

Abstract
Alterations in thalamic T-type Ca2+ channels are thought to contribute to the pathogenesis of absence seizures. Here, we found that mice with a null mutation for the pore-forming α1A subunits of P/Q-type channels (α1A–/– mice) were prone to absence seizures characterized by typical spike-and-wave discharges (SWDs) and behavioral arrests. Isolated thalamocortical relay (TC) neurons from these mice showed increased T-type Ca2+ currents in vitro. To examine the role of increased T-currents in α1A–/– TC neurons, we cross-bred α1A–/– mice with mice harboring a null mutation for the gene encoding α1G, a major isotype of T-type Ca2+ channels in TC neurons. α1A–/–/α1G–/– mice showed a complete loss of T-type Ca2+ currents in TC neurons and displayed no SWDs. Interestingly, α1A–/–/α1G+/– mice had 75% of the T-type Ca2+ currents in TC neurons observed in α1A+/+/α1G+/+ mice and showed SWD activity that was quantitatively similar to that in α1A–/–/α1G+/+ mice. Similar results were obtained using double-mutant mice harboring the α1G mutation plus another mutation also used as a model for absence seizures, i.e., lethargic (β4lh/lh), tottering (α1Atg/tg), or stargazer (γ2stg/stg). The present results reveal that α1G T-type Ca2+ channels play a critical role in the genesis of spontaneous absence seizures resulting from hypofunctioning P/Q-type channels, but that the augmentation of thalamic T-type Ca2+ currents is not an essential step in the genesis of absence seizures.
Keywords: calcium, Ca, EEG, electroencephalogram, epilepsy, gene, mutant, thalamus
Introduction
Pharmacological studies suggest that low-voltage-activated T-type Ca2+ channels are involved in the genesis of absence seizures, which are characterized by spike-and-wave discharges (SWDs) (van Luijtelaar et al., 2000; Porcello et al., 2003). Antagonists of T-type Ca2+ channels suppress both slow intrathalamic rhythms in vitro (Porcello et al., 2003) and SWDs in human absence seizure patients and in rodent models of absence seizures (Heller et al., 1983; Hosford et al., 1992; van Luijtelaar et al., 2000). Previous genetic studies indicate that of the three α1 subunits of T-type Ca2+ channels (G, H, and I), the α1G subunit is critically involved in SWD genesis. Mice with a null mutation of the α1G gene lacked low-threshold burst firing in thalamocortical relay (TC) neurons in vitro (Kim et al., 2001) and in vivo (Kim et al., 2003) and were resistant to SWDs induced by GABAB-receptor agonists (Kim et al., 2001).
T-type Ca2+ channel activity has been studied in rodent models of absence seizures to determine the role of this channel in SWD genesis. The augmentation of T-type Ca2+ currents in the thalamus was first reported in studies using genetic absence epilepsy rats from Strasbourg (Tsakiridou et al., 1995; Talley et al., 2000). Computational modeling studies support the concept that augmented T-type Ca2+ currents increase the number of burst spikes and thereby enhance thalamic synchrony (Destexhe et al., 1996, 1998; Hughes et al., 1999; Thomas and Grisar, 2000). These studies imply that functional enhancement of the T-type Ca2+ channel can contribute to the development of absence seizures by enhancing the probability of thalamocortical hypersynchronization. Mice with mutations in various subunits of the high-voltage-activated (HVA) Ca2+ channels, namely tottering (α1Atg/tg), lethargic (β4lh/lh), and stargazer (γ2stg/stg) mice, display SWDs (Noebels and Sidman, 1979; Hosford et al., 1992; Qiao and Noebels, 1993). Recently, these mutant mice were shown to have higher T-type Ca2+ current levels in their TC neurons compared with normal mice, suggesting a possibility that the enhancement of T-currents might underlie the pathogenesis of absence seizures in those mutants (Tsakiridou et al., 1995; Zhang et al., 2002).
To address these issues, we examined absence seizures in mice with a null mutation in the gene coding for pore-forming α1A subunits (α1A–/– mice), which therefore lack the P/Q-type Ca2+ currents. We then explored the role of T-type Ca2+ channels in the genesis of absence seizures in these null mice and other spontaneous mutant mice. Our results provide in vivo evidence that baseline T-type Ca2+ currents but not their augmentation in TCs are necessary and sufficient to support absence seizures in various genetic mouse models.
Materials and Methods
Animals. The α1A mice used for EEG recording and patch-clamp analyses were F2 progeny derived from intercrossing heterozygotes of the F1 (129/sv×C57BL/6J) genetic background. Mice heterozygous for the α1A null mutation (Jun et al., 1999), lethargic (β4lh/lh), tottering (α1Atg/tg), or stargazer (γ2stg/stg) were mated with α1G–/– mice (Kim et al., 2001) to obtain α1A+/–/α1G+/–, α1Atg/+/α1G+/–, β4lh/+/α1G+/–, γ2stg/+/α1G+/– offspring. Double-heterozygous mice for the two mutations were intercrossed to obtain α1A–/–, β4lh/lh, α1Atg/tg, γ2stg/stg mice with different numbers of α1G gene alleles. These mice allowed examination of the effect of deleting α1G on the background of absence seizures in α1A–/– mice. In addition, the mice allowed investigation of the pathological role of T-current enhancement in absence seizures. The animals were housed at room temperature (22°C), fed ad libitum, and submitted to a 12 hr light/dark cycle. All handling of mice was in accordance with the regulations of the institute.
Electrode implantation and cortical EEG recording. Differential EEG recording was performed as described previously (Kim et al., 2001). Mice were anesthetized with avertin (tribromoethyl alcohol/tertiary amyl alcohol; Aldrich, Milwaukee, WI). Subdural tungsten electrodes (A-M Systems, Carlsborg, WA) were bilaterally or unilaterally implanted in the temporal lobe region, and a ground electrode was implanted in the occipital region of the brain (Schridde and van Luijtelaar, 2004). The head mount was secured using dental cement, and mice were allowed to recover for at least 24 hr before EEG recordings. EEG activity (sampling frequency, 200 μsec) was recorded during 0.5–1 hr samples for 1–2 hr using a pCLAMP8.0 program (Axon Instruments, Foster City, CA). Only SWDs with a minimum voltage amplitude of twice the background EEG and a minimum duration of 0.7 sec were included in analysis, and SWDs separated by <1 sec were regarded as a single SWD event.
To test the effects of drugs on absence seizures, EEGs of α1A–/– mice (3–4 weeks old) were recorded for 2 hr, starting 1 hr before drug administration. Valproic acid (Sigma, St. Louis, MO) or ethoxusimide (Sigma) was diluted in physiological saline (0.85% NaCl) and injected intraperitoneally. The selection of the drug dose was based on published data (Heller et al., 1983; Aizawa et al., 1997) and preliminary experiments.
Data analysis. EEG signals were amplified, filtered, and recorded using pCLAMP8 software (Axon Instruments). To assess the difference in EEG activity between each group of mice, we used the linear spectra of consecutive EEG data sections (duration over 1 min; range, 1–15 Hz) computed using the pCLAMP8 program using the fast Fourier transform.
Northern blot. Total RNA was isolated from the thalamic region of 3- to 4-week-old mice. RNA (20 μg per lane) was separated on 1.0% agarose gels containing 2.2 m formaldehyde and then transferred to nylon membranes by capillary blot. The hybridization solution comprised 7% SDS, 1% BSA, 0.5 m NaHPO4, 1 mm EDTA, and a random-primed rat cDNA probe corresponding to nucleotides 4699–6174 bp of the α1G clone. A glyceraldehyde 3-phosphate dehydrogenase probe was also hybridized to blots, and this signal was used to normalize for RNA loading. Signal detection and normalization were performed using the ImageQuant Image Analysis system (Amersham Biosciences, Arlington Heights, IL). Concentrations are expressed as “percentage of wild-type control” analyzed on the same blots.
Whole-cell voltage-clamp analysis. Patch-clamp analysis of thalamic relay neurons was performed as described previously (Kim et al., 2001). Thalamic relay neurons were acutely dissociated as described previously (Tsakiridou et al., 1995; Raman and Bean, 1999). Briefly, brains were cooled rapidly in ice-chilled slicing solution consisting of (in mm): 122 NaCl, 26 NaHCO3, 1.2 NaH2PO4, 2 MgCl2, 2 CaCl2, 3 KCl and 10 glucose, after which 300 μm sections were cut in the coronal plane using a Vibratome (Ted Pella, Redding, CA). Slices containing the ventrobasal complex were dissected with a scalpel to isolate the thalamus. Thalamic slices were incubated at 35°C for 6 min with protease XXIII (3 mg/ml; Sigma) in an oxygenated HEPES-buffered solution consisting of (in mm): 82 Na2SO4, 30KSO4, 5 MgCl2, 10 HEPES, 10 glucose, 0.01% phenol red, and adjusted to pH 7.4 with NaOH. The enzymatic reaction was stopped by adding BSA (1 mg/ml; Sigma) and trypsin inhibitor (1 mg/ml; Sigma). Each thalamic slice was triturated with fire-polished Pasteur pipettes and plated onto a recording chamber. Healthy-looking neurons of triangular or multipolar shapes with processed dendrites were used for patch-clamp recordings. Recordings were performed using electrodes (3.5–6.5 MΩ) fabricated from borosilicate glass (Warner Instruments, Hamden, CT) in an extracellular solution consisting of (in mm): 55 TEA-Cl, 3 CaCl2, 10 HEPES, adjusted to pH 7.4 with TEA-OH. Patch pipettes were filled with a solution containing (in mm): 110 TrisPO4 dibasic, 28 Tris-base, 11 EGTA, 2 MaCl2, 0.5 CaCl2, 4 Na2ATP, 0.3 GTP-Na, 0.001 TTX, pH 7.3. The series resistance compensation (>60%) was used routinely, and patch-recording data with access resistance (>20 MΩ) were discarded. The currents were leak-subtracted using a P/4 protocol. Signals were digitized using an Axopatch 200-B amplifier (Axon Instruments) and analyzed using pCLAMP8 software (Axon Instruments).
Results
Absence seizures with 3 Hz SWDs in α1A–/– mice
α1A–/– mice develop progressive neurological symptoms characterized specifically by ataxia and dystonia, before dying ∼4 weeks after birth (Jun et al., 1999). We recorded cortical EEG activities in α1A–/– and wild-type mice at 3–4 weeks of age. We found that α1A–/– mice (n = 10) exhibited spontaneous 3–5 Hz SWDs (Fig. 1A) and that each episode was accompanied by behavioral arrest, often with twitching of the vibrissa. These abnormal cortical activities were not observed in wild-type littermates (Fig. 1A). SWDs occurred ∼160 times per hour in α1A–/– mice (Table 1). We examined the effect of the anti-epileptic drugs ethoxusimide (150 mg/kg; n = 6) and valproic acid (10 mg/kg; n = 5) on the incidence of SWDs in α1A–/– mice. These drugs are documented to suppress absence seizure SWDs in humans and rodents (Heller et al., 1983; Hosford et al., 1992). Compared with vehicle-treated α1A–/– mice, we found that ethoxusimide decreased the incidence of SWDs from 161.7 ± 40.5 to 40.8 ± 18.6 per hour, whereas valproic acid reduced the incidence from 181.6 ± 28.9 to 64.4 ± 26.6 per hour (Fig. 1B). Taken together, the behavioral, electrographic, and pharmacological characters of seizures in α1A–/– mice are similar to those of mice absence seizures(Hosford et al., 1992; Aizawa et al., 1997).
Figure 1.

EEG recordings from the cortex of freely moving α1A–/– mice. A, Top, Representative EEG traces show no SWD activity from wild-type mice. Bottom, Spontaneous 3–5 Hz SWDs with high amplitude of cortical activity occurred in 3- to 4-week-old α1A–/– mice (n = 10). The α1A–/– mice displayed behavioral immobility and maintained a fixed posture throughout the SWDs. The black dot indicates SWDs. The thin scale bars are valid for all EEG recordings displayed. The thick-lined periods are expanded for detail. B, Effect of anti-epileptic drugs (white bars) and control vehicle (shaded bars) on the occurrence of absence seizures in α1A–/– mice. Bars represent the mean number of SWDs per hour for α1A–/– mice exposed to each drug. Both ethoxusimde (150 mg/kg; n = 6) and valproic acid (10 mg/kg; n = 5) were effective in reducing the occurrence of SWDs of α1A–/– mice (*p < 0.025; two-tailed t test). Error bars represent ±SEM.
Table 1.
Characteristic of SWDs in mutant mice
|
Mutant mice |
Frequency (Hz) |
Mean duration (sec) |
Number of incidents per hour |
|---|---|---|---|
| α1A-/- | |||
| α1G+/+ | 3-4 | 2.0 ± 0.2 (0.7-5) | 161.7 ± 40.5 |
| α1G+/- | 3-4 | 2.1 ± 0.2 (0.7-5) | 156.2 ± 22.0 |
| α1G-/- | — | — | 0** |
| α1Atg/tg | |||
| α1G+/+ | 6-7 | 2.2 ± 0.2 (0.7-6) | 98.3 ± 12.2 |
| α1G+/- | 6-7 | 2.1 ± 0.2 (0.7-5) | 90.2 ± 15.3 |
| α1G-/- | — | — | 0** |
| β4lh/lh | |||
| α1G+/+ | 5-6 | 1.5 ± 0.1 (0.7-5) | 164.6 ± 25.7 |
| α1G+/- | 5-6 | 1.3 ± 0.1 (0.7-5) | 204.8 ± 26.0 |
| α1G-/- | 5-6 | 1.6 ± 0.2 (0.7-5) | 7.1 ± 4.7** |
| γ2stg/stg | |||
| α1G+/+ | 5-7 | 1.9 ± 0.2 (0.7-10) | 132.0 ± 8.7 |
| α1G+/- | 5-7 | 1.8 ± 0.2 (0.7-10) | 135.6 ± 20.0 |
| α1G-/-
|
5-7 |
1.3 ± 0.1 (0.7-2)* |
9.4 ± 3.4** |
Values, except for frequency, are means ± SEM. Values in parentheses are ranges. —, Not found. *p <0.05, **p <0.005 (mutants compared with α1G+/+ within each subgroup; two-tailed t test).
Generation of α1A–/–mice with differing numbers of α1G alleles
α1G is one of the T-type Ca2+ channels highly expressed in thalamic relay neurons and is selectively involved in SWD seizures induced by GABAB receptor agonists (Kim et al., 2001). We sought to determine whether α1G T-type Ca2+ channels are pathophysiologically involved in the generation and expression of absence seizures in α1A–/– mice. α1G+/– and α1A+/– mice were cross-bred to generate double heterozygotes, α1A+/–/α1G+/– mice. A result of double heterozygote matings was α1A–/– mice with different numbers of α1G gene alleles, i.e., α1A–/–/α1G+/+, α1A–/–/α1G+/–, and α1A–/–/α1G–/–. On visual inspection, α1A–/–/α1G+/– and α1A–/–/α1G–/– mice exhibited severe ataxia and weakness, similar to that observed in α1A–/– single knock-out mice (Jun et al., 1999). In addition, α1A–/–/α1G+/– and α1A–/–/α1G–/– mice did not survive past weaning.
Decreased HVA Ca2+ currents in α1A–/– thalamic relay neurons
We performed whole-cell voltage-clamp experiments to examine the effect of α1A genetic deletion on HVA Ca2+ currents in acutely dissociated TC neurons, which are characterized by their large size and triangular or multipolar shape with truncated dendrites (Huguenard and Prince, 1992; Pape et al., 1994; Kim et al., 2001). HVA Ca2+ currents, supported by 3 mm Ca2+ as a charge carrier, were activated by step depolarization from a holding potential of –60 mV. As a result, large sustained voltage-dependent Ca2+ inward currents were evoked. Figure 2A shows a typical trace of total Ca2+ currents with a slowly inactivating component recorded from TC neurons. A significant difference was observed between neurons from α1A–/–/α1G+/+ mice compared with neurons of control (α1A+/+/α1G+/+) mice in terms of the amplitude of Ca2+ currents (Fig. 2A). The amplitudes of HVA Ca2+ currents at all command membrane potentials were smaller in α1A–/–/α1G+/+ (n = 12) and α1A–/–/α1G–/– (n = 11) TC neurons compared with controls (n = 10). This decrease in HVA Ca2+ current can be accounted for by the loss of P/Q-type Ca2+ currents in α1A–/– mice, consistent with previous results in which P/Q-type Ca2+ currents were shown to be a component of HVA Ca2+ currents in TC neurons (Pfrieger et al., 1992; Kammermeier and Jones, 1997).
Figure 2.
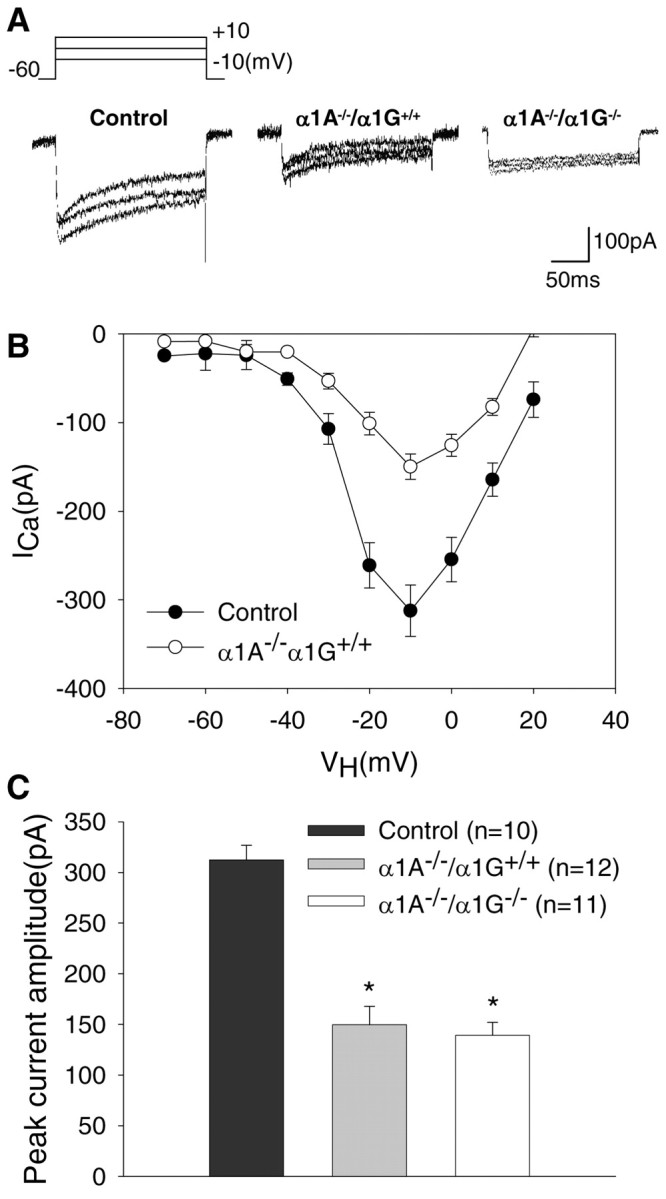
HVA Ca2+ currents in acutely isolated thalamic relay neurons. A, Representative traces of total Ca2+ currents of control, α1A–/–/α1G+/+, and α1A–/–/α1G–/– TC neurons evoked by stepping membrane potential voltages between –10 and +10 mV in 10 mV increments from a holding potential of –60 mV. Sustained HVA Ca2+ currents decayed slowly during the 200 msec step commands. B, Mean peak I–V curves for total Ca2+ currents in TC neurons show different features between the two groups. The I–V relationship at this voltage protocol shows that the HVA Ca2+ currents in α1A–/–/α1G+/+ cells were dramatically reduced at all testing voltage steps above –40 mV compared with those in control. Symbols represent pooled data from α1A–/–/α1G+/+ (open symbols; n = 8) and control mice (filled symbols; n = 10). C, The HVA Ca2+ histogram of peak amplitude is at –10 mV in control (black bars; n = 10), α1A–/–/α1G+/+ (gray bars; n = 12), and α1A–/–/α1G–/– (white bars; n = 11), with holding potential at –60 mV. Note that the peak amplitude of HVA Ca2+ currents in α1A–/–/α1G+/+ and α1A–/–/α1G–/– mice was decreased significantly more than in α1A+/+/α1G+/+ mice(*p<0.001; two-tailed t test). No statistically significant changes occurred in the HVA Ca2+ current of α1A–/–/α1G–/– mice compared with that from TC neurons of α1A–/–/α1G+/+ mice (p > 0.05). Control indicates α1A+/+/α1G+/+.
To determine the current–voltage (I–V) relationship of total HVA Ca2+ currents in neurons from α1A–/–/α1G+/+ (n = 10) and control (n = 8) mice, a series of voltage steps from –70 to +20 mV with +10 mV increments was delivered from a holding potential at –70 mV. The I–V relationship curve in control mice revealed a prominent shoulder at negative potentials, indicating channel activation at low voltages, with the peak of the I–V curve occurring at –10 mV (Fig. 2B). In contrast to these findings using control neurons, in neurons from α1A–/–/α1G+/+ mice, the I–V relationships showed a significant decrease in the amplitude of the HVA Ca2+ current in voltage steps from –30 to +20 mV, peaking near –10 mV without a change in the shape of the I–V curves (Fig. 2B). We examined the profile of HVA Ca2+ currents in TC cells from neurons of α1A–/–/α1G–/– and α1A–/–/α1G+/– (data not shown) mice and found no difference in HVA Ca2+ currents between these two genotypes and α1A–/–/α1G+/+ mice (Fig. 2A). These findings indicate that genetic reduction of T-type Ca2+ currents does not strongly modulate HVA Ca2+ currents for compensation. We quantitatively compared the peak amplitude of inward Ca2+ currents evoked by depolarization from –60 to –10 mV in TC neurons from control and α1A–/–/α1G+/+ mice. Pooled data showed that the total HVA Ca2+ current was 312.3 ± 14.5 pA in neurons of controls (n = 10) (Fig. 2C, black bars) and 149.8 ± 18 pA in neurons of α1A–/–/α1G+/+ (n = 12) (Fig. 2C, gray bars) mice (p < 0.001); however, the averaged peak amplitude of the HVA Ca2+ current in neurons of α1A–/–/α1G–/– mice at –10 mV (139.2 ± 12.9 pA; n = 11) (Fig. 2C, white bars) was similar to that of neurons of α1A–/–/α1G+/+ mice (149.8 ± 18 pA; n = 12) (Fig. 2C, gray bars) (p > 0.05). The differences in the peak amplitudes of HVA Ca2+ currents between neurons of α1A–/–/α1G+/+ and control mice were not caused by differences in the surface area of cells because the value of the whole-cell capacitance was not different between α1A–/–/α1G+/+ (11.5 ± 0.6 pF; n = 12) and control (11.5 ± 0.9 pF; n = 10) cells.
Increased T-type calcium currents in α1A–/– thalamic relay neurons
To examine T-type Ca2+ currents, we used a voltage protocol in which a voltage step from –110 to –45 mV activates transient T-currents (Huguenard and Prince, 1992) (Fig. 3A). This T-current reached peak amplitude ∼10 msec after onset of the 100 msec voltage step and then inactivated rapidly. The TC neurons from α1A–/–/α1G–/– mice displayed a near complete loss of T-type Ca2+ currents (n = 8) (Fig. 3A), consistent with previous data (Kim et al., 2001). In contrast, transient Ca2+ currents of larger amplitude were evoked from α1A–/–/α1G+/+ cells compared with control (α1A+/+/α1G+/+) cells (Fig. 3A). The data presented in Figure 3B show that the averaged peak value of T-type Ca2+ currents at –45 mV was significantly larger in α1A–/–/α1G+/+ neurons (264.4 ± 24.6 pA; n = 10) (Fig. 3B, gray bars) than in controls (165.2 ± 10.5 pA; n = 13) (black bars) (p < 0.0005). In contrast, the peak amplitude of T-currents from α1A–/–/α1G+/– neurons was 125.3 ± 10.0 pA (n = 18) (Fig. 3B, dark gray bars), which is ∼50% of that in α1A–/–/α1G+/+ and ∼75% of that in control TC cells (p < 0.005). These properties of T-type Ca2+ currents from each genotype were consistent with the current density histogram of peak T-currents (Fig. 3C). These data indicate that the enhancement of T-type Ca2+ currents was not caused by an increase in TC neuron membrane size. No significant difference in time-to-peak was observed between groups [8.2 ± 0.4 msec for control (n = 11), 8.4 ± 0.6 msec for α1A–/–/α1G+/+ (n = 11), and 8.1 ± 0.5 msec for α1A–/–/α1G+/– (n = 18) TC cells].
Figure 3.
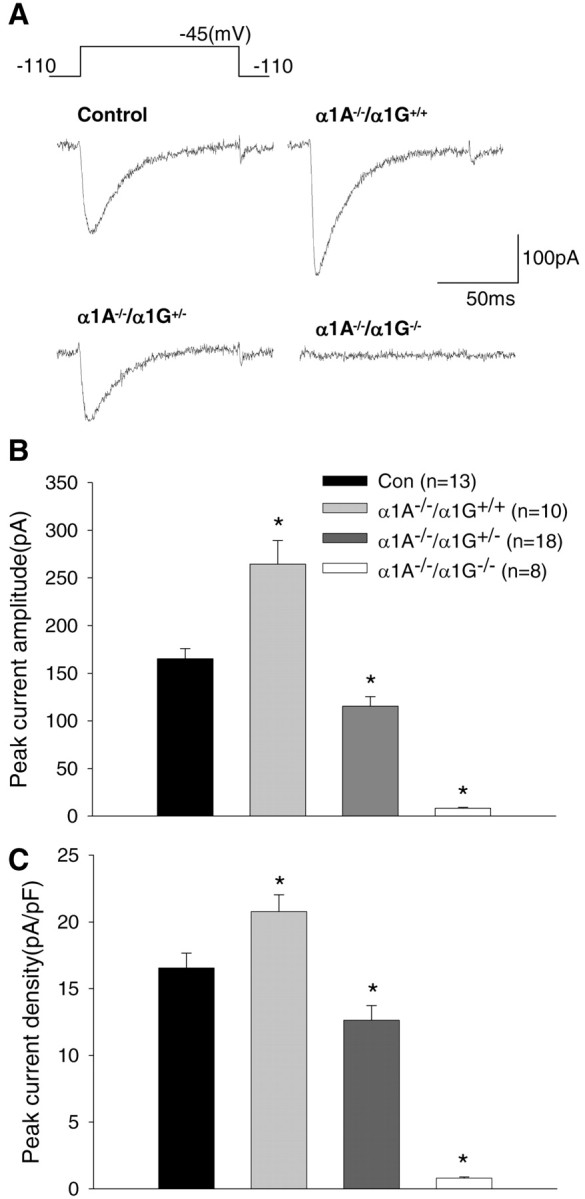
HVA Ca2+ currents in acutely isolated thalamic relay neurons. A, Much larger T-currents, which are rapidly inactivated, were obtained from TC neurons in α1A–/–/α1G+/+ than from control (α1A+/+/α1G+/+), whereas T-currents were nearly absent in the α1A–/–/α1G–/– TC neurons. B, The histogram shows the mean peak amplitude of T-type Ca2+ current in acutely isolated TC neurons from control (black bars), α1A–/–/α1G+/+ (gray bars), α1A–/–/α1G+/– (dark gray bars), and α1A–/–/α1G–/– (white bars). C, The histogram indicates the mean peak T-type Ca2+ current densities that were from the same cells used for the histogram (B) in each group. The asterisks indicate the significant difference between each mutant and control (*p < 0.05; two-tailed t test).
One explanation for augmented T-type Ca2+ currents in α1A–/– TC cells is a change in voltage dependence, such as steady-state inactivation to a more depolarized level. To obtain steady-state inactivation curves, we delivered a prepulse to various membrane potentials before a –40 mV test stimulus. Current traces of steady-state inactivation of T-currents are shown in Figure 4A. The data presented in Figure 4B show that despite increased T-type Ca2+ current amplitude, steady-state inactivation was similar for both groups when peak current values from the test pulses were normalized to the maximal current amplitude in each cell.
Figure 4.

Steady-state inactivation of T-currents. A, The membrane potential was stepped to –40 mV from holding potentials ranging from –110 to –40 mV. B, The normalized peak amplitude of the Ca2+ currents elicited by the test pulse at –40 mV was plotted as a function of the holding potential. The symbols represent pooled data from α1A+/+ (filled square; n = 6) and α1A–/– (open square; n = 7). Error bars represent ±SEM.
We examined the possibility that the alteration in T-current amplitudes was caused by an increase in the amount of T-type Ca2+ channels. Using rat α1G cDNA [nucleotide (nt) 4699–6174] as a probe in Northern blot analysis, we examined α1G gene expression in thalamus tissue isolated from α1A–/– and α1A+/+ mice. Although no signal was detected in α1G–/– thalamic tissues, visual examination of autoradiographs indicated similar α1G mRNA expression in thalamic tissues from both α1A–/– (n = 5) and α1A+/+ (n = 4) mice (Fig. 5). Quantitative image analysis confirmed that there was no significant difference in expression (94 ± 9 for α1A–/– and 100 ± 5% for α1A+/+; p > 0.5).
Figure 5.

Northern blot analysis of α1G transcripts from thalamus. For Northern blot analysis, mouse (α1G–/–, α1A+/+, and α1A–/–) thalamic tissue was probed with α1G (nt 4699–6174 bp) and then exposed for 3 d. Internal control was performed using GADPH. Thalamic α1G transcript expression that was not detected inα1G–/– mice did not differ between α1A+/+ and α1A–/–.
Effect of α1G allele number on SWD generation in α1A–/– mice
We investigated whether α1G genes were necessary for spontaneous absence seizures in α1A–/– mice, because they are functionally involved in drug-induced SWD seizures (Kim et al., 2001). Subdural EEG measurements were conducted on 3-week-old α1A–/–/α1G+/+ (n = 5) and α1A–/–/α1G–/– (n = 4) mice. We found that α1A–/–/α1G–/– mice did not exhibit the typical 3–4 Hz SWDs that were observed in α1A–/–/α1G+/+ littermates (Fig. 6A) (157.5 ± 43.3 per hour for α1A–/–/α1G+/+ and 0 ± 0 per hour for α1A–/–/α1G–/– mice; p < 0.005) (Table 1). A power spectrum analysis confirmed this alteration in cortical paroxysmal activity, i.e., disappearance of the 3 Hz peak frequency in α1A–/–/α1G–/– compared with α1A–/–/α1G+/+ mice (Fig. 7A). It appears that complete genetic deletion of α1G genes functionally abolishes generation of the spontaneous cortical SWD activity observed in α1A–/–/α1G+/+ mice.
Figure 6.

EEG recording of α1G genetic deletion in SWD activities of various genetic mutants. Three traces of EEG recordings from α1A–/– (A), α1Atg/tg (B), β4lh/lh (C), and γ2stg/stg(D) mice are illustrated according to αlG gene dosage. The dotted line shown within the 1 min trace (left) is expanded as 15 sec EEG trace (right). The asterisks indicate SWDs on the EEG traces in each genotype. A, Spontaneous SWD activities with high amplitude were frequently recorded in the α1A–/–/α1G+/+ (top) and α1A–/–/α1G+/– mice (middle). SWD activities that still remained in α1A–/–/α1G+/– could not be observed from α1A–/–/α1G–/– mice (bottom).
Figure 7.
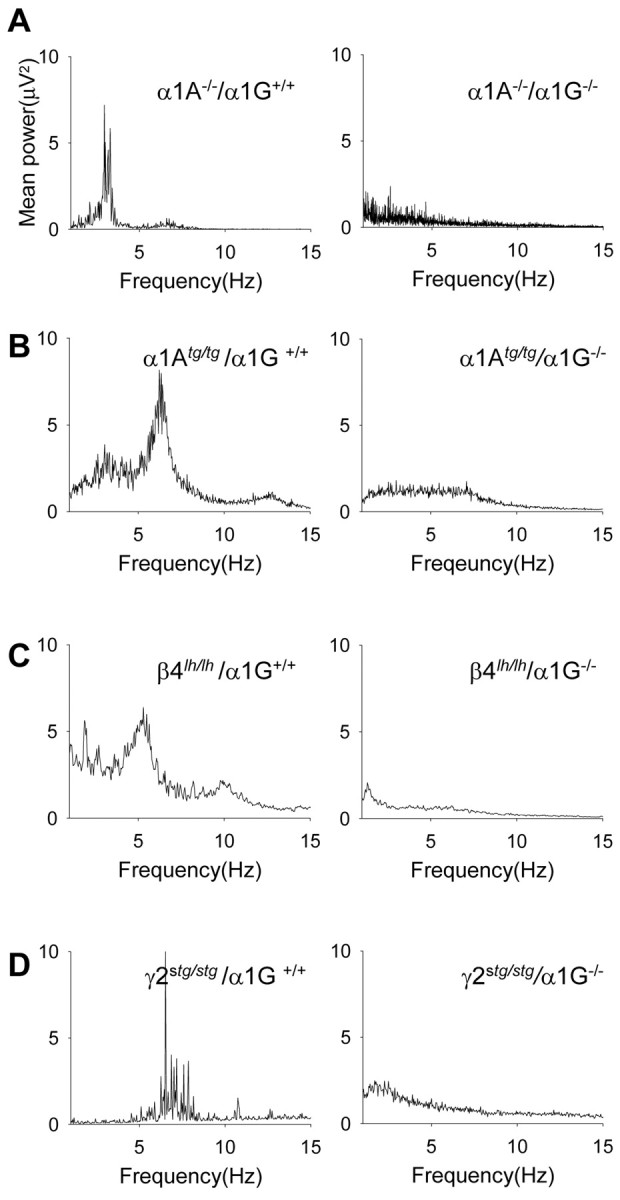
Power spectral analysis (1–15 Hz) of the filled potentials. A, Comparison of power spectra analysis between α1A–/–/α1G+/+ (left) and α1A–/–/α1G–/– (right) mice shows disappearance of the dominant frequency (3–4Hz) of SWDs in α1A–/–/α1G+/+. The alterations of major peak frequency in the power spectra analysis between the two groups of each spontaneous mutant line were clear as shown in α1A null mice (B–D). EEG traces >1 min taken from each of the double mutants were used for the power analysis (α1A–/–/α1G+/+, 6 traces from 4 mice; α1A–/–/α1G–/–, 5 traces from 3 mice; α1Atg/tg/α1G+/+, 20 traces from 3 mice; α1Atg/tg/α1G–/–, 20 traces from 5 mice; β4lh/lh/α1G+/+, 10 traces from 3 mice; β4lh/lh/α1G–/–, 10 traces from 3 mice; γ2stg/stg/α1G+/+, 9 traces from 3 mice; γ2stg/stg/α1G–/–, 9 traces from 3 mice).
We investigated whether augmentation of thalamic T-currents was necessary for absence seizures. We examined SWDs in α1G heterozygote mice on a α1A–/– background. These mice were shown previously to exhibit 75% of the T-currents observed in wild-type mice (Fig. 3). Such experiments present an opportunity to investigate the functional consequence of reduced T-current on absence seizures caused by lack of α1A subunits. If increased TC neuron T-current (compared with wild type) is a causative factor in SWD genesis in α1A–/– mice, mice with reduced or the same level of T-currents compared with nonepileptic mice should express no or altered SWD cortical activity. Contrary to our expectations, we found no significant difference between α1A–/–/α1G+/– and α1A–/–/α1G+/+ mice in terms of SWD duration or frequency (Table 1).
Role of α1G in SWD generation in various absence seizure mice models
Having found that SWDs in α1A/G double-mutant mice differed according to α1G allele “dose,” we examined the role of α1G in the genesis of SWDs in other mouse models of absence seizures. We again cross-bred heterozygote mice to produce double mutants containing α1G–/– and α1Atg/tg (Noebels and Sidman, 1979), β4lh/lh (Hosford et al., 1992), or γ2stg/stg (Noebels et al., 1990). Our study was on the basis of findings that compared with neurons of control mice, T-type Ca2+ currents are greater in thalamic relay neurons from α1Atg/tg, β4lh/lh, and γ2stg/stg mice (146, 151, and 145% of control, respectively) (Zhang et al., 2002). We performed EEG recordings in freely moving young (3–4 week) α1Atg/tg/α1G+/+ (n = 3), β4lh/lh/α1G+/+(n = 4), and γ2stg/stg/α1G+/+ (n = 3) mice on a mixed genetic background (129/sv×C57BL/6J). These mice exhibited 5–7 Hz SWDs with high amplitude, with the morphology and dominant frequency of the SWDs being slightly different among groups (Fig. 6B–D, Table 1). During a period of robust cortical paroxysmal activity, all mice exhibited a sudden behavioral arrest of movement and a fixed posture, indicating behavioral absence seizures. The mean seizure durations were 2.2 ± 0.2 sec (range, 0.7–6 sec) for α1Atg/tg/α1G+/+ mice, 1.5 ± 0.1 sec (0.7–5 sec) for β4lh/lh/α1G+/+ mice, and 1.9 ± 0.2 sec (0.7–10 sec) for γ2stg/stg/α1G+/+ mice (Table 1). The mean number of SWD events per hour were 98.3 ± 12.2 in α1Atg/tg/α1G+/+ (n = 3), 164 ± 25.7 in β4lh/lh/α1G+/+ (n = 4), and 132.0 ± 8.7 in γ2stg/stg/α1G+/+ (n = 3) mice (Table 1). The SWD patterns in these mutants were not different from those described previously (Noebels, 1984; Hosford et al., 1992; Qiao and Noebels, 1993). We took EEG measurements in double mutants completely lacking α1G genes. We found a complete suppression of SWDs in α1Atg/tg/α1G–/– mice (n = 5). Indeed, we did not observe any 6–7 Hz SWDs with minimum voltage amplitude of twice the EEG background and a minimum duration of 0.7 sec (Figs. 6B, 7B; Table 1). For both β4lh/lh/α1G–/– (n = 4) and γ2stg/stg/α1G–/– (n = 4) mice, cortical SWD paroxysmal activities were strongly suppressed (Fig. 6C,D; Table 1). The 5–7 Hz SWDs with very short duration (0.7–2 sec) were rare in γ2stg/stg/α1G–/– mice (9.4 ± 3.4 per hour; n = 4), and very few 5–6 Hz SWDs with a duration similar to those observed in β4lh/lh/α1G+/+ mice were detected on EEG recordings from β4lh/lh/α1G–/– mice (7.1 ± 4.7 per hour; n = 4) (Table 1). Interestingly, the results show that contrary to genetic ablation of α1G, which abolished cortical SWDs in mice harboring genetic dysfunction, in the α1A gene that encodes the main subunit of P/Q-type Ca2+ channel there was some paroxysmal cortical activity in α1G–/– mice, with a mutation in the regulatory subunit of the HVA Ca2+ channel (i.e., β4 and γ2). We used power spectrum analysis to simplify our data regarding changes in the dominant peak frequency. From this analysis, we conclude that there are few or no SWDs in any of these mutants as a result of homologous deletion of α1G genes (Fig. 7). Additionally, EEG analysis showed that SWDs were present in all double mutants that were heterozygous for α1G (i.e., α1G+/–) (Fig. 6B–D). Indeed, there was no difference in SWDs between these mutants and those with α1G+/+ (Table 1).
Discussion
P/Q-type Ca2+ channels and absence seizures
HVA Ca2+ channels (subdivided into L, N, P/Q, and R types) play critical roles in neuron function, such as neurotransmitter release (Wheeler et al., 1994), patterning of cell excitability (Cavelier et al., 2002; Park et al., 2003), and gene expression (Sutton et al., 1999). Spontaneous absence seizures are reported in mice with Ca2+ channelopathy caused by various mutations in the subunits of P/Q-type Ca2+ channels, which show a partial reduction in P/Q-type currents with no difference in mRNA and protein levels of the channels (Fletcher et al., 1996; Mori et al., 1996; Wakamori et al., 1998; Zwingman et al., 2001). Consistent with these findings, we found that α1A–/– mice, which lack P/Q-type channels, had SWD activity similar to that reported in mice with other α1A point mutations (Fletcher et al., 1996; Mori et al., 1996; Wakamori et al., 1998; Zwingman et al., 2001); however, in contrast to the reports showing that mutations in α1Atg/tg, β4lh/lh, and γ2stg/stg mice result in increased total HVA Ca2+ currents in TC neurons (Zhang et al., 2002), α1A null TC neurons had reduced total HVA Ca2+ currents, possibly because of the absence of P/Q-type Ca2+ currents. Thus, the present results suggest that the loss of P/Q-type current function contributes to the genesis of SWDs without any indirect changes to other HVA Ca2+ current types, at least in TC neurons. Consistent with this proposal, expression studies using a cloned α1A gene from a child patient with absence seizure and ataxia revealed a near-complete deletion of P/Q-type currents (Jouvenceau et al., 2001), similar to the findings obtained from α1A–/– mice (Kammermeier and Jones, 1997; Jun et al., 1999).
Heterogeneity of absence seizures: is α1G a common mediator for absence seizures?
Data from mouse models displaying various and nonoverlapping neurological abnormalities indicate the involvement of multiple mechanisms in the genesis of SWDs (Hosford et al., 1992; Di Pasquale et al., 1997; Zhang et al., 2002). Pharmacological studies support the complexity of SWDs. Although GABAB receptor-induced SWDs are associated with thalamocortical pathways (Caddick and Hosford, 1996; Kim et al., 2001), systemic administration of GABAA antagonists can induce SWDs in athalamic cats (Steriade and Contreras, 1998; Kim et al., 2001). Previously, we revealed that there were different mechanisms involved in GABAB receptor-mediated and GABAA antagonist-induced SWDs by showing that they are either dependent or independent of α1G T-type channels, respectively, because α1G–/– mice that lack thalamic burst firings are exclusively resistant to GABAB antagonist-induced SWDs (Kim et al., 2001). Thus, an aim of the present study was to characterize various mouse models of spontaneous absence seizures according to their dependence on α1G gene function. The present results show that α1G null mutation abolished SWDs of α1A–/– and α1Atg/tg mice and drastically reduced SWDs in β4lh/lh and γ2stg/stg mice. It is interesting to note that there were residual SWDs in double mutants with α1G–/– and γ2stg/stg or β4lh/lh. Considering that both γ2 and β4 are auxiliary subunits of α1 subunits, which have been known to modulate voltage dependence, kinetics, and amplitude of other types of Ca2+ channels as well as the P/Q-type (Kang et al., 2001; Schjott et al., 2003), the residual SWDs in γ2stg/stg/αlG–/– and β4lh/lh/αlG–/– mice appear independent of pathological interactions between α1A and dysfunctional γ2 and β4 subunits. This concept is supported by pharmacological studies using these mice in which absence seizures in γ2stg/stg mice were sensitive to MK-801, which is ineffective in the treatment of absence seizures in other mice (Heller et al., 1983; Aizawa et al., 1997); however, a common thread that weaves through the generation and propagation of absence seizures in α1A–/– mice, as well as other mutant mice, is critical dependence on the α1G gene.
Functional significance of T-currents in TC neurons
Many studies on Ca2+ channelopathy have reported altered Ca2+ current profiles as a result of Ca2+ channel mutations, indicating a strong correlation between a disease symptom and alteration in Ca2+ currents. For example, altered expression of the N-type Ca2+ channel α1B in β4lh/lh mice is suggested as a possible mechanism underlying absence seizures (McEnery et al., 1998), and increased expression of the α1C Ca2+ channel in cerebellar Purkinje cells in α1Atg/tg mice is associated with the dystonia in these mutant mice (Campbell and Hess, 1999). Similarly, the importance of T-currents in the development of absence seizures is underlined by recent studies using animal models of absence seizures (Tsakiridou et al., 1995; Zhang et al., 2002). These findings have motivated computational modeling studies to describe how augmented T-currents in thalamic neurons contribute to either physiological or pathophysiological synchrony in thalamocortical networks (Destexhe et al., 1996, 1998; Hughes et al., 1999; Thomas and Grisar, 2000). Somewhat unexpectedly, however, the present study using double-mutant mice (epileptic mice on a α1G–/– background) revealed that the basal level of T-currents in TC neurons was enough to support SWD generation. There was no quantitative difference in the severity of SWDs between 75 and 150% of wild-type dosage of T-currents in TC neurons isolated from the double mutants, suggesting that an increase in thalamic T-currents might not contribute to SWD genesis in vivo. At this point it cannot be ruled out that the increase of T-currents in other brain regions may support absence seizure development in these mutants, considering that the expression of α1G is also detected in other regions of brain, including cortex, olfactory bulb, and cerebellum (Talley et al., 1999). The question arises as to how normal levels of T-type Ca2+ currents in TC neurons contribute to SWDs generation. It is interesting to note that the hyperpolarizing shift in the resting membrane potential of hyperpolarization-activated cation channel 2-deficient thalamic relay neurons removes inactivation of T-type Ca2+ channels and thereby promotes burst rather than tonic firing in response to depolarizing inputs resulting in increased susceptibility to oscillations (Ludwig et al., 2003). Reduced excitatory but normal inhibitory synaptic transmission in β4lh/lh and α1Atg/tg mice thalami (Caddick et al., 1999) and enhanced GABAB receptor expression (Hosford et al., 1992) would result in relatively enhanced GABAergic input in β4lh/lh and α1Atg/tg thalamic neurons. Thus, more effective hyperpolarization per se could increase the likelihood of a T-type Ca2+ channel opening in TC neurons, enough to support SWDs without an increase in T-currents.
Finally, the present results suggest that a shift in research direction is required to determine the mechanisms underlying absence seizures. Beyond the issue of augmentation of T-type Ca2+ channels in TC neurons, studies are required to elucidate how hyperpolarizing inputs are overloaded in these neurons. The relationship between hypofunctioning P/Q-type channels and hyperpolarization of TC neurons sheds light on a possible novel therapeutic strategy for absence seizures.
Footnotes
This work was supported by a grant from the Chemoinformatics Program of the Korea Institute of Science and Technology and by a Creative Research Initiative grant from the Ministry of Science and Technology, Korea. We thank Dr. Donghyun Park for advice on analysis of EEG data and Heekyung Lee for reading this manuscript.
Correspondence should be addressed to Hee-Sup Shin, Center for Calcium and Learning, Korea Institute of Science and Technology, P.O. Box 131, Cheongryang, Seoul, 136-791, Korea. E-mail: shin@kist.re.kr.
DOI:10.1523/JNEUROSCI.5546-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/245249-09$15.00/0
References
- Aizawa M, Ito Y, Fukuda H (1997) Pharmacological profiles of generalized absence seizures in lethargic, stargazer and gamma-hydroxybutyrate-treated model mice. Neurosci Res 29: 17–25. [DOI] [PubMed] [Google Scholar]
- Caddick SJ, Hosford DA (1996) The role of GABAB mechanisms in animal models of absence seizures. Mol Neurobiol 13: 23–32. [DOI] [PubMed] [Google Scholar]
- Caddick SJ, Wang C, Fletcher CF, Jenkins NA, Copeland NG, Hosford DA (1999) Excitatory but not inhibitory synaptic transmission is reduced in lethargic (Cacnb4(lh)) and tottering (Cacna 1atg) mouse thalami. J Neurophysiol 81: 2066–2074. [DOI] [PubMed] [Google Scholar]
- Campbell DB, Hess EJ (1999) L-type calcium channels contribute to the tottering mouse dystonic episodes. Mol Pharmacol 55: 23–31. [DOI] [PubMed] [Google Scholar]
- Cavelier P, Pouille F, Desplantez T, Beekenkamp H, Bossu JL (2002) Control of the propagation of dendritic low-threshold Ca(2+) spikes in Purkinje cells from rat cerebellar slice cultures. J Physiol (Lond) 540: 57–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M, Sejnowski TJ, Huguenard JR (1996) In vivo, in vitro, and computational analysis of dendritic calcium currents in thalamic reticular neurons. J Neurosci 16: 169–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Neubig M, Ulrich D, Huguenard J (1998) Dendritic low-threshold calcium currents in thalamic relay cells. J Neurosci 18: 3574–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pasquale E, Keegan KD, Noebels JL (1997) Increased excitability and inward rectification in layer V cortical pyramidal neurons in the epileptic mutant mouse Stargazer. J Neurophysiol 77: 621–631. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy Jr JD, Hawkes R, Frankel WN, Copeland NG, Jenkins NA (1996) Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell 87: 607–617. [DOI] [PubMed] [Google Scholar]
- Heller AH, Dichter MA, Sidman RL (1983) Anticonvulsant sensitivity of absence seizures in the tottering mutant mouse. Epilepsia 24: 25–34. [DOI] [PubMed] [Google Scholar]
- Hosford DA, Clark S, Cao Z, Wilson Jr WA, Lin FH, Morrisett RA, Huin A (1992) The role of GABAB receptor activation in absence seizures of lethargic (lh/lh) mice. Science 257: 398–401. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Cope DW, Toth TI, Williams SR, Crunelli V (1999) All thalamocortical neurones possess a T-type Ca2+ “window” current that enables the expression of bistability-mediated activities. J Physiol (Lond) 517: 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA (1992) A novel T-type current underlies prolonged Ca(2+)-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci 12: 3804–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenceau A, Eunson LH, Spauschus A, Ramesh V, Zuberi SM, Kullmann DM, Hanna MG (2001) Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet 358: 801–807. [DOI] [PubMed] [Google Scholar]
- Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, Lee TG, Chin H, Adams ME, Scheller RH, Tsien RW, Shin HS (1999) Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc Natl Acad Sci USA 96: 15245–15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammermeier PJ, Jones SW (1997) High-voltage-activated calcium currents in neurons acutely isolated from the ventrobasal nucleus of the rat thalamus. J Neurophysiol 77: 465–475. [DOI] [PubMed] [Google Scholar]
- Kang MG, Chen CC, Felix R, Letts VA, Frankel WN, Mori Y, Campbell KP (2001) Biochemical and biophysical evidence for gamma 2 subunit association with neuronal voltage-activated Ca2+ channels. J Biol Chem 276: 32917–32924. [DOI] [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW, Shin HS (2001) Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron 31: 35–45. [DOI] [PubMed] [Google Scholar]
- Kim D, Park D, Choi S, Lee S, Sun M, Kim C, Shin HS (2003) Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science 302: 117–119. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M, Hofmann F (2003) Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J 22: 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEnery MW, Copeland TD, Vance CL (1998) Altered expression and assembly of N-type calcium channel alpha 1B and beta subunits in epileptic lethargic (lh/lh) mouse. J Biol Chem 273: 21435–21438. [DOI] [PubMed] [Google Scholar]
- Mori Y, Mikala G, Varadi G, Kobayashi T, Koch S, Wakamori M, Schwartz A (1996) Molecular pharmacology of voltage-dependent calcium channels. Jpn J Pharmacol 72: 83–109. [DOI] [PubMed] [Google Scholar]
- Noebels JL (1984) A single gene error of noradrenergic axon growth synchronizes central neurones. Nature 310: 409–411. [DOI] [PubMed] [Google Scholar]
- Noebels JL, Sidman RL (1979) Inherited epilepsy: spike-wave and focal motor seizures in the mutant mouse tottering. Science 204: 1334–1336. [DOI] [PubMed] [Google Scholar]
- Noebels JL, Qiao X, Bronson RT, Spencer C, Davisson MT (1990) Stargazer: a new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Res 7: 129–135. [DOI] [PubMed] [Google Scholar]
- Pape HC, Budde T, Mager R, Kisvarday ZF (1994) Prevention of Ca(2+)-mediated action potentials in GABAergic local circuit neurones of rat thalamus by a transient K+ current. J Physiol (Lond) 478: 403–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Hwang IK, An SJ, Won MH, Kang TC (2003) Elevated P/Q type (alpha 1A) and L2 type (alpha 1D) Purkinje cell voltage-gated calcium channels in the cerebella of seizure prone gerbils. Mol Cells 16: 297–301. [PubMed] [Google Scholar]
- Pfrieger FW, Veselovsky NS, Gottmann K, Lux HD (1992) Pharmacological characterization of calcium currents and synaptic transmission between thalamic neurons in vitro J Neurosci 12: 4347–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcello DM, Smith SD, Huguenard JR (2003) Actions of U-92032, a T-type Ca2+ channel antagonist, support a functional linkage between I(T) and slow intrathalamic rhythms. J Neurophysiol 89: 177–185. [DOI] [PubMed] [Google Scholar]
- Qiao X, Noebels JL (1993) Developmental analysis of hippocampal mossy fiber outgrowth in a mutant mouse with inherited spike-wave seizures. J Neurosci 13: 4622–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP (1999) Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci 19: 1663–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schjott JM, Hsu SC, Plummer MR (2003) The neuronal beta 4 subunit increases the unitary conductance of L-type voltage-gated calcium channels in PC12 cells. J Biol Chem 278: 33936–33942. [DOI] [PubMed] [Google Scholar]
- Schridde U, van Luijtelaar G (2004) The influence of strain and housing on two types of spike-wave discharges in rats. Genes Brain Behav 3: 1–7. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D (1998) Spike-wave complexes and fast components of cortically generated seizures. I. Role of neocortex and thalamus. J Neurophysiol 80: 1439–1455. [DOI] [PubMed] [Google Scholar]
- Sutton KG, McRory JE, Guthrie H, Murphy TH, Snutch TP (1999) P/Q-type calcium channels mediate the activity-dependent feedback of syntaxin-1A. Nature 401: 800–804. [DOI] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA (1999) Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci 19: 1895–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Solorzano G, Depaulis A, Perez-Reyes E, Bayliss DA (2000) Low-voltage-activated calcium channel subunit expression in a genetic model of absence epilepsy in the rat. Brain Res Mol Brain Res 75: 159–165. [DOI] [PubMed] [Google Scholar]
- Thomas E, Grisar T (2000) Increased synchrony with increase of a low-threshold calcium conductance in a model thalamic network: a phase-shift mechanism. Neural Comput 12: 1553–1571. [DOI] [PubMed] [Google Scholar]
- Tsakiridou E, Bertollini L, de Curtis M, Avanzini G, Pape HC (1995) Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J Neurosci 15: 3110–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Luijtelaar G, Wiaderna D, Elants C, Scheenen W (2000) Opposite effects of T- and L-type Ca(2+) channels blockers in generalized absence epilepsy. Eur J Pharmacol 406: 381–389. [DOI] [PubMed] [Google Scholar]
- Wakamori M, Yamazaki K, Matsunodaira H, Teramoto T, Tanaka I, Niidome T, Sawada K, Nishizawa Y, Sekiguchi N, Mori E, Mori Y, Imoto K (1998) Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem 273: 34857–34867. [DOI] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW (1994) Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 264: 107–111. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Mori M, Burgess DL, Noebels JL (2002) Mutations in high-voltage-activated calcium channel genes stimulate low-voltage-activated currents in mouse thalamic relay neurons. J Neurosci 22: 6362–6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwingman TA, Neumann PE, Noebels JL, Herrup K (2001) Rocker is a new variant of the voltage-dependent calcium channel gene Cacna 1a. J Neurosci 21: 1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]