RGCs and DRGs
mGluR1 is localized to major targets of glutamatergic inputs, and its expression increases in the rat between E18 through postnatal day 11 (Shigemoto et al., 1992). In the adult rat brain, it is expressed in Purkinje cells, the olfactory bulb, and neurons in the hippocampus (Shigemoto et al., 1992). GABA-positive cells in the inner nuclear layer and some RGCs express mGluR1 in the adult chick retina (Kreimborg et al., 2001). We examined the expression of mGluR1 by in situ hybridization with antisense probes against the mouse mGluR1 sequence (Zhu et al., 1999) in relatively early embryonic mouse tissues when primary axons are known to be extending (Young, 1985; Cepko et al., 1996). Retinal ganglion cells express mGluR1 in sections of E13.5 retinal tissue. An in situ for mouse mGluR1 is shown on a cross section through the embryonic eye in Figure 3A. RGCs can be visualized in the same section with an islet-1/2 antibody (Fig. 3B) (Erskine et al., 2000). The merged image in Figure 3C shows staining for mGluR1 (dark reaction product) surrounding islet-positive nuclei in RGCs (red). In E13.5 spinal cords, mGluR1 expression is seen also seen in cross sections through spinal cords of E13.5 embryos (Fig. 3D). The islet-1 antibody identifies motor neurons and DRG neurons (Fig. 3E), and the merged image shows mGluR1 expression within motor pools in the ventral cord and in the DRGs (Fig. 3F).
Figure 3.
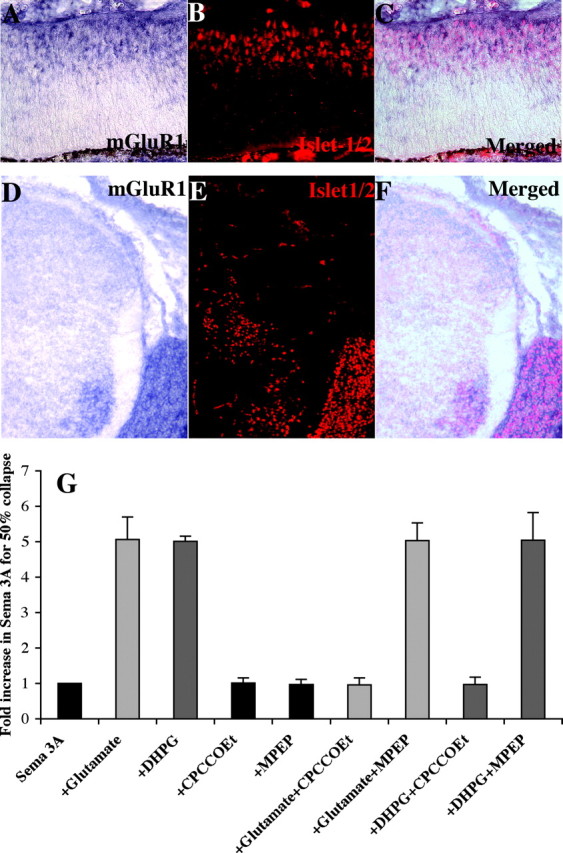
mGluR1 is expressed by mouse embryonic RGC and DRG neurons. A, In situ for mouse mGluR1 is shown on a cross section of E13.5 retina. B, The same section is stained with anti-islet-1/2, identifying RGCs. C, The merged image is shown identifying mGluR1 expression in islet-1-positive RGCs. D, In situ for mouse mGluR1 is shown on a cross section of E13.5 mouse spinal cord. E, The same section is stained with anti-islet-1/2, identifying motor and DRG neurons. F, A merged image showing islet-1/2-positive motor neurons and DRGs expressing mGluR1. G, Fold increase in amount of repellent required to cause 50% collapse of mouse DRG cultures is shown. Higher values indicate a reduction in repellent response. All cultures with glutamate are shown in light gray, whereas those with DHPG are in dark gray. Glutamate (columns 1, 2) and DHPG (columns 1, 3) reduce the effectiveness of sema 3A on mouse DRGs. CPCCOEt, an mGluR1 antagonist, has no effect on sema 3A activity (columns 1, 4) but blocks glutamate-induced (columns 2, 6) and DHPG-induced (columns 3, 8) modulation. MPEP, an mGluR5 antagonist, has no effect on sema 3A activity (columns 1, 5) or glutamate-induced (columns 2, 7) and DHPG-induced (columns 3, 9) modulation.
We tested whether mGluR1 activation reduces axonal repellent activities in mouse tissues. Like their chick counterparts, mouse DRG growth cone responses to sema 3A are reduced in the presence of either glutamate or DHPG. Cultured E13.5 mouse DRGs respond to recombinant sema 3A with a dose-dependent increase in the percentage of collapsed growth cones. Glutamate or DHPG reduced the responsiveness of mouse DRGs by approximately fivefold to sema 3A (Fig. 3G, compare columns 1 with columns 2, 3). The mGluR1 antagonist CPCCOEt has no effect on sema 3A repellent activity by itself (Fig. 3G, compare columns 1, 4) but blocks glutamate-induced (Fig. 3G, compare columns 2, 6) or DHPG-induced (Fig. 3G, compare columns 3, 8) modulation of sema 3A responsiveness. The mGluR5 antagonist MPEP has no effect on sema 3A repellent activity by itself (Fig. 3G, columns 1, 5) nor on the modulation of sema 3A responsiveness by glutamate (Fig. 3G, columns 2, 7) or DHPG (Fig. 3G, columns 3, 9). These data confirm that embryonic mouse RGCs and DRGs express mGluR1 and show that embryonic mouse DRG axon responsiveness to sema 3A, as in chick tissues, is reduced in the presence of glutamate or DHPG.
mGluR1 and CXCR4 activation are not additive
The activation of mGluR1 reduces axonal responsiveness to multiple repellents in a manner analogous to SDF-1/CXCR4 signaling (Chalasani et al., 2003a). If both SDF-1/CXCR4 and glutamate/mGluR1 modulate repellent signaling through pathways that converge on some of the same intermediates, then their modulatory effects should fail to augment those of the other. To determine whether this is the case, we measured the modulatory effects of SDF-1 alone, DHPG alone, and SDF-1 and DHPG together. Slit-2 induces a dose-dependent collapse of RGC growth cones (Fig. 4A, black diamonds). The addition of DHPG shifts the dose-response curve to the right (Fig. 4A, gray triangles). The addition of SDF-1 also shifts the curve in a similar manner (light gray squares). When both of these modulators are added to RGC cultures, there is a shift to the right that does not exceed the effect of SDF-1 alone (Fig. 4A, open circles). The addition of these modulators in the combinations described causes an 8- to 10-fold shift in the normal dose-response curve to slit-2 (Fig. 4B). These findings suggest that modulation evoked by either SDF-1/CXCR4 or by glutamate/mGluR1 signaling could share key downstream components.
Figure 4.

Modulation mediated by SDF-1 and DHPG on RGC axons is not additive. A, Dose-response curves showing the response of retinal growth cones to increasing concentrations of slit-2 (black diamonds). SDF-1A (light gray squares) or DHPG (gray triangles) shifts the dose-response curve to the right, indicating that more slit is required to give the same response in their presence. The addition of SDF-1A and DHPG together (open circles) has no marked effect compared with SDF-1A alone. B, The fold increase in the amount of slit-2 required to give 50% collapse is shown for each condition.
mGluR1-induced modulation of Slit-2 responsiveness requires PKA activation
mGluR1 is a Gi/o protein-coupled receptor that has been shown to induce phosphoinositide (PI) hydrolysis and cAMP elevation in Chinese hamster ovary (CHO) cells (Aramori and Nakanishi, 1992). We used PTX to test whether the Gi/o family of second messengers mediates DHPG-induced reduction in slit responsiveness (Kaslow et al., 1987). PTX by itself has no effect on RGC responsiveness to slit-2 (Fig. 5A, compare columns 1, 3). DHPG reduces RGC responsiveness to slit-2 (Fig. 5A, compare columns 1, 2), and PTX completely blocks DHPG-induced modulation of slit responsiveness (Fig. 5A, compare columns 2, 6). These results are consistent with the hypothesis that DHPG uses a Gi/o-coupled PTX-sensitive receptor (mGluR1) to modulate growth cone responsiveness to slit-2.
Figure 5.
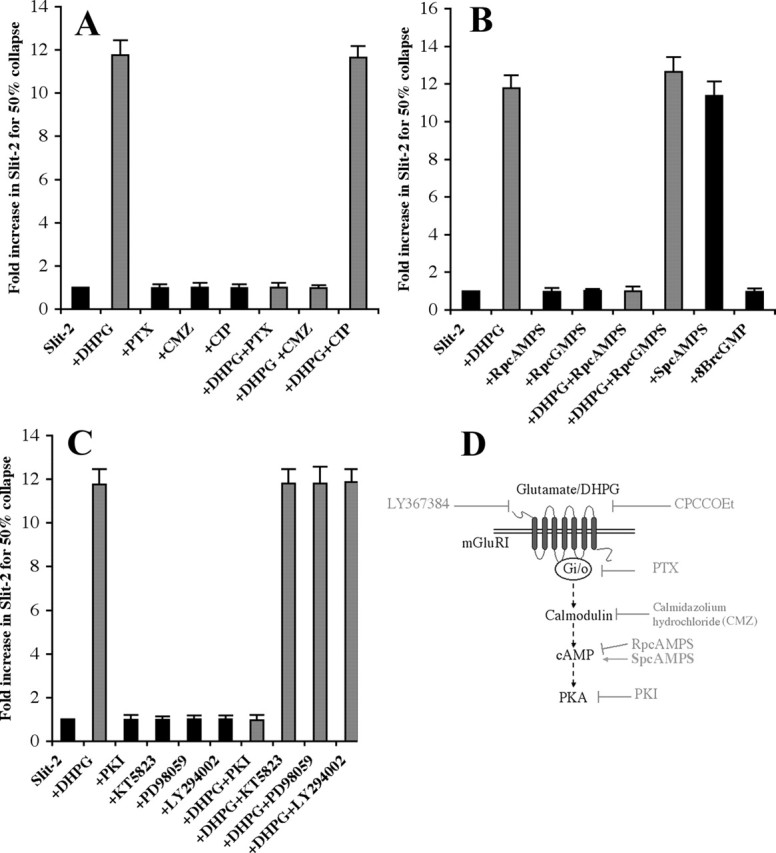
DHPG activates Gi/o-coupled mGluR1, elevating cAMP via calmodulin, and activates PKA to modulate Slit-2 responsiveness. Specific inhibitors and agonists were used with or without DHPG to identify signaling components involved in modulating the activity of slit-2. Larger values indicate a greater amount of slit-2 (fold increase) required to cause 50% collapse of retinal growth cones. In all panels, DHPG reduces the activity of slit-2 (columns 1, 2), causing an ∼12-fold increase in the amount of slit-2 to give 50% collapse. A, PTX, Gi/o blocker, has no effect on the activity of slit-2 (columns 1, 3) but can block DHPG-induced modulation (columns 2, 6). CMZ, a calmodulin inhibitor, has no effect on slit-2 (column 1, 4) but blocks DHPG-induced modulation (columns 2, 7). A peptide (calmodulin inhibitory peptide) that can block calmodulin activation of MLCK has no effect on slit-2-induced (columns 1, 5) or DHPG-induced (columns 2, 8) modulation. B, cAMP and cGMP agonists and antagonists were used to identify the cyclic nucleotide, if any, being elevated by DHPG. RpcAMPS, a cAMP antagonist, has no effect on slit-2 activity (columns 1, 3) but blocks the DHPG-induced modulation (columns 2, 5). 8-Br-cGMP, a cGMP antagonist, has no effect on slit-2-induced (columns 1, 4) or DHPG-induced (columns 2, 6) modulation. SpcAMPS, a cAMP agonist, can modulate slit even in the absence of DHPG (columns 1, 7), whereas 8-BrcGMP, a cGMP agonist, has no effect on the activity of slit-2 (columns 1, 8). C, Inhibitors against four kinases were used to determine whether any of these proteins had a role in the DHPG-induced signaling pathway. PKI, a PKA inhibitor, has no effect on slit-2 (columns 1, 3) but can abolish the DHPG-induced modulation (columns 2, 7). None of the other inhibitors (KT5823, a protein kinase G inhibitor; or PD98059, a MAPK inhibitor; or LY294002, a PI-3 kinase inhibitor) have any effect on the activity of slit-2-induced (compare column 1 with columns 4-6) or on DHPG-induced (compare column 2 with columns 8-10) modulation. D, A model consistent with our inhibitor experiments is shown. The signaling components we have identified are shown in black, whereas the inhibitors or antagonists used are in gray. SpcAMPS, a cAMP antagonist, which can mimic the DHPG effect, is shown with an arrow pointing toward cAMP.
Previous studies in CHO cells and BHK 570 cells have suggested that mGluR1 activation induces PI hydrolysis, increases in cAMP levels (Aramori and Nakanishi, 1992), and an elevation of intracellular calcium via a PTX-sensitive mechanism (Pickering et al., 1993). Calmodulin is a key intermediary in many signaling pathways activated by a rise in intracellular calcium. Its activity can be blocked by a specific antagonist, CMZ (Gietzen, 1983; Akiyama et al., 2001; Takano et al., 2003). Low concentrations of CMZ have no effect on RGC responsiveness to slit-2 (Fig. 5A, compare columns 1, 5). However, CMZ abolishes the DHPG-induced modulation of slit-2 (Fig. 5A, compare columns 2, 7). Calmodulin, in turn, can act on several different targets including adenylate cyclase type I, CAM kinase II+IV, and myosin light chain kinase (MLCK) (Gopalakrishna and Anderson, 1983; Bagchi et al., 1992; Hanson and Schulman, 1992; Schulman, 1993; Menini, 1999). We next tested a specific peptide inhibitor (calmodulin inhibitory peptide) that can block calmodulin-mediated activation of MLCK (Torok and Trentham, 1994). This peptide has no effect on the activity of slit-2 by itself (Fig. 5A, compare columns 1, 5), nor does it block DHPG-induced modulation of slit-2 responsiveness (Fig. 5A, compare columns 2, 8). These results indicate that elevated intracellular calcium levels and calmodulin activation are key downstream signaling components of mGluR1 activation-mediated modulation of axonal responsiveness to slit-2.
Agonists and antagonists of cAMP and cGMP were used to study the potential roles of cyclic nucleotides in DHPG-induced modulation of slit-2 responsiveness. The cAMP antagonist RpcAMPS has no effect on the activity of slit-2 by itself (Fig. 5B, compare columns 1, 3) but can block DHPG-induced modulation of slit-2 responsiveness (Fig. 5B, compare columns 2, 5). The cGMP antagonist RpcGMPS has no effect on either the activity of slit-2 (Fig. 5B, compare columns 1, 4) or on DHPG-induced modulation of slit-2 responsiveness (Fig. 5B, compare columns 2, 6). The cAMP agonist SpcAMPS mimics effects of DHPG by reducing the response of retinal growth cones to slit-2 (Fig. 5B, compare columns 1, 2 with column 7). The cGMP agonist 8-Br-cGMP does not reduce the effectiveness of slit-2 (Fig. 5B, compare columns 1, 8). These results are consistent with our previous finding that SDF-1 reduces the responsiveness of retinal axons to slit-2, sensory axons to sema 3A, and sympathetic axons to sema 3C through a cAMP-dependent pathway (Chalasani et al., 2003a). This is in contrast to some other reports that cGMP agonists can have a similar modulatory effect (Schmidt et al., 2002; Mann et al., 2003; Nishiyama et al., 2003). We also observed a significant cGMP-mediated effect on DRG growth cones responding to sema 3A if we expose them to cGMP agonists for longer periods before testing them for collapse. However, this cGMP-mediated effect is pharmacologically separable from the cAMP-dependent pathway we describe here (K. A. Sabelko, E. N. Twery, and J. A. Raper, unpublished observations). In summary, our results show that a cAMP antagonist blocks the DHPG-induced modulation of slit-2 and that a cAMP agonist mimics the DHPG effect. cGMP agonists and antagonists have an insignificant effect under these experimental conditions. These results suggest that mGluR1 activation modulates slit-2 responsiveness through an elevation in cAMP.
Specific inhibitors against PKA, protein kinase G, MAPK, and PI-3 kinase were tested to determine whether any of these kinases are required for DHPG-induced modulation of slit-2 responsiveness. The PKA inhibitor PKI does not affect the activity of slit-2 (Fig. 5C, compare columns 1, 3) but does block DHPG-induced modulation of slit-2 responsiveness (Fig. 5C, compare columns 2, 7). The protein kinase G inhibitor KT5823, the MAPK inhibitor PD98059, and the PI-3 kinase inhibitor LY294002 do not interfere with DHPG-induced modulation of slit-2 (Fig. 5C, compare column 2 with columns 8-10). These inhibitors were applied at concentrations shown to be effective in other in vitro assays (Vlahos et al., 1994; Nishiyama et al., 2003). We have previously tested the MAPK inhibitor PD98059 and found it to be effective in blocking SDF-1-induced cell survival in longer-term retinal neuron cultures (Chalasani et al., 2003b). The positive results we have obtained, however, indicate that PKA activation is an important intermediate step in modulating slit-2 responsiveness. Again, the effects of these pharmacological reagents on DHPG-mediated modulation are identical to those previously found for SDF-1-mediated modulation (Chalasani et al., 2003a). A model for some of the signaling steps consistent with these inhibitor data are shown in Figure 5D. We propose that glutamate signals via the activation of mGluR1 using a Gi/o intermediary. This, in turn, induces the activation of calmodulin, leading to an elevation of cAMP levels and the activation of PKA.
Activation of mGluR1 leads to phosphorylation and nuclear translocation of CREB
The actions of selected pharmacological agents on this signaling pathway were visualized through the phosphorylation of the CREB at serine 133 and its translocation into the nucleus (Gonzalez and Mont-miny, 1989; Ginty et al., 1993; Hagiwara et al., 1993). Dissociated sympathetic neurons cultured in defined minimal medium for 6 hr have little detectable phospho-CREB (P-CREB) in their nuclei (Fig. 6A1, sympathetic neurons visualized with anti-islet-1 stained green; A2, phospho-CREB stained red; A3, a merged image of the red and green signals). In contrast, DHPG-treated cultures show a dramatic increase in nuclear P-CREB (Fig. 6B1-B3). This DHPG-induced phosphorylation is blocked by the PKA inhibitor PKI (Fig. 6G1-G3). CREB can be activated by a variety of pathways (Shaywitz and Greenberg, 1999; Lonze and Ginty, 2002), but the requirement for PKA is consistent with DHPG inducing an elevation of cAMP that activates PKA, which in turn phosphorylates CREB. DHPG-induced CREB phosphorylation is blocked by the competitive mGluR1 antagonist LY367385 (Fig. 6C1-C3). In contrast, the competitive mGluR5 antagonist SIB1893 does not block CREB phosphorylation after DHPG treatment (Fig. 6D1-D3). These results support the bioassays that show that DHPG-induced signaling in neurons is mediated by mGluR1. Similarly, PTX (Fig. 6E1-E3) and the calmodulin inhibitor CMZ (Fig. 6F1-F3) both block DHPG-induced modulation of slit-2 responsiveness and also block DHPG-induced phosphorylation of CREB. These results are consistent with the signaling pathway outlined in Figure 6D: glutamate acts via mGluR1 to activate a Gi/o-coupled intermediary that, in turn, activates calmodulin, elevates cAMP, and activates PKA.
Figure 6.
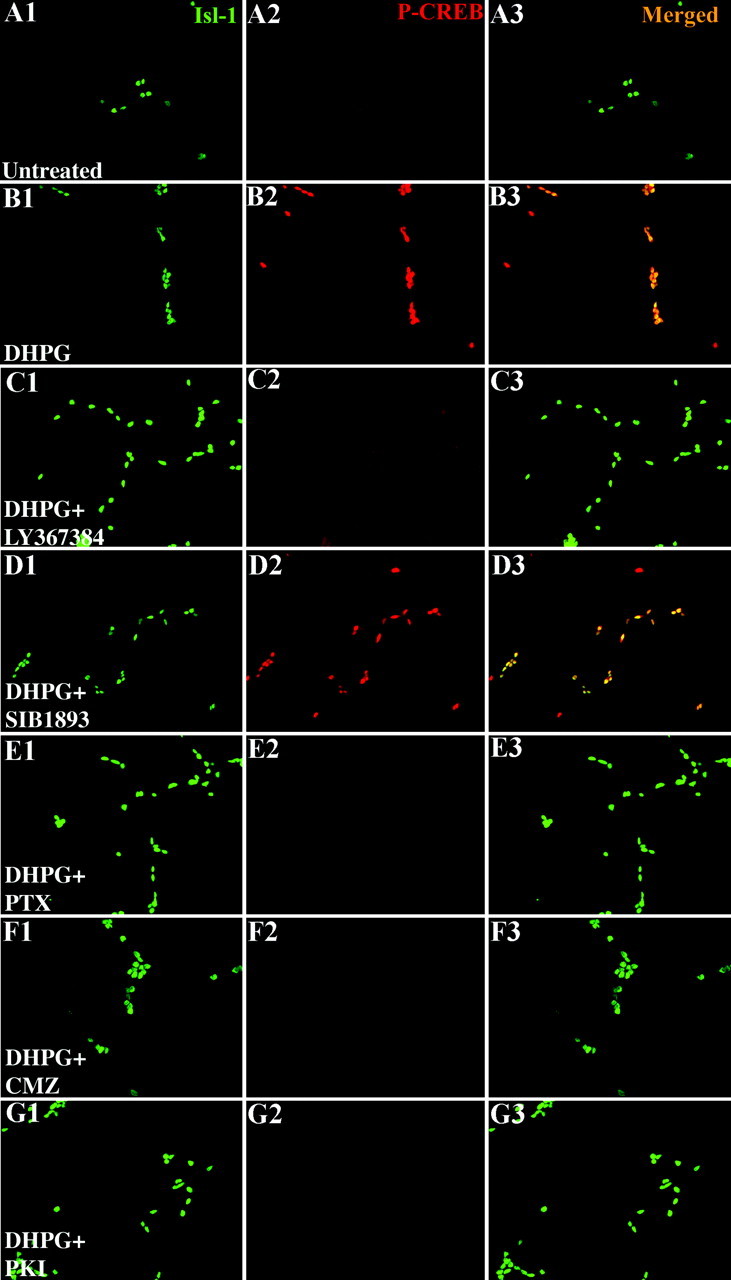
mGluR1 activation induces CREB phosphorylation. Dissociated sympathetic cultures from E8 chicks were set up in defined minimal medium. In all the conditions, nuclei are identified by anti-islet-1 in green (A1, B1, C1, D1, E1, F1, G1), P-CREB is identified by anti-phospho-specific CREB in red (A2, B2, C2, D2, E2, F2, G2), and the merged image is shown in A3, B3, C3, D3, E3, F3, and G3. Under these conditions very little P-CREB is identified (A1-A3). After 30 min of DHPG stimulation, CREB is phosphorylated and translocated into the nucleus (B1-B3). A competitive antagonist of the mGluR1, LY367385, can block the DHPG-induced CREB phosphorylation (C1-C3), but SIB1893, a competitive mGluR5 antagonist, has no effect on the DHPG effect (D1-D3). Three inhibitors, PTX (a Gi/o blocker; E1-E3), CMZ (a calmodulin antagonist; F1-F3), and PKI (PKA inhibitor;G1-G3), all of whom block DHPG-induced modulation, also block DHPG-induced phosphorylation of CREB. In all the conditions, nuclei are identified by anti-islet-1 in green (A1, B1, C1, D1, E1, F1, G1), P-CREB is identified by anti-phospho-specific CREB in red (A2, B2, C2, D2, E2, F2, G2), and the merged image is shown in A3, B3, C3, D3, E3, F3, and G3.
Activation of mGluR1 inactivates Rho
PKA can inactivate the small GTPase Rho by phosphorylating ser-188 in both neuronal and non-neuronal cells (Dong et al., 1998). Our PKI and P-CREB assays suggest that mGluR1 stimulation activates PKA. DHPG activation of mGluR1 might therefore induce the inactivation of Rho. If so, then activators of Rho should block DHPG modulation of slit-2 responsiveness, whereas inhibitors of Rho should mimic DHPG modulation of slit-2 responsiveness. Low concentrations of cytotoxic necrotizing factor 1 (CNF1), a toxin purified from E. coli, have been shown to activate Rho specifically (Olson et al., 1998). At higher doses, CNF1 activates Rac and Cdc42 as well (Flatau et al., 1997; Schmidt et al., 1997). Conversely, tat-C3, a fusion of the HIV-tat sequence to the C3 toxin, has been shown to block the activation of Rho (Aktories et al., 1992; Coleman et al., 2001). CNF1, at a low dose that should specifically activate Rho, has no effect on the activity of slit-2 (Fig. 7A, compare columns 1, 3) but partially blocks DHPG-induced modulation of slit-2 responsiveness (Fig. 7A, compare columns 2, 5). Conversely, Tat-C3 mimics the ability of DHPG to modulate slit-2 responsiveness (Fig. 7A, compare columns 1, 2 with column 4). There is no additional effect of adding DHPG to cultures treated with Tat-C3 and slit-2 (Fig. 7A, compare columns 4, 6). Thus, a Rho blocker mimics the addition of DHPG, whereas a Rho activator partially blocks the DHPG effect. This is consistent with a signaling pathway in which mGluR1 activation reduces slit-2 responsiveness through the inactivation of Rho.
Figure 7.

mGluR1 activation inactivates Rho. Downstream of PKA activation, DHPG seems to work via the inactivation of the small GTPase Rho. Larger values indicate a greater amount of slit-2 (fold increase) is required to cause 50% collapse of retinal growth cones. A, CNF1, a toxin that can activate Rho at low concentrations, has no effect on the activity of slit-2 (columns 1, 3) but can partially block the DHPG-induced modulation (columns 2, 5). Tat-C3, a cell-permeable Rho blocker, can reduce the activity of slit-2 by itself (columns 1, 4), and there is no additional effect on DHPG-induced modulation (columns 2, 6). B, Y-27632, a Rho kinase inhibitor, can modulate slit-2 by itself (columns 1, 3) and has no additive effect on DHPG-induced modulation (columns 2, 4). C, ML-7, a MLCK inhibitor, can reduce the activity of slit-2 (columns 1, 8) and has no synergistic effect on DHPG-induced modulation (columns 2, 9). Reagents that block Rho (Tat-C3) or downstream targets of Rho (Rho kinase and MLCK) mimic the DHPG effect by reducing slit-2 activity, whereas Rho activator (CNF1) blocks the DHPG effect, suggesting that DHPG might be inactivating Rho in our cultures. Four different inhibitors, PTX, CMZ, RpcAMPS, and PKI, all of which can block the DHPG-induced modulation (compare column 2 with columns 3-6), suggest that the molecules they block are downstream of DHPG. However, they cannot block ML-7-induced modulation (compare column 8 with columns 10-13), suggesting that molecules they block are upstream of MLCK. A calmodulin antagonist cannot block SpcAMPS-induced modulation (compare columns 1, 2 with column 7), suggesting that calmodulin is upstream of cAMP in the signaling pathway. D, A model consistent with inhibitor data is shown. Tat-C3 is shown to inactivate Rho, whereas CNF1 can activate it. Two inhibitors, Y-27632 and ML-7, are shown with arrows pointing toward their targets because they can mimic the DHPG-induced modulation of slit-2.
Additional evidence supporting this proposed signaling pathway was obtained using inhibitors against known downstream targets of Rho. If activation of mGluR1 inactivates Rho, then blocking the downstream targets of Rho should mimic the effects of mGluR1 activation. We tested inhibitors against Rho kinase and MLCK (Kimura et al., 1996; Bishop and Hall, 2000). The Rho kinase inhibitor Y-27632 reduces RGC responsiveness to slit-2 (Fig. 7B, compare columns 1, 3), and there is no additional effect of adding DHPG (Fig. 7B, compare columns 3, 4). The MLCK inhibitor ML-7 also reduces responsiveness to slit-2 (Fig. 7C, compare columns 1, 8), and, again, there is no additional effect of adding DHPG (Fig. 7C, compare columns 8, 9). These results are consistent with a model in which activation of mGluR1 inactivates Rho to reduce responsiveness to slit-2.
The relative positions of four key steps in the proposed signaling pathway were checked using combinations of selected inhibitors, specifically PTX (Gi/o blocker), CMZ (calmodulin blocker), RpcAMPS (cAMP antagonist), and PKI (PKA blocker). Each blocks DHPG-induced modulation of slit responsiveness (Fig. 7C, compare column 2 with columns 3-6). However, none of these inhibitors blocks ML-7-induced modulation of slit-2 responsiveness (Fig. 7C, compare column 8 with columns 10-13), suggesting that MLCK is the most downstream step in the signaling pathway. The antagonist of calmodulin blocks DHPG-induced modulation of slit-2 responsiveness (Fig. 7C, compare columns 2, 4) but does not block modulation induced by either a cAMP agonist (Fig. 7C, compare columns 2, 4 with column 7) or a MLCK inhibitor (Fig. 7A, compare columns 2, 4 with column 11). These data show that calmodulin acts upstream from both cAMP and MLCK. A proposed model showing the signaling steps induced by activation of mGluR1 downstream of PKA is shown in Figure 7D.
Activation of mGluR1 promotes the survival of cultured RGCs
mGluR1 activation reduces the effectiveness of multiple repellents in a manner analogous to SDF-1/CXCR4 signaling, and SDF-1/CXCR4 signaling promotes the survival of dissociated embryonic RGCs (Chalasani et al., 2003b). We therefore tested whether mGluR1 activation also promotes the survival of cultured RGCs (Fig. 8A). Dissociated E6 retinal cells were cultured on laminin in a minimal, fully defined medium without added neurotrophins for 48 hr. RGCs were identified by islet-1 staining. The highest level of RGC survival is obtained in medium containing SDF-1 (Fig. 8A, column 2). In contrast, only 60% as many RGCs survive in minimal medium alone (Fig. 7A, column 1). Approximately 80% as many RGCs survive in medium containing the class I mGluR agonist DHPG (Fig. 8A, column 3). This survival-promoting effect of DHPG is blocked by the mGluR1 antagonist CPCCOEt and is not affected by the mGluR5 antagonist MPEP (Fig. 8A, compare column 3 with columns 6, 7). Inhibitors of PKA (PKI) and MAPK 1/2 (PD98059) have no detectable effect on cell survival in the absence of mGluR1 stimulation under these conditions (Fig. 8B, compare column 1 with columns 4, 5) but they do block DHPG-induced survival (Fig. 8B, compare column 3 with columns 6, 7). These results suggest that mGluR1 activation promotes the survival of cultured embryonic RGCs via activation of PKA and MAPK 1/2.
Figure 8.

mGluR1 activation promotes survival of embryonic RGCs. Dissociated RGCs cultures from E6 chick retinas were set up in minimal medium for 48 hr with or without DHPG. The average numbers of live RGCs were determined in different conditions as islet-1-positive staining. A, The number of RGCs that survived in SDF-1-treated cultures was set at 100, and those in other conditions were compared with that number. DHPG promotes survival of RGCs (compare columns 1, 3; p < 0.05), but not as robustly as those with SDF-1 (compare columns 2, 3). Noncompetitive antagonists of class I mGluRs [CPCCOEt (CPC), a specific mGluR1 antagonist, and MPEP chloride, a specific mGluR5 antagonist] have no effect on RGC survival by themselves (compare column 1 with columns 4, 5). CPCCOEt blocks the DHPG-induced survival (compare columns 3, 6; p < 0.05), whereas MPEP has no effect (compare columns 3, 7). B, Specific inhibitors of PKA (PKI; compare columns 1, 4) and MAPK [PD98059 (PD); compare columns 1, 5] have no effect on RGC survival. Both inhibitors can block the DHPG-induced RGC survival (compare column 3 with columns 6, 7; p < 0.01).
Discussion
These data demonstrate that the excitatory neurotransmitter glutamate can alter growth cone responsiveness to multiple axonal repellents. This effect is mediated through its activation of the class I metabotropic receptor mGluR1 because the modulatory activity of either glutamate or the class I metabotropic agonist DHPG can be blocked by specific mGluR1 antagonists. Our data are consistent with a pathway in which PTX-sensitive activation of Gi/Go activates calmodulin, adenylate cyclase, and PKA. Although our analysis is limited to pharmacological manipulations, the pathway we propose is one that is well described (Tong et al., 1996; Linden and Ahn, 1999), the reagents we have used are well characterized, and their actions at several different steps in the pathway support each other. Elevated levels of cAMP and activation of PKA have been shown to facilitate sensory axon outgrowth on repellent substrata, and PKA activation has recently been implicated in reducing the responsiveness of Drosophila motor neurons to semaphorins (Neumann et al., 2002; Qiu et al., 2002; Bashaw, 2004; Terman and Kolodkin, 2004). Our findings further suggest that activation of PKA leads to the inactivation of the small GTPase Rho, as has previously been described in neuronal cell lines (Dong et al., 1998). Inactivation of Rho is expected to counteract the repellent effects of repulsive signaling molecules like those we tested (Liu and Strittmatter, 2001; Oinuma et al., 2003; Yuan et al., 2003).
We have previously shown that the chemokine SDF-1 also reduces RGC responses to slit-2, DRG responses to sema3A, and sympathetic responses to sema3C (Chalasani et al., 2003a). These modulatory effects resemble those of mGluR1 activation, and the effects of CXCR4 and mGluR1 activation are not additive in RGC axons. These findings suggested that the signaling pathways of SDF-1/CXCR4- and glutamate/mGluR1-mediated modulation of repellent responsiveness might converge on some of the same intermediaries. Just like mGluR1 activation, SDF-1 activation of CXCR4 stimulates a PTX-sensitive pathway that is also blocked by an antagonist of cAMP and an inhibitor of PKA. Both mGluR1 and CXCR4 activation are mimicked by a cAMP agonist, TAT-C3-induced inactivation of Rho, or the blockade of either Rho kinase or MLCK. That the same pharmacological reagents reduce axonal responses to the same repellents in multiple systems argues strongly that SDF-1 and glutamate converge onto the same common signaling pathway.
In principle, glutamate could be acting as an attractant, the effects of which counteract those of repellents when they are summed together, or as a modulator that interferes with repellent signaling without having attractive activities of its own. Because mGluR1 activates the same signaling pathway as SDF-1/CXCR4 signaling, and because SDF-1 appears to act as a modulator rather than an attractant in the same neurons in which we studied glutamate activity (Chalasani et al., 2003a), it seems most likely that glutamate acts as a modulator that reduces repellent responsiveness rather than an outright attractant.
Neurotransmitters have previously been reported to directly affect axon outgrowth in vitro. ACh inhibits the outgrowth of cultured postnatal rat RGC neurites (Lipton et al., 1988). It can also both promote the outgrowth, as well as attract, embryonic Xenopus spinal neurites in vitro (Zheng et al., 1994; Erskine and McCaig, 1995). Baclofen, an agonist of metabotropic GABAB receptors, was recently shown to repel Xenopus spinal neurons (Xiang et al., 2002). Glutamate itself has been found to have direct effects on neuronal morphology. Dendrite extension is inhibited in isolated cultured motor neurons through glutamate-induced stimulation of AMPA/KA glutamatergic receptors (Metzger et al., 1998). In contrast to these previous studies that show a direct effect of neurotransmitters on neurite motility, our results show that neurotransmitters alter axonal responses to traditional axonal guidance cues.
Many studies have identified a prominent role for ionotropic glutamate receptors, particularly the NMDA receptor, in neuronal plasticity (Riedel et al., 2003; Waltereit and Weller, 2003). However, other studies have also suggested a vital role for metabotropic signaling. Long-term potentiation (LTP) at hippocampal mossy fibers is independent of NMDA function and involves a presynaptic glutamatergic component (Castillo et al., 1997). Activation of group II mGluRs induces long-term depression (LTD) in rat prefrontal cortex (Otani et al., 2002). Interestingly, dendritic spines have been reported to elongate when stimulated by the class I mGluR agonist DHPG (Vanderklish and Edelman, 2002). Several neurological impairments have been noted in mice lacking mGluR1. They are viable and fertile but have an ataxic gait with an intention tremor (Aiba et al., 1994b). These animals have impaired motor learning, are deficient in cerebellar LTD, and show impaired hippocampal LTP and associative learning (Aiba et al., 1994a). An interesting structural defect has also been noted in these mice. Because of impaired regression of climbing fibers in the cerebellar cortex, multiple climbing fibers innervate individual Purkinje cells (Kano et al., 1997). This axonal guidance defect raises the possibility that some of the abnormal behaviors observed in these mice could be the consequence of additional guidance errors occurring during development. This warrants attention in future studies.
Although we observed a modulatory effect of glutamate on the responsiveness of primary axons to repellents, we speculate that the modulatory mechanism we describe is most relevant to activity-dependent remodeling of neuronal connectivity that takes place later in development. During this phase, synaptic connections are thought to be refined in part by the advance and withdrawal of axonal and dendritic processes (Jackson and Parks, 1982; Sanes and Lichtman, 1999; Hashimoto and Kano, 2003), and these morphological changes are controlled by neuronal firing patterns. Although some progress has been made in identifying rules that relate firing patterns to changes in morphology (Constantine-Paton et al., 1990; Katz and Shatz, 1996; Milner et al., 1998; Wong et al., 1998), it has been much more challenging to identify specific molecular components that provide the biochemical mechanisms for these changes. It is logical to assume that neurotransmitters released by neural activity are likely to play an important signaling role in activity-dependent changes in neuronal morphology, but how?
Our results demonstrate that glutamate acting through mGluR1 can reduce the activity of repellents. Other neurotransmitters may potentially activate the same signaling pathway through their action on select metabotropic receptors. We speculate that such mechanisms help promote the extension of active processes through environments rich in repellent signals. Active axons are known to release neurotransmitter (Hume et al., 1983; Rane, 1987; Sun and Poo, 1987). Growing presynaptic processes respond to repellent cues in their surrounding environment; for example, retinal axons confront the repellents ephrinA2 and A5 in the optic tectum and the lateral geniculate (Cheng et al., 1995; Feldheim et al., 1998, 2000) and are known to be electrically active from very early embryonic times (Ziv and Smith, 1996; Wong et al., 1998; Ritzenthaler et al., 2000). We hypothesize that the release of glutamate from active axons could reduce their own response to target-derived repellents. This autocrine modulatory mechanism would give active axons a competitive advantage in extending within their target. Once axons have acquired their targets, we hypothesize that the release of glutamate from presynaptic elements could make postsynaptic elements less responsive to repellent cues and thereby facilitate synapse formation. There is growing evidence that the postsynaptic cell is not a passive partner in synapse formation. Both muscle cells and neurons have been observed to advance fine filopodial-like processes toward potential synaptic partners (Ziv and Smith, 1996; Ritzenthaler et al., 2000). The release of neurotransmitters from active presynaptic elements could attract postsynaptic elements by making them resistant to repellents expressed either in the general environment or by the presynaptic cell itself.
High concentrations of glutamate are generally thought of as excitotoxic to neurons (Lucas and Newhouse, 1957; Olney, 1989). Calcium entry through NMDA receptors has been shown to mediate glutamate-induced cell death (Sattler and Tymianski, 2001). In contrast, there are mixed reports on the effects of mGluR activation on cell death and survival (Spillson and Russell, 2003). On balance, most support a survival-enhancing effect. For example, activation of class I mGluRs promote the differentiation and survival of cerebellar Purkinje cells (Catania et al., 2001), protect rat spinal motor neurons against a kainate insult (Pizzi et al., 2000), and can be neuroprotective against NMDA excitotoxicity in hippocampal slice cultures (Blaabjerg et al., 2003). Previous groups have reported that neuroprotection mediated by class II (D'Onofrio et al., 2001) and class III (Iacovelli et al., 2002) mGluRs requires MAPK activation. Our data suggest that selectively activating mGluR1 can promote embryonic neuronal survival through MAPK activation and suggest a mechanism by which the establishment of synaptic connectivity could support the survival of the target cell.
We have now shown that two extracellular signals, the chemokine SDF-1 and the neurotransmitter glutamate, modulate the responsiveness of multiple axon types to several different guidance cues. Both of these modulatory cues achieve their effects through the stimulation of G-protein-coupled receptors and their activation of a common signaling pathway. Given the tremendous number and diversity of G-protein-coupled receptors, it seems likely that other ligand-receptor pairs will be identified that activate the same pathway. Some of these may be other metabotropic receptors for neurotransmitters like dopamine, serotonin, or norepinephrine that are known to activate PKA (Roseboom and Klein, 1995; Barbas et al., 2003; Jay, 2003). Based on these findings, we propose a specific, novel mechanism by which neuronal activity could influence the advance and withdrawal of neuritic processes. Activation of specific metabotropic receptors by activity-driven release of neurotransmitter would reduce the responsiveness of neuronal processes to repellent guidance cues. This signaling mechanism might provide a mechanistic link between patterned neuronal activity and morphological changes observed during activity-dependent remodeling of neuronal circuitry.
Footnotes
This work was supported by National Institutes of Health Grant RO1-NS26527 to J.A.R. We gratefully acknowledge Jared Pisapia, Cynthia Ito, and Radhia Ben-Mohamed for technical help. We thank Drs. Chris Burd and Michael Olson for help with the tat-c3 purification and Drs. Klaus Aktories and Gudula Schmidt for providing CNF1. We also thank Drs. Judith Meinkoth and Thomas Jessell for the kind gifts of anti-P-CREB and anti-islet-1/2 antibodies. We are grateful to Cynthia Ito, Dr. Andrea Webber, and Dr. Kimberly Sabelko for help with the mice. We thank Drs. Judith Meinkoth, David Manning, and Jeffery Masuda-Robbens for advice. We also thank Arati Sadalge Kreibich and Dr. Matthew Dalva for help with this manuscript.
Correspondence should be addressed to Dr. Jonathan A. Raper, University of Pennsylvania School of Medicine, 1115 Biomedical Research Building II/III, 421 Curie Boulevard, Philadelphia, PA 19104. E-mail: raperj@mail.med.upenn.edu.
S. H. Chalasani's present address: Department of Anatomy, Howard Hughes Medical Institute, University of California San Francisco, S1457C, San Francisco, CA 94143-0452.
Copyright © 2004 Society for Neuroscience 0270-6474/04/247085-11$15.00/0
T.A.K. and S.H.C. contributed equally to this work.
References
- Aiba A, Chen C, Herrup K, Rosenmund C, Stevens CF, Tonegawa S (1994a) Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in mGluR1 mutant mice. Cell 79: 365-375. [DOI] [PubMed] [Google Scholar]
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S (1994b) Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell 79: 377-388. [PubMed] [Google Scholar]
- Akiyama M, Minami Y, Nakajima T, Moriya T, Shibata S (2001) Calcium and pituitary adenylate cyclase-activating polypeptide induced expression of circadian clock gene mPer1 in the mouse cerebellar granule cell culture. J Neurochem 78: 499-508. [DOI] [PubMed] [Google Scholar]
- Aktories K, Mohr C, Koch G (1992) Clostridium botulinum C3 ADP-ribosyltransferase. Curr Top Microbiol Immunol 175: 115-131. [DOI] [PubMed] [Google Scholar]
- Aramori I, Nakanishi S (1992) Signal transduction and pharmacological characteristics of a metabotropic glutamate receptor, mGluR1, in transfected CHO cells. Neuron 8: 757-765. [DOI] [PubMed] [Google Scholar]
- Bagchi IC, Huang QH, Means AR (1992) Identification of amino acids essential for calmodulin binding and activation of smooth muscle myosin light chain kinase. J Biol Chem 267: 3024-3029. [PubMed] [Google Scholar]
- Barbas D, DesGroseillers L, Castellucci VF, Carew TJ, Marinesco S (2003) Multiple serotonergic mechanisms contributing to sensitization in aplysia: evidence of diverse serotonin receptor subtypes. Learn Mem 10: 373-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashaw GJ (2004) Semaphorin signaling unplugged; a nervy AKAP cAMP(s) out on plexin. Neuron 42: 363-366. [DOI] [PubMed] [Google Scholar]
- Bishop AL, Hall A (2000) Rho GTPases and their effector proteins. Biochem J 348: 241-255. [PMC free article] [PubMed] [Google Scholar]
- Blaabjerg M, Fang L, Zimmer J, Baskys A (2003) Neuroprotection against NMDA excitotoxicity by group I metabotropic glutamate receptors is associated with reduction of NMDA stimulated currents. Exp Neurol 183: 573-580. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Janz R, Sudhof TC, Tzounopoulos T, Malenka RC, Nicoll RA (1997) Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature 388: 590-593. [DOI] [PubMed] [Google Scholar]
- Catania MV, Bellomo M, Di Giorgi-Gerevini V, Seminara G, Giuffrida R, Romeo R, De Blasi A, Nicoletti F (2001) Endogenous activation of group-I metabotropic glutamate receptors is required for differentiation and survival of cerebellar Purkinje cells. J Neurosci 21: 7664-7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepko CL, Austin CP, Yang X, Alexiades M, Ezzeddine D (1996) Cell fate determination in the vertebrate retina. Proc Natl Acad Sci USA 93: 589-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalasani SH, Sabelko KA, Sunshine MJ, Littman DR, Raper JA (2003a) A chemokine, SDF-1, reduces the effectiveness of multiple axonal repellents and is required for normal axon pathfinding. J Neurosci 23: 1360-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalasani SH, Baribaud F, Coughlan CM, Sunshine MJ, Lee VM, Doms RW, Littman DR, Raper JA (2003b) The chemokine stromal cell-derived factor-1 promotes the survival of embryonic retinal ganglion cells. J Neurosci 23: 4601-4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HJ, Nakamoto M, Bergemann AD, Flanagan JG (1995) Complementary gradients in expression and binding of ELF-1 and Mek4 in development of the topographic retinotectal projection map. Cell 82: 371-381. [DOI] [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF (2001) Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol 3: 339-345. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205-237. [DOI] [PubMed] [Google Scholar]
- Constantine-Paton M, Cline HT, Debski E (1990) Patterned activity, synaptic convergence, and the NMDA receptor in developing visual pathways. Annu Rev Neurosci 13: 129-154. [DOI] [PubMed] [Google Scholar]
- Dong JM, Leung T, Manser E, Lim L (1998) cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKalpha. J Biol Chem 273: 22554-22562. [DOI] [PubMed] [Google Scholar]
- D'Onofrio M, Cuomo L, Battaglia G, Ngomba RT, Storto M, Kingston AE, Orzi F, De Blasi A, Di Iorio P, Nicoletti F, Bruno V (2001) Neuroprotection mediated by glial group-II metabotropic glutamate receptors requires the activation of the MAP kinase and the phosphatidylinositol-3-kinase pathways. J Neurochem 78: 435-445. [DOI] [PubMed] [Google Scholar]
- Erskine L, McCaig CD (1995) Growth cone neurotransmitter receptor activation modulates electric field-guided nerve growth. Dev Biol 171: 330-339. [DOI] [PubMed] [Google Scholar]
- Erskine L, Williams SE, Brose K, Kidd T, Rachel RA, Goodman CS, Tessier-Lavigne M, Mason CA (2000) Retinal ganglion cell axon guidance in the mouse optic chiasm: expression and function of robos and slits. J Neurosci 20: 4975-4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldheim DA, Vanderhaeghen P, Hansen MJ, Frisen J, Lu Q, Barbacid M, Flanagan JG (1998) Topographic guidance labels in a sensory projection to the forebrain. Neuron 21: 1303-1313. [DOI] [PubMed] [Google Scholar]
- Feldheim DA, Kim YI, Bergemann AD, Frisen J, Barbacid M, Flanagan JG (2000) Genetic analysis of ephrin-A2 and ephrin-A5 shows their requirement in multiple aspects of retinocollicular mapping. Neuron 25: 563-574. [DOI] [PubMed] [Google Scholar]
- Flatau G, Lemichez E, Gauthier M, Chardin P, Paris S, Fiorentini C, Boquet P (1997) Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 387: 729-733. [DOI] [PubMed] [Google Scholar]
- Gietzen K (1983) Comparison of the calmodulin antagonists compound 48/80 and calmidazolium. Biochem J 216: 611-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME (1993) Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science 260: 238-241. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR (1989) Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59: 675-680. [DOI] [PubMed] [Google Scholar]
- Gopalakrishna R, Anderson WB (1983) Calmodulin interacts with cyclic nucleotide phosphodiesterase and calcineurin by binding to a metal ion-independent hydrophobic region on these proteins. J Biol Chem 258: 2405-2409. [PubMed] [Google Scholar]
- Hagiwara M, Brindle P, Harootunian A, Armstrong R, Rivier J, Vale W, Tsien R, Montminy MR (1993) Coupling of hormonal stimulation and transcription via the cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol Cell Biol 13: 4852-4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Schulman H (1992) Neuronal Ca2+/calmodulin-dependent protein kinases. Annu Rev Biochem 61: 559-601. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Kano M (2003) Functional differentiation of multiple climbing fiber inputs during synapse elimination in the developing cerebellum. Neuron 38: 785-796. [DOI] [PubMed] [Google Scholar]
- Hermans E, Challiss RA (2001) Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: prototypic family C G-protein-coupled receptors. Biochem J 359: 465-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopker VH, Shewan D, Tessier-Lavigne M, Poo M, Holt C (1999) Growth-cone attraction to netrin-1 is converted to repulsion by laminin-1. Nature 401: 69-73. [DOI] [PubMed] [Google Scholar]
- Hume RI, Role LW, Fischbach GD (1983) Acetylcholine release from growth cones detected with patches of acetylcholine receptor-rich membranes. Nature 305: 632-634. [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Bruno V, Salvatore L, Melchiorri D, Gradini R, Caricasole A, Barletta E, De Blasi A, Nicoletti F (2002) Native group-III metabotropic glutamate receptors are coupled to the mitogen-activated protein kinase/phosphatidylinositol-3-kinase pathways. J Neurochem 82: 216-223. [DOI] [PubMed] [Google Scholar]
- Jackson H, Parks TN (1982) Functional synapse elimination in the developing avian cochlear nucleus with simultaneous reduction in cochlear nerve axon branching. J Neurosci 2: 1736-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TM (2003) Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog Neurobiol 69: 375-390. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Kurihara H, Watanabe M, Inoue Y, Aiba A, Tonegawa S (1997) Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron 18: 71-79. [DOI] [PubMed] [Google Scholar]
- Kapfhammer JP, Raper JA (1987) Collapse of growth cone structure on contact with specific neurites in culture. J Neurosci 7: 201-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaslow HR, Lim LK, Moss J, Lesikar DD (1987) Structure-activity analysis of the activation of pertussis toxin. Biochemistry 26: 123-127. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ (1996) Synaptic activity and the construction of cortical circuits. Science 274: 1133-1138. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K (1996) Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273: 245-248. [DOI] [PubMed] [Google Scholar]
- Koppel AM, Feiner L, Kobayashi H, Raper JA (1997) A 70 amino acid region within the semaphorin domain activates specific cellular response of semaphorin family members. Neuron 19: 531-537. [DOI] [PubMed] [Google Scholar]
- Kreimborg KM, Lester ML, Medler KF, Gleason EL (2001) Group I metabotropic glutamate receptors are expressed in the chicken retina and by cultured retinal amacrine cells. J Neurochem 77: 452-465. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Ahn S (1999) Activation of presynaptic cAMP-dependent protein kinase is required for induction of cerebellar long-term potentiation. J Neurosci 19: 10221-10227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Frosch MP, Phillips MD, Tauck DL, Aizenman E (1988) Nicotinic antagonists enhance process outgrowth by rat retinal ganglion cells in culture. Science 239: 1293-1296. [DOI] [PubMed] [Google Scholar]
- Liu BP, Strittmatter SM (2001) Semaphorin-mediated axonal guidance via Rho-related G proteins. Curr Opin Cell Biol 13: 619-626. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605-623. [DOI] [PubMed] [Google Scholar]
- Lucas DR, Newhouse JP (1957) The toxic effect of sodium l-glutamate on the inner layers of the retina. Am Arch Opthalmol 58: 193-201. [DOI] [PubMed] [Google Scholar]
- Luo Y, Raible D, Raper JA (1993) Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell 75: 217-227. [DOI] [PubMed] [Google Scholar]
- Mann F, Miranda E, Weinl C, Harmer E, Holt CE (2003) B-type Eph receptors and ephrins induce growth cone collapse through distinct intracellular pathways. J Neurobiol 57: 323-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menini A (1999) Calcium signalling and regulation in olfactory neurons. Curr Opin Neurobiol 9: 419-426. [DOI] [PubMed] [Google Scholar]
- Metzger F, Wiese S, Sendtner M (1998) Effect of glutamate on dendritic growth in embryonic rat motoneurons. J Neurosci 18: 1735-1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner B, Squire LR, Kandel ER (1998) Cognitive neuroscience and the study of memory. Neuron 20: 445-468. [DOI] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI (2002) Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34: 885-893. [DOI] [PubMed] [Google Scholar]
- Niclou SP, Jia L, Raper JA (2000) Slit2 is a repellent for retinal ganglion cell axons. J Neurosci 20: 4962-4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Hoshino A, Tsai L, Henley JR, Goshima Y, Tessier-Lavigne M, Poo MM, Hong K (2003) Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature 424: 990-995. [DOI] [PubMed] [Google Scholar]
- Oinuma I, Katoh H, Harada A, Negishi M (2003) Direct interaction of Rnd1 with Plexin-B1 regulates PDZ-RhoGEF-mediated Rho activation by Plexin-B1 and induces cell contraction in COS-7 cells. J Biol Chem 278: 25671-25677. [DOI] [PubMed] [Google Scholar]
- Olney JW (1989) Excitatory amino acids and neuropsychiatric disorders. Biol Psychiatry 26: 505-525. [DOI] [PubMed] [Google Scholar]
- Olson MF, Paterson HF, Marshall CJ (1998) Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature 394: 295-299. [DOI] [PubMed] [Google Scholar]
- Otani S, Daniel H, Takita M, Crepel F (2002) Long-term depression induced by postsynaptic group II metabotropic glutamate receptors linked to phospholipase C and intracellular calcium rises in rat prefrontal cortex. J Neurosci 22: 3434-3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering DS, Thomsen C, Suzdak PD, Fletcher EJ, Robitaille R, Salter MW, MacDonald JF, Huang XP, Hampson DR (1993) A comparison of two alternatively spliced forms of a metabotropic glutamate receptor coupled to phosphoinositide turnover. J Neurochem 61: 85-92. [DOI] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R (1995) The metabotropic glutamate receptors: structure and functions. Neuropharmacology 34: 1-26. [DOI] [PubMed] [Google Scholar]
- Pizzi M, Benarese M, Boroni F, Goffi F, Valerio A, Spano PF (2000) Neuro-protection by metabotropic glutamate receptor agonists on kainate-induced degeneration of motor neurons in spinal cord slices from adult rat. Neuropharmacology 39: 903-910. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT (2002) Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34: 895-903. [DOI] [PubMed] [Google Scholar]
- Rane SG (1987) Neuromuscular synaptic transmission in Limulus polyphemus-II. Release of amino acid putative transmitters from the neuromuscular preparation. Comp Biochem Physiol C 87: 131-137. [DOI] [PubMed] [Google Scholar]
- Riedel G, Platt B, Micheau J (2003) Glutamate receptor function in learning and memory. Behav Brain Res 140: 1-47. [DOI] [PubMed] [Google Scholar]
- Ritzenthaler S, Suzuki E, Chiba A (2000) Postsynaptic filopodia in muscle cells interact with innervating motoneuron axons. Nat Neurosci 3: 1012-1017. [DOI] [PubMed] [Google Scholar]
- Roseboom PH, Klein DC (1995) Norepinephrine stimulation of pineal cyclic AMP response element-binding protein phosphorylation: primary role of a beta-adrenergic receptor/cyclic AMP mechanism. Mol Pharmacol 47: 439-449. [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22: 389-442. [DOI] [PubMed] [Google Scholar]
- Sattler R, Tymianski M (2001) Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol 24: 107-129. [DOI] [PubMed] [Google Scholar]
- Schmidt G, Sehr P, Wilm M, Selzer J, Mann M, Aktories K (1997) Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 387: 725-729. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Werner M, Heppenstall PA, Henning M, More MI, Kuhbandner S, Lewin GR, Hofmann F, Feil R, Rathjen FG (2002) cGMP-mediated signaling via cGKIalpha is required for the guidance and connectivity of sensory axons. J Cell Biol 159: 489-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA (1999) Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 38: 1431-1476. [DOI] [PubMed] [Google Scholar]
- Schulman H (1993) The multifunctional Ca2+/calmodulin-dependent protein kinases. Curr Opin Cell Biol 5: 247-253. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68: 821-861. [DOI] [PubMed] [Google Scholar]
- Shewan D, Dwivedy A, Anderson R, Holt CE (2002) Age-related changes underlie switch in netrin-1 responsiveness as growth cones advance along visual pathway. Nat Neurosci 5: 955-962. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Nakanishi S, Mizuno N (1992) Distribution of the mRNA for a metabotropic glutamate receptor (mGluR1) in the central nervous system: an in situ hybridization study in adult and developing rat. J Comp Neurol 322: 121-135. [DOI] [PubMed] [Google Scholar]
- Song H, Ming G, He Z, Lehmann M, McKerracher L, Tessier-Lavigne M, Poo M (1998) Conversion of neuronal growth cone responses from repulsion to attraction by cyclic nucleotides. Science 281: 1515-1518. [DOI] [PubMed] [Google Scholar]
- Song HJ, Ming GL, Poo MM (1997) cAMP-induced switching in turning direction of nerve growth cones. Nature 388: 275-279. [DOI] [PubMed] [Google Scholar]
- Spillson AB, Russell JW (2003) Metabotropic glutamate receptor regulation of neuronal cell death. Exp Neurol 184 [Suppl 1]: S97-S105. [DOI] [PubMed] [Google Scholar]
- Sun YA, Poo MM (1987) Evoked release of acetylcholine from the growing embryonic neuron. Proc Natl Acad Sci USA 84: 2540-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano H, Fukushi H, Morishima Y, Shirasaki Y (2003) Calmodulin and calmodulin-dependent kinase II mediate neuronal cell death induced by depolarization. Brain Res 962: 41-47. [DOI] [PubMed] [Google Scholar]
- Terman JR, Kolodkin AL (2004) Nervy links protein kinase a to plexin-mediated semaphorin repulsion. Science 303: 1204-1207. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Goodman CS (1996) The molecular biology of axon guidance. Science 274: 1123-1133. [DOI] [PubMed] [Google Scholar]
- Toms NJ, Jane DE, Tse HW, Roberts PJ (1995) Characterization of metabotropic glutamate receptor-stimulated phosphoinositide hydrolysis in rat cultured cerebellar granule cells. Br J Pharmacol 116: 2824-2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Malenka RC, Nicoll RA (1996) Long-term potentiation in cultures of single hippocampal granule cells: a presynaptic form of plasticity. Neuron 16: 1147-1157. [DOI] [PubMed] [Google Scholar]
- Torok K, Trentham DR (1994) Mechanism of 2-chloro-(epsilon-amino-Lys75)-[6-[4-(N,N-diethylamino)phenyl]-1,3,5-triazin-4-yl]calmodulin interactions with smooth muscle myosin light chain kinase and derived peptides. Biochemistry 33: 12807-12820. [DOI] [PubMed] [Google Scholar]
- Vanderklish PW, Edelman GM (2002) Dendritic spines elongate after stimulation of group 1 metabotropic glutamate receptors in cultured hippocampal neurons. Proc Natl Acad Sci USA 99: 1639-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 269: 5241-5248. [PubMed] [Google Scholar]
- Waltereit R, Weller M (2003) Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol Neurobiol 27: 99-106. [DOI] [PubMed] [Google Scholar]
- Wong WT, Sanes JR, Wong RO (1998) Developmentally regulated spontaneous activity in the embryonic chick retina. J Neurosci 18: 8839-8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Li Y, Zhang Z, Cui K, Wang S, Yuan XB, Wu CP, Poo MM, Duan S (2002) Nerve growth cone guidance mediated by G protein-coupled receptors. Nat Neurosci 5: 843-848. [DOI] [PubMed] [Google Scholar]
- Young RW (1985) Cell differentiation in the retina of the mouse. Anat Rec 212: 199-205. [DOI] [PubMed] [Google Scholar]
- Yu TW, Bargmann CI (2001) Dynamic regulation of axon guidance. Nat Neurosci [Suppl] 4: 1169-1176. [DOI] [PubMed] [Google Scholar]
- Yuan XB, Jin M, Xu X, Song YQ, Wu CP, Poo MM, Duan S (2003) Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat Cell Biol 5: 38-45. [DOI] [PubMed] [Google Scholar]
- Zheng JQ, Felder M, Connor JA, Poo MM (1994) Turning of nerve growth cones induced by neurotransmitters. Nature 368: 140-144. [DOI] [PubMed] [Google Scholar]
- Zhu H, Ryan K, Chen S (1999) Cloning of novel splice variants of mouse mGluR1. Brain Res Mol Brain Res 73: 93-103. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ (1996) Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 17: 91-102. [DOI] [PubMed] [Google Scholar]