

Abstract
Although the importance of dopamine (DA) for prefrontal cortical (PFC) cognitive functions is widely recognized, the nature of DA actions in the PFC remains controversial. A critical component in DA actions is its modulation of glutamate transmission, which can be different when specific receptors are activated. To obtain a clear picture of cellular mechanisms involved in these interactions, we studied the effects of DA–glutamate coactivation on pyramidal cell excitability in brain slices obtained from developmentally mature rats using whole-cell patch-clamp recordings. Bath application of NMDA, AMPA, and the D1 agonist SKF38393 induced concentration-dependent excitability increases, whereas bath application of the D2 receptor agonist quinpirole induced a concentration-dependent excitability decrease. The NMDA-mediated response was potentiated by SKF38393. This NMDA–D1 synergism required postsynaptic intracellular Ca2+ and protein kinase A (PKA) and was independent of membrane depolarization. On the other hand, the excitatory effects of both NMDA and AMPA were attenuated by a D2 agonist. Surprisingly, the D2–NMDA interaction was also blocked by the GABAA antagonists bicuculline and picrotoxin, suggesting that the inhibitory action of D2 receptors on NMDA-induced responses in the PFC may be mediated by GABAergic interneurons. In contrast, the D2–AMPA interaction involves inhibition of PKA and activation of phospholipase lipase C–IP3 and intracellular Ca2+ at a postsynaptic level. Thus, the modulatory actions of D1 and D2 receptors on PFC pyramidal cell excitability are mediated by multiple intracellular mechanisms and by activation of GABAA receptors, depending on the glutamate receptor subtypes involved.
Keywords: prefrontal cortex, dopamine receptors, AMPA, NMDA, whole-cell patch clamp, electrophysiology
Introduction
The prefrontal cortex (PFC) receives widespread inputs from cortical and subcortical areas involved in sensorimotor and limbic functions. The integration of these glutamatergic inputs is essential for the PFC role in executive functions and goal-directed behavior (Miller, 2000). It is also well known that PFC activity is shaped by a number of neuromodulators, most notably monoamines. Among these, dopamine (DA) stands out as having an important role in PFC cognitive functions, including working memory, reward, and attention (Schultz, 2002). DA-containing neurons are located in the ventral tegmental area (Lindvall et al., 1974) and project to the PFC. Several studies have highlighted the need of DA–glutamate coactivation for a number of PFC functions (Gurden et al., 1999; Baldwin et al., 2002; Jay, 2003). Thus, the interactions between DA and glutamate receptors may be essential for a proper PFC function, and a thorough understanding of the cellular underpinnings of those interactions needs to be obtained.
DA affects glutamate function with different effects according to the receptors involved (Cepeda et al., 1993; Nicola et al., 2000), the cell type in which this interaction occurs (Nicola et al., 2000), and the membrane potential of the neuron (O'Donnell et al., 1999), as well as several other factors (Nicola et al., 2000; O'Donnell, 2003). In the striatum, for instance, D1 receptors potentiate responses mediated by NMDA receptors, whereas D2 receptors depress AMPA responses (Cepeda et al., 1993). A similar pattern has been reported in human neocortical neurons (Cepeda et al., 1992), and a D1 enhancement of NMDA responses has been observed in the PFC of young rats (Wang and O'Donnell, 2001). Despite the wealth of data indicating that DA–glutamate interactions are essential for mature cognitive functions, the few studies conducted to address cellular mechanisms of these interactions have been performed in slices from prepubertal (typically <40 d old) animals. Thus, the cellular mechanisms of DA–glutamate interactions that control pyramidal cell activity remain to be established in the mature PFC. This becomes essential because the effects observed in prepubertal animals may not necessarily reflect the function in adulthood. Neocortical pyramidal neurons undergo several morphological and physiological changes during postnatal development until postnatal day (P) 42 (Petit et al., 1988; Zhu, 2000). To confidently address DA–glutamate interactions as relevant to adult cognitive functions, we performed whole-cell patch-clamp recordings in brain slices obtained from developmentally mature (P42–65) rats. We examined the cellular mechanisms involved in the modulation of NMDA and AMPA responses by D1 and D2 receptors in medial PFC pyramidal neurons, measuring changes in cell excitability induced by combinations of DA and glutamate agonists and antagonists.
Materials and Methods
All experimental procedures were performed according to the United States Public Health Service Guide for Care and Use of Laboratory Animals and were approved by the Albany Medical College Institutional Animal Care and Use Committee. Male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) were maintained on a 12 hr light/dark cycle with food and tap water available ad libitum until the time of the experiment. Rats were anesthetized with chloral hydrate (400 mg/kg, i.p.) before being decapitated. Brains were rapidly removed into ice-cold artificial CSF (ACSF) containing (in mm): 125 NaCl, 25 NaHCO3, 10 glucose, 3.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 3 MgCl2, pH 7.45; osmolarity 295 ± 5 mOsm. Coronal slices (300 μm) containing the medial PFC were cut on a Vibratome in ice-cold ACSF, transferred, and incubated in warm (±35°C) ACSF solution constantly oxygenated with 95% O2–5% CO2 for at least 90 min before recording.
Whole-cell patch-clamp recordings were performed in slices obtained from developmentally mature (P42–65) rats. Layers V–VI medial PFC pyramidal neurons were identified under visual guidance using infrared-differential interference contrast (IR-DIC) video microscopy with a 40× water immersion objective. The image was detected with an IR-sensitive CCD camera and displayed on a monitor. All experiments were conducted at 33–35°C. In the recording ACSF (2 ml/min), CaCl2 was increased to 2 mm and MgCl2 was decreased to 1 mm. Patch pipettes (5–8 MΩ) were filled with (in mm): 115 K-gluconate, 10 HEPES, 2 MgCl2, 20 KCl, 2 MgATP, 2 Na2-ATP, 0.3 GTP, pH = 7.3; 280 ± 5 mOsm. All drugs were mixed into oxygenated ACSF and applied in the recording solution in known concentrations. Both control and drug-containing ACSF were oxygenated continuously throughout the experiments. SKF38393, quinpirole, eticlopride, and AMPA were purchased from Sigma (St. Louis, MO), and NMDA and SCH23390 were obtained from Research Biochemicals (Natick, MA). In some experiments, 10 μm bicuculline (Sigma) or picrotoxin (Sigma) was applied in the bath to block GABAA receptors. For intracellular Ca2+ chelation, BAPTA (Sigma) was included in the recording micropipette at a concentration of 2 mm. To block protein kinase A activity, 20 μm peptide protein kinase inhibitor (PKI)-[5–24] (Calbiochem, La Jolla, CA) was included in the recording electrode, whereas 10 μm U73122 and 2 μm Xestospongin C (XeC) (Calbiochem) were used to block PLC and IP3, respectively.
Whole-cell current-clamp recordings were performed with a computer-controlled amplifier (MultiClamp 700A; Axon Instruments, Foster City, CA) and acquired with Axoscope 8.1 (Axon Instruments) at a sampling rate of 10 kHz. Electrode potentials were adjusted to zero before recording without correcting the liquid junction potential. In each cell, input resistance (measured by 200–300 msec duration hyperpolarizing pulses), membrane potential, the number of evoked spikes, and the latency to the first spike evoked by a 500 msec duration depolarizing current pulse were analyzed before and after drug treatment. Typically, baseline recordings were conducted for ∼10 min before perfusing with a solution containing the different mixture of drugs for 5–6 min. The effects of drug treatment were evident during the first 2–3 min and became stable during the remainder of the drug application. All neurons recorded in the present study showed a similar pattern of responses, and all data shown were analyzed from the stable 3–5 min period of drug administration. The effects observed with NMDA or AMPA required ∼5–10 min to wash out. In contrast, 15–20 min were required to partially wash out (they never reversed completely) the effects obtained with D1 and D2 agonists. Values of all measures are expressed as mean ± SD. Drug effects were compared using repeated measures ANOVA, and the differences between experimental conditions were considered statistically significant when p < 0.05.
Results
Whole-cell current-clamp recordings of 242 pyramidal neurons located in deep layers of the medial PFC were conducted in brain slices obtained from young adult (P50.4 ± 7.7; mean ± SD) rats. Recordings were made using visual guidance under differential interface contrast in the infrared range, allowing selection of pyramidal neurons (Fig. 1A). All neurons recorded were silent at rest, and action potentials were observed only in response to depolarizing somatic current injection (Fig. 1B). The membrane potential of these neurons was –69.8 ± 3.2 mV, and their input resistance was 151.0 ± 44.5 MΩ calculated from the linear portion of the I–V curve in the hyperpolarized direction (Fig. 1C). Most pyramidal neurons recorded (∼85%) exhibited an initial spike doublet followed by spike frequency accommodation in response to suprathreshold current steps (Fig. 1B) and typically showed inward rectification in response to a series of current pulses in both the hyperpolarizing and depolarizing directions (Fig. 1C). As measures of cell excitability, the number of spikes and the latency to the first spike evoked by the depolarizing current pulses were quantified. Changes in cell excitability induced by drugs (DA and glutamate agonists and antagonists) were assessed by repeated delivery of similar-intensity depolarizing pulses while the agents were added to the bath. In some experiments (n = 83 cells), neurons were filled with Neurobiotin (0.125%) to further confirm the cell type as well as the recording site. All cells filled with Neurobiotin were identified as pyramidal neurons located in layer V or VI of the medial PFC.
Figure 1.

Whole-cell recording from PFC pyramidal neurons. A, IR-DIC image of a layer V medial PFC pyramidal neuron recorded from a 300-μm-thick PFC slice (P52). The arrows point to the shadow of the recording electrode. B, Typical response to depolarizing and hyperpolarizing somatic current pulses (200 msec; from –300 to +100 pA). C, Current–voltage plot from the traces shown in B. Currents larger than –100 pA yielded a marked inward rectification.
D1 and D2 receptors have opposite effects on PFC pyramidal neuron excitability
D1 and D2 agonists affected differentially the number of spikes and latency to the first spike evoked by constant-amplitude depolarizing current pulses. Bath application of the D1 agonist SKF38393 resulted in a concentration-dependent excitability increase (n = 5–10 cells per dose). The number of spikes increased significantly from 1.7 ± 0.4 spikes (baseline) to 3.5 ± 0.5 spikes (4 μm SKF38393) and 5.0 ± 0.8 spikes (8 μm SKF38393) (Fig. 2A). Similarly, the latency to the first evoked spike was reduced from 111.9 ± 21.8 msec (baseline) to 60.1 ± 14.0 and 33.3 ± 13.5 msec with 4 and 8 μm SKF38393, respectively (Fig. 2A). This effect was independent of membrane potential changes and was blocked by the D1 antagonist SCH23390 (10 μm; n = 4) (Fig. 2B). No significant changes were observed with SKF38393 on input resistance or action potential kinetics (spike threshold and duration; data not shown). On the other hand, bath application of the D2 agonist quinpirole induced a decrease in excitability without changes in membrane potential or action potential threshold or kinetics (n = 5–6 cells per dose). Input resistance increased from 140.6 ± 38.2 (baseline) to 162.7 ± 36.4 MΩ in the presence of 1 μm quinpirole (p < 0.05; paired t test). The number of evoked spikes was significantly reduced from 4.0 ± 0.6 (baseline) to 2.1 ± 0.4 spikes, and the latency to the first evoked spike was increased from 62.1 ± 17.2 msec (baseline) to 92.6 ± 16.5 msec in presence of 1 μm quinpirole (Fig. 3A). This effect was prevented by pre-exposing the slice to the D2 antagonist eticlopride (20 μm; n = 4) (Fig. 3B). These results indicate that pharmacological activation of D1 receptors increase, whereas D2 receptors decrease, PFC pyramidal cell excitability.
Figure 2.
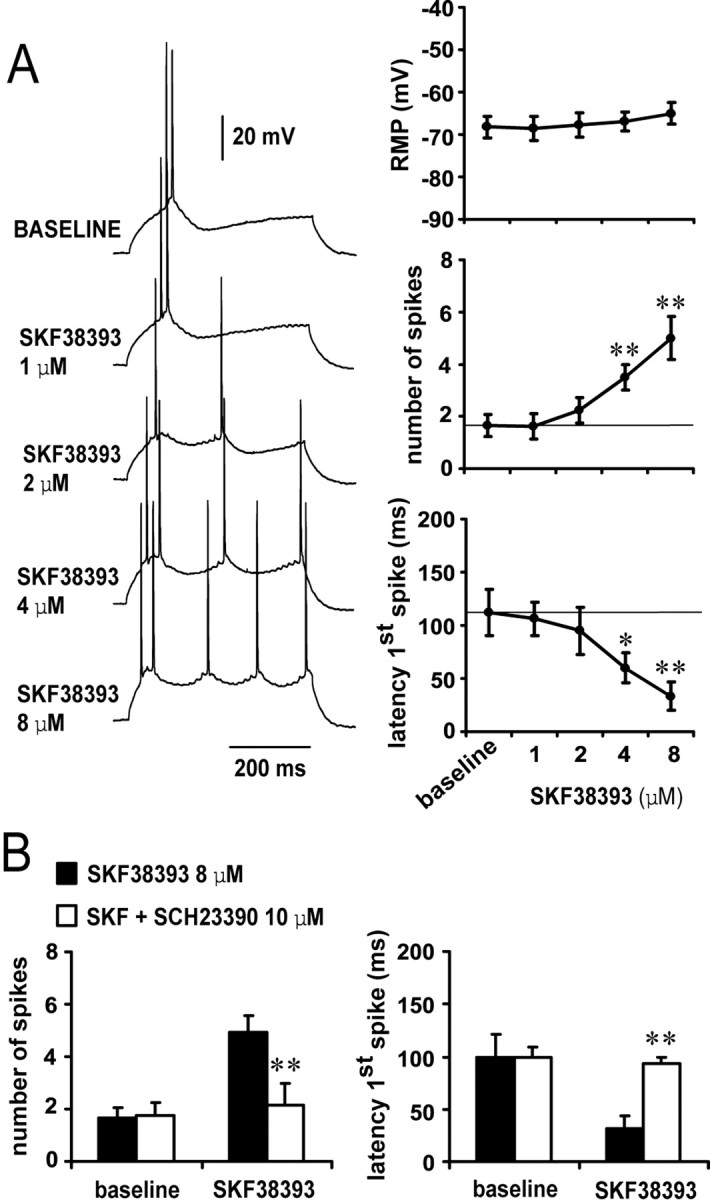
D1 activation enhances pyramidal cell excitability. A, Concentration-dependent excitability increases induced by bath application of SKF38393 in the PFC. Left panel, Representative tracings showing the increased action potential firing evoked by depolarizing current injection in the presence of 1, 2, 4, and 8 μm SKF38393 concentrations. Right panel, Dose-dependent effect of SKF38393 on pyramidal cell excitability (measured as the latency to the first evoked spike and the number of spikes evoked by depolarizing current injection). The resting membrane potential was not significantly affected by bath application of SKF38393; however, the number of spikes was increased significantly and the latency to the first spike decreased with 4 and 8 μm SKF38393 (compared with baseline; *p < 0.005, **p < 0.0002; repeated measures ANOVA).B, Bar graphs illustrating that the effects of 8 μm SKF38393 on pyramidal cell excitability were blocked by the D1 antagonist SCH23390 (10 μm). The excitatory action of 8 μm SKF38393 on both the number of evoked spikes (left) and first spike latency were prevented by SCH23390 (compared with SKF38393 alone; **p < 0.005; repeated measures ANOVA).
Figure 3.
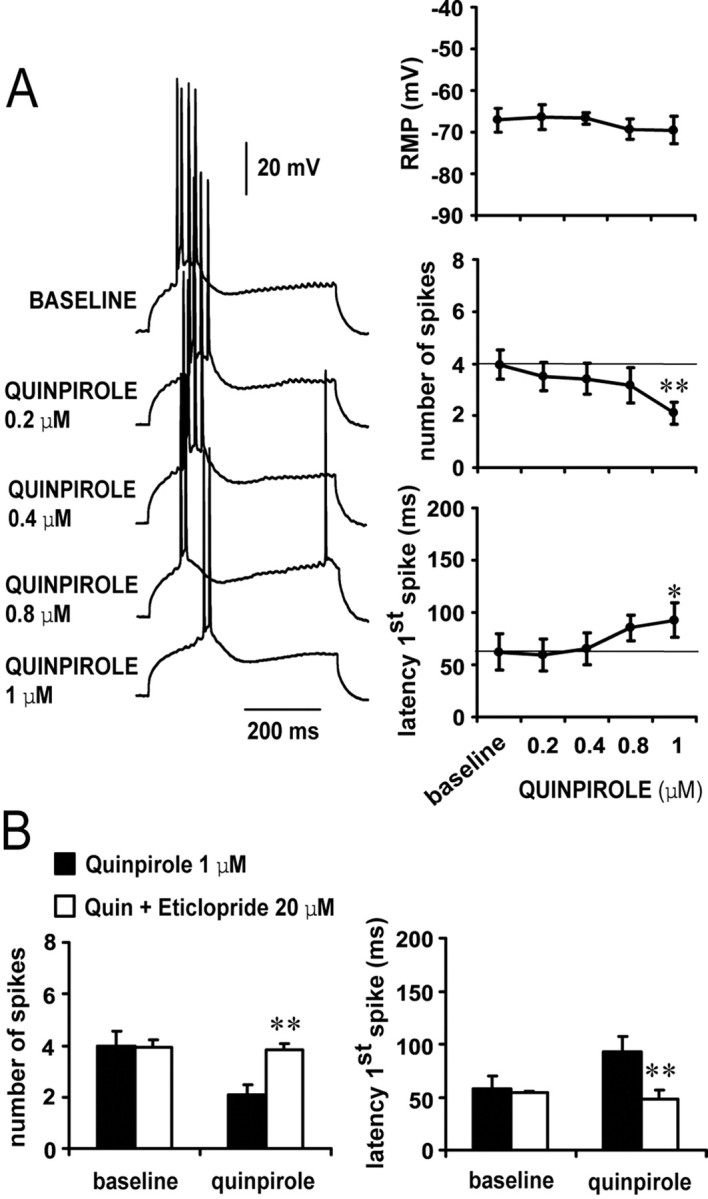
D2 receptors decrease pyramidal cell excitability. A, Concentration-dependent excitability decreases induced by bath application of quinpirole in the PFC. Left panel, Traces recorded from a single neuron illustrating the effects of increasing quinpirole concentrations on action potential firing evoked by depolarizing current injection. Right panel, Dose-dependent effect of quinpirole on PFC pyramidal cell excitability. A significant decrease in the number of evoked spikes and increased first spike latency were observed only with 1 μm quinpirole (compared with baseline; **p < 0.001, *p < 0.01; repeated measured ANOVA). No changes were observed in membrane potential. B, The inhibitory action of 1 μm quinpirole on PFC pyramidal cell excitability was blocked by 20 μm eticlopride (compared with quinpirole alone; **p < 0.001; repeated measures ANOVA).
Opposite action of D1 and D2 receptors on NMDA enhancement of cell excitability
Bath application of NMDA resulted in a concentration-dependent excitability increase in PFC pyramidal neurons. Significant increases in the number of evoked spikes and decreases in the latency to the first evoked spike were observed with 2, 4, and 8 μm, but not 1 μm, NMDA (n = 7–8 cells per dose). No changes in input resistance, action potential threshold, or duration were observed. The number of evoked spikes increased significantly from 1.7 ± 0.5 (baseline) to 4.0 ± 1.4, 7.0 ± 1.4, and 10.2 ± 1.1 spikes, and the latency to the first evoked spike decreased from 106.0 ± 26.2 msec (baseline) to 55.1 ± 16.8, 22.1 ± 12.8, and 7.9 ± 6.6 msec with 2, 4, and 8 μm SKF38393, respectively (Fig. 4). Adding an ineffective dose of the D1 agonist (2 μm) (Fig. 2A) potentiated the excitatory effects of NMDA on both number of spikes and latency to the first evoked spike (n = 8–10 cells per dose). A D1 enhancement of NMDA-mediated responses was observed for 1 and 2 μm but not 4 and 8 μm NMDA (Fig. 4). In contrast, NMDA-mediated excitability increase was attenuated by coadministration of the D2 agonist quinpirole (0.4 μm; n = 6–8 cells per dose) (Fig. 4), which was ineffective when applied alone at this concentration (Fig. 3A). This inhibitory action of quinpirole was effective for 2, 4, and 8 μm NMDA. These results indicate that D1 receptors facilitate and D2 receptors depress NMDA-mediated responses in mature PFC pyramidal neurons.
Figure 4.

D1 and D2 modulation of NMDA effects on PFC cell excitability. Top, Overlay of tracings obtained with increasing NMDA concentrations (bottom to top) illustrating the effect of NMDA alone (left), NMDA plus SKF38393 (center), and NMDA plus quinpirole (right). Bottom, Graphs summarizing the effects of increasing NMDA concentrations on PFC pyramidal cell resting membrane potential (left), number of evoked spikes (center), and first spike latency (right). SKF38393 (2 μm) significantly enhanced the effects of 1 and 2 μm NMDA. In contrast, the effect of NMDA was significantly reduced in presence of quinpirole (0.4 μm). *p < 0.03; **p < 0.002; Tukey post hoc test after significant ANOVA.
D2 receptors attenuate the AMPA-mediated excitability increase in PFC pyramidal neurons
Bath application of AMPA also induced concentration-dependent increases in PFC pyramidal cell excitability. The number of spikes significantly increased from 1.8 ± 0.4 (baseline) to 5.5 ± 1.5, 7.5 ± 0.8, and 10.1 ± 1.4 spikes (n = 6–7 per dose) with 0.1, 0.2, and 0.4 μm AMPA, respectively (Fig. 5). No changes in input resistance were detected. These effects were accompanied by significant decreases in the first spike latency from 94.2 ± 36.6 msec to 23.7 ± 10.2, 18.4 ± 9.0, and 8.0 ± 4.5 msec, respectively (Fig. 5). This excitatory action of AMPA was not affected by 2 μm SKF38393. The D1 agonist failed to modify any electrophysiological effect of AMPA (Fig. 5) (n = 5–6 cells per dose). In contrast, the increases in cell excitability brought by 0.1, 0.2, and 0.4 μm AMPA were significantly attenuated by 0.4 μm quinpirole (Fig. 5) (n = 7–9 cells per dose), which was ineffective when applied alone (Fig. 3A). Input resistance and spike kinetics remained unchanged across doses (data not shown). These results indicate that AMPA effects on PFC pyramidal cell excitability are not affected by D1 but are strongly inhibited by D2 receptors.
Figure 5.

D1 and D2 modulation of AMPA effects on PFC pyramidal cell excitability. Top, Overlays of traces obtained with increasing AMPA concentrations (bottom to top) illustrating the effects of AMPA alone (left), AMPA plus SKF38393 (center), and AMPA plus quinpirole (right). Bottom, Plots summarizing the effects of increasing AMPA concentrations (alone and in the presence of either a D1 or a D2 agonist) on PFC pyramidal cell resting membrane potential, number of evoked spikes, and latency to the first evoked spike. The excitatory effect of AMPA was attenuated in the presence of 0.4 μm quinpirole. SKF38393 failed to change any AMPA-mediated electrophysiological action. *p < 0.03; **p < 0.001; Tukey post hoc test after significant ANOVA.
The D1–NMDA synergism is independent of membrane depolarization and requires both intracellular Ca2+ and protein kinase A activation
The D1 enhancement of NMDA responses can be prevented by administration of a D1 antagonist. As shown above, coadministration of SKF38393 (2 μm) and NMDA (1 μm) (n = 8) significantly increased the number of spikes and decreased the latency to the first evoked spike when compared with NMDA alone (n = 8) (Fig. 6). This potentiation was blocked by the D1 antagonist SCH23390 (10 μm; n = 7) (Fig. 6) and by the NMDA blocker APV (50 μm; n = 5; data not shown), confirming that it results from D1–NMDA interactions. To account for any role of the slight depolarization elicited by this combination of drugs (∼3.5 mV), cell excitability was determined in some cases (n = 6) after adjusting the membrane potential to baseline value with intracellular current injection. The D1 agonist was still able to potentiate NMDA effects, suggesting that the D1–NMDA synergism on PFC pyramidal cell excitability is independent of membrane depolarization (Fig. 6).
Figure 6.

The D1 enhancement of NMDA response is independent of membrane depolarization and requires intracellular Ca2+ and PKA activation. Left, Representative traces illustrating the response of a PFC pyramidal neuron to intracellular current injection after (from top to bottom) NMDA (1 μm), NMDA plus SKF38393 (2 μm), NMDA plus SKF38393 plus SCH23390 (10 μm), NMDA plus SKF38393 plus the Ca2+ chelator BAPTA (2 mm), and NMDA plus SKF38393 plus the PKA blocker PKI-[5–24] (20 μm). Right, Bar graphs summarizing the changes in number of evoked spikes and first spike latency observed in each experimental condition. The synergistic D1–NMDA effect on the number of evoked spikes and first spike latency was not affected by holding the resting membrane potential at baseline value (Vm adj). This potentiation was blocked by SCH23390 (10 μm) and in recordings with electrodes containing BAPTA (2 mm) or PKI-[5–24] (20 μm). *p < 0.001; Tukey post hoc test after significant ANOVA.
It has been shown that D1–NMDA interactions involve intracellular Ca2+ and protein kinase A (PKA) (Cepeda and Levine, 1998; Cepeda et al., 1998; Wang and O'Donnell, 2001). To determine whether this is also the case in slices from mature animals, a subset of recordings was conducted with electrodes containing the Ca2+ chelator BAPTA (2 mm; n = 5) or the PKA inhibitor peptide PKI-[5–24] (20 μm; n = 5). In those experiments, the D1 agonist failed to potentiate NMDA effects (Fig. 6). No apparent changes in input resistance or action potential threshold were observed. These results indicate that D1–NMDA interactions on PFC pyramidal cell excitability require intracellular Ca2+ and activation of a PKA cascade at a postsynaptic level.
D2–NMDA interactions require activation of GABAA receptors
To study the cellular mechanisms involved in D2–NMDA interactions, a low dose of the D2 agonist quinpirole (0.4 μm) (Fig. 3) was combined with 4 μm NMDA (Fig. 4), and different intracellular cascades were blocked with selective antagonists. Although bath application of 0.4 μm quinpirole failed to modify pyramidal cell excitability when applied alone (Fig. 3), it significantly reduced the excitability increase induced by 4 μm NMDA (Fig. 7) (n = 7). This interaction was blocked by the D2 antagonist eticlopride (20 μm; n = 5) (Fig. 7), confirming that it is a D2-mediated effect. Surprisingly, the interaction was also blocked by the GABAA antagonist bicuculline (10 μm; n = 6) or picrotoxin (10 μm; n = 6) (Fig. 7) without significant changes in cellular input resistance and action potential kinetics. Recordings with electrodes containing the Ca2+ chelator BAPTA (2 mm; n = 6) failed to prevent the inhibitory action of quinpirole on NMDA responses.
Figure 7.

D2–NMDA interactions involve GABAA receptors. Left, Traces of responses to current injection illustrating changes in action potential firing observed in the different experimental conditions. From top to bottom, the traces are representative recordings obtained with NMDA alone (4 μm), NMDA plus quinpirole (0.4 μm), NMDA plus quinpirole plus the D2 antagonist eticlopride (20 μm; eticlo), NMDA plus quinpirole plus the GABAA antagonist bicuculline (10 μm; bic), NMDA plus quinpirole plus the GABAA antagonist picrotoxin (10 μm; ptx), NMDA plus quinpirole plus BAPTA-containing electrode (2 mm), and NMDA plus PKI [5–24]-containing electrode (20 mm). Right, Bars graphs summarizing the changes in number of evoked spikes and first spike latency in each experimental condition. The inhibitory effect of quinpirole on NMDA responses was blocked by eticlopride (20 μm; n = 5), by bicuculline (10 μm; n = 6), and by picrotoxin (10 μm; n = 6) but not in recordings with electrodes containing the calcium chelator BAPTA (n = 6). Intracellular application of the PKA inhibitor PKI-[5–24] (n = 5) failed to mimic the effects of quinpirole. *p < 0.001; Tukey post hoc test after significant ANOVA.
It is well known that D2 receptors inhibit adenylyl cyclase, reducing cytosolic cAMP levels and PKA activity (Lachowicz and Sibley, 1997). Therefore, a subset of experiments was conducted to investigate the role of PKA by including the PKA inhibitor PKI-[5–24] (20 μm; n = 5) in the recording electrodes instead of bath application of quinpirole. Intracellular application of the PKA inhibitor peptide PKI-[5–24] was unable to mimic the effects of quinpirole on NMDA-mediated excitability increase (Fig. 7). These results suggest that the inhibitory effect of D2 receptors on NMDA-mediated responses in PFC pyramidal neurons requires GABAA receptor activation but does not involve postsynaptic intracellular Ca2+ or blockade of a PKA cascade via activation of the G-protein Gi.
It is therefore possible that the inhibitory D2 action on NMDA responses is mediated by activation of GABAergic interneurons. To test this hypothesis, recordings were conducted in interneurons to investigate the impact of the D2 agonist on their activity. To minimize the impact of interneuron diversity, we selected only interneurons located in layers III–V that had a fast spiking response to current pulse injection. Bath application of 1 μm quinpirole enhanced PFC interneuron excitability. The number of spikes evoked by somatic current injection increased significantly from 8.3 ± 2.6 (baseline) to 11.5 ± 2.7 with quinpirole (n = 6; p < 0.001; paired t test) (Fig. 8A,B). Input resistance increased from 282.0 ± 18.4 to 340.8 ± 28.4 MΩ (p < 0.001; paired t test), and the neurons were depolarized from –63.2 ± 1.4 to –59.9 ± 2.7 mV (p = 0.029; paired t test). In those cells showing spontaneous action potential firing (n = 3), the depolarization was accompanied by an increase in firing rate (Fig. 8C). These results reinforce the notion that D2–NMDA interactions in the PFC may involve activation of GABAergic interneurons.
Figure 8.

Quinpirole enhances interneuron excitability. A, Representative traces of responses to current injection showing the effect of quinpirole (1 μm) on interneuron excitability. The number of evoked spikes increased from 12 to 15. B, Scatter graph summarizing the excitatory effect of quinpirole on PFC interneurons. The number of spikes evoked by intracellular current injection increased significantly in the presence of quinpirole 1 μm (n = 6; **p < 0.001; paired t test). C, A pair of traces illustrating the membrane depolarization with increased firing rate induced by quinpirole.
D2–AMPA interactions require activation of a postsynaptic PLC–IP3–Ca2+ cascade
Bath application of 0.2 μm AMPA and 0.4 μm quinpirole was used to examine the cellular mechanisms involved in D2–AMPA interactions. The excitatory effects of 0.2 μm AMPA on both the number of evoked spikes and the latency to the first spike were significantly attenuated by 0.4 μm quinpirole (Fig. 9) (n = 9). These inhibitory actions of quinpirole on AMPA responses were blocked by the D2 receptor antagonist eticlopride (20 μm; n = 5) but not by the GABAA antagonist bicuculline (10 μm; n = 6) (Fig. 9). In contrast to the modulation of NMDA responses by D2 receptors, a D2–AMPA interaction could not be observed in recordings with electrodes containing the Ca2+ chelator BAPTA (2 mm; n = 6) (Fig. 9). This suggests that the D2 reduction of AMPA responses occurs at a postsynaptic level and requires intracellular Ca2+. Intracellular administration of 20 μm PKI-[5–24] significantly reduced the excitatory effects of 0.2 μm AMPA (n = 6) but fell short of completely mimicking the effects of quinpirole (Fig. 9). This suggests that blockade of PKA pathways may be a mechanism by which D2 receptors downregulate AMPA activity, but it also indicates that another intracellular pathway may be involved in this interaction.
Figure 9.

D2–AMPA interactions require intracellular Ca2+, PLC–IP3 pathway, and PKA inactivation. Left panel, Traces of responses to current injection showing the effects of AMPA on PFC pyramidal cell action potential firing obtained in different experimental conditions. The pairs of traces (baseline and treatment) are representative of (from top to bottom) experiments conducted with AMPA (0.2 μm), AMPA plus quinpirole (0.4 μm), AMPA plus quinpirole plus eticlopride (20 μm; eticlo), AMPA plus quinpirole plus bicuculline (10 μm; bic), AMPA plus quinpirole plus BAPTA (2 mm), AMPA plus PKI-[5–24] (20 μm), AMPA plus quinpirole plus PKI-[5–24] (20 μm), AMPA plus quinpirole plus the PLC blocker U73122 (10 μm), and AMPA plus quinpirole plus the IP3 inhibitor Xec (2 μm). Right, Bar graphs summarizing the changes in number of evoked spikes and first spike latency observed in each experimental condition. The effect of quinpirole on AMPA responses was blocked by eticlopride but not bicuculline. In the presence of PKI-[5–24], the excitatory effect of AMPA on PFC pyramidal cell excitability was partially attenuated. Recordings with electrodes containing BAPTA, U-73122, or Xestospongin-C were able to block the inhibitory action of D2 receptors on AMPA-mediated effects. Intracellular application of 0.15% DMSO did not alter the effects of quinpirole on AMPA responses (n = 5). *p < 0.001; Tukey post hoc test after significant ANOVA.
Given that D2–AMPA interactions require intracellular Ca2+, it is possible that the inhibitory action of D2 receptors on AMPA responses also involves activation of the PLC–IP3 pathway (Sternweis and Smrcka, 1992; Hernandez-Lopez et al., 2000; Kotecha et al., 2002). To test this hypothesis, the effects of the PLC blocker U73122 (Kotecha et al., 2002) and the IP3 inhibitor XeC (Kotecha et al., 2002) were examined in the presence of 0.4 μm quinpirole and 0.2 μm AMPA. Intracellular application of 10 μm U73122 (n = 5) or 2 μm XeC (n = 5) prevented the inhibitory effect of quinpirole on AMPA-mediated excitability increase (Fig. 9), without apparent changes in input resistance or action potential threshold and duration. Together, these results indicate that D2–AMPA interactions in the PFC involve several mechanisms, including intracellular Ca2+, activation of a PLC–IP3 pathway, and inhibition of PKA at a postsynaptic level.
Discussion
The modulatory actions of D1 and D2 receptors on NMDA and AMPA responses of PFC pyramidal neurons were studied in brain slices obtained from developmentally mature rats. Because recordings were not voltage clamped, the interactions examined here may reflect the natural behavior of the neurons.
D1 receptors enhance NMDA responses via PKA activation. It is well known that D1 agonists increase cAMP levels and PKA activity (Surmeier et al., 1995; Nicola et al., 2000), activating the phosphoprotein DARPP-32 (dopamine receptor-activated phospho protein 32 kDa (Snyder et al., 1998), which can enhance NMDA currents in striatal medium spiny neurons (Flores-Hernandez et al., 2002). We have reported previously a PKA-dependent D1 potentiation of NMDA effects in PFC slices from young (P24–28) animals (Wang and O'Donnell, 2001). This signaling cascade could phosphorylate a number of proteins involved in NMDA responses, including the NMDA receptors themselves (Snyder et al., 1998). Other cellular mechanisms that could be involved in a D1 potentiation of NMDA responses include NMDA receptor trafficking to the postsynaptic membrane, as observed in striatal neurons (Dunah and Standaert, 2001), or actions on voltage-gated ion channels. A D1 modulation of NMDA receptor trafficking in PFC pyramidal neurons remains to be determined. Thus, the ability of D1 receptors to enhance NMDA-mediated excitability via PKA signaling could take place via different factors, including glutamate receptors or voltage-gated ion channels activated downstream of NMDA receptors.
The modulatory D1 action on AMPA responses is complex, with evidence supporting both positive and negative interactions. In the striatum, D1 receptors increased AMPA-evoked cell firing (Cepeda et al., 1993) but produced little net change on synaptic responses (Levine et al., 1996). A D1 enhancement of AMPA responses has been reported in layer II–III PFC pyramidal neurons (Gonzalez-Islas and Hablitz, 2003). In layer V, however, D1 receptors can attenuate AMPA synaptic responses (Gao et al., 2001). Here we failed to find consistent D1 modulation of AMPA effects. Because cell excitability measured in response to somatic current injection reflects essentially postsynaptic effects, a presynaptic D1 action (Gao et al., 2001) would certainly be missed. At a postsynaptic level, however, there appears not to be a D1–AMPA interaction.
D2 receptors can modulate neuronal activity in many cortical and subcortical brain regions. For example, D2 receptors depress glutamate responses in striatal (Cepeda et al., 1993; O'Donnell and Grace, 1994) and hippocampal (Kotecha et al., 2002) neurons. A similar D2 inhibitory action on PFC pyramidal cell excitability has been observed in immature animals (Gulledge and Jaffe, 1998). The cellular mechanisms yielding D2 inhibitory actions may include multiple intracellular cascades (Cepeda et al., 1998; Hernandez-Lopez et al., 2000; Kotecha et al., 2002) (for review, see Nicola et al., 2000). For instance, D2 receptors inhibit adenylyl cyclase via Gi, thereby reducing cellular levels of cAMP and PKA activity (for review, see Lachowicz and Sibley, 1997). Also, D2 receptors can modulate neuronal excitability by activating the PLC–IP3–Ca2+ cascade (Hernandez-Lopez et al., 2000). Here we showed that a D2 attenuation of NMDA effects does not require intracellular Ca2+ or PKA inhibition. This interaction, however, was blocked by bicuculline or picrotoxin, suggesting that the D2 inhibitory effect on NMDA responses requires GABAA receptors. We also observed a D2-mediated increase in firing rate and excitability of fast-spiking PFC interneurons. These findings are consistent with reports of the inhibitory effect of DA on PFC pyramidal cell firing being mediated, in part, by an increase in GABAergic transmission (Gulledge and Jaffe, 1998, 2001). Moreover, in vivo microdialysis from mature animals revealed a dose-dependent increase in extracellular GABA by intra-PFC quinpirole, an effect blocked by D2 but not D1 antagonists (Grobin and Deutch, 1998). Thus, it is possible that D2 receptors attenuate PFC NMDA responses by exciting GABA interneurons.
An intriguing observation is that this D2-dependent GABA activation affects NMDA but not AMPA responses. Although speculative, a few potential explanations can be advanced. First, interneurons could be selectively activated by NMDA. This is unlikely because spontaneous and evoked IPSCs in pyramidal neurons are blocked by AMPA and not NMDA antagonists (Salin and Prince, 1996a; Ling and Benardo, 1999). Another possibility is that D2-activated interneurons release GABA acting on receptors located on NMDA-rich areas of the pyramidal cell membrane, enabling a GABA shunting effect only when NMDA receptors are activated. A spatial selectivity of NMDA and AMPA receptors in pyramidal neurons has been suggested, with NMDA preferentially located in the soma and proximal dendrites and AMPA in distal dendrites (Dodt et al., 1998). It has also been observed that GABA IPSCs are stronger in the soma (Salin and Prince, 1996b) compared with distal regions of pyramidal neurons. This would allow a stronger impact on membrane regions that express primarily NMDA receptors. Further experiments are necessary to elucidate how D2–GABA interactions control NMDA responses.
In contrast, the D2 inhibitory action on AMPA responses does not involve GABAA receptors. D2 effects on AMPA responses were blocked by intracellular BAPTA and by PLC and IP3 antagonists, suggesting that the interactions require intracellular Ca2+ and the PLC–IP3 cascade. Because this pathway is one of the primary mechanisms for receptor-mediated mobilization of intracellular Ca2+ (Sternweis and Smrcka, 1992), D2 receptors could promote intracellular Ca2+ release, enhancing Ca2+-dependent AMPA inactivation (Kotecha et al., 2002). Alternatively, D2 receptors could reduce L-type Ca2+ currents via the PLC–IP3–calcineurin signaling pathway, thereby decreasing cell excitability (Hernandez-Lopez et al., 1997, 2000). In addition to the PLC–IP3–Ca2+ cascade, we also observed that intracellular administration of a PKA blocker was able to reduce the excitability increase induced by AMPA. This suggests that the D2 inhibitory effect can be mimicked, albeit partially, by inactivation of the cAMP–PKA cascade. Thus, D2 receptors attenuate AMPA-mediated responses in PFC pyramidal neurons through at least two different postsynaptic mechanisms.
A DA modulation of glutamate activity may be important for PFC function (Jay, 2003; O'Donnell, 2003). For instance, an intact mesocortical projection is necessary for hippocampal→ PFC long-term potentiation (LTP) (Gurden et al., 1999), which is enhanced by D1 agonists (Gurden et al., 2000). Similarly, PFC D1 receptors improve memory retrieval (Floresco and Phillips, 2001), and coincident D1–NMDA activation in the PFC is required for appetitive instrumental learning (Baldwin et al., 2002). Because the cAMP–PKA pathway seems to play a key role in the early stages of hippocampal→ PFC LTP (Gurden et al., 2000; Jay, 2003) and D1–NMDA interactions are PKA dependent, it is possible that their synergistic action on pyramidal cell excitability facilitates cortical plasticity mechanisms, reinforcing ongoing activity in the PFC. Thus, a D1 enhancement of NMDA responses in PFC pyramidal cell excitability may provide a cellular mechanism by which DA sustains prolonged depolarizations, acting as a state-dependent stabilizer of the appropriate neural ensembles (O'Donnell et al., 1999; O'Donnell 2003).
In contrast to D1 receptors, the D2 role in PFC function is less clear. There is evidence suggesting a D2 modulation of working memory (Druzin et al., 2000) and age-related PFC cognitive functions (Arnsten et al., 1995). The inhibitory D2 effects on pyramidal cell excitability may constitute important cellular mechanisms for selection of relevant information in the PFC. Both the direct (D2–AMPA) and indirect (D2–NMDA via GABA activation) interactions can result in dampening of excitatory inputs, which would contribute to fine tuning of PFC cell firing. Indeed, it has been shown that GABAA activation improves spatial selectivity of PFC pyramidal cell firing (Rao et al., 2000), suggesting that interactions between pyramidal and nonpyramidal neurons are important in shaping neuronal activity engaged in working memory processes.
During conditions eliciting DA cell burst firing (i.e., salient stimuli) (Schultz, 2002), the phasic DA increase in the PFC activates both D1 and D2 receptors. It is possible that the inhibitory D2 effect on AMPA-mediated responses may increase the signal-to-noise ratio by attenuating weak signals, whereas the concurrent dampening of NMDA effects by D2 receptors via an enhanced GABA transmission may be a critical element in the control of timing and spatial selectivity of pyramidal cell firing (Rao et al., 2000). For instance, in vivo intracellular studies assessing responses of PFC neurons to ventral tegmental area stimulation have revealed a prolonged plateau depolarization along with suppression of action potential firing (Lewis and O'Donnell, 2000). This persistent depolarization was reduced by a D1 antagonist; thus, it could represent a D1 enhancement of NMDA function. The decrease in firing could be the result of both direct and indirect (via GABA interneurons) D2 actions. Such prolonged depolarization with reduced firing was proposed to reflect a gating mechanism of the appropriate PFC neural ensemble, although it reduced irrelevant information. Thus, combined activation of D1, D2, GABA, and glutamate receptors may allow a “gating with filtering” phenomenon by virtue of the interactions reported here. Disruption of these interactions in the PFC may contribute to abnormal coordination of pyramidal neuron firing and may yield the cognitive deficits observed in schizophrenia and related neuropsychiatric disorders (O'Donnell and Grace, 1998; Laruelle, 2000).
Footnotes
This work was supported by National Institutes of Health Grant MH57683 and a National Alliance for Research on Schizophrenia and Depression Independent Investigator Award (P.O.).
Correspondence should be addressed to Kuei-Yuan Tseng, Center for Neuropharmacology and Neuroscience, Albany Medical College (MC-136), 47 New Scotland Avenue, Albany, NY 12208. E-mail: tsengky@mail.amc.edu.
DOI:10.1523/JNEUROSCI.1021-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/245131-09$15.00/0
References
- Arnsten AF, Cai JX, Steere JC, Goldman-Rakic PS (1995) Dopamine D2 receptor mechanisms contribute to age-related cognitive decline: the effects of quinpirole on memory and motor performance in monkeys. J Neurosci 15: 3429–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AE, Sadeghian K, Kelley AE (2002) Appetitive instrumental learning requires coincident activation of NMDA and dopamine D1 receptors within the medial prefrontal cortex. J Neurosci 22: 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Levine MS (1998) Dopamine and N-methyl-d-aspartate receptor interactions in the neostriatum. Dev Neurosci 20: 1–18. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Radisavljevic Z, Peacock W, Levine MS, Buchwald NA (1992) Differential modulation by dopamine of responses evoked by excitatory amino acids in human cortex. Synapse 11: 330–341. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Buchwald NA, Levine MS (1993) Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci USA 90: 9576–9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Colwell CS, Itri JN, Chandler SH, Levine MS (1998) Dopaminergic modulation of NMDA-induced whole cell currents in neostriatal neurons in slices: contribution of calcium conductances. J Neurophysiol 79: 82–94. [DOI] [PubMed] [Google Scholar]
- Dodt H-U, Frick A, Kampe K, Zieglgänsberger W (1998) NMDA and AMPA receptors on neocortical neurons are differentially distributed. Eur J Neurosci 10: 3351–3357. [DOI] [PubMed] [Google Scholar]
- Druzin MY, Kurzina NP, Malinina EP, Kozlov AP (2000) The effects of local application of D2 selective dopaminergic drugs into the medial prefrontal cortex of rats in a delayed spatial choice task. Behav Brain Res 109: 99–111. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Standaert DG (2001) Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J Neurosci 21: 5546–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, Phillips AG (2001) Delay-dependent modulation of memory retrieval by infusion of a dopamine D1 agonist into the rat medial prefrontal cortex. Behav Neurosci 115: 934–939. [PubMed] [Google Scholar]
- Flores-Hernandez J, Cepeda C, Hernandez-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS (2002) Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J Neurophysiol 88: 3010–3020. [DOI] [PubMed] [Google Scholar]
- Gao WJ, Krimer LS, Goldman-Rakic PS (2001) Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci USA 98: 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Hablitz JJ (2003) Dopamine enhances EPSCs in layer II–III pyramidal neurons in rat prefrontal cortex. J Neurosci 23: 867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobin AC, Deutch AY (1998) Dopaminergic regulation of extracellular gamma-aminobutyric acid levels in the prefrontal cortex of the rat. J Pharmacol Exp Ther 285: 350–357. [PubMed] [Google Scholar]
- Gulledge AT, Jaffe DB (1998) Dopamine decreases the excitability of layer V pyramidal cells in the rat prefrontal cortex. J Neurosci 18: 9139–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Jaffe DB (2001) Multiple effects of dopamine on layer V pyramidal cell excitability in rat prefrontal cortex. J Neurophysiol 86: 586–595. [DOI] [PubMed] [Google Scholar]
- Gurden H, Tassin JP, Jay TM (1999) Integrity of the mesocortical dopaminergic system is necessary for complete expression of in vivo hippocampal-prefrontal cortex long-term potentiation. Neuroscience 94: 1019–1027. [DOI] [PubMed] [Google Scholar]
- Gurden H, Takita M, Jay TM (2000) Essential role of D1 but not D2 receptors in the NMDA receptor-dependent long-term potentiation at hippocampal-prefrontal cortex synapses in vivo. J Neurosci 20: RC106(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Bargas J, Surmeier DJ, Reyes A, Galarraga E (1997) D1 receptor activation enhances evoked discharge in neostriatal medium spiny neurons by modulating an L-type Ca2+ conductance. J Neurosci 17: 3334–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ (2000) D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLCβ1-IP3-calcineurin-signaling cascade. J Neurosci 20: 8987–8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TM (2003) Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog Neurobiol 69: 375–390. [DOI] [PubMed] [Google Scholar]
- Kotecha SA, Oak JN, Jackson MF, Perez Y, Orser BA, Van Tol HH, MacDonald JF (2002) A D2 class dopamine receptor transactivates a receptor tyrosine kinase to inhibit NMDA receptor transmission. Neuron 35: 1111–1122. [DOI] [PubMed] [Google Scholar]
- Lachowicz JE, Sibley DR (1997) Molecular characteristics of mammalian dopamine receptors. Pharmacol Toxicol 81: 105–113. [DOI] [PubMed] [Google Scholar]
- Laruelle M (2000) The role of endogenous sensitization in the pathophysiology of schizophrenia: implications from recent brain imaging studies. Brain Res Brain Res Rev 31: 371–384. [DOI] [PubMed] [Google Scholar]
- Levine MS, Li Z, Cepeda C, Cromwell HC, Altemus KL (1996) Neuromodulatory actions of dopamine on synaptically-evoked neostriatal responses in slices. Synapse 24: 65–78. [DOI] [PubMed] [Google Scholar]
- Lewis BL, O'Donnell P (2000) Ventral tegmental area afferents to the prefrontal cortex maintain membrane potential “up” states in pyramidal neurons via D1 dopamine receptors. Cereb Cortex 10: 1168–1175. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Bjorklund A, Moore RY, Stenevi U (1974) Mesencephalic dopamine neurons projecting to neocortex. Brain Res 81: 325–331. [DOI] [PubMed] [Google Scholar]
- Ling DS, Benardo LS (1999) Restrictions on inhibitory circuits contribute to limited recruitment of fast inhibition in rat neocortical pyramidal cells. J Neurophysiol 82: 1793–1807. [DOI] [PubMed] [Google Scholar]
- Miller EK (2000) The prefrontal cortex and cognitive control. Nat Rev Neurosci 1: 59–65. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Surmeier J, Malenka RC (2000) Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci 23: 185–215. [DOI] [PubMed] [Google Scholar]
- O'Donnell P (2003) Dopamine gating of forebrain neural ensembles. Eur J Neurosci 17: 429–435. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA (1994) Tonic D2-mediated attenuation of cortical excitation in nucleus accumbens neurons recorded in vitro. Brain Res 634: 105–112. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA (1998) Dysfunctions in multiple interrelated systems as the neurobiological bases of schizophrenic symptom clusters. Schizophr Bull 24: 267–283. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Greene J, Pabello N, Lewis BL, Grace AA (1999) Modulation of cell firing in the nucleus accumbens. Ann NY Acad Sci 877: 157–175. [DOI] [PubMed] [Google Scholar]
- Petit TL, LeBoutillier JC, Gregorio A, Libstug H (1988) The pattern of dendritic development in the cerebral cortex of the rat. Brain Res 469: 209–219. [DOI] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS (2000) Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci 20: 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Prince DA (1996a) Spontaneous GABAA receptor-mediated inhibitory currents in adult rat somatosensory cortex. J Neurophysiol 75: 1573–1588. [DOI] [PubMed] [Google Scholar]
- Salin PA, Prince DA (1996b) Electrophysiological mapping of GABAA receptor-mediated inhibition in adult rat somatosensory cortex. J Neurophysiol 75: 1589–1600. [DOI] [PubMed] [Google Scholar]
- Schultz W (2002) Getting formal with dopamine and reward. Neuron 36: 241–263. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P (1998) A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci 18: 10297–10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternweis PC, Smrcka AV (1992) Regulation of phospholipase C by G proteins. Trends Biochem Sci 17: 502–506. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC, Nairn AC, Greengard P (1995) Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 14: 385–397. [DOI] [PubMed] [Google Scholar]
- Wang J, O'Donnell P (2001) D1 dopamine receptors potentiate NMDA-mediated excitability increase in layer V prefrontal cortical pyramidal neurons. Cereb Cortex 11: 452–462. [DOI] [PubMed] [Google Scholar]
- Zhu JJ (2000) Maturation of layer 5 neocortical pyramidal neurons: amplifying salient layer 1 and layer 4 inputs by Ca2+ action potentials in adult rat tuft dendrites. J Physiol (Lond) 526: 571–587. [DOI] [PMC free article] [PubMed] [Google Scholar]