

Abstract
Glutamate transporters limit cross talk between excitatory synapses by removing synaptically released glutamate. However, the role of glutamate transporters in limiting the action of synaptically released glutamate at inhibitory synapses remains unknown. Single and paired whole-cell patch-clamp recordings were obtained from Purkinje neurons and Bergmann glia in mouse cerebellar slices to determine the function of neuronal and glial glutamate transporters at GABAergic synapses onto Purkinje neurons. NMDA and inhibition of glutamate transporters with dl-threo-β-benzyloxyaspartic acid (TBOA) increased the frequency of miniature IPSCs (mIPSCs) by enhancing presynaptic NMDA receptor (NMDAR) activation, whereas inhibition of cystine-glutamate antiporters had no effect on mIPSCs. Selective inhibition of glutamate transporters in an individual Bergmann glial cell enhanced mIPSC frequency recorded in an adjacent Purkinje neuron significantly more than did postsynaptic transporter inhibition. TBOA did not change the frequency of spontaneous IPSCs (sIPSCs) but decreased their amplitude, as well as that of evoked IPSCs (eIPSCs), and enhanced the paired-pulse ratio. The action of NMDAR activation on eIPSCs but not that on mIPSCs was prevented by 100 μm ryanodine. Repetitive stimulations of climbing fibers resulted in a NMDAR-dependent reduction of sIPSC amplitude, and this effect was enhanced by TBOA even when postsynaptic glutamate transporters were blocked. These data indicate that glial glutamate transporters limit glutamate spillover from excitatory to inhibitory synapses and strongly influence synaptic inhibition of Purkinje neurons by controlling NMDAR activation on GABAergic terminals.
Keywords: astrocyte, glutamate uptake, cerebellum, patch-clamp, glial cells, slice
Introduction
Extracellular glutamate levels are regulated by high-affinity transporters located in neurons and glial cells. Five subtypes of glutamate transporters have been cloned and named excitatory amino acid transporter EAAT1 [GLAST (glutamate-aspartate transporter)], EAAT2 [GLT-1 (glutamate transporter)], EAAT3 [EAAC1 (excitatory amino acid carrier)], EAAT4, and EAAT5 in humans (rodent homologs) (Palacin et al., 1998). The neuronal glutamate transporters EAAC1 and EAAT4 are confined to dendritic membranes and are essential in terminating signals at excitatory synapses (Furuta et al., 1997; Otis et al., 1997; Dehnes et al., 1998; Auger and Attwell, 2000; He et al., 2000) and in limiting extrasynaptic receptor activation (Brasnjo and Otis, 2001). The glutamate transporters in glial cells, GLAST and GLT-1, are directly apposed to glutamatergic terminals (Chaudhry et al., 1995; Lehre and Danbolt, 1998; Minelli et al., 2001), sense synaptically released glutamate, and are important in both removing synaptically released glutamate (Bergles et al., 1997; Clark and Barbour, 1997; Oliet et al., 2001) and limiting cross talk between excitatory synapses (Marcaggi et al., 2003). Glial membranes containing high levels of glutamate transporters also ensheath GABAergic terminals (Chaudhry et al., 1995; Minelli et al., 2001), suggesting that glial transporters could limit cross talk between excitatory and inhibitory synapses. Furthermore, glutamate activation of presynaptic ionotropic glutamate receptors increases GABA release at central synapses (Khakh and Henderson, 2000; Kullmann, 2001), and some studies have reported that presynaptic kainate receptors could be activated by synaptically released glutamate, diffusing from excitatory terminals to GABAergic terminals (Min et al., 1999; Cossart et al., 2001; Jiang et al., 2001). However, a function of neuronal and/or glial glutamate transporters at limiting presynaptic glutamate receptor activation and glutamate spillover from excitatory to inhibitory synapses, and at controlling GABA release, has not been demonstrated.
In the cerebellum, exogenous NMDA and ambient glutamate enhance spontaneous GABA release onto Purkinje neurons via presynaptic NMDA receptor (NMDAR) activation on GABAergic interneurons (Glitsch and Marty, 1999). However, it remains unknown whether synaptically released glutamate from excitatory terminals onto Purkinje neurons enhances GABA release via NMDAR activation. Furthermore, the function of GLAST and EAAT4, highly expressed in Bergmann glial processes (Lehre and Danbolt, 1998; Yamada et al., 2000) and in Purkinje neuron dendrites (Furuta et al., 1997; Dehnes et al., 1998), respectively, on glutamate spillover and NMDAR-dependent GABA release remains unknown.
Here we used patch-clamp recordings of Purkinje neurons and paired recordings of Purkinje neurons and adjacent Bergmann glial cells to examine the physiological impact of inhibiting neuronal or glial glutamate transporters on the action of endogenous, synaptically released glutamate on GABA release in cerebellar slices. We found that NMDA or inhibition of glial glutamate transporters and, to a lesser extent, postsynaptic glutamate transporters significantly increased the activation of NMDARs on GABAergic terminals as well as the frequency of miniature IPSCs (mIPSCs) in Purkinje neurons. Furthermore, glutamate uptake inhibition and synaptically released glutamate from climbing fiber stimulation decreased the amplitude of action potential-dependent IPSCs; this effect on evoked IPSCs (eIPSCs) was abolished with ryanodine. Dynamic changes in glial glutamate transporter activity that is observed during physiological and pathological brain functioning (Anderson and Swanson, 2000; Danbolt, 2001; Gegelashvili et al., 2001) could thus profoundly affect tonic synaptic inhibition exerted on Purkinje neurons.
Materials and Methods
Preparation of slices. Sagittal cerebellar slices were prepared as described previously for coronal slices (Bordey and Sontheimer, 2003). Briefly, 10- to 20-d-old mice were anesthetized using pentobarbital (50 mg/kg) and decapitated. A rapid craniotomy that removed the occipital bone and mastoid processes allowed the cerebellum to be quickly detached, removed, and chilled (0-4°C) in 95% O2-5% CO2-saturated artificial CSF (ACSF). The standard external solution (ACSF) contained (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 NaHCO3, and 10 glucose, pH 7.4 (when equilibrated with a mixture of 95% O2 and 5% CO2). Next, the cerebellum was glued (using cyanoacrylate glue) to the stage of a vibratome, and sagittal slices (200 μm thick) were cut in cold oxygenated ACSF. After a recovery period of at least 1 hr in ACSF, slices were placed in a flow-through chamber, held in position by a nylon mesh that was glued to a U-shaped platinum wire, and continuously superfused with oxygenated ACSF. The chamber was mounted on the stage of an upright microscope (BX50; Olympus Optical, Melville, NY), equipped with a 60× water immersion objective and infrared optics.
Electrophysiology. Whole-cell patch-clamp recordings were obtained as described previously (Edwards et al., 1989; Bordey and Sontheimer, 2000). Patch pipettes were pulled from thin-walled borosilicate glass (outer diameter, 1.55 mm; inner diameter, 1.2 mm; TW150F-40; World Precision Instruments, Sarasota, FL) on a PP-83 puller (Narishige, Tokyo, Japan). Pipettes had resistances of 2-4 MΩ (Cs+-containing internal solution) or 5-7 MΩ (K+-containing internal solution). The intracellular solution consisted of the following (in mm): 150 CsCl for Purkinje neurons and 150 KCl for Bergmann glia and basket cells; 0.1 CaCl2, 4.0 MgCl2, 1 EGTA, 10 HEPES, 4 Na2ATP, and 0.5 NaGTP for both Cs+- and K+-containing internal solutions. The pH was adjusted to 7.3 with CsOH or KOH, respectively. A junction potential of 3-4 mV with the internal solutions was not corrected. Whole-cell recordings were performed using an Axopatch-200B amplifier (Axon Instruments, Foster City, CA). To obtain pair recordings of a neuron and a glial cell, whole-cell recordings were first obtained from a Purkinje neuron using an Axopatch-200B (Axon Instruments). After 2-4 min of recording to obtain baseline synaptic activity at a holding potential of -70 mV, a whole-cell recording of an adjacent Bergmann glial cell (not >30 μm away) was established using another Axopatch-200B amplifier. Current signals were low-pass filtered at 2-5 kHz and digitized on-line at 5-20 kHz using a Digidata 1320 digitizing board (Axon Instruments), interfaced with an IBM-compatible computer system. Data acquisition, storage, and analysis were performed using pClamp version 8.0.2 (Axon Instruments). Settings were determined by compensating the transients of a small (5 mV), 10 msec hyperpolarizing voltage step. The capacitance reading of the amplifier was used as value for the whole-cell capacitance. Capacitive and leak conductances were not subtracted. During paired recordings, different dyes (Lucifer yellow or an Alexa fluor dye) were used. After the experiments, the slices were fixed, mounted, and coverslipped. The fluorescence image of the dye-filled basket cell, glial cell, and Purkinje neuron was captured on a confocal scanning laser microscope (MRC600; Bio-Rad, Hercules, CA). The recording chamber was superfused at a rate of 1-1.5 ml/min with ACSF. Receptor and transporter inhibitors were diluted in ACSF and applied by a rapid bath application system composed of a six-channel mini-valve perfusion system (Warner Instruments, New Haven, CT). To induce glutamate transporter currents in Bergmann glial cells, a computer-driven Picospritzer II (General Valve, Fairfield, NJ) was used to pressure apply d-aspartate (2-3 psi) for 5 sec.
The experiments were performed at room temperature (22-24°C), except when indicated. The higher recording temperature (33-36°C) was achieved by controlling the temperature of the perfusion solution and of the chamber with a dual-channel heater controller (Warner Instruments). dl-Threo-β-benzyloxyaspartic acid (TBOA) at 100 μm (4 μl in intracellular solution) was injected intracellularly in a Bergmann glial cell for 2 min, using a Microsyringe controller (World Precision Instruments). TBOA, dl-threo-β-hydroxyaspartic acid (THA), 6-nitro-7-sulfamoylbenzo-quinoxaline-2, 3-dione (NBQX), d(-)-2-amino-5-phosphonopentanoic acid (d-APV), CGP55845 [(2S)-3-{[(15)-1-(3,4-dichlorophenyl) ethyl]amino-2-hydroxypropyl) (phenylmethyl)phosphinic acid], (RS)-α-ethyl-4-carboxyphenylglycine (E4CPG), (S)-4-carboxyphenylglycine (S-4-CPG), and AM-251 [N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,3-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] were purchased from Tocris Cookson (Ellisville, MO). All other drugs and chemicals were purchased from Sigma (St. Louis, MO).
eIPSCs in Purkinje neurons and climbing fiber stimulation were obtained using ACSF-filled patch pipettes as stimulation electrodes. To evoke IPSCs, bipolar stimulation electrodes were positioned in the molecular layer at ∼150 μm away from the recorded Purkinje neurons. Voltage pulses (20-200 μA; duration, 200 μsec; rate, 0.33-0.5 Hz) were applied through the electrode. For the paired-pulse experiments, the onsets of the two pulses were separated by 30 msec. This double stimulation was repeated every 5 sec. For stimulating climbing fibers, stimulation electrodes were positioned in the granule cell layer, ∼150-200 μm away from the recording site. The climbing fibers were activated by repetitive stimulations at 10 Hz for 4 sec (Satake et al., 2000).
Data analysis. Miniature IPSCs were analyzed off-line using an event-detection routine (Minianalysis 5; Synaptosoft, Decatur, GA). mIPSCs were reversibly blocked by bath application of GABAA receptor (GABAAR) blockers, picrotoxin or SR95531 [2-(3-carboxypropyl)-3-amino-6-(4-methoxyphenyl)-pyridazinium bromide] (n = 6), identifying these synaptic currents as GABAAR-mediated currents. mIPSCs occurred randomly with no bursts, and the intervals between mIPSCs were exponentially distributed in all cells studied (data not shown). Detection threshold was adjusted between 6 and 15 pA, depending on the noise of the recording. The decay time constant was determined by fitting the 10-90% decay of mIPSCs with one or two exponential curves. Analyzed recordings were between 100 and 120 sec under each experimental condition. The paired-pulse ratio was measured as the ratio between the averaged amplitude of the second eIPSCs over that of the first for groups of paired evoked currents. For experiments with climbing fiber stimulations, spontaneous IPSC (sIPSC) amplitude and frequency were analyzed during the 4-sec-long stimulation. Only one cell per slice was used to circumvent possible long-term effects of the drugs used. Data are given as mean ± SEM, with n being the number of experiments. Levels of significance were determined by Student's t test.
Results
Whole-cell recordings were obtained from 136 Purkinje neurons and eight Purkinje neuron-Bergmann glial cell pairs in cerebellar slices from postnatal day 10 (P10) to P20 mice. To isolate GABAAR-mediated mIPSCs, antagonists of AMPA-kainate receptors (NBQX, 20 μm) and GABAB receptors (CGP55845, 5 μm) were routinely included in the bath solution. When applied in the presence of a GABAAR blocker (bicuculline, 50 μm), NBQX completely abolished miniature postsynaptic currents (n = 5; data not shown). This is consistent with the lack of functional postsynaptic NMDARs in Purkinje neurons at the age studied (Rosenmund et al., 1992) and the lack of inward current during bath applications of NMDA (Fig. 1A).
Figure 1.
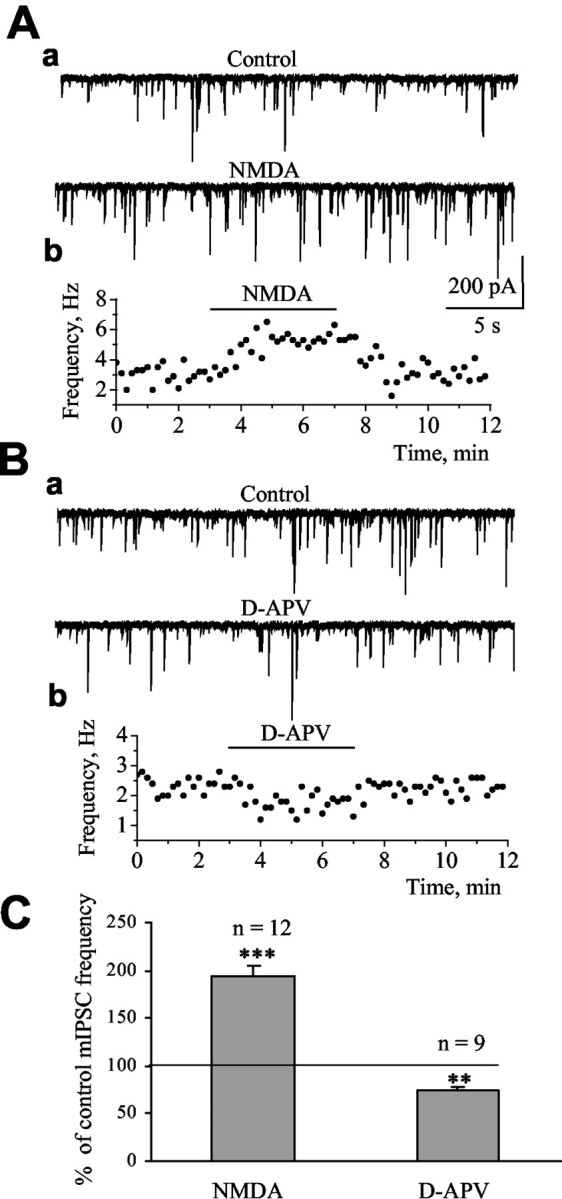
Tonic NMDAR activation enhances spontaneous GABA release. Aa, Ba, Records of mIPSCs under control and in the presence of 30 μm NMDA (A) and 50 μm d-APV (B). Ab, Bb, Frequency of mIPSCs as a function of the recording time. C, Average enhancement and decrease of mIPSC frequency by NMDA and d-APV, respectively. The asterisks indicate significant enhancement above control levels of 100% (**p < 0.01; ***p < 0.001).
Tonic presynaptic NMDAR activation enhances spontaneous GABA release onto Purkinje neurons
A previous study performed in young rats reported the presence of presynaptic NMDARs, presumably located on GABAergic terminals synapsing on Purkinje neurons (Glitsch and Marty, 1999). To examine the effect of NMDA on action potential-independent GABA release from GABAergic terminals, mIPSCs were recorded in Purkinje neurons in the presence of tetrodotoxin (TTX) (0.5-1.0 μm). In the present study performed in P10-P20 mice, rapid bath application of NMDA (30 μm) led to a significant increase in the frequency of mIPSCs to 193.7 ± 12.5% of control (n = 12; p < 0.001) (Fig. 1A, C). The action of NMDA was specific to GABAergic synapses because the frequency of mEPSCs was not affected by NMDA (30 μm) application (n = 5; data not shown). For these experiments, picrotoxin (100 μm) or bicuculline (50 μm) replaced DNQX. Importantly, bath application of the specific antagonist of NMDARs, d-APV (50 μm), significantly decreased mIPSC frequency to 74.0 ± 4.3% of control (n = 9; p < 0.01) (Fig. 1B, C). When tested at 33 ± 1°C, 50 μm d-APV also significantly decreased mIPSC frequency to 79.6 ± 4.5% of control (n = 5; p < 0.05; data not shown). The effect of NMDA on mIPSC frequency was not accompanied by a significant change in the amplitude, 10-90% rise time, or decay time constant of mIPSCs (Table 1), consistent with a presynaptic but not a postsynaptic site of action of NMDA at the age studied (Rosenmund et al., 1992).
Table 1.
Effect of NMDA and TBOA on the frequency, amplitude, rise time, and decay time constant (τ) of mIPSCs
|
|
Frequency (Hz) |
Amplitude (pA) |
Rise time (msec) |
Decay time constants |
||||
|---|---|---|---|---|---|---|---|---|
|
|
τ (msec) |
τ1 (msec) |
τ2 (msec) |
|||||
| Room temperature (n = 12) | ||||||||
| Control | 1.66 ± 0.39 | 67.7 ± 15.5 | 1.52 ± 0.21 | 9.15 ± 0.62 (8) | 3.59 ± 0.33 (4) | 14.50 ± 0.54 (4) | ||
| NMDA | 3.02 ± 0.62 ** | 64.3 ± 13.7 | 1.60 ± 0.22 | 9.24 ± 0.57 (8) | 3.71 ± 0.38 (4) | 14.00 ± 0.41 (4) | ||
| Room temperature (n = 19) | ||||||||
| Control | 2.40 ± 0.30 | 88.8 ± 10.8 | 1.59 ± 0.12 | 9.07 ± 0.52 (16) | 3.94 ± 0.40 (3) | 13.50 ± 0.24 (3) | ||
| TBOA | 3.90 ± 0.50 ** | 86.0 ± 9.5 | 1.64 ± 0.11 | 8.89 ± 0.49 (16) | 4.06 ± 0.51 (3) | 13.20 ± 0.12 (3) | ||
| 33°C (n = 4) | ||||||||
| Control | 5.03 ± 1.14 | 84.7 ± 17.4 | 0.86 ± 0.07 | 4.44 ± 0.48 (2) | 2.82 ± 0.11 (2) | 10.62 ± 0.37 (2) | ||
| TBOA |
6.54 ± 1.39 *
|
80.3 ± 15.4 |
0.92 ± 0.10 |
4.54 ± 0.21 (2) |
2.71 ± 0.27 (2) |
11.53 ± 0.82 (2) |
||
In 26 of 35 cells, the decay could be well fitted with one exponential, and, in the remaining nine cells, it could be fitted with a double exponential. The number of cells is indicated in parentheses. NMDA and TBOA significantly increased the frequency of mIPSCs, although they had no significant effect on the amplitude, rise time, and decay of mIPSCs.
*p<0.05, ** p<0.0018, compared with control.
Inhibition of glutamate transporters but not of cystine-glutamate antiporters enhances action potential-independent GABA release onto Purkinje neurons by increasing tonic activation of presynaptic NMDARs
We investigated the contribution of glutamate transporters to spontaneous, action potential-independent GABA release by examining the effect of neuronal and glial transporter inhibition on mIPSC properties. Glutamate uptake was inhibited by bath applications of TBOA, a nontransportable neuronal and glial transporter antagonist (Shimamoto et al., 1998; Shigeri et al., 2001). For some experiments with TBOA and for subsequent experiments, a blocker of metabotropic glutamate receptors (E4CPG, 500 μm) was included in the bath solution. However, data with and without E4CPG were not significantly different, and the data were pooled. Bath application of TBOA (100 μm) significantly increased mIPSC frequency to 156.8 ± 5.3% of control (from 2.4 ± 0.3 to 3.9 ± 0.5 Hz; n = 19; p < 0.001) (Fig. 2A, B). This effect persisted throughout TBOA application and reversed after washout (data not shown). Furthermore, the effect of TBOA was reversed by the application of 50 μm d-APV in the presence of TBOA (89.6 ± 11.8% of control; n = 8) (Fig. 2A, B), suggesting that increased GABA release in the presence of TBOA resulted from enhancement of NMDAR activation. There were no significant changes in the amplitude, rise time, or decay of mIPSCs in the presence of TBOA (Fig. 2Ab, Ca, Table 1), suggesting that TBOA did not significantly affect the postsynaptic cell membrane properties. In addition, TBOA did not alter the series (access) resistance, the input resistance, or the cell capacitance calculated from a 10 mV hyperpolarizing pulse (Fig. 2Cb). Similar to the effect of NMDA, the action of TBOA was specific to GABAergic synapses because the frequency of mEPSCs was not affected by TBOA or by THA, a transportable blocker of glutamate transporters known to induce glutamate release via heteroexchange (data not shown; n = 6). At 33 ± 1°C, TBOA (100 μm) also significantly increased the frequency of mIPSCs to 132.0 ± 4.0% of control without affecting the amplitude, rise time, or decay of mIPSCs (n = 4; p < 0.05) (Table 1). The effect of TBOA was reversed to 103.2 ± 7.6% of control (n = 4) by the application of 50 μm d-APV (in the presence of TBOA). Glitsch and Marty (1999) reported that an 11 mV depolarization of basket or stellate cells is sufficient to significantly increase mIPSC frequency in Purkinje neurons. To assess whether GABAergic interneurons were depolarized during glutamate uptake inhibition with TBOA, presumed basket cells (filled with Lucifer yellow during the recording) were recorded under current clamp in the absence and the presence of TBOA. Bath application of TBOA (100 μm) had no significant effect on the membrane potential of presumed basket cells (mean depolarization of 0.2 ± 0.7 mV; n = 5) (Fig. 2D). These results suggest that the increase in mIPSC frequency by TBOA was mediated by the activation of NMDARs on GABAergic terminals or possibly axons but not of those on the somata or dendrites of GABAergic interneurons.
Figure 2.

Inhibition of glial and neuronal glutamate transporters enhances mIPSC frequency. Aa, Records of mIPSCs under control, in the presence of 100 μm TBOA, and in the presence of TBOA plus 50 μm d-APV. Ab, Frequency (f), 10-90% rise time (rT), and amplitude (Amp) of mIPSCs as a function of the recording time. TBOA enhanced the frequency of mIPSCs but did not alter their rise time or amplitude. B, The enhancement of mIPSC frequency by TBOA is blocked during d-APV application (***p < 0.001 compared with control of 100%). Ca, Traces represent the averaged mIPSCs in control and during TBOA application (n = 40-50 events in both cases). The decay time constant (τ) of mIPSCs was not affected by TBOA. Cb, Traces represent the responses of the cell to a 10 mV hyperpolarizing pulse applied before and with TBOA to show that the access resistance did not change during TBOA application. D, Membrane potential (VM) traces under current clamp in control and 2 min after TBOA application from a presumed basket cell. The upward deflections represent IPSPs. The membrane potential of presumed basket cells did not change during TBOA application (n = 5).
Whereas glutamate transporters remove glutamate from the extracellular space, cystine-glutamate antiporters have been shown to induce glutamate release in exchange of extracellular cystine uptake (Baker et al., 2002). To determine whether cystine-glutamate antiporters expressed in cerebellar cells (Warr et al., 1999) contributed to endogenous glutamate accumulation around GABAergic synapses, mIPSCs were recorded in the presence of S-4-CPG, an inhibitor of the cystine-glutamate antiporter (Baker et al., 2002). Bath application of 100 μm S-4-CPG had no significant effect on mIPSC frequency (99.5 ± 2.2% of control; n = 4; p > 0.5) (Fig. 3B), suggesting that the antiporter did not contribute to the levels of ambient glutamate around GABAergic synapses. Furthermore, TBOA increased mIPSC frequency to 140.6 ± 2.7% of control (n = 3) in the presence of S-4-CPG, which is similar to the increase without S-4-CPG (data not shown).
Figure 3.

Selective inhibition of postsynaptic glutamate uptake enhances the frequency of mIPSCs. A, Records of mIPSCs under control and in the presence of 500 μm l-α-AA, a competitive blocker of EAAT4 glutamate transporters expressed in Purkinje neurons. B, Average enhancement of mIPSC frequency by l-α-AA, S-4-CPG, and l-α-AA in the presence of S-4-CPG (**p < 0.01 compared with control of 100%).
Glial glutamate transporters control tonic synaptic inhibition of Purkinje neurons significantly more than postsynaptic transporters do
EAAT4 is highly expressed on the dendritic shafts of Purkinje neurons (Dehnes et al., 1998). We therefore tested the effect of l-α-amino adipate (l-α-AA), a transportable blocker of EAAT4 (Fairman et al., 1995; Brasnjo and Otis, 2001), on the properties of mIPSCs. Bath application of l-α-AA (500 μm) enhanced the frequency of mIPSCs to 121.5 ± 1.3% of control in five of six Purkinje neurons (n = 5; p < 0.01) (Fig. 3A, B). The amplitude and the 10-90% rise time of mIPSCs were not affected by l-α-AA (101.8 ± 4.1 and 99.4 ± 1.2% of control, respectively). Because l-α-AA is also a substrate agonist of the cystine-glutamate anti-porter (Tsai et al., 1996; Bender et al., 2000), application of l-α-AA can induce glutamate release by exchange through this antiporter. We therefore investigated the effect of l-α-AA on mIPSC frequency in the presence of S-4-CPG, which blocks the cystine-glutamate antiporter. In the presence of S-4-CPG, bath application of l-α-AA increased mIPSC frequency to 113.2 ± 1.8% of control (n = 6; p < 0.01) (Fig. 3B), which is a significantly smaller increase than that without S-4-CPG in the bath (p < 0.01) (Fig. 3B). The latter result suggests that part of the l-α-AA-induced increase in mIPSC frequency without S-4-CPG was caused by glutamate being released into the extracellular space in exchange for l-α-AA.
To determine whether glial glutamate transporters are important for controlling spontaneous GABA release, two strategies were used. In the first set of experiments, glutamate transporters were blocked in the recorded Purkinje neurons by including the competitive blocker d-aspartate in the pipette solution as reported previously (Takahashi et al., 1996). With 10 mm d-aspartate in the pipette solution for >15 min in the whole-cell mode, bath application of 500 μm l-α-AA produced no significant effect on mIPSC frequency (to 102.6 ± 2.9% of control; n = 5; p > 0.5) (Fig. 4A). This result suggests that intracellular d-aspartate is efficient at blocking EAAT4 in Purkinje neurons. However, in the same recording conditions, TBOA (100 μm) induced an increase in mIPSC frequency to 125.7 ± 3.1% of control (n = 4; p < 0.05) (Fig. 4A). The effect of TBOA was likely underestimated because of a progressive increase in the series (access) resistance during the recording with an intracellular solution containing d-aspartate. Nevertheless, these data suggest that glial glutamate transporters are involved in limiting NMDAR activation and GABA release. In a second series of experiments, we set out to investigate directly whether the glial glutamate transporters GLAST and, to a lesser extent, GLT-1 regulate GABA release, using two approaches. Although there is no available blocker for GLAST, glutamate uptake via GLT-1 can be blocked with dihydrokainate (DHK). We investigated whether the glial transporter GLT-1 controls spontaneous GABA release onto Purkinje neurons by testing the effect of DHK on mIPSC frequency. Bath application of 300 μm DHK had no significant effect on the frequency of mIPSCs (100.8 ± 1.52% of control; n = 4; data not shown). We then investigated whether glial glutamate transporters regulate GABA release by intracellular injection of TBOA during the recording to block transporters in a Bergmann glial cell and simultaneously recorded mIPSCs in an adjacent Purkinje neuron. When recording Bergmann glial cells with a solution containing 20 μm TBOA, the amplitudes of d-aspartate-induced transporter currents were either completely blocked (n = 2; recordings with a KCl internal solution) or reduced to 38.0 ± 2.5% of control (time 0 of recording) after 6 min of recording (n = 6; recording with a KNO3 internal solution) (Fig. 4B) in the presence of AMPA-kainate and NMDA receptor antagonists (20 μm NBQX and 50 μm d-APV) and a GABAA receptor antagonist (20 μm SR95531). Glutamate transporter currents, recorded in Bergmann glial cells without internal TBOA, display stable amplitudes during recording (∼45 min; n = 7; three and four cells recorded with KCl and KNO3, respectively; data not shown). Intracellular d-aspartate could not be used with glial cell recordings because it led to a progressive and significant increase in the series (access) resistance during the glial cell recordings. A typical neuron-glial cell-pair recording and photograph are shown in Figure 4, C and D, respectively. The photograph illustrates the close contact between the Bergmann glial cell and the Purkinje neuron. During paired recordings, intracellular injection of TBOA (final pipette concentration of 20 μm achieved in 2 min) increased the frequency of mIPSCs to 125.0 ± 0.9% of control (p < 0.01) in four of five Purkinje neurons at 7-9 min after TBOA injection (Fig. 4E). Injection of intracellular solution without TBOA produced no significant effect at 7-9 min after the injection (98.7 ± 2.9% of control; n = 3) (Fig. 4E). Our results indicate that the glial glutamate transporter GLAST plays an essential role in limiting presynaptic NMDAR activation and synaptic inhibition of Purkinje neurons.
Figure 4.
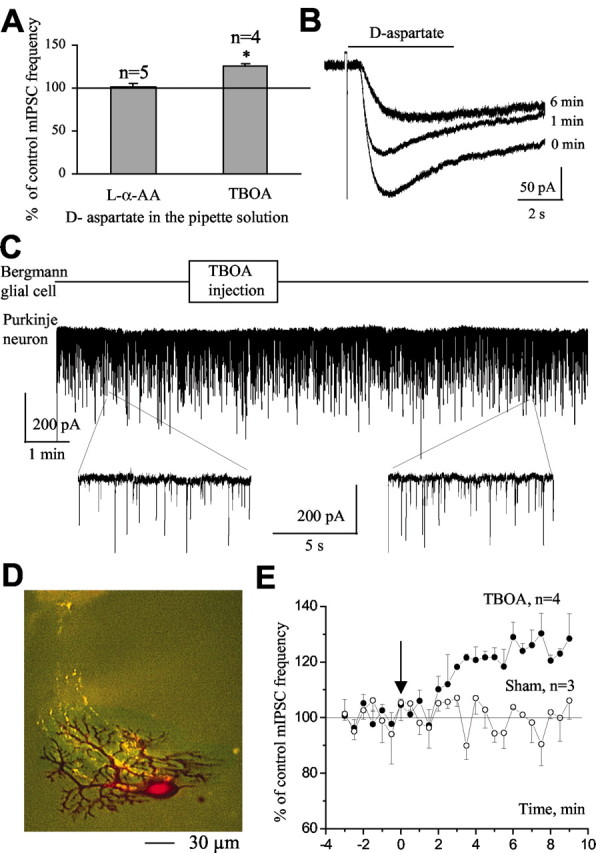
Inhibition of glutamate uptake into Bergmann glial cells significantly enhances mIPSC frequency. A, Selective inhibition of postsynaptic glutamate uptake by blocking glutamate transporters in only the recorded Purkinje neuron was obtained by including d-aspartate in the pipette solution. Average enhancements of mIPSC frequency by bath-appliedl-α-AA and TBOA when d-aspartate was included in the recording solution. The asterisks indicate significant enhancement above control levels of 100% (*p < 0.05 compared with control of 100%). B, d-aspartate-induced currents recorded in a Bergmann glial cell in the presence of glutamate receptor blockers (20 μm NBQX and 50 μm d-APV) and the GABAA receptor blocker SR95531 (20 μm) were significantly reduced by addition of TBOA in the pipette solution. C-E, A Purkinje neuron and a Bergmann glial cell were recorded simultaneously, allowing us to monitor mIPSCs during selective inhibition of glial glutamate transporters by injecting TBOA into the glial cell during the recording. C, Records of mIPSCs under control, during, and after the intracellular injection of TBOA into a single Bergmann glial cell. Bottom, Records of mIPSCs on an expanded time scale. D, Photograph of a closely associated Purkinje neuron (red)-Bergmann glial cell (green) pair. E, A plot of mIPSC frequency (percentage of control) against time in experiments in which TBOA (filled circles) or intracellular solution alone (open circles) was intracellularly injected at time 0 (as indicated by the arrow).
To rule out the possibility that an inadequate space clamp affected the measured effects of transporter blockers on mIPSC frequency, we determined whether we could analyze the effects of transporter blockers on mIPSCs that originated at synapses close to the soma. We found that the distribution of 10-90% rise times (range, 0.2-4.5 msec) of mIPSCs contain two populations of events. A Gaussian population of events has fast 10-90% rise times (mean 10-90% rise time, 0.6 msec) (Fig. 5Aa), and these events will be referred to as fast events. A second, non-Gaussian population of events has slower 10-90% rise times (between 1 and 4.5 msec), and these events will be referred to as slow events. Fast events covered the whole range of amplitudes (Fig. 5Ab), suggesting that they were not significantly distorted by dendritic filtering (Rall, 1977; Spruston et al., 1993; Soltesz et al., 1995). Fast events likely originated at synapses on or near the soma that receive synaptic inputs from basket cells (King et al., 1993), in which an adequate space clamp should be achieved. Slow events have small amplitudes (Fig. 5Ab), suggesting that these events were affected by dendritic filtering. Slow events likely originated at nonsomatic sites on Purkinje neuron dendrites receiving synapses from basket cells and stellate cells (King et al., 1993). The frequency of fast synaptic events was increased significantly more by intracellular TBOA injection (124.9 ± 1.3% of control; range, 122-128%) in a single Bergmann glial cell than by bath application of l-α-AA (108.8%; range, 106-115%; p < 0.01) in the presence of S-4-CPG (Fig. 5B).
Figure 5.

Blocking glutamate transporters in a Bergmann glial cell enhances spontaneous GABA release at sites close to the soma more than postsynaptic transporter inhibition does. Aa, Mean distributions of the 10-90% rise times of mIPSCs in cells from P14 mice. The recordings were obtained at a holding potential of -70 mV and were selected on the basis of similar access resistances (10-14 MΩ before compensation). The events under the first Gaussian had fast 10-90% rise time with a mean of ∼0.6 msec and were called fast synaptic events. The remaining events were termed slow synaptic events. Ab, Scatter plots of the amplitude as a function of 10-90% rise time at the same age group. Fast events cover the whole range of amplitudes, whereas slow events have small amplitudes. B, Average enhancements of fast mIPSC frequency by bath-applied l-α-AA in the presence of S-4-CPG (n = 6) and intracellular injection of TBOA in a single Bergmann glial cell (n = 4).
Inhibition of glutamate transporters decreases action potential-dependent GABA release onto Purkinje neurons via activation of presynaptic NMDARs and ryanodine-sensitive Ca2+ stores
To determine whether inhibition of glutamate transporters influences action potential-dependent GABA release, the effect of TBOA was examined on spontaneous and evoked IPSCs. TBOA did not significantly affect sIPSC frequency (88.8 ± 5.5% of control; from 14.9 ± 1.6 to 13.0 ± 1.4 Hz; n = 6; p > 0.05; data not shown), whereas Glitsch and Marty (1999) and Farrant and Cull-Candy (1991) reported a large increase in sIPSC frequency by NMDA that was presumably attributable to an increase in the firing rate of GABAergic interneurons. However, TBOA decreased the amplitude of sIPSCs to 82.0 ± 2.7% of control (n = 6; p < 0.05) (Fig. 6A). This effect was reversed to 93.0 ± 3.7% of control (p > 0.5; n = 6) (Fig. 6A) by further application of 50 μm d-APV in the presence of TBOA, consistent with the activation of NMDARs during TBOA applications. Consistent with its effect on sIPSC amplitudes, TBOA decreased the amplitude of eIPSCs to 64.5 ± 8.3% of control (p < 0.05; n = 7; data not shown). Evoked IPSCs were obtained by minimal electrical stimulation in the molecular layer. We then compared the paired-pulse ratio before and during the application of TBOA. The paired-pulse ratio was significantly increased from 1.07 ± 0.04 to 1.70 ± 0.15 (p < 0.01; n = 5) (Fig. 6B, C) during glutamate transporter inhibition with TBOA. For all experiments, TBOA did not alter the series resistance, the input resistance, or the cell capacitance calculated from a 10 mV hyperpolarizing pulse (data not shown). These data suggest that glutamate acts presynaptically to reduce the probability of action potential-dependent GABA release.
Figure 6.
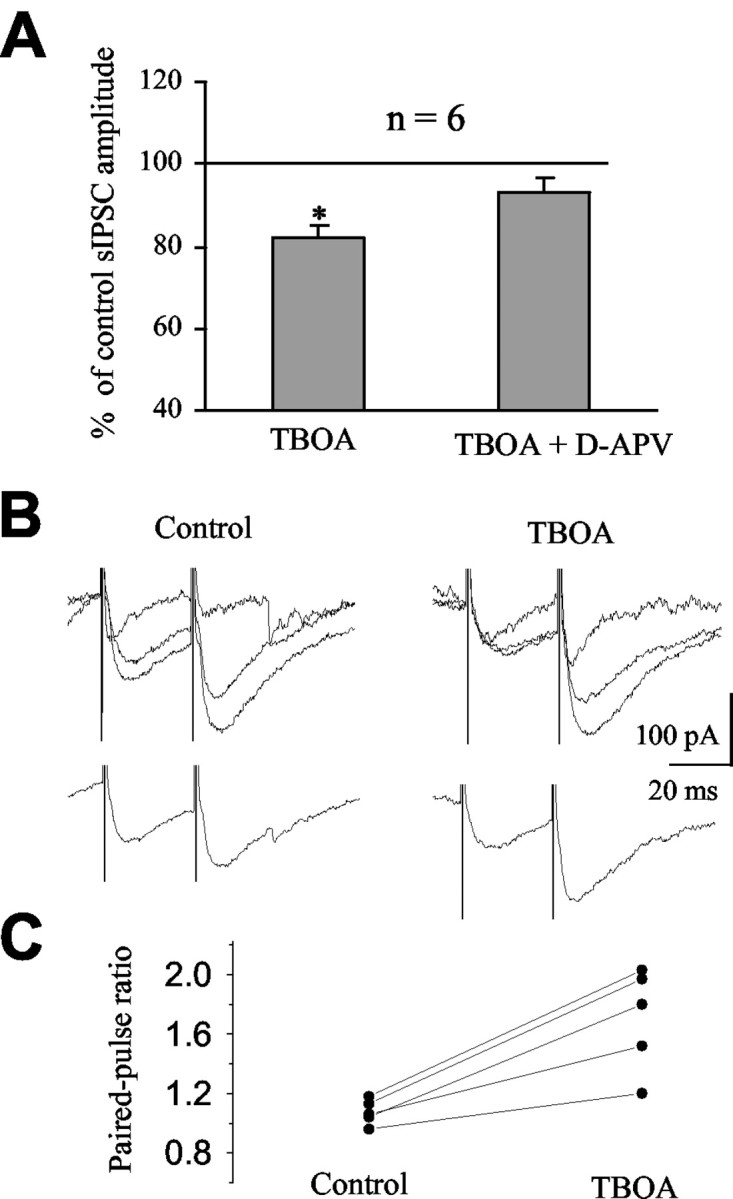
Inhibition of glutamate transporters reduces spontaneous and evoked IPSCs. A, Average reductions of sIPSC amplitude by bath-applied TBOA and TBOA in the presence of d-APV. B, Superimposed traces in control conditions and in the presence of TBOA (top). Each trace represents the response to two successive stimulations separated by 30 msec. Average responses under control conditions and in the presence of TBOA are shown in the bottom. C, Paired-pulse ratios under control conditions and in the presence of TBOA.
Presynaptic ryanodine-sensitive Ca2+ stores in cerebellar GABAergic interneurons contribute to both action potential-dependent and action potential-independent GABA release onto Purkinje neurons (Llano et al., 2000; Bardo et al., 2002; Galante and Marty, 2003). Thus, we examined the effect of 100 μm ryanodine, which blocks ryanodine-sensitive Ca2+ stores in presynaptic terminals (Llano et al., 2000), on mIPSC frequency and eIPSC amplitude. As shown previously (Llano et al., 2000; Galante and Marty, 2003), bath application of 100 μm ryanodine for 10 min significantly reduced mIPSC frequency to 81.0 ± 5.2% of control (p < 0.05; n = 3/4) (Fig. 7A) and eIPSC amplitude to 83.0 ± 2.1% of control (p < 0.05; n = 4) (Fig. 7B). Because NMDAR activation leads to Ca2+ increase in neurons and, in some cases, involves Ca2+-induced Ca2+ release from ryanodine-sensitive Ca2+ stores (Simpson et al., 1995; Emptage et al., 1999), we tested whether NMDAR activation by endogenous glutamate during TBOA application involved ryanodine-sensitive Ca2+ stores. In the presence of ryanodine, TBOA increased mIPSC frequency to the same extent as without ryanodine when tested in the same cells. TBOA increased mIPSC frequency to 148.0 ± 3.8% of control without ryanodine. Whereas ryanodine decreased mIPSC frequency to 81% of control, further application of TBOA in the presence of ryanodine increased mIPSC frequency to 125.0 ± 1.6% of control (n = 4), which corresponds to a 45% increase in mIPSC frequency by TBOA in the presence of ryanodine (Fig. 7A). However, ryanodine (100 μm) prevented the effect of TBOA on eIPSC amplitude, which was 83.0 ± 2.1% of control with ryanodine and 81.0 ± 4.3% of control with ryanodine and TBOA (n = 4) (Fig. 7B). These data indicate that presynaptic NMDAR activation involves ryanodine-sensitive Ca2+ stores in the modulation of action potential-dependent but not action potential-independent GABA release. These data further support the notion that NMDARs are located on GABAergic terminals in which ryanodine-sensitive Ca2+ stores are present and modulate GABA release (Llano et al., 2000).
Figure 7.
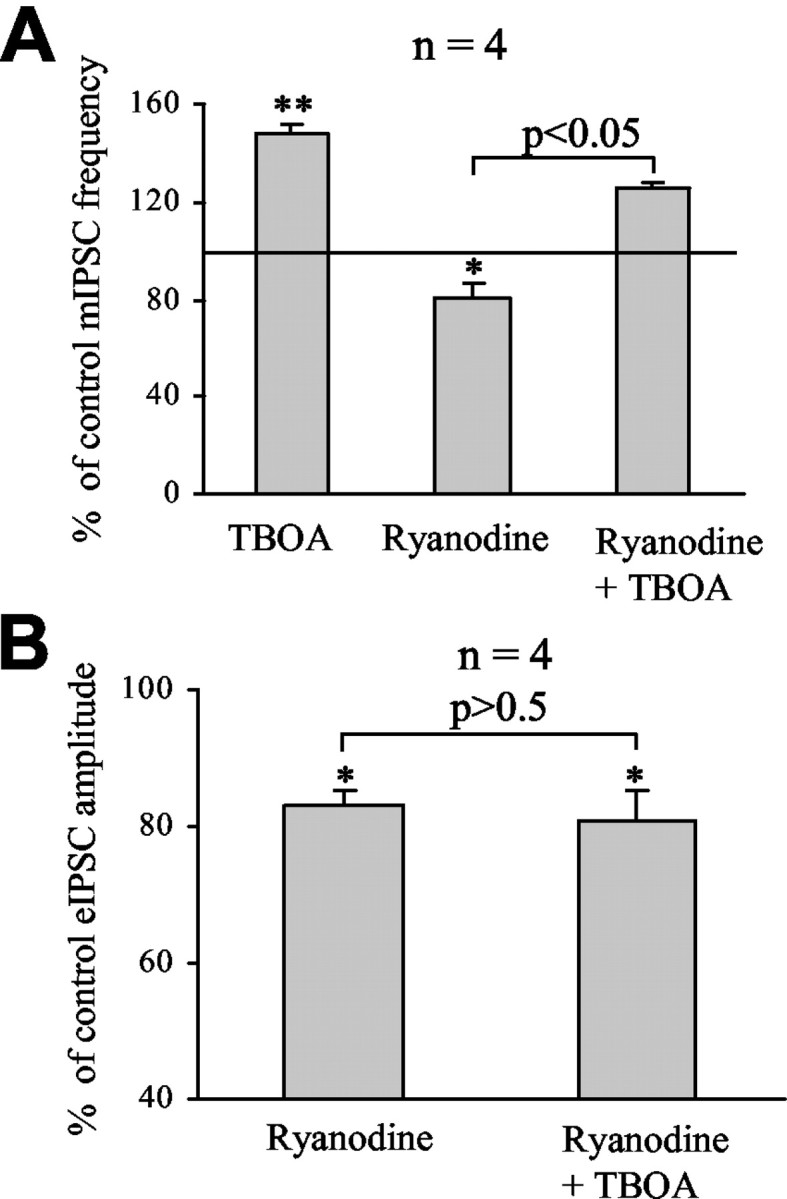
The action of NMDAR activation on evoked IPSCs but not that on mIPSCs was prevented by 100 μm ryanodine. A, Average changes of mIPSC frequency induced by successive applications of TBOA (100 μm) and ryanodine (100 μm) after washing out TBOA and of TBOA in the presence of ryanodine. B, Average decreases of eIPSC amplitude induced by successive applications of ryanodine (100 μm) and TBOA (100 μm) in the presence of ryanodine. *p < 0.05; **p < 0.01.
Glutamate transporters limit presynaptic NMDA receptor activation by glutamate spillover from climbing fibers
To determine whether synaptically released glutamate from excitatory inputs contributes to presynaptic NMDAR activation, climbing fibers were electrically stimulated at 10 Hz (40 pulses) at near-physiological temperature (35-36°C). The frequency and amplitude of sIPSCs were analyzed during the 4-sec-long stimulation of climbing fibers. During the stimulation, sIPSC amplitude was decreased to 79.0 ± 4.2% of control (p < 0.05; n = 8) (Fig. 8A, B), and the frequency was not significantly changed (to 107.0 ± 3.4% of control; p > 0.05; n = 8; data not shown). These experiments and the following experiments were performed in the presence of the AMPA-kainate receptor antagonist NBQX (20 μm) and in the presence of a type 1 cannabinoid receptor antagonist (1 μm AM-251) to prevent a possible suppression of inhibition at GABAergic interneurons or excitation climbing fibers (Takahashi and Linden, 2000; Kreitzer and Regehr, 2001a,b; Diana et al., 2002). In the presence of d-APV, stimulation of climbing fibers had no effect on sIPSC amplitude (to 103.0 ± 3.6% of control; n = 7; p > 0.5) or frequency (to 98.2 ± 2.7% of control; n = 7; p > 0.8) (Fig. 8B). These data suggest that synaptically released glutamate diffuses from climbing fibers to GABAergic synapses in which it activates presynaptic NMDARs and reduces GABA release. We then examined the effect of glutamate transporter inhibition on spillover-induced decrease of spontaneous GABA release. Inhibition of glutamate uptake with TBOA (100 μm) significantly increased the effect of climbing fiber stimulation on sIPSC amplitude by 6% (from a decrease to 82.1 ± 1.7 to 76.2 ± 1.9% of control with and without TBOA, respectively; n = 4; p < 0.01) (Fig. 8C). However, TBOA did not change the effect of climbing fiber stimulation on sIPSC frequency (from 104.0 ± 2.3 to 106.0 ± 4.7% of control with and without TBOA, respectively; n = 4). When Purkinje neurons were recorded with 10 mm d-aspartate in the internal solution to block postsynaptic glutamate transporters, TBOA increased the effect of climbing fiber stimulation on sIPSC amplitude by ∼6% (from a decrease to 80.8 ± 0.7 to 75.1 ± 0.9% of control with and without TBOA, respectively; n = 4; p < 0.01) (Fig. 8C), which is similar to the increase without inhibition of postsynaptic glutamate transporters. These data strongly suggest that glial glutamate transporters limit spillover of synaptically released glutamate from climbing fibers to GABAergic synapses, where glutamate activates pre-synaptic NMDARs and reduces action potential-dependent GABA release.
Figure 8.

Glial glutamate transporters limit glutamate spillover from excitatory to inhibitory synapses. A, Spontaneous IPSC recorded before (Control) and during repetitive stimulation (Stimulation) of climbing fibers at 10 Hz (40 pulses). B, Average decreases of sIPSC amplitude during climbing fiber stimulation under control conditions and in the presence of d-APV. *p < 0.05. C, Average decreases of sIPSC amplitude induced by stimulation under control conditions (light gray bars) and in the presence of TBOA (dark gray bars) either without or with d-aspartate (d-ASP) in the recording solution. All of these experiments were performed in the presence of the AMPA-kainate receptor antagonist NBQX (20 μm) and in the presence of a type 1 cannabinoid receptor antagonist (1 μm AM-251).
Discussion
We demonstrate here that inhibition of glutamate transporters increases tonic and spillover activation of presynaptic NMDARs expressed on GABAergic terminals and influences spontaneous GABA release onto Purkinje neurons, the principal neurons of the cerebellum. Furthermore, glutamate transporters in Bergmann glial cells play an essential role in controlling synaptic inhibition, in particular at synapses near Purkinje neuron somata in which sodium action potentials are generated (Stuart and Hausser, 1994).
Inhibition of glial and, to lesser extent, postsynaptic glutamate transporters enhances tonic activation of NMDARs on GABAergic interneurons synapsing onto Purkinje neurons
In the present study, we first confirmed that presynaptic NMDARs on GABAergic interneurons are tonically activated by ambient glutamate, resulting in increased spontaneous GABA release as d-APV reduced mIPSC frequency, as shown previously by Glitsch and Marty (1999). Then, by inhibiting glutamate transporters with TBOA, we found that glutamate transporters are important in limiting the activation of presynaptic NMDARs by preventing accumulation of glutamate around GABAergic synapses. Inhibiting the cystine-glutamate antiporter did not alter mIPSC frequency, suggesting that the antiporter does not contribute to glutamate accumulation around GABAergic synapses in our recording condition. However, this antiporter was functional in our cerebellar slices because part of the effect of l-α-AA, a substrate agonist of this antiporter, was prevented by blocking the antiporter. Because glutamate uptake is enhanced at higher temperature (Schwartz and Tachibana, 1990; Wadiche et al., 1995), we expected that uptake inhibition would have a greater effect on mIPSC frequency at 33°C than at room temperature, as reported previously for the action of glutamate uptake on glutamate spillover between glutamatergic synapses (Asztely et al., 1997). However, TBOA had a smaller effect on mIPSC frequency at 33°C than at room temperature. It is possible that, during inhibition of glutamate uptake, glutamate is cleared more easily from the extracellular space of our relatively thin slices (200 μm) because of a decrease in the diffusional barriers at 33°C. NMDA- and TBOA-induced enhancement of mIPSC frequency could result from the activation of NMDARs on the terminals or axons of basket cells (Petralia et al., 1994; Duguid and Smart, 2004) and, to a lesser extent, of NMDARs on the soma or dendrites of GABAergic interneurons (Akazawa et al., 1994; Thompson et al., 2000), because basket cell depolarization in the presence of TTX enhances GABA release onto Purkinje neurons (Glitsch and Marty, 1999). Because bath application of TBOA did not depolarize presumed basket cells, enhancement of GABA release resulting from glutamate uptake inhibition likely results from activation of NMDARs along the axons or more likely on GABAergic terminals, as suggested recently (Duguid and Smart, 2004).
Both Purkinje neurons and Bergmann glial cells express high levels of glutamate transporters. Our results show that inhibition of postsynaptic transporters with l-α-AA and glial transporters with intracellular TBOA injection in a single Bergmann glial cell corresponded to approximately one-fourth and one-half of the effect of bath-applied TBOA on mIPSC frequency, respectively. These results suggest an essential role of glial glutamate transporters and a more modest role of neuronal EAAT4 in limiting presynaptic NMDAR activation and action potential-independent GABA release onto Purkinje neurons. It is possible that part of the effect of l-α-AA also results from glutamate release attributable to heteroexchange for l-α-AA, which is a substrate agonist of EAAT4 (Fairman et al., 1995). In that case, the contribution of postsynaptic transporters to the regulation of glutamate levels at GABAergic synapses would be overestimated in the present study. Increased mIPSC frequency by intracellular TBOA injection into a single glial cell likely results from inhibition of GLAST because inhibition of GLT-1 with DHK had no effect on mIPSC frequency. Intracellular TBOA injection into a single cell likely underestimates the actual contribution of glial glutamate transporters to the control of NMDAR activation. TBOA injection into a single Bergmann glial cell may diffuse into one or two adjacent glial cells via gap junctions. However, the Purkinje neuron dendritic tree can expand to the domains of up to 7-10 Bergmann glial cells in the transverse plane of the folium (de Blas, 1984). Processes from approximately three to four Bergmann glial cells may completely ensheath proximal synapses that likely represent most of the recorded mIPSCs. Although the role of glial glutamate transporters may be underestimated, their role is significantly greater than that of postsynaptic transporters in reducing synaptic inhibition of Purkinje neurons, particularly in inhibition targeted at somatic synapses and at synapses near the somata.
Inhibition of glutamate transporters decreases action potential-dependent GABA release onto Purkinje neurons via activation of ryanodine-sensitive Ca2+ stores
TBOA significantly decreased sIPSC amplitude via NMDAR activation but did not alter the frequency of sIPSCs. These data suggest that TBOA did not alter the firing rate of GABAergic interneurons. TBOA-induced increase in glutamate levels may be confined at synapses or may not be sufficient to activate somatic or axonal NMDARs on GABAergic interneurons. In contrast, applications of NMDA likely activated somatic NMDARs on GABAergic interneurons resulting in increased firing of these interneurons (Farrant and Cull-Candy, 1991; Glitsch and Marty, 1999). The lack of effect of TBOA on sIPSC frequency was surprising at first because TBOA increased mIPSC frequency. However, some sIPSCs may fall below the detection level during TBOA application because the amplitudes of sIPSCs are decreased by 20%. Such an apparent loss of sIPSCs could therefore counteract the TBOA-induced increase in mIPSC frequency. TBOA also decreased the amplitude of eIPSCs and induced an increase in paired-pulse ratio, suggesting that TBOA induced activation of presynaptic NMDARs presumably located on GABAergic terminals. Glitsch and Marty (1999) did not observe a change in paired-pulse ratio. The reason for this discrepancy is unclear but may be attributable to a species difference (mouse vs rat). A recent study by Duguid and Smart (2004) reported that presynaptic NMDAR activation after Purkinje neuron depolarization, by either applying voltage steps or by stimulating climbing fibers, increased mIPSC and sIPSC frequencies and decreased paired-pulse ratio, which is different from our results during TBOA application or climbing fiber stimulation. The reason for this discrepancy is unclear. However, it is possible that another factor is coreleased with glutamate during Purkinje neuron depolarization that results in a delayed and relatively long (∼8 min) increase in GABA release compared with a transient decrease in action potential-dependent GABA release in our study. We found that 100 μm ryanodine, which irreversibly blocks ryanodine-sensitive Ca2+ stores (Llano et al., 2000), prevented the effect of TBOA on eIPSC amplitude but not on mIPSC frequency, although ryanodine affected the basal mIPSC frequency and evoked IPSC amplitude, as reported previously (Llano et al., 2000; Bardo et al., 2002; Galante and Marty, 2003). Duguid and Smart (2004) also reported that blockers of voltage-gated Ca2+ channels did not reduce exogenous NMDA-induced increase in mIPSC frequency in Purkinje neurons. These data suggest that, in our study, only Ca2+ influx through NMDARs contributed to the increased action potential-independent GABA release. Furthermore, to explain the differential effect of ryanodine on mIPSC and eIPSC, we suggest that, although Ca2+ influx through NMDARs was not sufficient to enhance Ca2+ release from intracellular stores, a larger Ca2+ influx through both NMDARs and voltage-gated Ca2+ channels after an action potential became sufficient to promote Ca2+-induced Ca2+ release from intracellular Ca2+ stores. Ca2+ that is released from these intracellular stores may activate an intracellular pathway that reduces action potential-dependent GABA release, as suggested previously for induction of long-term depression at glutamatergic synapses (Reyes and Stanton, 1996). Presynaptic ryanodine-sensitive Ca2+ stores have also been involved in long-term depression at GABAergic synapses via an unknown pathway (Caillard et al., 2000). Identifying the intracellular mechanism of action of NMDAR activation on GABA release is outside the scope of the present study.
Glial glutamate transporters limit activation of NMDARs on GABAergic synapses by glutamate spillover from climbing fibers
Stimulation of climbing fibers decreased sIPSC amplitude via NMDAR activation, suggesting that synaptically released glutamate from climbing fibers diffused to GABAergic synapses and activated presynaptic NMDARs on GABAergic interneurons. In Duguid and Smart's study (2004), when glutamate release from Purkinje neurons was prevented, the authors did not find any change in sIPCS frequency and presumably amplitude after climbing fiber stimulation, whereas we observed a reversible 21% decrease in sIPSC amplitude during stimulation. This discrepancy can be explained by performing the analysis at different time periods (during or after the stimulation). Inhibition of glutamate uptake with TBOA significantly enhanced the effect of climbing fiber stimulation on sIPSC amplitude. Intracellular inhibition of postsynaptic glutamate transporters did not alter the effect of TBOA on glutamate spillover from climbing fibers, suggesting a major contribution of glial glutamate transporters at limiting glutamate spillover from climbing fibers to GABAergic synapses. These experiments on glutamate spillover were performed at near-physiological temperature (35-36°C) because glutamate uptake and spillover among other processes are strongly influenced by temperature (Schwartz and Tachibana, 1990; Wadiche et al., 1995; Asztely et al., 1997). However, glutamate spillover may be underestimated because glutamate can be cleared from the extracellular space from thin slices (as mentioned previously), and sectioned climbing fibers are expected to release less glutamate than they release in vivo. This notion of glutamate diffusion from excitatory to inhibitory synapses and its regulation by glial transporters are supported by the architecture of the inhibitory and excitatory inputs onto Purkinje neurons and of glial processes around synapses. Whereas axo-axonic synapses onto basket cell axons have not been reported (Palay and Chan-Palay, 1974b), basket cell synapses can be found in close proximity to excitatory synapses from climbing fibers (Palay and Chan-Palay, 1974a). Furthermore, only glial processes separate these excitatory and inhibitory axon terminals, suggesting that glutamate is free to diffuse between synapses during uptake inhibition. The extent of glial coverage of synapses could therefore determine the degree to which the activity of glutamatergic synapses depresses neighboring GABAergic transmission.
In conclusion, glutamate transporters exert a strong influence on synaptic inhibition of Purkinje neurons by limiting glutamate spillover and affecting the degree of activation of presynaptic NMDARs presumably expressed on GABAergic terminals. The activity and expression of neuronal and glial glutamate transporters are subject to regulation by glutamate itself (Duan et al., 1999) and by several endogenous substances released by excitatory granule cells or Purkinje neurons in an activity-dependent manner (Kataoka et al., 1997; Swanson and Duan, 1999), suggesting that glutamate transporters dynamically regulate GABA release onto Purkinje neurons. Such a dynamic mechanism may also apply in other brain areas, such as the hippocampus, in which presynaptic kainate-receptor activation modulates GABA release (Kullmann, 2001). Importantly, synaptic inhibition that is concentrated at synapses near the soma is strongly influenced by glial glutamate transporters, which may thus affect the output pattern of Purkinje neurons.
Footnotes
This work was supported by National Institutes of Health Grant NS042189-01A2 (A.B.). We thank Stephan Krueger, Stéphane Dieudonné, Anthony van den Pol, Anne Williamson, and Keith Gipson for valuable comments on this manuscript.
Correspondence should be addressed to Dr. Angélique Bordey, Department of Neurosurgery, Yale University, 333 Cedar Street, Laboratory for Surgery, Obstetrics, and Gynecology Building 228, New Haven, CT 06520-8082. E-mail: angelique.bordey@yale.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/245659-11$15.00/0
References
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N (1994) Differential expression of five N-methyl-d-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol 347: 150-160. [DOI] [PubMed] [Google Scholar]
- Anderson CM, Swanson RA (2000) Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32: 1-14. [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann DM (1997) Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron 18: 281-293. [DOI] [PubMed] [Google Scholar]
- Auger C, Attwell D (2000) Fast removal of synaptic glutamate by postsynaptic transporters. Neuron 28: 547-558. [DOI] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW (2002) The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci 22: 9134-9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardo S, Robertson B, Stephens GJ (2002) Presynaptic internal Ca2+ stores contribute to inhibitory neurotransmitter release onto mouse cerebellar Purkinje cells. Br J Pharmacol 137: 529-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AS, Reichelt W, Norenberg MD (2000) Characterization of cystine uptake in cultured astrocytes. Neurochem Int 37: 269-276. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Dzubay JA, Jahr CE (1997) Glutamate transporter currents in bergmann glial cells follow the time course of extrasynaptic glutamate. Proc Natl Acad Sci USA 94: 14821-14825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H (2000) Ion channel expression by astrocytes in situ: comparison of different CNS regions. Glia 30: 27-38. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H (2003) Modulation of glutamatergic transmission by bergmann glial cells in rat cerebellum in situ. J Neurophysiol 89: 979-988. [DOI] [PubMed] [Google Scholar]
- Brasnjo G, Otis TS (2001) Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron 31: 607-616. [DOI] [PubMed] [Google Scholar]
- Caillard O, Ben Ari Y, Gaiarsa JL (2000) Activation of presynaptic and postsynaptic ryanodine-sensitive calcium stores is required for the induction of long-term depression at GABAergic synapses in the neonatal rat hippocampus amphetamine. J Neurosci 20: RC94(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J (1995) Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15: 711-720. [DOI] [PubMed] [Google Scholar]
- Clark BA, Barbour B (1997) Currents evoked in Bergmann glial cells by parallel fibre stimulation in rat cerebellar slices. J Physiol (Lond) 502: 335-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Tyzio R, Dinocourt C, Esclapez M, Hirsch JC, Ben Ari Y, Bernard C (2001) Presynaptic kainate receptors that enhance the release of GABA on CA1 hippocampal interneurons. Neuron 29: 497-508. [DOI] [PubMed] [Google Scholar]
- Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65: 1-105. [DOI] [PubMed] [Google Scholar]
- de Blas AL (1984) Monoclonal antibodies to specific astroglial and neuronal antigens reveal the cytoarchitecture of the Bergmann glia fibers in the cerebellum. J Neurosci 4: 265-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC (1998) The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci 18: 3606-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana MA, Levenes C, Mackie K, Marty A (2002) Short-term retrograde inhibition of GABAergic synaptic currents in rat Purkinje cells is mediated by endogenous cannabinoids. J Neurosci 22: 200-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Stein BA, Swanson RA (1999) Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci 19: 10193-10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguid IC, Smart TG (2004) Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat Neurosci 7: 525-533. [DOI] [PubMed] [Google Scholar]
- Edwards FA, Konnerth A, Sakmann B, Takahashi T (1989) A thin slice preparation for patch clamp recordings from neurones of the mammalian central nervous system. Pflügers Arch 414: 600-612. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, Fine A (1999) Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 22: 115-124. [DOI] [PubMed] [Google Scholar]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG (1995) An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375: 599-603. [DOI] [PubMed] [Google Scholar]
- Farrant M, Cull-Candy SG (1991) Excitatory amino acid receptor-channels in Purkinje cells in thin cerebellar slices. Proc R Soc Lond B Biol Sci 244: 179-184. [DOI] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ (1997) Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci 17: 8363-8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galante M, Marty A (2003) Presynaptic ryanodine-sensitive calcium stores contribute to evoked neurotransmitter release at the basket cell-Purkinje cell synapse. J Neurosci 23: 11229-11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegelashvili G, Robinson MB, Trotti D, Rauen T (2001) Regulation of glutamate transporters in health and disease. Prog Brain Res 132: 267-286. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Marty A (1999) Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci 19: 511-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Janssen WG, Rothstein JD, Morrison JH (2000) Differential synaptic localization of the glutamate transporter EAAC1 and glutamate receptor subunit GluR2 in the rat hippocampus. J Comp Neurol 418: 255-269. [PubMed] [Google Scholar]
- Jiang L, Xu J, Nedergaard M, Kang J (2001) A kainate receptor increases the efficacy of GABAergic synapses. Neuron 30: 503-513. [DOI] [PubMed] [Google Scholar]
- Kataoka Y, Morii H, Watanabe Y, Ohmori H (1997) A postsynaptic excitatory amino acid transporter with chloride conductance functionally regulated by neuronal activity in cerebellar Purkinje cells. J Neurosci 17: 7017-7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Henderson G (2000) Modulation of fast synaptic transmission by presynaptic ligand-gated cation channels. J Auton Nerv Syst 81: 110-121. [DOI] [PubMed] [Google Scholar]
- King JS, Chen YF, Bishop GA (1993) An analysis of HRP-filled basket cell axons in the cat's cerebellum. II. Axonal distribution. Anat Embryol (Berl) 188: 299-305. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001a) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29: 717-727. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001b) Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci 21: RC174(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM (2001) Presynaptic kainate receptors in the hippocampus: slowly emerging from obscurity. Neuron 32: 561-564. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC (1998) The number of glutamate transporter sub-type molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 18: 8751-8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Gonzalez J, Caputo C, Lai FA, Blayney LM, Tan YP, Marty A (2000) Presynaptic calcium stores underlie large-amplitude miniature IPSCs and spontaneous calcium transients. Nat Neurosci 3: 1256-1265. [DOI] [PubMed] [Google Scholar]
- Marcaggi P, Billups D, Attwell D (2003) The role of glial glutamate transporters in maintaining the independent operation of juvenile mouse cerebellar parallel fibre synapses. J Physiol (Lond) 552: 89-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min MY, Melyan Z, Kullmann DM (1999) Synaptically released glutamate reduces gamma-aminobutyric acid (GABA)ergic inhibition in the hippocampus via kainate receptors. Proc Natl Acad Sci USA 96: 9932-9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minelli A, Barbaresi P, Reimer RJ, Edwards RH, Conti F (2001) The glial glutamate transporter GLT-1 is localized both in the vicinity of and at distance from axon terminals in the rat cerebral cortex. Neuroscience 108: 51-59. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA (2001) Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292: 923-926. [DOI] [PubMed] [Google Scholar]
- Otis TS, Kavanaugh MP, Jahr CE (1997) Postsynaptic glutamate transport at the climbing fiber-Purkinje cell synapse. Science 277: 1515-1518. [DOI] [PubMed] [Google Scholar]
- Palacin M, Estevez R, Bertran J, Zorzano A (1998) Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev 78: 969-1054. [DOI] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V (1974a) The basket cell. In: Cerebellar cortex, cytology and organization (Palay SL, Chan-Palay V, eds), pp 180-215. New York: Springer.
- Palay SL, Chan-Palay V (1974b) The climbing fiber. In: Cerebellar cortex, cytology and organization (Palay SL, Chan-Palay V, eds), pp 236. New York: Springer.
- Petralia RS, Wang YX, Wenthold RJ (1994) The NMDA receptor subunits NR2A and NR2B show histological and ultrastructural localization patterns similar to those of NR1. J Neurosci 14: 6102-6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall W (1977) Core conductor theory and cable properties of neurons. In: Handbook of physiology, Sec I, The nervous system (Kandel ER, ed), pp 39-97. Bethesda, MD: American Physiological Society.
- Reyes M, Stanton PK (1996) Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J Neurosci 16: 5951-5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Legendre P, Westbrook GL (1992) Expression of NMDA channels on cerebellar Purkinje cells acutely dissociated from newborn rats. J Neurophysiol 68: 1901-1905. [DOI] [PubMed] [Google Scholar]
- Satake S, Saitow F, Yamada J, Konishi S (2000) Synaptic activation of AMPA receptors inhibits GABA release from cerebellar interneurons. Nat Neurosci 3: 551-558. [DOI] [PubMed] [Google Scholar]
- Schwartz EA, Tachibana M (1990) Electrophysiology of glutamate and sodium co-transport in a glial cell of the salamander retina. J Physiol (Lond) 426: 43-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeri Y, Shimamoto K, Yasuda-Kamatani Y, Seal RP, Yumoto N, Nakajima T, Amara SG (2001) Effects of threo-beta-hydroxyaspartate derivatives on excitatory amino acid transporters (EAAT4 and EAAT5). J Neurochem 79: 297-302. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T (1998) dl-threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol 53: 195-201. [DOI] [PubMed] [Google Scholar]
- Simpson PB, Challiss RA, Nahorski SR (1995) Neuronal Ca2+ stores: activation and function. Trends Neurosci 18: 299-306. [DOI] [PubMed] [Google Scholar]
- Soltesz I, Smetters DK, Mody I (1995) Tonic inhibition originates from synapses close to the soma. Neuron 14: 1273-1283. [DOI] [PubMed] [Google Scholar]
- Spruston N, Jaffe DB, Williams SH, Johnston D (1993) Voltage-and space-clamp errors associated with the measurement of electrotonically remote synaptic events. J Neurophysiol 70: 781-802. [DOI] [PubMed] [Google Scholar]
- Stuart G, Hausser M (1994) Initiation and spread of sodium action potentials in cerebellar Purkinje cells. Neuron 13: 703-712. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Duan S (1999) Regulation of glutamate transporter function. The Neuroscientist 5: 280-282. [Google Scholar]
- Takahashi KA, Linden DJ (2000) Cannabinoid receptor modulation of synapses received by cerebellar Purkinje cells. J Neurophysiol 83: 1167-1180. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Sarantis M, Attwell D (1996) Postsynaptic glutamate uptake in rat cerebellar purkinje cells. J Physiol (Lond) 497.2: 523-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CL, Drewery DL, Atkins HD, Stephenson FA, Chazot PL (2000) Immunohistochemical localization of N-methyl-d-aspartate receptor NR1, NR2A, NR2B and NR2C/D subunits in the adult mammalian cerebellum. Neurosci Lett 283: 85-88. [DOI] [PubMed] [Google Scholar]
- Tsai MJ, Chang YF, Schwarcz R, Brookes N (1996) Characterization of l-alpha-aminoadipic acid transport in cultured rat astrocytes. Brain Res 741: 166-173. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP (1995) Kinetics of a human glutamate transporter. Neuron 14: 1019-1027. [DOI] [PubMed] [Google Scholar]
- Warr O, Takahashi M, Attwell D (1999) Modulation of extracellular glutamate concentration in rat brain slices by cystine-glutamate exchange. J Physiol (Lond) 514: 783-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Fukaya M, Shibata T, Kurihara H, Tanaka K, Inoue Y, Watanabe M (2000) Dynamic transformation of Bergmann glial fibers proceeds in correlation with dendritic outgrowth and synapse formation of cerebellar Purkinje cells. J Comp Neurol 418: 106-120. [PubMed] [Google Scholar]