

Abstract
Our data suggest a novel mechanism whereby pathological-length polyglutamine (polyQ) proteins promote the spermine synthetic pathway, increasing polyQ-aggregation and cell death. As detected in a cell-free turbidity assay, spermine promotes aggregation of thio-polyQ62 in a dose-dependent manner. Using a stable neuronal cell line expressing pathological-length [polyQ57-yellow fluorescent protein (YFP) (Q57)] or non-pathological-length [polyQ19-YFP (Q19)] polyglutamine protein, we show that multiple steps in the production of polyamines are affected in Q57 cells, suggesting dysfunctional spermine homeostasis. As the building block for spermine synthesis, arginine transport is significantly increased in neuronal cell lines stably expressing Q57. Q57 lines displayed upregulated basal and inducible arginase I activities that were not seen in polyQ19-YFP lines. Normal induction of spermidine/spermine N-acetyltransferase in Q19 lines regulating back-conversion of spermine, thereby reducing spermine levels, however, was not observed in Q57 lines. Pharmacological activation of ornithine decarboxylase (ODC), a key enzyme of the polyamine synthetic pathway, increased cellular aggregates and increased cell death in Q57 cells not observed in Q19 cells. Inhibition of ODC by difluoromethylornithine prevented basal and induced cell death in Q57 cells, demonstrating a central role for polyamines in this process.
Keywords: spermine, polyglutamine, Huntington's disease, difluoromethylornithine, protein aggregation, nitric oxide
Introduction
Polyglutamine-repeat diseases, a group of at least eight disorders including Huntington's disease (HD), result from the expression of mutant proteins containing an expanded polyglutamine domain (Feigin and Zgaljardic, 2002; Zoghbi and Botas, 2002). The pathological-length protein in HD, huntingtin (Htt), aggregates in the cytoplasm and nucleus and kill cells. Among the neuronal populations affected in HD, the medium spiny neurons in the striatum are most susceptible, and >50% of this class of neuron die (Sieradzan and Mann, 2001). The specific loss of these neurons appears paradoxical because the mutant Htt is expressed widely throughout the CNS and in peripheral, non-neuronal tissues (Ferrante et al., 1997; Sathasivam et al., 1999; Sieradzan and Mann, 2001; Luthi-Carter et al., 2002).
Glutamate-mediated excitotoxicity is an accepted mechanism for the death of medium spiny neurons in HD (Petersen et al., 1999; Sieradzan and Mann, 2001). These neurons receive input from glutamatergic cortical neurons projecting into the striatum from cortical layers V and VI (Calabresi et al., 2000; Sieradzan and Mann, 2001; Zeron et al., 2001). They express ionotropic glutamate receptors, which, when excessively activated, result in excitotoxic cell death (Bruyn and Stoof, 1990; Stefani et al., 1998; Chen et al., 1999; Levine et al., 1999; Cicchetti et al., 2000; Cepeda et al., 2001; Zeron et al., 2001; VanDamme et al., 2002). Glutamate receptor excitotoxicity is modulated by spermine and nitric oxide (NO) through the regulation of specific subtypes of glutamate receptors (Arandea et al., 1999; Gbadegesin et al., 1999). These same glutamate receptor subtypes are implicated in the striatal and cortical neuronal loss in HD (Stefani et al., 1998; Zeron et al., 2001).
Our study explores the involvement of spermine and NO in modulating neuronal loss in the polyglutamine-repeat diseases. Interestingly, NO and spermine synthesis are linked through a common precursor, arginine. Arginine is the sole substrate for both nitric oxide synthase (NOS), in NO synthesis, and arginase I and II, in spermine synthesis. Because intracellular arginine levels are rate-limiting for the production of ornithine by arginase and of NO by NOS (Wu and Morris, 1998; Boucher et al., 1999), competition for intracellular arginine is an important mechanism regulating these two enzymatic pathways. To study polyglutamine (polyQ) protein-mediated toxicity, CAD cells, a tyrosine hydroxylase-positive neuronal cell line, were transduced with trans-lentiviral vectors using the cellular polypeptide chain elongation factor 1 α (EF1α) promoter to express either polyQ19-yellow fluorescent protein (YFP) (a nonpathological polyQ domain) or polyQ57-YFP (a pathological-length domain).
The presence of pathological-length polyglutamine protein in cells expressing the pathological-length polyglutamine [polyQ57 (Q57)] protein shifts arginine use to arginase I (and the polyamine synthetic pathway) and away from NOS (reducing production of NO). Furthermore, we find that cell death is increased when this polyamine production pathway is activated and is inhibited by a specific antagonist of ornithine decarboxylase, a regulatory enzyme critical to the production of spermine. Activation of the polyamine synthetic pathway is also associated with increased aggregation of polyQ proteins in situ and promotes pathological-length polyQ protein aggregation in vitro. These changes we observe in the pathway producing spermine provide novel insights into the mechanism for cell death in cells expressing pathological-length polyQ protein.
Materials and Methods
Cell culture. CAD cells were a generous gift from Dr. Dona Chikaraishi (Neurobiology Department, Duke University Medical Center, Durham, NC) and were cultured at 37°C in DMEM and F-12 medium supplemented with 8% fetal bovine serum (Hyclone, Logan, UT), 100 U/ml penicillin, and 100 μg/ml streptomycin (Qi et al., 1997). CAD cell derivation, phenotypic characterization, and NO production have been described previously (Qi et al., 1997; Wang and Oxford, 2000; Xu and Colton, 2004).
Cell transduction. CAD cells were plated into 24-well dishes and incubated for 4 hr with a trans-lentiviral vector [Tranzyme vector (TZV)] containing a polyQn-YFP fusion construct (1-5 × 105 transforming units per well). The construct has been previously described (Nagai et al., 2000) and incorporates XhoI and NotI sites to facilitate insertion into the gene transfer component of the split-function lentivector system designed by Tranzyme Inc. This gene transfer component also contains an internal ribosome entry site-driven puromycin resistance gene making the trans-lentiviral vector medium of TZV-EF1α-polyQn-YFP capable of bicistronic message expression and allowing the selection of the transduced cells. Gene expression was visible within 48 hr after transduction in the form of YFP fluorescence. The transduced cells were replated and cultured under normal growth conditions. After the third replating, puromycin (2 μg/ml) was included in the normal growth media to select for transduced cells.
NO production. NO production was determined from the supernatant level of nitrite, the stable oxidation product of NO in biological solutions, using a Seivers 280 nitric oxide analyzer (Seivers Instruments Inc., Boulder, CO) as previously described (Xu and Colton, 2004). Protein content was determined using a BCA assay (Pierce, Rockford, IL), and the data were normalized to nanomolar concentration of nitrite per microgram of protein. Average values were obtained from four to six wells from each plate for a minimum of three different culture groups. Significance was determined using a Student's t test or ANOVA with the Bonferroni correction (GraphPad Software Inc., San Diego, CA).
Arginine uptake. Arginine uptake was measured essentially as described by Colton et al. (2001). CAD cells were plated into 24-well dishes and preequilibrated in serum-free media containing low arginine (10 μm). After 30 min, the low-arginine media were replaced with uptake buffer (in mm: 137 NaCl, 2.7 KCl, 1.5 CaCl2, 1.2 KH2PO4, 1.0 MgSO4, and 20 HEPES, pH7.4) containing a defined arginine concentration (from 3 μm to1mm) plus [3H]arginine (1 μCi/ml). Uptake was followed for 4 min at 37°C and stopped by washing with ice-cold uptake buffer containing 10 mm unlabeled arginine. Cells were then lysed in 1.0% Triton X-100, and radioactivity was measured by liquid scintillation counting. Specific arginine uptake was determined from the difference between uptake at 4°C (nonspecific) and uptake at 37°C (total uptake). At least six wells from three different culture groups were assayed, and uptake values were normalized to microgram of protein.
Arginase activity. Total cellular arginase enzymatic activity was determined as described by Corraliza et al. (1994). Cells plated into 100 mm dishes were lysed in 50 mm Tris HCl, pH 7.5, and 0.1% Triton X-100 containing 1× protease inhibitor mixture (Sigma, St. Louis, MO). Ten micromolar MnCl2 in 50 mm Tris-HCl, pH 7.5, was added to each cell sample, and the arginase was activated by heating to 55°C for 10 min. After the addition of 0.5 m arginine, pH 9.7, the reaction mixture was incubated at 37°C for 60 min, and the reaction was stopped by the addition of H2SO4, H3PO4, and H2O (1:3:7, v/v). Nine percent α-isonitrosopropio phenone was then added, and the samples were heated for 45 min at 100°C. After additional incubation for 10 min in the dark at 25°C, the absorbance at 540 nm was measured, and the content of urea was determined from a standard curve using varying concentrations of urea. Data are presented as the average micromole urea per microgram of protein per minute ± SEM.
Reverse transcription-PCR and Western blot. Total RNA was isolated for cell samples using an RNeasy kit (Qiagen, Valencia, CA). RNA was reverse-transcribed to cDNA using a High-Capacity cDNA archive kit (Applied Biosystems, Foster City, CA) with MultiScribe reverse transcriptase and random primers. The resulting cDNA templates were then mixed with target Assay-on-Demand primer/probe sets for murine arginase I (AGI), AGII, NOS1, or spermidine/spermine acetyltransferase (SSAT) and Taqman universal PCR master mix (Applied Biosystems). YFP mRNA expression was analyzed by reverse transcription (RT)-PCR using a YFP PCR primer set (BD Biosciences, San Jose, CA). The PCR amplification was performed using an Applied Biosystems ABI 7000HT sequence detection system, and the threshold cycle (CT, the cycle number at which the amount of amplified target gene reaches a fixed threshold) was determined for each sample. Relative quantitation of mRNA expression was calculated by the comparative CT method with the amount of target = 2-ΔΔCT (Livak and Schmittgen, 2001). Data are presented as the average fold change for triplicate samples from three different culture groups per experimental condition in which the value of the target was normalized to that of the endogenous control (18s) and relative to a calibrator. The calibrator in this case was untreated cells expressing Q19.
For Western blot of nNOS and arginase I, CAD cells were harvested directly into lysis buffer and boiled in 2× Laemmli sample buffer, and the protein amount was quantified by the BCA assay (Pierce). Equal amounts of lysate proteins were size-separated by Tris-glycine PAGE, electroblotted to an Immobilon-P membrane (Millipore, Bedford, MA), and probed with monoclonal antibodies against nNOS (BD Biosciences; 1:2000), arginase 1 (BD Biosciences; 1:1000) or α-tubulin (Sigma; 1:2500). After incubation with the appropriate secondary antibody coupled to peroxidase, immunoreactive bands were revealed by ECL. Band density was analyzed using Kodak 1D image analysis software (Eastman Kodak Co., Rochester, NY)
In vitro aggregation assay. Aggregation of a thioredoxin-polyglutamine fusion protein, thio-Qn, for which n = 62 glutamines, was determined by a turbidometric assay. Incubations were performed in a low-protein-binding 96-well plate in a cell-free system in varying concentrations of spermine (100 μm-1 mm) or other polyamines and a concentration of thio-Q62 previously shown to form aggregates (17 μm) (Nagai et al., 2000; Ren et al., 2001). Incubations were performed at 37°C in PBS and were assayed continuously over time using a Thermo Max plate reader (Molecular Devices, Sunnyvale, CA).
Aggregate filter assay. Total cellular aggregate was measured using a filter retardation assay as described by Heiser et al. (2002). Cells were homogenized into PBS and then passed through a cellulose acetate membrane (0.2 μm, OE66; Schleicher & Schuell, Keene, NH) using vacuum filtration. Multiple-sized aggregates were retained on the filter, whereas nonaggregated polyQ proteins passed through the filter. The aggregate level was measured using an indirect immunoassay for YFP, and immunoreactivity was detected using ECL. Band density was determined as described above for the Western blot.
Cell proliferation. Q19 and Q57 cell proliferation was determined at specific time points after plating by measuring total DNA as an index of cell number (FluoReporter blue fluorometric double-stranded DNA quantitation kit; Molecular Probes, Eugene, OR) and bromodeoxyuridine (BrdU) incorporation into DNA (cell proliferation ELISA; Roche Applied Science, Penzberg, Germany) following the manufacturer's instructions. Significance was determined using a repeated measures (twoway) ANOVA for n = 12-24 wells analyzed (GraphPad Software Inc.).
Cell toxicity. Cell viability was determined using propidium iodide (PI). Cells were treated for 15 min at 37°C with 50 μg/ml PI added to the medium, washed with PBS, and then either viewed under fluorescent microscopy or cell-sorted using a fluorescence-activated cell sorter (FACS). For microscopy, PI-stained cells were counted as a percentage of the total cells for a minimum of 10 random fields per coverslip using a minimum of three coverslips per experimental condition for at least triplicate experiments. For cytometry, 10,000 cells per experimental sample were loaded into the FACS, and PI fluorescence was determined as a percentage of total cells. Data are presented as percentage dead cells ± SEM. Significance was determined using a Student's t test or ANOVA with the Bonferroni correction for at least three different culture groups per experimental condition (GraphPad Software Inc.).
Results
Pathological-length polyQ proteins aggregate and kill cells
Stable expression of polyQ19-YFP or polyQ57-YFP was observed in separate CAD cell lines, as demonstrated by the presence of YFP-mediated cellular fluorescence (Fig. 1A-E). Highly fluorescent aggregates were observed in both nuclear (Fig. 1D) and cytoplasmic (Fig. 1E) locations in CAD cells transduced with polyQ57-YFP. In contrast, aggregates were not visible in polyQ19-YFP cells (Fig. 1A,B). Analysis of YFP mRNA demonstrated approximately equal expression of the polyQ19-YFP and polyQ57-YFP protein mRNA (Fig. 1F), suggesting that the level of viral transduction between the two cell lines was equivalent. Expression of polyQ57-YFP protein altered cell proliferation and was toxic to CAD cells. Q57 cell number as measured by total DNA was significantly depressed compared with Q19 cells up to ∼36 hr after initial equal plating (Fig. 1G). The change in cell number was attributable both to increased cell death (Fig. 1H) and to decreased proliferation, as measured by incorporation of labeled BrdU into DNA (Fig. 1 I) in Q57 cells compared with Q19 cells.
Figure 1.
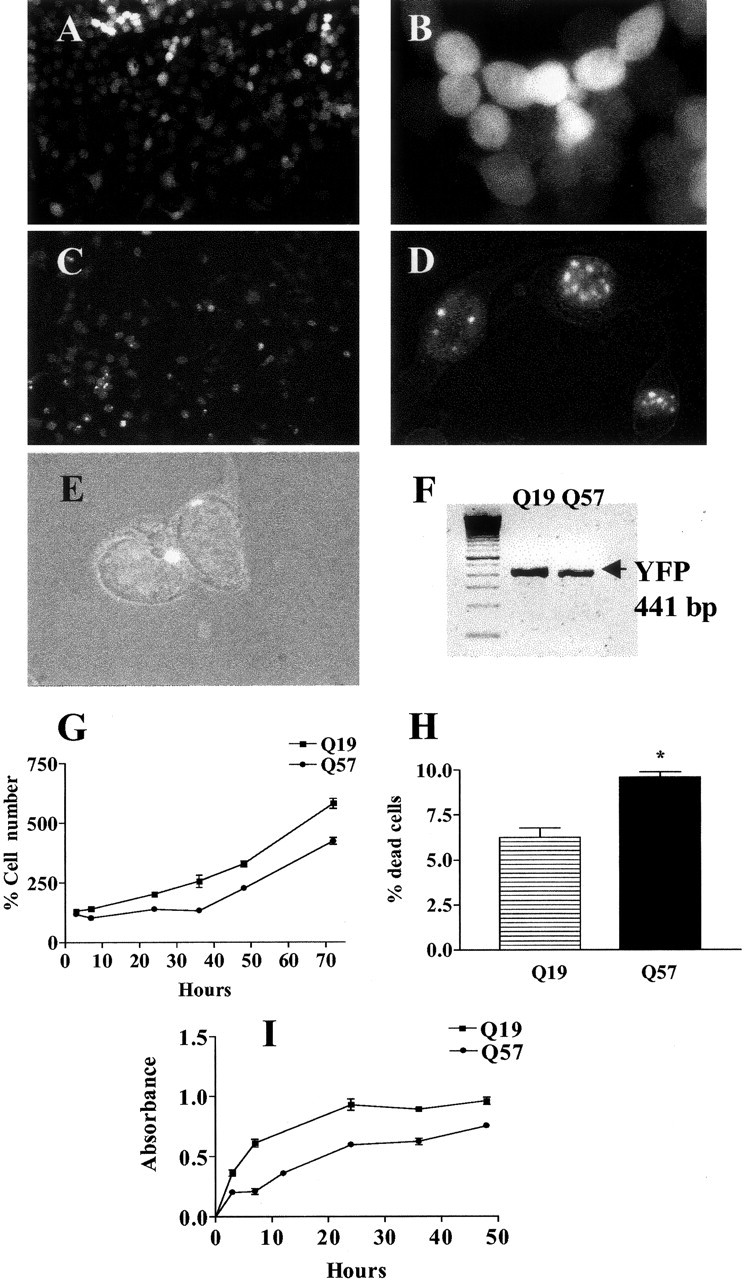
Properties of CAD cells transduced with a lentiviral vector containing polyQ19-YFP or polyQ57-YFP fusion constructs. Low-magnification (10×) views of CAD cells expressing polyQ19 YFP (A) and polyQ57-YFP (C) are shown. Highly fluorescent, visible intracellular inclusions are observed only in cells expressing polyQ57-YFP (C-E) and not in cells expressing polyQ19-YFP (A, B). As viewed by fluorescent microscopy or the superimposition of fluorescence and Hoffman optics, aggregates are localized to nuclear (D) or perinuclear (E) locations in Q57 cells (magnification, 40×). F, YFP mRNA expression in polyQ19-YFP and polyQ57-YFP cells by RT-PCR. G, The percentage cell number was determined for polyQ57-YFP and polyQ19-YFP cells at various time points after initial equal plating (t = 0) using a fluorometric measurement of DNA. Average percentage ± SEM was determined from the ratio of DNA content at 3-72 hr to DNA content at t = 0 × 100. Curves are significantly different at p < 0.0001 using two-way ANOVA; n = 12-24 wells analyzed. H, Average percentage cell death of Q57 and Q19 cells obtainedby fluorescence cytometry using PI at 36 hr after plating. Percentage death = number of PI+ cells/total amount of cells × 100. *p < 0.006 using the Student's t test with n = 3 separate culture groups. I, BrdU incorporation into Q19 and Q57 cells over time. Curves are significantly different at p < 0.0001 using two-way ANOVA; n = 12-24 wells analyzed.
Pathological-length polyQ cells do not increase NO production, whereas wild-type polyQ cells do
To determine the effect of the pathological-length polyQ57-YFP protein on NO production, nNOS activity was stimulated with ionomycin (3 μm). Ionomycin promotes Ca2+ entry into cells. The resultant increase in intracellular Ca2+ activates nNOS and initiates NO production by the enzymatic conversion of arginine to citrulline and NO (Kiedrowski et al., 1992; Weikert et al., 1997; Xu and Colton, 2004). As shown in Figure 2A, ionomycin increased supernatant nitrite levels in polyQ19-YFP cells but not in polyQ57-YFP cells. Decreased NO levels in polyQ57 cells were not attributable to decreased NOS1 levels. No significant difference was observed in NOS1 mRNA when using quantitative RT PCR to compare polyQ19-YFP and polyQ57-YFP cells (Fig. 2B). In addition, Western blot analysis demonstrated equivalent amounts of nNOS in polyQ57-YFP and polyQ19-YFP cells (Fig. 2C).
Figure 2.
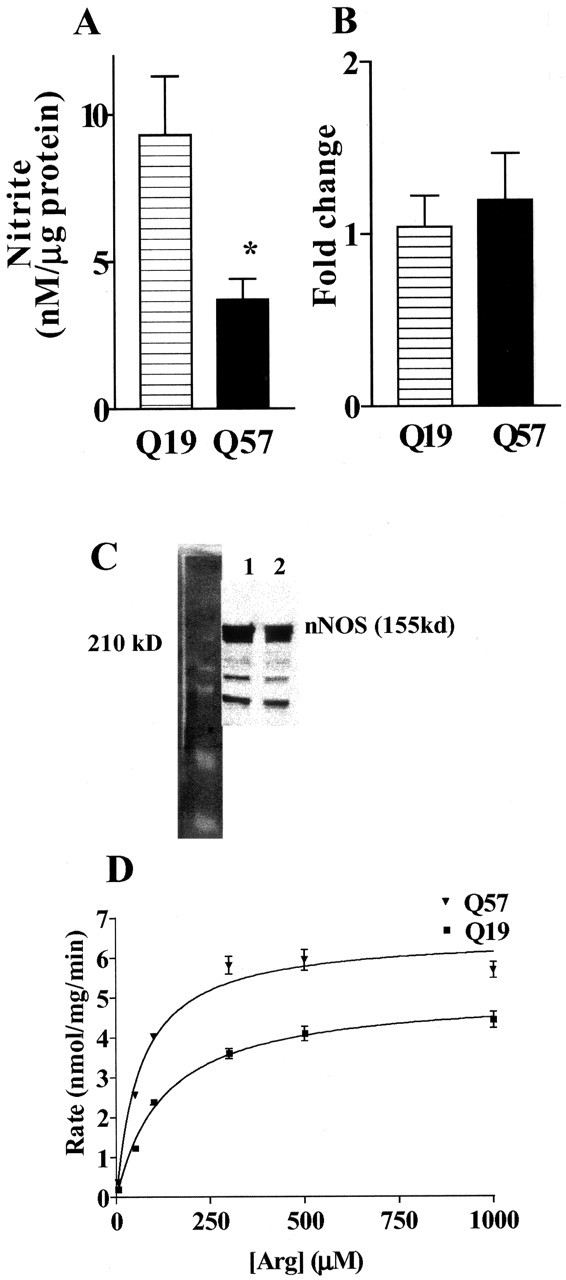
NO production and arginine uptake are different in polyQ57-YFP cells. A, Q19 and Q57 cells were plated for 72 hr and then treated with 3 μm ionomycin to initiate NO production by nNOS. Supernatant nitrite values were measured after 5 hr of treatment. Background (untreated) nitrite values were subtracted from ionomycin-treated values. *p < 0.04 compared with polyQ57-YFP cells; n = 12 wells assayed for three different culture groups. B, C, NOS1 mRNA (B) and nNOS protein (C) levels were determined for separate sister cultures. Fold changes in mRNA expression/18s was determined using quantitative real-time RT-PCR and the comparative CT method in which Q57 is compared with Q19. Lane 1, Q19; lane 2, Q57. D, Specific [3H]arginine uptake was determined from the difference between total [3H]arginine uptake at 4 min at 37°C minus the nonspecific uptake under the same conditions at 4°C. p < 0.001; n = 16-24 wells analyzed.
Arginine transport is increased in pathological-length polyQ cells
Decreased availability of intracellular arginine may limit NO production in neurons. Intracellular arginine pools are primarily supplied by arginine transporters that move arginine across the plasma membrane to the interior of the cell. Although arginine transport can regulate NO production in macrophages (Kakuda et al., 1999; Colton et al., 2001), some neuronal populations maintain intracellular arginine pools by recycling citrulline through the citrulline-arginine-NO cycle, thus bypassing arginine uptake (Braissant et al., 1999; Heneka et al., 2001). To determine whether CAD cells synthesize arginine via this mechanism, we examined the expression of argininosuccinate lyase (AL) and argininosuccinate synthase (AS), the two enzymes necessary for the citrulline-arginine-NO cycle. CAD neurons express AL mRNA but do not express AS mRNA (data not shown), indicating that this cell line lacks a functional citrulline-arginine-NO cycle. Thus, arginine transport is a rate-limiting step in the production of NO in cells expressing polyQ57-YFP. To determine whether the decrease in NO production we observed in polyQ57-YFP cells was attributable to inhibition of arginine transport, we measured total arginine uptake using [3H]arginine. As shown in Figure 2D, [3H]arginine uptake in CAD cells expressing polyQ57-YFP protein was significantly higher than in cells expressing polyQ19-YFP protein. Therefore, the decreased NO production we observed in cells expressing pathological-length polyQ is not a result of decreased arginine transport, leading to decreased intracellular arginine pools.
Polyamine synthesis is upregulated in pathological-length polyQ cells
Decreased NO production, despite higher arginine uptake, suggests that arginine is used in polyQ57-YFP cells differently than in polyQ19-YFP cells. Thus, we measured mRNA and protein levels for AGI and AGII, enzymes that compete with nNOS for intracellular arginine and initiate polyamine synthesis (Jenkinson et al., 1996; Mori et al., 1998; Boucher et al., 1999; Kepka-Lenhart et al., 2000; Li et al., 2001; de Jonge et al., 2002; Que et al., 2002). Using quantitative RT-PCR, we observed a significant increase in AGI mRNA in CAD cells expressing the pathological-length polyQ protein compared with cells expressing the non-toxic-length polyQ (Figs. 3A, 4A). No significant difference between polyQ19-YFP and polyQ57-YFP cells was observed for AGII mRNA (data not shown). Increased AGI mRNA correlated with increased arginase I protein expression (Fig. 3B) and a significant increase in the basal levels of total arginase activity in polyQ57-YFP cells compared with polyQ19-YFP cells (Fig. 3C). To determine whether the effect of polyQ protein was unique to CAD cells, we also examined arginase I protein expression in transduced COS-7 cells expressing polyQ proteins. COS-7 cells, derived from green monkey kidney, were transduced with polyQ57-YFP or polyQ19-YFP using lentiviral vectors. As shown in Figure 3D, the level of arginase 1 protein in COS-7 cells expressing polyQ57-YFP was greater than in cells expressing polyQ19-YFP.
Figure 3.

Arginase I is increased by pathological-length polyQ protein. A, Quantitative real-time RT-PCR demonstrates a significant fold increase in mRNA expression/18s for AGI; *p < 0.01; n = 4 separate culture groups of each cell type. B, Total basal arginase activity determined for Q57 compared with Q19. *p < 0.002; n = 4 separate culture groups of each cell type. C, Representative Western blot for arginase I. Ratios are calculated as the intensity of arginase I to α-tubulin intensity in which α-tubulin is used as a loading control. D, Western blot for arginase I immunoreactivity in three separate cell lysate groups prepared from COS7 cells expressing polyQ57-YFP or polyQ19-YFP. Ratios are calculated as the intensity of arginase I to α-tubulin intensity. Average fold increase for Q57 compared with Q19 = 2.6 ± 0.07; p < 0.0001.
Figure 4.

AGI and SSAT mRNA expression in polyQ 57 cells stimulated with 8-Br cAMP and l-vNIO. Quantitative real-time RT-PCR was used to detect significant differences in AGI and SSAT mRNA at 2, 6, and 17 hr after treatment of Q19 and Q57 cells with 8-Br cAMP (0.5 mm) and l-vNIO (10 μm) to enhance polyamine synthesis. Fold changes are determined using the comparative CT method with Q19 untreated (unt) cells as the reference value. A, AGI mRNA expression was significantly higher in Q57 cells. *p < 0. 02; **p < 0.04; n = 3 or 4 separate culture groups. B, C, Fold changes in SSAT mRNA expression in Q19 (B) and Q57 (C) cells with 8-Br-cAMP (0.5 mm) and l-vNIO (10 μm) treatment. Inhibition of ODC using DMFO (100 μm) was used to determine specificity of activation. *p < 0.01; **p = 0.05.
Enhanced expression of arginase I in polyQ57-YFP cells was further demonstrated by measuring levels of AGI mRNA after induction with 8-bromo-cAMP (8 Br-cAMP) (Fig. 4). To stimulate arginase expression, CAD cells were treated with 8 Br-cAMP and (N5-1-imino-3-butenyl)-l-ornithine (l-vNIO), a specific nNOS inhibitor (Babu and Griffith, 1998). 8-Br-cAMP increases AGI mRNA and arginase I protein expression, whereas l-vNIO inhibits NOS, thereby decreasing feedback inhibition of arginase 1 with the enzymatic products of NOS, N-hydroxy arginine and NO (Buga et al., 1996; Bauer et al., 1999; Kepka-Lenhart et al., 2000). Separate control studies demonstrated that l-vNIO does not alter arginine uptake (data not shown). AG1 mRNA expression was measured using quantitative RT-PCR at 2, 6, and 17 hr after treatment with 8-Br-cAMP (0.5 mm) and l-vNIO (10 μm). As shown in Figure 4A, AG1 mRNA increased slightly by 6 hr of treatment in cells expressing polyQ19-YFP but did not reach significance. However, a significant increase was observed in polyQ57-YFP cells at 2 and 6 hr of treatment that returned to the baseline values for polyQ57 cells by 17 hr. These data demonstrate that treatment with 8-Br-cAMP and l-vNIO induces AGI above baseline expression.
Our data suggest that polyamine production increases in CAD cells expressing polyQ57-YFP compared with cells expressing polyQ19-YFP. To determine whether polyamine metabolism is altered in polyQ57 cells, we next examined SSAT, the enzyme that catalyzes the back-conversion of spermine to N-acetylspermine (Pohjanpelto et al., 1985; Shappell et al., 1993; Suppola et al., 2001; Wallace et al., 2003). SSAT mRNA and protein expression are tightly regulated by intracellular polyamine levels and are part of a feedback system controlling cytosolic spermine concentrations (Porter et al., 1990). Thus, SSAT mRNA increases with increased cytosolic spermine concentration (Shappell et al., 1993; Fogel-Petrovic et al., 1996; Shantz and Pegg, 1999; Suppola et al., 2001). No significant differences were observed in the basal level of SSAT mRNA between polyQ19-YFP and polyQ57-YFP CAD cells. However, when polyQ19-YFP cells were treated with 8-Br-cAMP and l-vNIO for 2, 6, and 17 hr, SSAT mRNA rapidly increased and returned to baseline values after 6 hr (Fig. 4B). In contrast, no changes in SSAT mRNA levels were observed in cells expressing polyQ57-YFP (Fig. 4C). To corroborate that changes in SSAT mRNA expression in polyQ19-YFP cells are attributable to increased ornithine decarboxylase (ODC) activity, the enzyme that catalyzes the synthesis of the first step in higher polyamine production, we used difluoromethylornithine (DFMO). DFMO is a highly selective suicide inhibitor of ODC and decreases the production of polyamines (Slotkin et al., 1982; Porter et al., 1990; Jacoby et al., 2003). DFMO inhibited the increase in SSAT mRNA produced by 8-Br-cAMP and l-vNIO (Fig. 4B). These data suggest that the rise in SSAT expression in polyQ19-YFP cells was attributable to an increase in spermine synthesis. This change was not observed in polyQ57-YFP cells.
Polyamines potentiate pathological-length polyQ protein aggregation in vitro and in situ
Recent data demonstrate that the polyamines spermine and spermidine bind with, and accelerate the aggregation of α-synuclein, the protein that forms abnormal intracellular deposits within neurons in Parkinson's disease (Antony et al., 2003; Goers et al., 2003). We first determined whether polyamines have similar effects on polyQ protein aggregation in vitro using a turbidometic assay. Aggregation of thio-polyQ62 in the presence and absence of varying concentrations of spermine (100-1000 μm) is shown in Figure 5A. Spermine dramatically increased the aggregation of thio-polyQ62 in a dose-dependent manner. Other polyamines were examined for their effect on polyQ62 aggregation. At 250 μm, spermine was the most effective aggregating agent. Similar concentrations of spermidine, N-acetylspermine, N-acetylspermidine, and putrescine had no effect (Fig. 5B). Theeffect of spermine was specific to polyglutamine aggregation because no aggregation of bovine serum albumin plus spermine was observed or of a thioredoxin protein without the polyglutamine repeat domain (data not shown).
Figure 5.

Spermine increases aggregation of a thioredoxin-polyglutamine fusion protein, thio-Q62, in a cell-free assay. Data points represent relative turbidity changes at 37°C in media containing 17 μm thio-Q62 in the presence and absence of increasing doses of spermine (100-1000 μm; A) or in the presence of 250 μm spermine, spermidine, putrescine, N-acetylspermine (Spm), or N-acetylspermidine (Spd) (B). Relative turbidity is determined using the maximum turbidity value at 50 × 103 sec for 1000 μm spermine (A) or at 50 × 103 sec for 250 μm spermine (B) as the comparator.
Our data demonstrate that the pathway for polyamine synthesis is enhanced in cells expressing pathological-length polyQ proteins. Because the polyamines spermine and spermidine promote polyQ aggregation in vitro, we determined whether increased activity of the arginase-ODC pathway alters aggregation of the polyQ57-YFP protein within cells. We counted the number of cells with visible fluorescent polyQ57-YFP aggregates and compared the number after 15 hr of treatment with 8-Br-cAMP and l-vNIO to untreated cells. As shown in Figure 6A, the percentage of cells with aggregates significantly increased with 8-Br-cAMP alone and increased further with the combination of 8-Br-cAMP and l-vNIO. The increase in aggregation was confirmed by a filter assay that detects polyglutamine aggregates in cell samples (Fig. 6 B).
Figure 6.

Increasing polyamine synthesis in CAD cells expressing polyQ57-YFP increases intracellular aggregate. A, The number of polyQ57-YFP cells with any size of visible fluorescent aggregates was counted, and the percentage of total cells was determined for cells treated with 8-Br-cAMP (0.5 mm) alone or in combination with l-vNIO (10 μm). *p < 0.005; **p < 0.003; n = triplicate coverslips counted (8-10 fields per coverslip) for at least three different culture groups. B, A filter assay was used to trap aggregate from cell lysates prepared from untreated and treated CAD cells. Cells were treated for 15 hr with either 8-Br-cAMP (0.5 mm) alone or in combination with l-vNIO (10 μm). Immunoreactivity for YFP was used to detect the polyQ aggregate.
Increasing polyamine synthesis increases polyQ-mediated cell death
Our data confirm previously published findings that cell death is greater in cells expressing pathological-length polyQ proteins (Figs. 1, 7) (Ferrante et al., 1997; Petersen et al., 1999; Ross, 2000; Wyttenbach et al., 2001). To examine whether stimulating the polyamine production pathway increased cell death caused by polyQ57-YFP, we measured the viability of cells when arginase and ODC activity was further enhanced by treatment with 8-Br-cAMP and l-vNIO. Using PI as an indicator of cell death, the number of dead cells significantly increased after 15 hr of 8-Br-cAMP treatment in polyQ57 cells but not in polyQ19 cells (Fig. 7). In addition, simultaneous treatment with 100 μm DFMO reduced both the overall baseline level of cell death and the 8-Br-cAMP-stimulated increase in cell death in cells expressing polyQ57. No change in cell death was observed in polyQ19 cells with DFMO.
Figure 7.

Cell death in polyQ57 cells is higher when polyamine synthesis is activated. Q57 and Q19 cells were treated for 15 hr with 8 Br-cAMP (0.5 mm) and l-vNIO (10 μm) in the presence and absence of 100 μm DFMO. PI was used to determine the percentage of dead cells of the total cells for two or three coverslips (8-10 fields per coverslip) for each of four separate culture groups. Data points represent population means ± SEM *p < 0.02 using an unpaired Student's t test; **p ≤ 0.054 using a paired Student's t test. Unt, Untreated.
Discussion
Our data suggest a novel mechanism by which pathological-length polyQ proteins promote their own aggregation and cell death by increasing the polyamine production pathway (Fig. 8). We propose that the tightly controlled feedback mechanisms regulating intracellular spermine levels are disrupted by pathological-length polyQ57-YFP protein but not by polyQ19-YFP protein. This disruption may result from spermine interaction with the polyQ57 protein, leading to a reduction of free intracellular spermine levels and promoting polyQ aggregation. Compensation by feedback pathways then leads to feed forward effects, increasing arginine use by the cell and increasing the activity of arginase I, thereby increasing the polyamine synthetic pathway at the expense of other arginine-dependent pathways such as nNOS.
Figure 8.
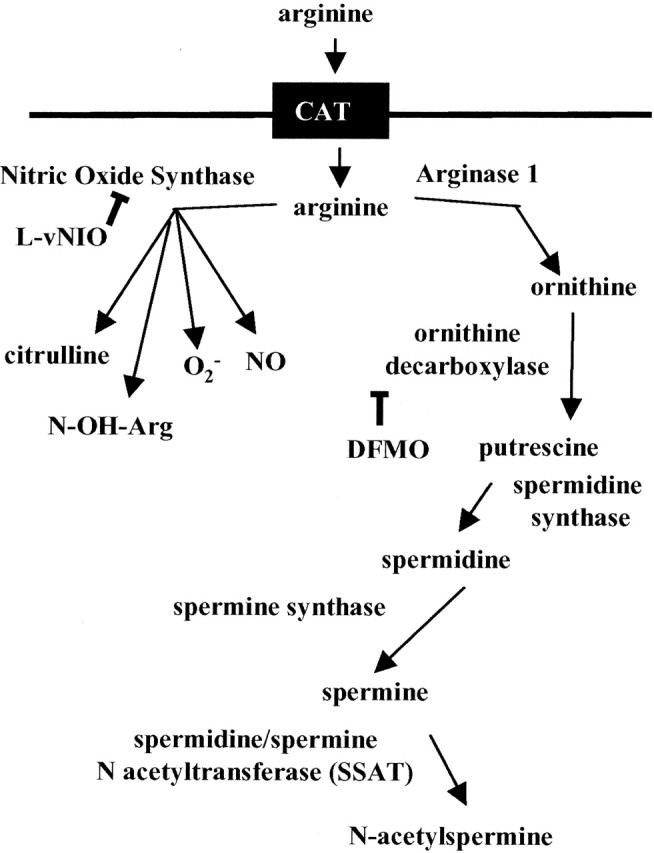
Schematic representation of arginine use by nitric oxide synthase and polyamine synthesis. CAT, Cationic amino acid transporter.
CNS arginase and nNOS
Arginase I and NOS are the primary enzymes in brain that require arginine as a substrate (Heneka et al., 2001). In cells that express both nNOS and arginase I, competition for arginine by the overexpression of arginase decreases NO production (Wu and Morris, 1998; Singh et al., 2000). Our data demonstrate that NO production is reduced in polyQ57 CAD cells, with no apparent change in NOS1 mRNA or nNOS protein levels and despite increased arginine uptake that provides more substrate to fuel NOS activity. Coupled with a significant increase in arginase I protein expression and activity, these findings suggest that arginase I successfully outcompetes nNOS for arginine in cells expressing the pathological-length polyQ protein (Boucher et al., 1999; Yu et al., 2001). Reduction in neuronal NO production is likely to significantly affect striatal function and may play a critical role in the observed motor deficits in HD. NO affects the normal physiological function of the striatum by setting the level of activity and responsiveness of neurons in the striatum (West and Grace, 2000; Saka et al., 2002). The importance of NO in the disease process has been confirmed by Deckel et al. (2001, 2002) using the R6/2 HD mouse HD model crossed with an nNOS knock-out mouse. Accelerated disease progression and a younger age of death were observed in this mouse, reinforcing an important role for nNOS in striatal function and in HD neuropathophysiology.
Arginase, ODC, and polyamine production
As shown in Figure 8, activation of arginase I and ornithine decarboxylase are initiating steps in polyamine production (Wallace et al., 2003). Both arginase I and ODC are feedback-controlled, regulatory enzymes for polyamine synthesis whose cellular overexpression increases production of polyamines and polyamine-mediated cellular effects (Kepka-Lenhart et al., 2000; Wei et al., 2001; Cai et al., 2002). The sustained increase in arginase I in polyQ57-YFP cells suggests dysfunctional homeostasis of polyamines not observed in polyQ19-YFP cells. This effect is not limited solely to neuronal cells coexpressing nNOS or to neurons, per se. A similar increase in arginase is observed in polyQ57-YFP COS-7 cells, a monkey kidney cell line that does not express nNOS. Although selectivity to specific neuronal subpopulations is a hallmark of the polyQ diseases, non-neuronal cells are also damaged by pathological Htt in Huntington's disease (Sathasivam et al., 1999; Luthi-Carter et al., 2002).
Polyamine regulation
Spermine is formed by sequential addition of aminopropyl groups derived from decarboxylated S-adenosylmethionine onto the core diamine (Fig. 8). Despite disruption of synthetic pathways, changes in intracellular spermine levels are difficult to observe because of multilevel homeostatic mechanisms controlling spermine concentrations (Porter et al., 1990; Halmekyto et al., 1991; Fogel-Petrovic et al., 1996; Hayashi et al., 1996; Pietila et al., 1997; Morrison et al., 1998; Kepka-Lenhart et al., 2000; Suppola et al., 2001). SSAT, the enzyme that catalyzes the back-conversion of spermine to N-acetyl forms, is tightly regulated by intracellular spermine. Under normal physiological conditions, increased cytosolic spermine induces a rapid, transient rise in SSAT mRNA and protein (Shappell et al., 1993; Fogel-Petrovic et al., 1996; Shantz and Pegg, 1999; Suppola et al., 1999). As a consequence, spermine levels are lowered by the back-conversion of spermine to N-acetylspermine that is either transported out of the cells or recycled to lower polyamines. This tight feedback regulation of SSAT mRNA by intracellular spermine levels can be used to indicate cellular spermine production (Shappell et al., 1993; Pietila et al., 1997). CAD cells expressing polyQ19-YFP have a functional feedback control system because activation of polyamine production by 8-Br-cAMP plus l-vNIO resulted in increased and then decreased SSAT mRNA as predicted from its role in the homeostatic control of free spermine levels inside the cell. In addition, the specific suicide inhibitor of ODC, DFMO, reduced the level of SSAT mRNA, demonstrating that the increase in SSAT mRNA in poly19Q cells was attributable to the increased spermine production initiated by 8-Br-cAMP plus l-vNIO. In contrast, SSAT mRNA levels did not change in polyQ57-YFP cells under the same conditions. This lack of induction of SSAT suggests that the normal homeostatic mechanisms designed to control spermine levels were not initiated in Q57 cells. One possible explanation is that effective intracellular spermine levels are reduced by interaction of spermine with pathological-length polyQ protein but are not reduced by the presence of non-pathological-length polyQ proteins. Vivo et al. (2001) found a reduction in striatal spermine concentrations in brain from HD patients. This change was not attributable solely to neuronal loss because putrescine concentrations were not decreased.
Polyamines and polyglutamine aggregation
Polyamines are multivalent cations that participate in cell proliferation and differentiation, are tightly regulated in most tissues, and play key roles in cell physiology (Williams, 1997; Childs et al., 2003). Polyamines stabilize DNA, promote binding of transcription factors, and enhance coactivator function (Thomas and Thomas, 2001; Childs et al., 2003). Both increased and decreased polyamine levels affect nuclear function. Spermine regulates ion channels such as the NMDA, glutamate receptor 1 (GluR1) and immunoreactive potassium channels (Williams, 1997; Dingledine et al., 1999), alters redox balance of the cell, and modulates immune responsiveness (Petersen et al., 1999; Soulet and Rivest, 2003).
Importantly, spermine complexes with and accelerates the aggregation of α-synuclein, a protein that forms abnormal intracellular deposits within neurons in Parkinson's disease (Antony et al., 2003; Goers et al., 2003). We demonstrate a similar phenomenon with pathological-length polyQ proteins. Addition of physiological concentrations of spermine to thioredoxin-polyQ62 protein in vitro increased aggregation compared with thioredoxinpolyQ62 protein alone. Interaction of spermine with pathological-length polyamine proteins not only may promote toxic aggregate formation but also may disrupt the normal homeostasis of polyamines within the cell. We postulate that the resultant fall in polyamines in certain cells may initiate a series of physiological changes that damage cell function. These actions are not mutually exclusive from the effects of pathological length polyQ proteins.
Conditions that increased polyamine production enhanced formation of aggregates in cells expressing polyQ57 proteins. Treatment of polyQ57 cells with 8-Br-cAMP plus l-vNIO significantly increased the number of cells with visible aggregates and the amount of aggregate detected by a filter assay. This is likely to reflect a true increase in aggregate formation (rather than preservation of cells with aggregate) because 8-Br-cAMP plus l-vNIO increased cell death.
Polyamines, polyglutamine, and cell viability
Although cell viability is reduced by pathological-length polyQ, we cannot distinguish between cell toxicity initiated directly by PolyQ57-YFP from cell toxicity secondarily initiated by the dysregulation of polyamine homeostasis. The increase in cell death by 8-Br-cAMP plus l-vNIO and the reduction of cell death by DFMO strongly suggest a direct role for intracellular polyamines in the toxicity of polyQ57. High levels of spermine induce apoptosis, via oxidative mechanisms involving the increased production of H2O2 by polyamine oxidase, a key enzyme in the back-conversion of polyamines (Porter et al., 1990; Wallace et al., 2003). Our data, however, suggest that intracellular levels of free spermine are reduced by pathological-length polyQ proteins, rather than increased. Low intracellular levels of spermine are also associated with increased cell death. Mutant Gy male mice lack functional spermine synthase, have low spermine levels, and show increased sensitivity to the chloroethlyating agent 1,2-bis (2-chloroethyl)-N-nitrosourea (Meyer et al., 1998; Mackintosh and Pegg, 2003). Also, caspase activation and apoptotic cell death initiated by UV radiation was greatly increased in the Gy mouse. For medium spiny neurons in the HD striatum, changes in spermine levels are likely to alter glutamate-mediated synaptic currents. Spermine regulates specific NMDA channel subunit combinations and GluR1 AMPA receptors (Williams et al., 1994; Donevan and Rogawski, 1995; Washburn and Dingledine, 1996; Arandea et al., 1999; Pellegrini-Giampietro, 2003). These same subunit combinations are expressed in the striatum (Kuppenbender et al., 1999, 2000) and are implicated in the striatal and cortical neuronal loss in HD (Zeron et al., 2001, 2002; Li et al., 2003). The profound change in polyamine synthesis in cells expressing pathological-length polyQ provides a novel approach to deciphering differential neuronal dysfunction in polyglutamine diseases.
Footnotes
This work was supported in part by National Institutes of Health Grants AG19740 (C.A.C.), AG19780 (M.P.V.), and NS40540 (J.R.B.). There is no financial conflict of interest for any author that might be construed to influence the results or interpretation of this manuscript. We thank Dr. John Whitesides (Department of Medicine, Human Vaccine Institute, Duke University Medical Center, Durham, NC) for assistance in analyzing the flow cytometry data, Dr. R. Ramabhadran (Tranzyme Inc., Research Triangle Park, NC) for trans-lentiviral vectors, Tim Tucker for technical assistance, and Dr. Kasturi Puranam for preparing the transduced cell lines.
Correspondence should be addressed to Dr. Carol A. Colton, Division of Neurology, Box 2900, Bryan Research Building, Duke University Medical Center, Durham, NC 27710. E-mail: glia01@aol.com.
Copyright © 2004 Society for Neuroscience 0270-6474/04/247118-10$15.00/0
References
- Antony T, Hoyer W, Chernyl W, Heim G, Jovin T, Subramaniam V (2003) Cellular polyamines promote the aggregation of α-synuclein. J Biol Chem 278: 3235-3240. [DOI] [PubMed] [Google Scholar]
- Arandea R, Lan J-Y, Zheng X, Zukin S, Bennett MVL (1999) Spermine and arcaine block and permeate N-Methyl-d-aspartate receptor channels. Biophys J 76: 2899-2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu B, Griffith O (1998) N5-(1-imino-3-butenyl)-l-ornithine: a neuronal isoform selective mechanism based inactivator of nitric oxide synthase. J Biol Chem 273: 8882-8889. [DOI] [PubMed] [Google Scholar]
- Bauer P, Fukuto J, Buga G, Pegg A, Ignarro L (1999) Nitric oxide inhibits ornithine decarboxylase by S-nitrosylation. Biochem Biophys Res Commun 262: 355-358. [DOI] [PubMed] [Google Scholar]
- Boucher JL, Moali C, Tenu JP (1999) Nitric oxide biosynthesis, nitric oxide synthase inhibitors and arginase competition for l-arginine utilization Cell Mol Life Sci 55: 1015-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braissant O, Gotoh T, Loup M, Mori M, Bachmann C (1999) l-Arginine uptake, the citrulline-NO cycle and arginase II in the rat brain, an in situ hybridization study. Brain Res Mol Brain Res 70: 231-241. [DOI] [PubMed] [Google Scholar]
- Bruyn R, Stoof J (1990) The quinolinic acid hypothesis in Huntington's chorea. J Neurol Sci 95: 29-38. [DOI] [PubMed] [Google Scholar]
- Buga G, Singh R, Pervin S, Rogers N, Schmitz DA, Jenkinson C, Cederbaum SD, Ignarro LJ (1996) Arginase activity in endothelial cells: inhibition by NG-hydroxy l-arginine during high output NO production. Am J Physiol 271: H1988-H1998. [DOI] [PubMed] [Google Scholar]
- Cai D, Deng K, Mellado W, Lee J, Ratan R, Filbin M (2002) Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron 35: 711-719. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Marfia GA, Pisani A, Sancesario G, Bernardi G (2000) Synaptic transmission in the striatum: from plasticity to neurodegeneration. Prog Neurobiol 61: 231-265. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Ariano M, Calvert CR, Flores-Hernandez J, Chandler S, Leavitt R, Hayden M, Levine M (2001) NMDA receptor function in mouse models of Huntington disease. J Neurosci Res 66: 525-539. [DOI] [PubMed] [Google Scholar]
- Chen N, Luo T, Wellington C, Metzler M, McCutcheon K, Hayden MR, Raymond LA (1999) Subtype specific enhancement of NMDA receptor currents by mutant Huntingtin. J Neurochem 72: 1890-1898. [DOI] [PubMed] [Google Scholar]
- Childs A, Mehta DJ, Gerner EW (2003) Polyamine-dependent gene expression. Cell Mol Life Sci 60: 1394-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti F, Prensa L, Wu Y, Parent A (2000) Chemical anatomy of striatal interneurons in normal individuals and in patients with Huntington's disease. Brain Res Rev 34: 80-101. [DOI] [PubMed] [Google Scholar]
- Colton CA, Czapiga M, Snell-Callanan J, Chernyshev O, Vitek MP (2001) Apolipoprotein E acts to increase nitric oxide production in macrophages by stimulating arginine transport. Biochim Biophys Acta 1535: 134-144. [DOI] [PubMed] [Google Scholar]
- Corraliza I, Campo M, Soler G, Modolell M (1994) Determination of arginase activity in macrophages: a micromethod. J Immunol Methods 174: 231-235. [DOI] [PubMed] [Google Scholar]
- Deckel W, Gordinier A, Nuttal D, Tang D, Kuwada C, Frietas R, Gary K (2001) Reduced activity and protein expression of NOS in R6/2 HD transgenic mice: effects of l-NAME on symptom progression. Brain Res 919: 70-81. [DOI] [PubMed] [Google Scholar]
- Deckel W, Tang V, Nuttal D, Gary K, Elder R (2002) Altered neuronal nitric oxide synthase expression contributes to disease progression in Huntington's disease transgenic mice. Brain Res 939: 76-86. [DOI] [PubMed] [Google Scholar]
- de Jonge WJ, Hallemeesch MM, Kwikkers KL, Ruitjer JM, de Gier-de Vries C, van Roon MA, Meijer AJ, Marescau B, de Deyn PP, Deutz NE, Lamers WH (2002) Overexpression of arginase I in enterocytes of transgenic mice elicits a selective arginine deficiency and affects skin, muscle and lymphoid development. Am J Clin Nutr 76: 128-140. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Donevan S, Rogawski M (1995) Intracellular polyamines mediate inward rectification of Ca2+ permeable α-amino-3-hydroxy-5-methyl-4-isoxaleproprionic acid receptors. Proc Natl Acad Sci USA 92: 9298-9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigin A, Zgaljardic D (2002) Recent advances in Huntington's disease: implication for experimental therapeutics. Curr Opin Neurol 15: 483-489. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Gutekunst CA, Perischetti F, McNeil SM, Kowall NW, Gusella JF, MacDonald ME, Beal MF, Hersch SM (1997) Heterogenous topographic and cellular distribution of huntingtin expression in the normal human neostriatum. J Neurosci 17: 3052-3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel-Petrovic M, Vujcic S, Miller J, Porter C (1996) Differential post-transcriptional control of ornithine decarboxylase and spermidinespermine N1-acetyltransferase by polyamines. FEBS Lett 391: 89-94. [DOI] [PubMed] [Google Scholar]
- Gbadegesin M, Vicini S, Hewett S, Wink D, Espey M, Pluta R, Colton C (1999) Hypoxia modulates nitric oxide-induced regulation of NMDA receptor currents and neuronal cell death. Am J Physiol 277: C673-C683. [DOI] [PubMed] [Google Scholar]
- Goers J, Uversky V, Fink A (2003) Polycation induced oligomerization and accelerated fibrillation of human α-synuclein in vitro. Protein Sci 12: 702-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halmekyto M, Alhonen L, Wahlfors J, Sinervirta R, Eloranta T, Janne J (1991) Characterization of a transgenic mouse line over-expressing the human ornithine decarboxylase gene. Biochem J 278: 895-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Murkai Y, Matsufuji S (1996) Ornithine decarboxylase antizyme: a novel type of regulatory protein. Trends Biochem Sci 21: 27-30. [PubMed] [Google Scholar]
- Heiser V, Engemann S, Brocker W, Dunkel I, Boeddrich A, Waelter S, Nordhoff E, Lurz R, Schugardt N, Rautenberg S, Herhaus C, Barnickel G, Bottcher H, Lehrach H, Wanker EE (2002) Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of Huntington's disease by using an automated filter retardation assay. Proc Natl Acad Sci USA 99: 16400-16406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M, Weisinger H, Dumitrescu-Ozimek L, Riederer P, Feinstein D, Klockgether T (2001) Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer disease. J Neuropathol Exp Neurol 60: 906-916. [DOI] [PubMed] [Google Scholar]
- Jacoby R, Cole C, Tutsch K, Nwton M, Kelloff G, Hawk E, Lubert R (2003) Chemopreventive efficacy of combined piroxicam and difluoromethylornithine treatment of Apc mutant Min mouse adenomas and selective toxicity against Apc mutant embryos. Cancer Res 60: 1864-1870. [PubMed] [Google Scholar]
- Jenkinson C, Grody W, Cederbaum S (1996) Comparative properties of arginases. Comp Biochem Physiol 114B: 107-132. [DOI] [PubMed] [Google Scholar]
- Kakuda D, Sweet MJ, MacLeod C, Hume D, Markovich D (1999) CAT2-mediated l-arginine transport and nitric oxide production in activated macrophages. Biochem J 340: 549-553. [PMC free article] [PubMed] [Google Scholar]
- Kepka-Lenhart D, Mistry S, Wu G, Morris S (2000) Arginase I: a limiting factor for nitric oxide and polyamine synthesis by activated macrophages. Am J Physiol 279: R2237-R2242. [DOI] [PubMed] [Google Scholar]
- Kiedrowski L, Costa E, Wroblewski J (1992) Glutamate receptor agonists stimulate nitric oxide synthase in primary cultures of cerebellar granule cells. J Neurochem 58: 335-342. [DOI] [PubMed] [Google Scholar]
- Kuppenbender K, Albers D, Iadarola MJ, Landwehrmeyer G, Standaert DG (1999) Localization of alternatively spliced NMDAR1 glutamate receptor isoforms in rat striatal neurons. J Comp Neurol 415: 204-217. [PubMed] [Google Scholar]
- Kuppenbender K, Standaert D, Feuerstein T, Penny J, Young A, Landwehrmeyer G (2000) Expression of NMDA receptor subunit mRNAs in neuro-chemically identified projection and interneurons in the human striatum. J Comp Neurol 419: 407-421. [DOI] [PubMed] [Google Scholar]
- Levine MS, Klapstein G, Koppel A, Gruen E, Cepeda C, Vargas M, Jokel E, Carpender E, Zanjani H, Hurst R, Efstratiadis A, Zeitlin S, Chesselet MF (1999) Enhanced sensitivity to N-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res 58: 515-532. [PubMed] [Google Scholar]
- Li H, Meininger CJ, Hawker Jr JR, Haynes TE, Kepka-Lenhart D, Mistry SK, Morris Jr SM, Wu G (2001) Regulatory role of arginase I and II in nitric oxide, polyamine and proline syntheses in endothelial cells. Am J Physiol 280: E75-E82. [DOI] [PubMed] [Google Scholar]
- Li L, Fan M, Icton C, Chen N, Leavitt B, Hayden M, Murphy T, Raymond L (2003) Role of NR2B-type NMDA receptors in selective neurodegeneration in Huntington disease. Neurobiol Aging 24: 1113-1121. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen T (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-(delta delta C(T)) method. Methods 25: 402-408. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM (2002) Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet 11: 1911-1926. [DOI] [PubMed] [Google Scholar]
- Mackintosh C, Pegg A (2003) Effect of spermine synthase deficiency on polyamine biosynthesis and content in mice and embryonic fibroblasts, and the sensitivity of fibroblasts to 1,3-bis-(2-chloroethyl)-N-nitrosourea. Biochem J 351: 439-447. [PMC free article] [PubMed] [Google Scholar]
- Meyer R, Henley C, Meyer M, Morgan P, McDonald A, Mills C, Price DK (1998) Partial deletion of both the spermine synthase gene and Pex gene in the X-linked hypophosphatemic, Gyro (Gy) mouse. Genomics 48: 289-295. [DOI] [PubMed] [Google Scholar]
- Mori M, Gotoh T, Nagasaki A, Takiguchi M, Sonoki T (1998) Regulation of the urea cycle enzyme genes in nitric oxide synthesis. J Inherit Metab Dis 21 [Suppl 1]: 59-71. [DOI] [PubMed] [Google Scholar]
- Morrison L, Cao X-C, Kish S (1998) Ornithine decarboxylase in human brain: influence of aging, regional distribution and Alzheimer's disease. J Neurochem 71: 288-294. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Tucker T, Ren H, Kenan D, Henderson B, Keene J, Strittmatter W, Burke J (2000) Inhibition of polyglutamine protein aggregation and cell death by novel peptides identified by phage display screening. J Biol Chem 275: 10437-10442. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro D (2003) An activity dependent spermine mediated mechanism that modulates glutamate transmission. Trends Neurosci 26: 9-11. [DOI] [PubMed] [Google Scholar]
- Petersen A, Mani K, Brundin P (1999) Recent advances on the pathogenesis of Huntington's disease. Exp Neurol 157: 1-18. [DOI] [PubMed] [Google Scholar]
- Pietila M, Alhonen L, Halmekyto M, Kanter P, Janne J, Porter C (1997) Activation of polyamine catabolism profoundly alters tissue polyamine pools and affects hair growth and female fertility in transgenic mice overexpressing spermidine/spermine acetyltransferase. J Biol Chem 272: 18746-18751. [DOI] [PubMed] [Google Scholar]
- Pohjanpelto P, Holtta E, Janne OA, Knuutila S, Alitalo K (1985) Amplification of ornithine decarboxylase gene in response to polyamine deprivation in Chinese hamster ovary cells. J Biol Chem 260: 8532-8537. [PubMed] [Google Scholar]
- Porter C, Pegg A, Ganis B, Madhabala R, Bergeron R (1990) Combined regulation of ornithine and S-adenosylmethionine decarboxylases by spermine and the spermine analogue N1 N12 bis ethyl spermine. Biochem J 268: 207-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Wang J, McMillian M, Chikaraishi D (1997) Characterization of a NS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J Neurosci 17: 1217-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que L, George S, Gotoh T, Mori M, Huang Y (2002) Effects of arginase isoforms on NO production by nNOS. Nitric Oxide 6: 1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Nagai Y, Tucker T, Strittmatter W, Burke J (2001) Amino acid sequence requirements of peptides that inhibit polyglutamine protein aggregation and cell death. Biochem Biophys Res Commun 288: 703-710. [DOI] [PubMed] [Google Scholar]
- Ross CA (2000) Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders Neuron 35: 819-822. [DOI] [PubMed] [Google Scholar]
- Saka E, Iadarola M, Fitzgerald D, Graybiel A (2002) Local circuit neurons in the striatum regulate neural and behavioral responses to dopaminergic stimulation. Proc Natl Acad Sci USA 99: 9004-9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathasivam K, Hobbs C, Turmaine M, Mangiarini L, Mahal A, Bertaux F, Wanker EE, Doherty P, Davies SW, Bates GP (1999) Formation of polyglutamine inclusion in non CNS tissue. Hum Mol Genet 8: 813-822. [DOI] [PubMed] [Google Scholar]
- Shantz LM, Pegg AE (1999) Translational regulation of ornithine decarboxylase and other enzymes of the polyamine pathway. Int J Biochem Cell Biol 31: 107-122. [DOI] [PubMed] [Google Scholar]
- Shappell N, Fogel-Petrovic M, Porter C (1993) Regulation of spermidine/spermine N1-acetyltransferase by intracellular polyamine pools. FEBS Lett 321: 179-183. [DOI] [PubMed] [Google Scholar]
- Sieradzan K, Mann D (2001) The selective vulnerability of nerve cells in Huntington's disease. Neuropathol Appl Neurobiol 27: 1-21. [DOI] [PubMed] [Google Scholar]
- Singh R, Pervin S, Karimi A, Cederbaum S, Chaudhuri G (2000) Arginase activity in human breast cancer cell lines: N(omega)-hydroxy-l-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res 60: 3305-3312. [PubMed] [Google Scholar]
- Slotkin T, Sidler F, Trepanier P, Whitmore W, Lerea L, Barnes G, Weigel S, Bartolome J (1982) Ornthine decarboxylase and polyamines in tissues of the neonatal rat: effects of α-difluoromethylornithine, a specific irreversible inhibitor of ornithine decarboxylase. Pharmacol Exp Ther 222: 741-745. [PubMed] [Google Scholar]
- Soulet D, Rivest S (2003) Polyamines play a critical role in the control of the innate immune response in the mouse central nervous system. J Cell Biol 162: 257-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani A, Chen Q, Flores-Hernandez J, Jiao Y, Reiner A, Surmeier J (1998) Physiological and molecular properties of AMPA/kainate receptors expressed by striatal medium spiny neurons. Dev Neurosci 20: 242-252. [DOI] [PubMed] [Google Scholar]
- Suppola S, Pietila M, Parkkinen JJ, Korhonen VP, Alhonen L, Halmekyto M, Porter CW, Janne J (1999) Overexpression of spermidine/spermine N1-acetyltransferase under the control of mouse metallothionein I promoter in transgenic mice: evidence for a striking post-transcriptional regulation of transgene expression by a polyamine analogue. Biochem J 338: 311-316. [PMC free article] [PubMed] [Google Scholar]
- Suppola S, Heikkinen S, Parkkinen JJ, Uusi-Oukari M, Korhonen VP, Keinanen T, Alhonen L, Janne J (2001) Concurrent overexpression of ornithine decarboxylase and spermidine/spermine N1-acetyltransferase further accelerates the catabolism of hepatic polyamines in transgenic mice. Biochem J 3358: 343-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Thomas TJ (2001) Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications. Cell Mol Life Sci 58: 244-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDamme P, Van Den Bosch L, Van Houtte E, Callewaert G, Robberecht W (2002) GluR2-dependent properties of AMPA receptors determine the selective vulnerability of motor neurons to excitotoxicity. J Neurophysiol 88: 1279-1287. [DOI] [PubMed] [Google Scholar]
- Vivo M, deVera N, Cortes R, Mengod G, Camon L, Marinez E (2001) Polyamines in the basal ganglia of human brain. Influence of aging and degenerative movement disorders. Neurosci Lett 304: 107-111. [DOI] [PubMed] [Google Scholar]
- Wallace H, Fraser AV, Hughes A (2003) A perspective of polyamine metabolism. Biochem J 376: 1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Oxford G (2000) Voltage dependent ion channels in CAD cells: a catecholaminergic neuronal line that exhibits inducible differentiation. J Neurophysiol 84: 2888-2895. [DOI] [PubMed] [Google Scholar]
- Washburn M, Dingledine R (1996) Block of α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptors by polyamines and polyamine toxins. J Pharmacol Exp Ther 278: 669-678. [PubMed] [Google Scholar]
- Wei L, Wu G, Morris S, Ignarro LJ (2001) Elevated arginase I expression in rat aortic smooth muscle cells increases cell proliferation. Proc Natl Acad Sci USA 98: 9260-9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weikert S, Fryer D, Weih M, Isaev N, Busch C, Schultze J, Megow D, Dirnagl U (1997) Rapid Ca2+-dependent NO-production from central nervous system cells in culture measured by NO-nitrite/ozone chemoluminescence. Brain Res 748: 1-11. [DOI] [PubMed] [Google Scholar]
- West A, Grace A (2000) Striatal nitric oxide signaling regulates the neuronal activity of midbrain dopamine neurons in vivo. J Neurophysiol 83: 1796-1808. [DOI] [PubMed] [Google Scholar]
- Williams K (1997) Interactions of polyamines with ion channels. Biochem J 325: 289-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Zappia AM, Pritchett D, Shen YM, Molinoff P (1994) Sensitivity of the N-methyl-d-aspartate receptor to polyamines is controlled by NR2 subunits. Pharmacol Exp Ther 45: 803-809. [PubMed] [Google Scholar]
- Wu G, Morris Jr SM (1998) Arginine metabolism: nitric oxide and beyond. Biochem J 336: 1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, Bradley J, Brown R, Maxwell M, Schapira A, Orntoft TF, Kato K, Rubinsztein DC (2001) Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntingon's disease. Hum Mol Genet 10: 1829-1845. [DOI] [PubMed] [Google Scholar]
- Xu Q, Colton C (2004) Nitric oxide production and regulation of neuronal NOS in tyrosine hydroxylase containing neurons. J Exp Neurol 188: 341-350. [DOI] [PubMed] [Google Scholar]
- Yu H, Iyer R, Kern R, Rodriguez W, Grody W, Cederbaum S (2001) Expression of arginase isozymes in mouse brain. J Neurosci Res 66: 406-422. [DOI] [PubMed] [Google Scholar]
- Zeron M, Chen N, Moshaver A, Lee A, Wellington C, Hayden M, Raymond L (2001) Mutant huningtin enhances excitotoxic cell death. Mol Cell Neurosci 17: 41-53. [DOI] [PubMed] [Google Scholar]
- Zeron M, Hansson O, Chen N, Wellington C, Leavitt B, Brudin P, Hayden M, Raymond L (2002) Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron 33: 849-860. [DOI] [PubMed] [Google Scholar]
- Zoghbi H, Botas J (2002) Mouse and fly models of neurodegeneration. Trends Genet 18: 463-471. [DOI] [PubMed] [Google Scholar]